

Abstract
Pearson syndrome (PS) is a very rare and often fatal multisystemic mitochondrial disorder involving the liver, kidney, pancreas, and hematopoietic and central nervous system. It is characterized principally by a transfusion-dependent anemia that usually improves over time, a tendency to develop severe infections, and a high mortality rate. We describe a group of 11 PS patients diagnosed in Italy in the period 1993–2014. The analysis of this reasonably sized cohort of patients contributes to the clinical profile of the disease and highlights a rough incidence of 1 case/million newborns. Furthermore, it seems that some biochemical parameters like increased serum alanine and urinary fumaric acid can help to address an early diagnosis.
Keywords: Anemia, Mitochondrial disorders, Pearson syndrome
Introduction
PS is a multisystem disorder caused by large‐scale rearrangements of mitochondrial DNA (mtDNA), with consequent defects in the mitochondrial respiratory chain (Rötig et al. 1995). PS typically consists of refractory, hypoplastic macrocytic anemia with vacuolated marrow precursors, lactic acidosis, and exocrine pancreatic dysfunction; anemia is frequently associated with a variable degree of thrombocytopenia and neutropenia (Rötig et al. 1995). Additional reported manifestations include proximal myopathy, neurologic symptoms (seizures, ataxia, movement disorders), skin lesions, and proximal renal tubular acidosis (Rötig et al. 1995; Atale et al. 2009). Most infants reportedly die before 3 years of age (Rötig et al. 1995). In some cases, a phenotypic transformation to Leigh syndrome (LS) is observed (Santorelli et al. 1996), characterized by dysphagia, hypotonia, ataxia, peripheral neuropathy, and ophthalmoparesis, or Kearns–Sayre syndrome (KSS) (Mcshane et al. 1991; Lee et al. 2007), characterized by progressive external ophthalmoplegia, retinopathy, ataxia, cardiac conduction abnormalities, and deafness.
Patients and Methods
This retrospective study was designed by the Bone Marrow Failure (BMF) Study Group of A.I.E.O.P. (Associazione Italiana Emato-Oncologia Pediatrica) and approved by the Ethics Committee of the Civico Hospital, Palermo. All procedures followed were in accordance with the ethical standards of the committee in charge of human experimentation (institutional and national) and the Helsinki Declaration of 1975, as revised in 2000. Informed consent for inclusion in the study and genetic studies was obtained from the parents or the legal guardians of all patients.
A case report form (CRF) with 167 questions was sent to all 55 A.I.E.O.P. centers. In some cases, a single patient was treated by more than one center in different periods, so we received 14 CRFs regarding 11 PS patients, all diagnosed on the basis of clinical phenotype and genetic testing; 4 patients had been more succinctly presented in another report (Tumino et al. 2011).
Overall survival (OS) was calculated from the date of birth to death from any cause, or date of last follow-up (FUP). Survival analysis was performed using the open source statistical software R (R Development Core Team 2011) and the R package survival (Therneau 2012).
Results
Eleven PS patients (M/F: 5/6) from healthy unrelated parents were studied: their principal characteristics are summarized in Tables 1 and 2. The median age at diagnosis was 299 days.
Table 1.
Clinics
| Pt | HM | SM | Ins. Ex.Pancreas | IDDM | GrowthImpair. | VWT | Neurol.Sympt. | KSS | EyeProblems | Transf.Indep. | Sev.Infect. | D/A (years) | Cause of death |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Yes | No | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes | D (6.4) | Severe acidosis |
| 2 | No | No | No | No | Yes | No | No | No | No | Yes | No | A (2.9) | / |
| 3 | Yes | No | No | No | No | NK | No | No | Yes | Yes | No | Lost at FUP (3.7) | / |
| 4 | Yes | No | Yes | No | Yes | Yes | Yes | No | No | No | Yes | D (5.7) | Renal failure |
| 5 | No | No | No | No | Yes | No | Yes | No | Yes | Yes | Yes | A (6.6) | / |
| 6 | No | No | No | No | Yes | Yes | Yes | No | NK | NP | No | D (0.33) | NK |
| 7 | Yes | Yes | No | No | Yes | No | No | No | Yes | Yes | Yes | D (8.0) | Sepsis |
| 8 | Yes | No | No | No | No | Yes | Yes | No | Yes | NP | Yes | D (0.53) | Sepsis |
| 9 | Yes | No | No | Yes | No | No | Yes | Yes | No | Yes | Yes | D (10.42) | AML |
| 10 | Yes | Yes | No | No | Yes | No | Yes | Yes | Yes | Yes | Yes | D (10.44) | Renal failure |
| 11 | Yes | Yes | Yes | No | No | Yes | No | No | NK | Yes | Yes | D (3.9) | Sepsis |
Pt patient, HM hepatomegaly, SM splenomegaly, IDDM type 1 insulin-dependent diabetes mellitus, VWT ventricular wall thickness, KSS Kearns–Sayre syndrome, Transf. Indep transfusion independency, Sev. Infect severe infections, D/A dead/alive, FUP follow-up, AML acute myeloid leukemia, NK not known, NP not pertinent
Table 2.
Laboratory
| Pt | mtDNA del. | ↑ Serum lactate | ↑ Serum alanine | ↑ Urine lactate | ↑ Urine fumarate | ↑ Urine malic acid | Fetal Hgb | EPO | Reticulocytes >60,000 μL | BM |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5,000 N.S. | Yes | Yes | Yes | Yes | No | ND | ND | Yes | Vacuoles and ↓ cellularity |
| 2 | 8,648–15,368 | Yes | Yes | Yes | Yes | Yes | ND | ↑ | Yes | Vacuoles and ↓ cellularity |
| 3 | 4,000 N.S | Yes | ND | No | No | No | ND | ↑ | Yes | Dyserythropoiesis |
| 4 | 5,000 N.S. | Yes | Yes | Yes | Yes | Yes | ↑ | ↑ | Yes | Vacuoles |
| 5 | 8,843–13,459 | No | No | ND | ND | ND | ↑ | ↑ | Yes | Vacuoles and ↓ cellularity |
| 6 | 8,400–13,500 | Yes | Yes | Yes | Yes | Yes | ND | ND | Yes | Vacuoles and ↓ cellularity |
| 7 | 6,634–9,935 | Yes | Yes | ND | ND | ND | ND | ↑ | Yes | Vacuoles |
| 8 | 10,049–15,088 | Yes | Yes | Yes | Yes | No | ND | ↑ | No | ↓ Cellularity |
| 9 | 19,447–15,395 | Yes | Yes | No | No | No | ↑ | ND | Yes | Vacuoles and ↓ cellularity |
| 10 | 7,000 N.S. | Yes | Yes | Yes | Yes | Yes | ↑ | ND | Yes | Vacuoles and ↓ cellularity |
| 11 | 5,000 N.S. | Yes | Yes | Yes | Yes | No | ↑ | ND | Yes | ↓ Cellularity |
Pt patient, mtDNA delet mtDNA deletion, ND not determined, EPO erythropoietin, BM bone marrow
Clinical Phenotype
All mothers were Caucasian except one who was of South American origin. The median age of the mothers of Italian origin was similar to the Italian population: 32.0 vs. 32.2 years. Only one mother had a previous miscarriage. All pregnancies were uneventful. No patient was born preterm, and only 3 out of 11 (27%) had a neonatal weight less than 2,500 g: the smallest was 2,230 g. In 6 pregnancies (54%), a cesarean section was performed, and in 3 of them, the cause was acute fetal illness. In these 3 newborns, metabolic acidosis and an extremely severe anemia were present: hemoglobin (Hgb) of 1.8, 2.9, and 5.5 g/dL, respectively, and in 2/3 hypoglycemia was associated.
All patients but one had a heart evaluation in the course of disease: ventricular wall thickness (VWT), depolarization abnormalities, and prolonged QT were observed in 4, 2, and 1 patient, respectively.
Only one of the patients suffered from skin abnormalities (diffuse hyperpigmentation and a large café au lait spot). Exocrine pancreatic impairment was found in 3 patients (27%), and 1 of them also presented type 1 insulin-dependent diabetes mellitus (IDDM). IDDM was also diagnosed in another patient without involvement of exocrine pancreas. Complete adrenal insufficiency was found in 2 children (18%) and hypoparathyroidism in another.
In 8/11 (72%) patients, hepatomegaly was observed, and in 5/8 it appeared at less than 6 months of age. Splenomegaly was seen in only 3/11 patients (27%): in one it was present at birth and in the other two appeared at 4 and 15 months of age, respectively. Abdominal ultrasound was performed during the course of disease in 10 patients, and an increase in kidney echogenicity was observed in 5 patients, two of whom developed tubular acidosis and later severe renal failure. Triglycerides, cholesterol, and transaminases were normal in all patients. Two children presented slightly elevated values of gamma-glutamyltransferase. Growth impairment was observed in 7 (63%) of our patients. Furthermore, 2 patients (18%) presented severe malabsorption with a need for total parental nutrition, and 1 child had a severe duodenal ulcer.
Neurological examination was normal at birth in all children, but an impairment, above all characterized by retarded speech development, hypotonia, and muscle hypotrophy, developed in 7 patients (63%), 3 of whom later evolved towards a full KKS phenotype. A neonatal cerebral ultrasound was performed on 6 patients, and it was normal in all of them. Magnetic resonance imaging (MRI) of the brain was performed on 6 patients. In 2 cases (one with KSS), an abnormally high signal at brainstem level was observed; a similar anomaly was noted in a brain computed tomography (CT) scan of another patient affected by KSS. In another two cases (one with KSS), subcortical white matter hyperintensities were observed. Visual evoked potentials, performed on four patients, were not informative; in contrast, the auditory evoked potentials, performed on five patients, were abnormal only in the two patients affected by KSS, showing a compromised brainstem. Electroencephalography, performed on 8 patients, was found altered only in the three patients with KSS. Eye examination was performed on 9/11 patients, and in 6 (66%) a variety of problems (principally of the cornea and retina) were present. Two of the three patients later developing KSS presented congenital ptosis. No case of hearing loss was reported.
From the hematological point of view at the first blood count, pancytopenia was present in five children, anemia associated with leucopenia and/or neutropenia in five patients, and isolated anemia in 1 child. The median value of Hgb was 5.7 g/dL, the lowest value of platelets was 72,000 μL, and neutropenia (present in 10 patients) was severe (absolute neutrophil count (ANC) <0.5 × 109/L) in 1, moderate (ANC 0.5–1 × 109/L) in 8, and mild (ANC of 1.3 × 109/L) in another. Interestingly, in the course of disease in 10/11, at least one count of reticulocytes higher than 60,000 μL and of ANC higher than 0.5 × 109/L were encountered. Anemia associated with other cytopenias or not was the most relevant onset symptom in all patients, even though in the three children in whom the transfusion dependency started at 12–25 months of life (see later), a growth impairment at that time had already been addressed.
Fetal Hgb was assessed in five patients after the neonatal period, and it was found elevated (>2%) in all of them. Erythropoietin (EPO), dosed in 6/11 patients, was always high. Immunoglobulin levels, lymphocyte count, and vitamin B12/folate were evaluated in 10, 5, and 9, respectively, and were normal in all cases. BM examination performed on all patients showed a precursor vacuolization associated with reduced cellularity in 6 children (54%), whereas vacuoles without decreased cellularity were observed in only two patients; in one patient, there was only a mild dyserythropoiesis, and in two other patients, only a reduction of cellularity. Perls staining revealed the presence of sideroblasts in six out of seven tested patients (85%).
BM colonies were investigated in seven patients, and in all tests burst-forming units (BFU), colony-forming unit-erythroid (CFU-E), and colony-forming unit-granulocyte macrophage (CFU-GM) were moderately to extremely reduced. In our population, there was one patient (n. 9) who evolved into acute myeloid leukemia (after allogenic bone marrow transplantation). No solid tumor was observed.
Biochemical Profile
Serum lactate was dosed in every patient and found elevated in all but one with a 1.34–4.77-fold increase: in three children the levels of lactate were intermittently high. Urine organic acids were analyzed in nine patients, and in 7/9 (77%) an increased excretion of lactate was noted: in these seven children, an elevated excretion of fumaric acid was found too. An increase of malic acid was found in 4/9 patients (44%), whereas a methylglutaconic aciduria mentioned in two papers (Gibson et al. 1992; Lichter-Konecki et al. 1993) could be found only in two children. A plasmatic amino acid analysis was performed on ten children: in 9/10 alanine was high. Constitutional karyotype was found normal in all patients; diepoxybutane test, performed on 7 patients, was negative in all, and adenosine deaminase (ADA), tested in four patients, showed increased values in two of them.
Treatments
All patients were transfused with packed red blood cells (PRBC), six with platelets, and two with plasma (during the course of severe sepsis). The PRBC transfusion dependency started in four patients at birth, in four patients before 4 months of life, in two patients at 12 months of age, and at 25 months of life in the remaining one. A spontaneous improvement of the Hgb values was noted in eight of the nine patients with a sufficiently long FUP (88%): the last PRBC transfusion was done at a median age of 2.01 years. Therapy with erythropoietin (EPO) was attempted in three children, and in no case did it prove to be beneficial. Granulocyte colony stimulating factor (GCSF) was administered in three patients and proved to be beneficial in one. Pancreatic extracts were effective in the three children with exocrine pancreatic deficiency.
Survival
After a median FUP of 5.7 years, 8/11 patients died (two at less than 6 months of life), 1 was lost at FUP at 45 months of age, and only 2/11 patients are alive (at the age of 2.9 and 6.6 years, respectively), accounting for crude survival rate of 20% (Fig. 1).
Fig. 1.
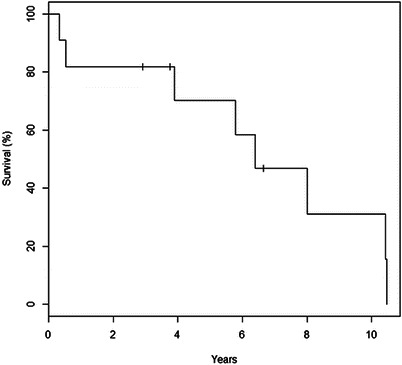
Kaplan Meier survival curve
Causes of death were sepsis (exclusive cause) in three cases, acute myeloid leukemia in one case, intractable metabolic acidosis in one case, and severe renal failure in two cases; in one patient (deceased at 4 months of age), it was impossible to clearly establish the cause of death. All three patients with KSS died.
Discussion
Mitochondrial abnormalities are causes of some severe human diseases. The full-blown picture of PS is typically characterized by refractory sideroblastic anemia with vacuolization of marrow precursors, lactic acidosis, neurological, renal, hepatic, and exocrine pancreatic dysfunction (Rötig et al. 1995; Atale et al. 2009). The incidence is unknown. Since the 55 units of the A.I.E.O.P. network are spread over the entire national territory, it seems unlikely that any child affected by BMF has never been observed in an A.I.E.O.P. center, and so this series is likely to represent the total or near total of PS children in Italy in the period from 1993 to 2014. Based on the average annual birthrate, we can deduce that the likely incidence of PS in Italy is approximately 1/1,000,000 newborns.
The complete mitochondrial genome is small (16.6 kb), and some copies (normal and mutated mtDNA) passed on to progeny via the cytoplasm and accounted for by maternal inheritance are present in the same cell (heteroplasmy). It is probable that a mitochondrial disease can appear only when the proportion of mutated mtDNA exceeds a given threshold. In PS the mtDNA mutations are rather homogeneous since the same 5.0 kb deletion constitutes the most common lesion (Rötig et al. 1995) and is typically sporadic. In our cohort, similar to other series (Rötig et al. 1995; Topaloğlu et al. 2008), no apparent correlation was found between the size and site of mtDNA deletion, clinical presentation, and final outcome.
Affected children are frequently reported born preterm, but all the patients in our series were born later than 38 weeks, and only three presented a birth weight under 2,500 g. In our analysis, apart from one case of bicameral right ventricle, and two of ptosis, no evident malformation or dysmorphism was observed. Splenic atrophy was reported in two patients of the original paper of Pearson et al. (1979), but no other case, including the present series, has yet been published.
The absence of exocrine pancreatic deficiency is reported in 23–63% of cases (Rötig et al. 1995; Atale et al. 2009; Broomfield et al. 2015): in our series the high proportion of 73% was free of this complication. Likewise and in contrast to the high incidence of hepatic failure (33%) reported by Rötig et al. (1995), none of our patients presented signs of serious liver involvement.
Two patients (18%) developed a severe tubulopathy progressing to severe renal failure, which is consistent with the 23–24% reported in another 2 series (Atale et al. 2009; Rötig et al. 1995). Both patients presented increased echogenicity of kidneys. The growth deficiency, described as typical of PS (Atale et al. 2009), was also observed in our series as it was seen in 63% of patients.
The neurological features of PS are quite variable, ranging from normal to highly severe. The neurological examination can be abnormal very precociously, and in some cases, it is possible to document progression in the neurological disability. Sometimes a final evolution to KSS (Mcshane et al. 1991; Lee et al. 2007) and LS (Santorelli et al. 1996) can be documented. Neuroimaging is quite variable: from completely normal to severely abnormal findings of white matter, deep gray nuclei, cerebellum, and brainstem (Lee et al. 2007; Morel et al. 2009). Neurologic problems are quite frequent in our series (8/11), above all seizures and hypotonia, and three patients (27%) developed full KSS. An abnormally high signal at brainstem level on MRI, or CT, was the most frequent finding in neuroimaging (three out of eight patients evaluated).
Echocardiography was performed on ten patients, and in four of them, a ventricular hypertrophy was observed. Occasionally, an impaired cardiac function has been described (Krauch et al. 2002; Broomfield et al. 2015), but to our knowledge, the frequent association between PS and VWT has never been reported previously even though hypertrophic cardiomyopathy is a well-known feature of many mitochondriopathies (Kopajtich et al. 2014; Wang et al. 2008; Kupari 1984). Elevated fetal Hgb, occasionally described in PS patients (Superti-Furga et al. 1993), and elevated EPO were observed in all patients evaluated.
All but one of the patients had elevated lactate, and interestingly, serum alanine was elevated in eight out of nine patients evaluated. This probably reflects, more than the altered NADH:NAD flux, the disruption of the Krebs cycle which causes the accumulation of pyruvate that can be reversibly converted to lactate or to alanine (Zschocke et al. 2004). Similarly, the increase of fumarate (7/9) and malic acid (4/9) can be explained by the fact that they are intermediate products generated by pyruvate carboxylase during pyruvate metabolism. Actually, although an increased urinary excretion of organic acids (independent of tubulopathy) has already been reported (Atale et al. 2009), this was not specifically focused on fumaric acid. Based on the above findings, it is important to highlight that both increased alanine serum levels and fumaric acid urinary excretion, even though not specific markers (since they can also be present in other mitochondriopathies (Broomfield et al. 2015), can be helpful above all for the differential diagnosis from other non-mitochondrial bone marrow failures.
Vacuoles of myeloid or erythroblastic progenitors in the BM, as well as the presence of ringed sideroblasts after Perls staining, are highly suggestive of PS (Pearson et al. 1979): in our series vacuolization was observed in 8/11 patients and Perls staining was positive in 6/7 patients.
For the 11 medical doctors taking care of the patients during the diagnostic phase, Diamond–Blackfan anemia (DBA) was the most frequent differential diagnosis (9/11 physicians); 2/11 initially suspected a “metabolic disease.” In a recent series of DBA patients (Gagne et al. 2014), a small percentage was found to be affected by PS: adenosine deaminase (ADA), typically high in DBA patients, can be of no help for differential diagnosis according to our data, since its elevation, already described in at least one report (Superti-Furga et al. 1993), was present in two out of four patients evaluated. Notwithstanding, we think that, also based on the findings of the present analysis, clinical and biochemical elements other than ADA can be helpful: above all, neurological symptoms and vacuoles in BM, together with increased levels of serum lactate, serum alanine, and urine fumarate, can be decisive in the differential diagnosis from DBA.
PRBC transfusion independency, occasionally reported in other papers (Muraki et al. 1997), was achieved by eight out of nine evaluable patients, and consequently it seems to be highly probable in the case of survival after the first 2–3 years of life.
PS is usually reported to lead to premature death even though survival up to young adulthood has occasionally been reported (Muraki et al. 1997). Our findings, with eight deaths/ten patients, confirm that the prognosis is dismal, with mortality more common at 5–11 years of age (five out of eight evaluable patients), whereas in other older series, more than 50% of patients died before 3.5 years of life (Rötig et al. 1995). Treatments other than supportive therapy and transfusions appear to be of limited utility: administration of bicarbonate can control metabolic acidosis, and episodic treatments with GCSF can reverse infections in the course of severe neutropenia. In our cohort, similarly to other recent series (Broomfield et al. 2015), the general improvement of supportive therapy probably explains the improvement in survival.
Conclusion
We report a reasonably sized cohort of PS, compared to those described previously. Our report is based on an accurate survey among physicians of the national A.I.E.O.P. network and enables us to estimate the likely incidence of this disease in our country as being about 1/million newborns. Moreover, our study highlights some clinical insights and a biochemical profile characterized by increased serum alanine and urinary fumaric acid that can be helpful in the initial diagnostic work-up.
Acknowledgments
Giuseppe Furfari is acknowledged for electronic CRF design. The Parents’ Association A.S.L.T.I – Liberi di crescere is acknowledged for supporting the activity of the Pediatric Onco-Hematology Unit of A.R.N.A.S. Ospedali Civico, Di Cristina e Benfratelli. No specific funding was received for this study.
Abbreviations
- ADA
Adenosine deaminase
- A.I.E.O.P.
Associazione Italiana di Ematologia ed Oncologia Pediatrica
- ANC
Absolute neutrophil count
- BFU
Burst-forming units
- BM
Bone marrow
- BMF
Bone marrow failure
- CFU-E
Colony forming unit-erythroid
- CFU-GM
Colony forming unit-granulocyte macrophage
- CRF
Case report form
- CT
Computed tomography
- DBA
Diamond–Blackfan anemia
- EPO
Erythropoietin
- FUP
Follow-up
- GCSF
Granulocyte colony stimulating factor
- Hgb
Hemoglobin
- IDDM
Insulin-dependent diabetes mellitus
- KSS
Kearns–Sayre syndrome
- LS
Leigh syndrome
- MRI
Magnetic resonance imaging
- mtDNA
Mitochondrial DNA
- PRBC
Packed red blood cells
- PS
Pearson syndrome
- VWT
Ventricular wall thickness
Take-Home Message
PS is a severe mitochondrial disorder characterized by increased alanine and lactate serum levels and fumaric acid urinary excretion and a frequent recovery of bone marrow (BM) in the case of survival after the first 2–3 years of life.
This article does not contain any studies with human or animal subjects performed by any of the authors.
Funding Source
No external funding was secured for this study.
Financial Disclosure
No authors have any financial relationships relevant to this article to disclose.
Compliance with Ethical Guidelines
Conflict of Interest
No authors have any conflicts of interest to disclose.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Contributor’s Statements
Drs. Farruggia and Di Cataldo conceptualized and designed the study and the data collection instruments. Drs. Farruggia, Pillon, and Dufour carried out the initial analyses and drafted the initial manuscript. Drs. Farruggia, Puccio, and Macaluso coordinated data collection and performed the statistical analysis. Drs. Macaluso, Palmisani, Pinto, Lo Valvo, Cantarini, Tornesello, Corti, Fioredda, Varotto, Martire, Russo, and Moroni contributed to the enrollment of patients, diagnosis, and sample collection.
All authors critically reviewed the paper, approved the final manuscript as submitted, and agreed to be accountable for all aspects of the work.
Footnotes
Competing interests: None declared
Contributor Information
Piero Farruggia, Email: farruggia.oep@ospedalecivicopa.org.
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Atale A, Bonneau-Amati P, Rötig A, Fischer A, Perez-Martin S, de Lonlay P, Niaudet P, De Parscau L, Mousson C, Thauvin-Robinet C, Munnich A, Huet F, Faivre L. Tubulopathy and pancytopaenia with normal pancreatic function: a variant of Pearson syndrome. Eur J Med Genet. 2009;52(1):23–26. doi: 10.1016/j.ejmg.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Broomfield A, Sweeney MG, Woodward CE, Fratter C, Morris AM, Leonard JV, Abulhoul L, Grunewald S, Clayton PT, Hanna MG, Poulton J, Rahman S. Paediatric single mitochondrial DNA deletion disorders: an overlapping spectrum of disease. J Inherit Metab Dis. 2015;38(3):445–457. doi: 10.1007/s10545-014-9778-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2011) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. ISBN 3-900051-07-0. http://www.R-project.org/
- Gagne KE, Ghazvinian R, Yuan, Zon RL, Storm K, Mazur-Popinska M, Andolina L, Bubala H, Golebiowska S, Higman MA, Kalwak K, Kurre P, Matysiak M, Niewiadomska E, Pels S, Petruzzi MJ, Pobudejska-Pieniazek A, Szczepanski T, Fleming MD, Gazda HT, Agarwal S (2014) Pearson marrow pancreas syndrome in patients suspected to have Diamond–Blackfan anemia. Blood 124(3):437–440 [DOI] [PMC free article] [PubMed]
- Gibson KM, Bennett MJ, Mize CE, Jakobs C, Rotig A, Munnich A, Lichter-Konecki U, Trefz FK. 3-Methylglutaconic aciduria associated with Pearson syndrome and respiratory chain defects. J Pediatr. 1992;121(6):940–942. doi: 10.1016/S0022-3476(05)80348-8. [DOI] [PubMed] [Google Scholar]
- Kopajtich R, Nicholls TJ, Rorbach J, Metodiev MD, Freisinger P, Mandel H, Vanlander A, Ghezzi D, Carrozzo R, Taylor RW, Marquard K, Murayama K, Wieland T, Schwarzmayr T, Mayr JA, Pearce SF, Powell CA, Saada A, Ohtake A, Invernizzi F, Lamantea E, Sommerville EW, Pyle A, Chinnery PF, Crushell E, Okazaki Y, Kohda M, Kishita Y, Tokuzawa Y, Assouline Z, Rio M, Feillet F, Mousson de Camaret B, Chretien D, Munnich A, Menten B, Sante T, Smet J, Régal L, Lorber A, Khoury A, Zeviani M, Strom TM, Meitinger T, Bertini ES, Van Coster R, Klopstock T, Rötig A, Haack TB, Minczuk M, Prokisch H (2014) Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am J Human Genet 95(6):708–720 [DOI] [PMC free article] [PubMed]
- Krauch G, Wilichowski E, Schmidt KG, Mayatepek E (2002) Pearson marrow-pancreas syndrome with worsening cardiac function caused by pleiotropic rearrangement of mitochondrial DNA. Am J Med Genet 110(1):57–61 [DOI] [PubMed]
- Kupari M. Asymmetric septal hypertrophy in Kearns-Sayre syndrome. Clin Cardiol. 1984;7(11):603–605. doi: 10.1002/clc.4960071109. [DOI] [PubMed] [Google Scholar]
- Lee HF, Lee HJ, Chi CS, Tsai CR, Chang TK, Wang CJ. The neurological evolution of Pearson syndrome: case report and literature review. Eur J Paediatr Neurol. 2007;11(4):208–214. doi: 10.1016/j.ejpn.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Lichter-Konecki U, Trefz FK, Rötig A, Munnich A, Pfeil A, Bremer HJ. 3-Methylglutaconic aciduria in a patient with Pearson syndrome. Eur J Pediatr. 1993;152(4):378. doi: 10.1007/BF01956761. [DOI] [PubMed] [Google Scholar]
- McShane MA, Hammans SR, Sweeney M, Holt IJ, Beattie TJ, Brett EM, Harding AE. Pearson syndrome and mitochondrial encephalomyopathy in a patient with a deletion of mtDNA. Am J Hum Genet. 1991;48(1):39–42. [PMC free article] [PubMed] [Google Scholar]
- Morel AS, Joris N, Meuli R, Jacquemont S, Ballhausen D, Bonafé L, Fattet S, Tolsa JF. Early neurological impairment and severe anemia in a newborn with Pearson syndrome. Eur J Pediatr. 2009;168(3):311–315. doi: 10.1007/s00431-008-0756-4. [DOI] [PubMed] [Google Scholar]
- Muraki K, Nishimura S, Goto Y, Nonaka I, Sakura N, Ueda K. The association between haematological manifestation and mtDNA deletions in Pearson syndrome. J Inherit Metab Dis. 1997;20(5):697–703. doi: 10.1023/A:1005378527077. [DOI] [PubMed] [Google Scholar]
- Pearson HA, Lobel JS, Kocoshis SA, Naiman JL, Windmiller J, Lammi AT, Hoffman R, Marsh JC. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr. 1979;95(6):976–984. doi: 10.1016/S0022-3476(79)80286-3. [DOI] [PubMed] [Google Scholar]
- Rötig A, Bourgeron T, Chretien D, Rustin P, Munnich A. Spectrum of mitochondrial DNA rearrangements in the Pearson marrow-pancreas syndrome. Hum Mol Genet. 1995;4(8):1327–1330. doi: 10.1093/hmg/4.8.1327. [DOI] [PubMed] [Google Scholar]
- Santorelli FM, Barmada MA, Pons R, Zhang LL, Di Mauro S. Leigh-type neuropathology in Pearson syndrome associated with impaired ATP production and a novel mtDNA deletion. Neurology. 1996;47(5):1320–1323. doi: 10.1212/WNL.47.5.1320. [DOI] [PubMed] [Google Scholar]
- Superti-Furga A, Schoenle E, Tuchschmid P, Caduff R, Sabato V, De Mattia D, Gitzelmann R, Steinmann B. Pearson bone marrow-pancreas syndrome with insulin-dependent diabetes, progressive renal tubulopathy, organic aciduria and elevated fetal haemoglobin caused by deletion and duplication of mitochondrial DNA. Eur J Pediatr. 1993;152(1):44–50. doi: 10.1007/BF02072515. [DOI] [PubMed] [Google Scholar]
- Therneau T (2012) A Package for Survival Analysis in S. R package version 2.37-2, http://CRAN.R-project.org/package=survival
- Topaloğlu R, Lebre AS, Demirkaya E, Kuşkonmaz B, Coşkun T, Orhan D, Gürgey A, Gümrük F. Two new cases with Pearson syndrome and review of Hacettepe experience. Turkish J Pediatr. 2008;50(6):572–576. [PubMed] [Google Scholar]
- Tumino M, Meli C, Farruggia P, La Spina M, Faraci M, Castana C, Di Raimondo V, Alfano M, Pittalà A, Lo Nigro L, Russo G, Di Cataldo A. Clinical manifestations and management of four children with Pearson syndrome. Am J Med Genet A. 2011;155A(12):3063–3066. doi: 10.1002/ajmg.a.34288. [DOI] [PubMed] [Google Scholar]
- Wang SB, Weng WC, Lee NC, Hwu WL, Fan PC, Lee WT. Mutation of mitochondrial DNA G13513A presenting with Leigh syndrome, Wolff-Parkinson-White syndrome and cardiomyopathy. Pediatr Neonatol. 2008;49(4):145–149. doi: 10.1016/S1875-9572(08)60030-3. [DOI] [PubMed] [Google Scholar]
- Zschocke J, Hoffmann GF, Burlina BA, Duran M, Leonard JV, Mayatepek E, Peters V, Smeitink JAM, Vockley J, Wendel U (2004)Vademecum metabolicum: manual of metabolic paediatrics. 2th edn. Schattauer Gmbh. p 100