

Abstract
Objective: To improve the efficacy of newborn screening (NBS) for very long chain acyl-CoA dehydrogenase deficiency (VLCADD).
Patients and Methods: Data on all dried blood spots collected by the Dutch NBS from October 2007 to 2010 (742.728) were included. Based solely on the C14:1 levels (cutoff ≥0.8 μmol/L), six newborns with VLCADD had been identified through NBS during this period. The ratio of C14:1 over C2 was calculated. DNA of all blood spots with a C14:1/C2 ratio of ≥0.020 was isolated and sequenced. Children homozygous or compound heterozygous for mutations in the ACADVL gene were traced back and invited for detailed clinical, biochemical, and genetic evaluation.
Results: Retrospective analysis based on the C14:1/C2 ratio with a cutoff of ≥0.020 identified an additional five children with known ACADVL mutations and low enzymatic activity. All were still asymptomatic at the time of diagnosis (age 2–5 years). Increasing the cutoff to ≥0.023 resulted in a sensitivity of 93% and a positive predictive value of 37%. The sensitivity of the previously used screening approach (C14:1 ≥0.8) was 50%.
Conclusion: This study shows that the ratio C14:1/C2 is a more sensitive marker than C14:1 for identifying VLCADD patients in NBS. However, as these patients were all asymptomatic at the time of diagnosis, this suggests that a more sensitive screening approach may also identify individuals who may never develop clinical disease. Long-term follow-up studies are needed to establish the risk of these VLCADD-deficient individuals for developing clinical signs and symptoms.
Keywords: Biomarker, C14:1, C2, Newborn screening, VLCADD
Introduction
Many newborn screening (NBS) programs in the world, including the Dutch NBS program, have very long chain acyl-CoA dehydrogenase deficiency (VLCADD) in their disease panel (Lindner et al. 2010; Loeber et al. 2012). VLCADD is a disorder of long-chain fatty acid beta-oxidation (OMIM 609575) that compromises energy homeostasis and leads to accumulation of long-chain fatty acids and derivatives. Patients may present with hypoglycemia, hepatomegaly, and cardiomyopathy in the neonatal period and rhabdomyolysis in early childhood. These features can be induced by fasting, exercise, illness, and fever (Vianey-Saban et al. 1998; Andresen et al. 1999; Laforêt et al. 2009; Baruteau et al. 2014). VLCADD is included in NBS programs mainly because life-threatening symptoms as hypoglycemia and cardiomyopathy can be prevented by dietary measures.
NBS for VLCADD is performed by measuring the concentration of accumulating long-chain acylcarnitines in blood spots, especially tetradecenoyl carnitine (C14:1). In the Netherlands, the cutoff level of C14:1 for referral of newborns was initially ≥0.80 μmol/L (2007). However, because one patient was missed (detected via screening of the family of an index patient), the cutoff level for referral was reduced to ≥0.60 μmol/L (2010). Results of the Region 4 database (McHugh et al. 2011), which contains collaborative data on the outcome of NBS programs worldwide (Houten et al. 2013), indicated that the ratio of C14:1 over acetylcarnitine (C2) might further improve the sensitivity of the screening procedure (Hall et al. 2014). C2 concentrations are often, and also in the Netherlands, measured in NBS screening programs as secondary markers for screening of isovaleric acidemia. In order to improve the NBS on VLCADD, we retrospectively investigated whether the ratio C14:1/C2 is a better marker for VLCADD than the original marker C14:1.
Patients and Methods
We retrospectively calculated the C14:1/C2 ratios and C14:1 levels of all 742.728 NBS blood spots from the Dutch NBS program taken in the period 2007–2010. NBS blood spots are taken within 72–144 h from birth. The levels were measured within 7 days after birth. All blood spots with a C14:1/C2 cutoff value of ≥0.020 and/or C14:1 ≥0.60 μmol/L were selected for further analysis. DNA of the selected blood spots was isolated using the NucleoSpin Tissue genomic DNA purification kit (Macherey-Nagel, Düren, Germany). All exons plus flanking intronic regions of the ACADVL gene were subsequently sequenced. Three proven VLCADD patients were included in a blinded manner as positive controls.
VLCAD enzymatic activity was measured in lymphocytes by using ferrocenium hexafluorophosphate as the electron acceptor, followed by UPLC, to separate the different acyl-CoA species (Wanders et al. 2010). Acylcarnitines were measured as described previously (Vreken et al. 1999).
Based on the duty of care principle (Sokol 2012), the patients who were originally classified as nonaffected but who turned out positive upon evaluation of the C14:1/C2 ratio were traced back and contacted for care. All were alive and all accepted the invitation for neurological, cardiological, biochemical, and genetic evaluation.
Results
Acylcarnitine Measurement and Mutation Analysis in Blood Spots
We found a C14:1/C2 ratio of ≥0.020 in 67 blood spots. Sequence analysis of the ACADVL gene in this group revealed five children who were either homozygous for a single mutation or compound heterozygous for two different mutations. These mutations were confirmed in independent samples (blood spot and blood). In addition, we identified 18 children who were carriers of one mutation in the ACADVL gene (Table 1).
Table 1.
Patient characteristics. Clinical, biochemical, and genetic details of each patient
| ID | Age (year) | Gender | Blood spot | Plasma | C14:1/C2 | Enzymatic activity | Genotype | Neurological examination | Cardiological examination | Other symptoms | Creatine kinase (normal <250) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [C14:1] (normal <0.60 μmol/L) | C14:1/C2 (normal <0.020 μmol/L) | [C14:1] (normal <0.26 μmol/L) | Lymphocytes (% of controls) | Allele 1 | Allele 2 | ||||||||
| 1 | 5 | M | 0.70 | 0.058 | 0.60 | 0.220 | <7 | p.G441D missense | p.A490P missense | Normal (MRC 5) | Normal (no echo/ECG abnormalities) | No hepatomegaly, fatigue, sometimes myalgia | 91 |
| 2 | 5 | M | 0.36 | 0.040 | 2.92 | 0.780 | <7 | p.P89HfsX28 frameshift | p.V283A missense | Normal | Normal | No hepatomegaly, no complaints | 198 |
| Sib of 2 | 11 | F | 11 | p.P89HfsX28 frameshift | p.V283A missense | No complaints | n.a. | ||||||
| 3 | 4 | F | 0.33 | 0.024 | 0.15 | 0.069 | 21 | p.V283A missense | p.V283A missense | Normal | Normal | No hepatomegaly, no complaints | 96 |
| 4 | 5 | M | 0.25 | 0.050 | 0.38 | 0.18 | 14 | p.V283A missense | p.V283A missense | Normal | Normal | No hepatomegaly, no complaints | 200 |
| 5 | 2 | M | 0.55 | 0.039 | 0.14 | 0.094 | 46 | p.G441D missense | p.R615Q missense | Normal | Normal | No hepatomegaly, no complaints | 120 |
| Sib of 5 | 2 | F | 40 | p.G441D missense | p.R615Q missense | Normal | Normal | No complaints | n.a. | ||||
The enzymatic activity of VLCAD in lymphocytes was severely deficient in two of the five detected children (PID 1 and 2) and mildly reduced in the other three patients (PID 3-5). The parents of individual 5 with 46% VLCAD activity were analyzed to check for heterozygosity. Both parents were heterozygotes for the found mutations in patient 5. VLCADD was subsequently also confirmed in two siblings (Table 1).
In the period 2007–2010, six VLCADD patients had been identified by the Dutch NBS program based on the original screening selection criteria: C14:1 ≥0.80 μmol/L. The five additional children detected in this study were not referred at the time. However, based on the current cutoff value of C14:1 ≥0.6 μmol/L (adopted 2013), individual 1 would have been referred.
Two of the three plasma acylcarnitine levels were below the age-adjusted reference value (95th percentile) of <0.26 μmol/L (patients 3 and 5).
Clinical Phenotype
All newly identified children with VLCADD were evaluated for clinical symptoms (Table 1). None of these children reported muscle-related symptoms and none had neurological or cardiological abnormalities. All were normoglycemic upon evaluation and none had suffered metabolic decompensation. Growth varied with a length range < −1.5 SD below the target height to appropriate to target height and a weight-length range of −2.18 to +1.94. The median creatine kinase level at the first evaluation was 120 U/L (range 91–200 U/L).
Sensitivity and Positive Predictive Value
Our results indicate that the sensitivity of C14:1 (≥0.8) is 50% and the sensitivity of C14:1 (≥0.6) is 58%, while the sensitivity of C14:1/C2 (≥0.020) is 93%. In addition, the positive predictive values of C14:1 (≥0.8 and ≥0.6) and C14:1/C2 (≥0.020) are 66%, 47%, and 16%, respectively (Fig. 1). With a C14:1/C2 cutoff value of ≥0.023, the sensitivity remains 93%, while the positive predictive value increases to 37% (Fig. 1).
Fig. 1.
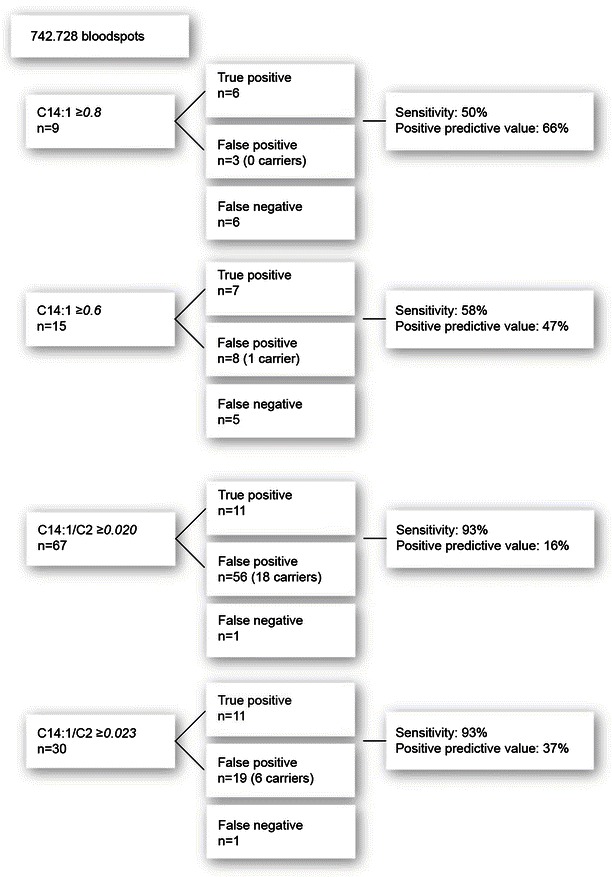
Sensitivity and positive predictive value. Sensitivity and positive predictive values of C14:1 and C14:1/C2 with the various cutoff values
Discussion
This study shows that inclusion of the ratio C14:1/C2 to the NBS increases the sensitivity to detect VLCADD. Accordingly, this ratio is now added to the Dutch NBS as primary marker for screening on VLCADD.
Introducing C14:1/C2 into the expanded NBS program has advantages as well as limitations. The inclusion of this ratio will increase the sensitivity from 50% to 93%, which leads to fewer false negative results and thus less missed patients. But, the increase of the sensitivity is at the expense of a lower positive predictive value. A false-positive NBS result may have great impact on the parents of newborns and the families involved (Waisbren et al. 2003; Gurian et al. 2006). Special care and a best practice for communication between healthcare providers and parents are therefore essential in mitigating the stress involved (Schmidt et al. 2012). Compared to other disease in the NBS, a positive predictive value of 37% is high (Hall et al. 2014).
Retrospective analyses allowed us to identify five children with reduced VLCAD activity who, on clinical evaluation, were all asymptomatic, but in whom the diagnosis VLCADD was confirmed by mutation analysis. These children may be at high risk of future metabolic crises and/or later-onset disease. However, it is not possible to define which outcomes are clinically relevant (Wilcken 2010; Bonham 2013). Children with a VLCAD activity of >20% appear to have no symptoms (Hoffmann et al. 2011). One could argue that individual 5 is therefore not a case. Even with a residual VLCAD activity <20%, the clinical outcome is not certain. Fatty acid oxidation flux might be a better biomarker to predict the clinical severity of VLCAD deficiency than enzyme activity (Diekman et al. 2015). With the current development rate of new techniques in genetics and biochemistry, sensitivity will probably increase even more in the coming years (Dixon et al. 2012; Bonham 2013). Although the introduction of worldwide NBS programs has offered significant health gain for many patients, it might be argued that too sensitive NBS methods can lead to “overdiagnosing” and as such may be harmful for patients and their families (Timmermans and Buchbinder 2010; Kwon and Steiner 2011).
Conclusion
In summary, we show that the biomarker C14:1/C2 (≥0.023) is a better marker (sensitivity 93%) compared to C14:1 (≥0.8, sensitivity 50%) to detect VLCADD patients and thus leads to fewer missed patients. However, the identified missed patients were all asymptomatic at the time of diagnosis. This suggests that a more sensitive screening approach may also identify individuals who may never develop clinical disease. Studies that evaluate the natural history of pre-NBS detected patients are needed to establish the risk of these VLCADD-deficient individuals for developing clinical signs and symptoms.
Acknowledgments
We are most grateful to Dr. B. Elvers of the National Institute for Public Health and the Environment (RIVM) for providing the selected blood spots for further analyses and to Dr. P. Verkerk of TNO (Applied Scientific Research Institute) and Prof. Dr. E.E.S. Nieuwenhuis of the Wilhelmina’s Children’s Hospital/University Medical Centre Utrecht (WKZ/UMCU) for their biochemical and ethical advice. They were not compensated for their efforts.
Synopsis
We show that the C14:1/C2 (≥0.023) ratio is a better biomarker (sensitivity 93%) to detect patients with VLCADD compared to C14:1 (≥0.8, sensitivity 50%) and identified five additional patients.
Conflict of Interest
Eugene Diekman has no conflict of interest, Monique de Sain-van der Velden has no conflict of interest, Hans Waterham has no conflict of interest, Leo Kluijtmans has no conflict of interest, Peter Schielen has no conflict of interest, Evert Ben van Veen has no conflict of interest, Sacha Ferdinandusse has no conflict of interest, Frits Wijburg has no conflict of interest, and Gepke Visser has no conflict of interest.
Funding Source
This research was supported by ZonMW (dossier 200320006), Metakids (www.metakids.nl), and the ESN-stimuleringsprijs.
Compliance with Ethics Guidelines
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Footnotes
Competing interests: None declared
Contributor Information
Gepke Visser, Email: gvisser4@umcutrecht.nl.
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Andresen BS, Olpin S, Poorthuis BJ, et al. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet. 1999;64:479–494. doi: 10.1086/302261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruteau J, Sachs P, Broué P, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study from 187 patients. Complementary data. J Inherit Metab Dis. 2014;37:137–139. doi: 10.1007/s10545-013-9628-9. [DOI] [PubMed] [Google Scholar]
- Bonham JR. Impact of new screening technologies: should we screen and does phenotype influence this decision? J Inherit Metab Dis. 2013;36:681–686. doi: 10.1007/s10545-013-9598-y. [DOI] [PubMed] [Google Scholar]
- Diekman EF, Ferdinandusse S, van der Pol WL, et al. Fatty acid oxidation flux predicts the clinical severity of VLCAD deficiency. Genet Med. 2015 doi: 10.1038/gim.2015.22. [DOI] [PubMed] [Google Scholar]
- Dixon S, Shackley P, Bonham J, Ibbotson R. Putting a value on the avoidance of false positive results when screening for inherited metabolic disease in the newborn. J Inherit Metab Dis. 2012;35:169–176. doi: 10.1007/s10545-011-9354-0. [DOI] [PubMed] [Google Scholar]
- Gurian EA, Kinnamon DD, Henry JJ, Waisbren SE. Expanded newborn screening for biochemical disorders: the effect of a false-positive result. Pediatrics. 2006;117:1915–1921. doi: 10.1542/peds.2005-2294. [DOI] [PubMed] [Google Scholar]
- Hall PL, Marquardt G, McHugh DMS, et al. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet Med. 2014 doi: 10.1038/gim.2014.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann L, Haussmann U, Mueller M, Spiekerkoetter U. VLCAD enzyme activity determinations in newborns identified by screening: a valuable tool for risk assessment. J Inherit Metab Dis. 2011 doi: 10.1007/s10545-011-9391-8. [DOI] [PubMed] [Google Scholar]
- Houten SM, Herrema H, te Brinke H, et al. Impaired amino acid metabolism contributes to fasting-induced hypoglycemia in fatty acid oxidation defects. Hum Mol Genet. 2013;22:5249–5261. doi: 10.1093/hmg/ddt382. [DOI] [PubMed] [Google Scholar]
- Kwon JM, Steiner RD. “I‘m fine; I’m just waiting for my disease”: the new and growing class of presymptomatic patients. Neurology. 2011;77:522–523. doi: 10.1212/WNL.0b013e318228c15f. [DOI] [PubMed] [Google Scholar]
- Laforêt P, Acquaviva-Bourdain C, Rigal O, et al. Diagnostic assessment and long-term follow-up of 13 patients with Very Long-Chain Acyl-Coenzyme A dehydrogenase (VLCAD) deficiency. Neuromuscul Disord. 2009;19:324–329. doi: 10.1016/j.nmd.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Lindner M, Hoffmann GF, Matern D. Newborn screening for disorders of fatty-acid oxidation: experience and recommendations from an expert meeting. J Inherit Metab Dis. 2010;33:521–526. doi: 10.1007/s10545-010-9076-8. [DOI] [PubMed] [Google Scholar]
- Loeber JG, Burgard P, Cornel MC, et al. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 1. From blood spot to screening result. J Inherit Metab Dis. 2012;35:603–611. doi: 10.1007/s10545-012-9483-0. [DOI] [PubMed] [Google Scholar]
- McHugh DMS, Cameron CA, Abdenur JE, et al. Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet Med. 2011;13:230–254. doi: 10.1097/GIM.0b013e31820d5e67. [DOI] [PubMed] [Google Scholar]
- Schmidt JL, Castellanos-Brown K, Childress S, et al. The impact of false-positive newborn screening results on families: a qualitative study. Genet Med. 2012;14:76–80. doi: 10.1038/gim.2011.5. [DOI] [PubMed] [Google Scholar]
- Sokol DK. Law, ethics, and the duty of care. BMJ. 2012;345 doi: 10.1136/bmj.e6804. [DOI] [PubMed] [Google Scholar]
- Timmermans S, Buchbinder M. Patients-in-waiting: Living between sickness and health in the genomics era. J Health Soc Behav. 2010;51:408–423. doi: 10.1177/0022146510386794. [DOI] [PubMed] [Google Scholar]
- Vianey-Saban C, Divry P, Brivet M, et al. Mitochondrial very-long-chain acyl-coenzyme A dehydrogenase deficiency: clinical characteristics and diagnostic considerations in 30 patients. Clin Chim Acta. 1998;269:43–62. doi: 10.1016/S0009-8981(97)00185-X. [DOI] [PubMed] [Google Scholar]
- Vreken P, van Lint AE, Bootsma AH, et al. Quantitative plasma acylcarnitine analysis using electrospray tandem mass spectrometry for the diagnosis of organic acidaemias and fatty acid oxidation defects. J Inherit Metab Dis. 1999;22:302–306. doi: 10.1023/A:1005587617745. [DOI] [PubMed] [Google Scholar]
- Waisbren SE, Albers S, Amato S, et al. Effect of expanded newborn screening for biochemical genetic disorders on child outcomes and parental stress. JAMA. 2003;290:2564–2572. doi: 10.1001/jama.290.19.2564. [DOI] [PubMed] [Google Scholar]
- Wanders RJA, Ruiter JPN, IJLst L, et al. (2010) The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J Inherit Metab Dis 33:479–494. doi: 10.1007/s10545-010-9104-8 [DOI] [PMC free article] [PubMed]
- Wilcken B. Fatty acid oxidation disorders: outcome and long-term prognosis. J Inherit Metab Dis. 2010;33:501–506. doi: 10.1007/s10545-009-9001-1. [DOI] [PubMed] [Google Scholar]