

Abstract
Classical galactosaemia (OMIM #230400) is a rare disorder of carbohydrate metabolism caused by deficiency of the galactose-1-phosphate uridyltransferase enzyme (EC 2.7.7.12). The cause of the long-term complications, including neurological, cognitive and fertility problems in females, remains poorly understood. The relatively small number of patients with galactosaemia and the lack of validated biomarkers pose a substantial challenge for determining prognosis and monitoring disease progression and responses to new therapies. We report an improved method of automated robotic hydrophilic interaction ultra-performance liquid chromatography N-glycan analysis for the measurement of IgG N-glycan galactose incorporation ratios applied to the monitoring of adult patients with classical galactosaemia. We analysed 40 affected adult patients and 81 matched healthy controls. Significant differences were noted between the G0/G1 and G0/G2 incorporation ratios between galactosaemia patients and controls (p < 0.001 and <0.01, respectively). Our data indicate that the use of IgG N-glycosylation galactose incorporation analysis may be now applicable for monitoring patient dietary compliance, determining prognosis and the evaluation of potential new therapies.
Introduction
Classical galactosaemia (OMIM #230400) is a rare disorder of carbohydrate metabolism caused by profound deficiency of the galactose-1-phosphate uridyltransferase (GALT) enzyme (EC 2.7.7.12). The disease is life-threatening if left untreated in neonates. The only current treatment available is a lifelong galactose-restricted diet. While this is life-saving in the neonate, long-term complications persist in treated patients despite early diagnosis, initiation of treatment and shared genotypes. These complications include cognitive impairment, neurological and speech abnormalities and fertility issues in female patients. The cause of the complications remains poorly understood (Fridovich-Keil and Walter 2008; Coss et al. 2014; Jumbo-Lucioni et al. 2012).
Reduced GALT activity results in decreased UDP-galactose bioavailability and the toxic build-up of intermediates of the galactose metabolism pathway. GALT maintains the balance between UDP-glucose (glc), UDP-galactose (gal), N-acetylgalactosamine (GalNAc) and N-acetylglucosamine (GlcNAc) (Frey 1996). These four UDP-hexoses are rate limiting for the biosynthesis of glycoproteins and proteoglycans, which form the foundation of the extracellular synaptomatrix of synaptic cleft and perisynaptic space (Dani and Broadie 2012).
Galactosaemia intoxicated neonates exhibit profound glycan assembly defects demonstrable in circulating transferrin and IgG. Galactosaemia patients (children and adults), on dietary galactose restriction, also have demonstrable defects in both assembly and processing of N-glycans ( Charlwood et al. 1998; Sturiale et al. 2005; Quintana et al. 2009; Coman et al. 2010; Coss et al. 2012, 2013). These biochemical defects resemble those observed in a number of congenital disorders of glycosylation (CDG) types I (N-glycan assembly defects) and II (N-glycan processing defects) (Freeze 2013). The glycan synthesis abnormalities in galactosaemia are proposed to result from a combination of decreased UDP-hexose substrates and increased levels of galactose-1-phosphate (gal-1-p) which cause ER stress and in turn disrupt glycosylation and subsequently cause systemic genomic dysregulation (Petry et al. 1991; Ornstein et al. 1992; Ng et al. 1989; Lai et al. 2003; Slepak et al. 2007; Coman et al. 2010; Jumbo-Lucioni et al. 2014; Coss et al. 2014). The proposed alternative pathways of galactose metabolism may be more active in some patients, offering a protective role against gal-1-p intoxication, e.g. UDP-glucose pyrophosphorylase (UGP), which has the ability to convert both gal-1-p and glucose-1-phosphate to UDP-galactose and UDP-glucose, respectively (Fridovich-Keil and Walter 2008).
In our previous studies, we have shown the presence of ongoing IgG N-glycan processing defects in adults and children with galactosaemia maintained on a galactose-restricted diet. It is accepted that dietary restriction of galactose is life-saving in the neonate. We have queried whether over-restriction of galactose after the initial perinatal galactose detoxication dietary treatment could have an additive effect on the ongoing pathophysiology on account of the continuing systemic glycosylation abnormalities observed (Hughes et al. 2009; Coman et al. 2010; Coss et al. 2013, 2014).
The absence of reliable sensitive markers to monitor the proposed cellular and systemic glycosylation abnormalities is problematic. Measuring the proximal metabolite markers, red blood cell (RBC) gal-1-p and urinary galactitol, is very informative in the initial neonatal confirmation of satisfactory treatment with a marked decrease noted in these metabolites. After the neonatal stage of galactosaemia, measuring red blood cell (RBC) gal-1-p and urinary galactitol, however, does not identify minor deviations in the diet or differentiate between patients. We (and others) have shown that RBC gal-1-p and urinary galactitol measurements do not distinguish optimum biochemical control in a gradual galactose diet liberalisation study (Hutchesson et al. 1999; Hughes et al. 2009; Bosch et al. 2004; Coss et al. 2012; Krabbi et al. 2011), whereas the analysis of IgG N-glycan profiles measured by normal-phase high-performance liquid chromatography (NP-HPLC) showed consistent individual responses to diet liberalisation (Coss et al. 2012).
We have recently reported an improved automated hydrophilic interaction ultra-performance liquid chromatography (HILIC-UPLC) method of glycan analysis which has allowed for more high-throughput processing of total IgG, with improved glycan peak (GP) resolution (Stöckmann et al. 2013). In our earlier preliminary studies reporting IgG N-glycan galactose incorporation ratios (G-ratios) as potential biomarkers for determining galactose tolerance in classical galactosaemia, we have applied this to small studies in children and adults (Coss et al. 2012, 2013). Of note, we have now reported this method in a moderate galactose relaxation dietary study in children (using galactose content of 300–500 mg/day). The method identified a number of favourable responders with improved glycosylation ratios with increasing galactose intake and an inverse relationship between the G0/G2 ratio and circulating leptin receptor (sOb-R) in the supplementation group (Knerr et al. 2015).
We now report the development and improvement of this rapid automated method of measuring IgG N-glycan galactose incorporation ratios (G-ratios), with a sample of 40 adult galactosaemia individuals from two populations and the characterisation of adult healthy control ratios from a large adult control dataset (81 matched healthy controls). We have also increased the efficiency and turnaround time of the test with development of the analysis now of undigested IgG glycans, which bypasses the time required for the glycan enzymatic digestion step.
Materials and Methods
Study Subjects and Characterisation
The study consisted of 32 classical Irish galactosaemia patients, 14 female and 18 males, 28 homozygous for Q188R, 2 heterozygous for Q188R/R333W and 2 heterozygous for Q188R/K285N, with age range 13 to 36 years and average age 21 years, and 8 Dutch patients, 5 female and 3 males, all homozygous for Q188R, with age range 14 to 26 years and average age 18 years (Table 1). The full-scale intelligence quotient (FSIQ) range for the Irish patients at last testing was FSIQ range 47 to 126, with an average FSIQ of 81. The FSIQ range for the Dutch group of patients at last testing was FSIQ range 56 to 97, with an average FSIQ of 71. The controls consisted of 81 healthy adults (25 Irish adults, average age 35 years, obtained from a healthy population health insurance screening panel and 56 Scottish healthy controls, average age 29 years, from an Orkney Islands, Scotland, healthy population epidemiological study). All galactosaemia adult study patients were maintained on a dietary galactose intake of less than 500 mg gal/day.
Table 1.
Galactosaemia patients’ clinical characteristics
| Ethnicity | Irish | Dutch |
|---|---|---|
| Patients | 32 | 8 |
| Age | 21 (13–36) | 18 (14–26) |
| Sex | 14 F, 18 M | 5 F, 3 M |
| Genotype | 28: Q188R/Q188R, 2: Q188R/R333W, 2: Q188R/K285N | Q188R/Q188R |
| FSIQ | 81 (47–126) | 71 (56–97) |
Ethical approval for this study was obtained from the Ethics Committee of the Children’s University Hospital, Dublin, Ireland, and the Ethics Committee of Maastricht University Hospital. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for inclusion in the study.
Biochemical N-Glycan IgG Analysis
Isolation of IgG from Whole Serum
Whole serum was prepared from freshly spun blood samples and immediately frozen at −20°C. IgG was isolated from patient serum samples using a Protein G 96-well Spin Plate for IgG Screening (Thermo Scientific, Dublin, Ireland), as previously described (Stöckmann et al. 2013). The purity of the isolated IgG was assessed using 10% reducing SDS–PAGE gel in an XCell SureLock Mini-Cell (Invitrogen, Carlsbad, CA) according to the manufacturer.
Removal of N-Linked Glycans from IgG
N-glycans were released from the total IgG glycoproteins (heavy and light chains) using the ultrafiltration plate high-throughput method previously described.
2-Aminobenzamide (2-AB) Labelling of N-Glycans
Released N-glycans were labelled via reductive amination with the fluorophore 2-AB as previously described. Tagging the glycans with the fluorescent label, 2-AB, allows for their detection at femtomole levels (Stöckmann et al. 2013).
Hydrophilic Interaction Ultra-performance Liquid Chromatography (HILIC-UPLC)
2-AB-labelled N-glycans were separated by HILIC-UPLC with fluorescence detection on a Waters ACQUITY™ UPLC H-Class instrument consisting of a binary solvent manager, sample manager and fluorescence detector under the control of Empower 3 chromatography workstation software (Waters, Milford, MA, USA), as previously described (Stöckmann et al. 2013). The HILIC separations were performed using Waters BEH Glycan column, 150 × 2.1 mm i.d., 1.7 μm BEH particles, using a linear gradient of 70–53% acetonitrile at 0.56 ml/min in 16.5 min for IgG separation and a linear gradient of 70–53% acetonitrile at 0.56 ml/min in 24.81 min. Solvent A was 50 mM formic acid adjusted to pH 4.4 with ammonia solution. Solvent B was acetonitrile. An injection volume of 10 μl sample prepared in 70% v/v acetonitrile was used throughout. Samples were maintained at 5°C prior to injection and the separation temperature was 40°C. The fluorescence detection excitation/emission wavelengths were λex = 330 nm and λem = 420 nm, respectively.
Method Validation The recovery of the total IgG is estimated to be 40%. The coefficient of variation for all the 28 IgG N-glycan peaks (with peak % areas greater than 1%) was based on quadruplicate assays performed on four different days and was below 10% for all major peaks as published previously (Stöckmann et al. 2013). To assess biological variation, samples were taken from one galactosaemia patient on three separate days. The biological replicate CV for the total G0 peaks was 2%, and this was 3% for G1 and 2% for G2.
The system was calibrated using an external standard of hydrolyzed and 2-AB-labelled glucose oligomers to create a dextran ladder, as described previously (Stöckmann et al. 2013; Royle et al. 2008).
IgG N-Glycan G-Ratios
G-ratios are informative indicators of the level of galactose incorporation into the N-glycan structures. The peak % areas of agalactosylated (G0), monogalactosylated (G1) and digalactosylated (G2) structures were determined to provide a quantitative measurement of the incorporation of galactose into IgG N-glycans as we have previously described (Coss et al. 2012, 2013).
Glycan Feature Statistical Analysis
A nonparametric, Wilcoxon–Mann–Whitney test was used to examine potential differences in the G0/G1, (G0/G1)/G2 and G0/G2 ratios between the galactosaemia cases and controls. The analysis was performed using R software (www.r-project.org) with the R base package. Box plots were constructed using GraphPad Prism.
Oxford Glycan Annotation System
The specific N-glycans were annotated according to the Oxford notation system representing N-linked glycan composition and structure. The assignment of glycan structures by HILIC-UPLC in serum was performed by comparison with an updated GlycoBase 3.2 (http://glycobase.nibrt.ie/), as well as assignments published (Royle et al. 2008; Stöckmann et al. 2013). Assignment of the IgG glycans in each peak was based on the analysis in Pučić et al. 2011.
Results
The characteristics of the 40 adult galactosaemia study patients for the serum IgG N-glycosylation are shown in Table 1. The study group consisted of 32 Irish and 8 Dutch adult galactosaemia patients. All patients have classical galactosaemia, and 36 of the 40 patients are homozygous for the severe GALT mutation Q188R. The mean FSIQ of the 32 Irish patients was 81 (range 47–126). The mean FSIQ of the Dutch patients was 71 (range 56–97).
Figure 1 illustrates the peak assignments of the IgG N-glycan profile as previously published (Stöckmann et al. 2013).
Fig. 1.
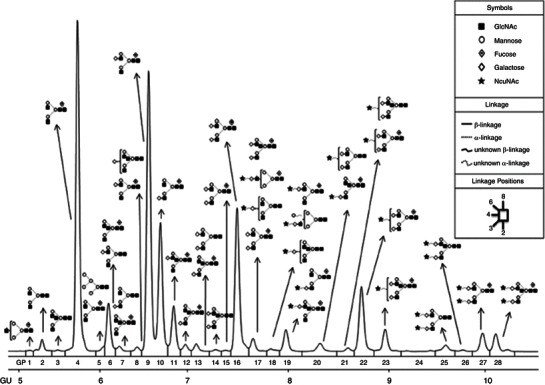
Representation of released undigested human IgG N-glycosylation profile and glycan structures. The linkages and abbreviations used are seen in the boxed inset, and the major structures assigned are based on the structural annotations for IgG described in Stöckmann et al. (2013). GP glycan peak number, GU glucose units. Reprinted with permission from Stöckmann et al. (2013). Copyright 2013 American Chemical Society
In Table 2, the G-ratios for galactosaemia patients and controls are shown with means and standard deviations. The differences in G0/G1 and G0/G2 ratios between patients and controls are all statistically significant (p < 0.001 and <0.01, respectively), indicating the presence of continuing N-glycan processing defects in these galactosaemia patients on dietary galactose restriction.
Table 2.
Adult undigested IgG N-glycan galactosylation average ratios and SD
| Group | n | G0/G1 | G0/G2 |
|---|---|---|---|
| Controls | 81 | 0.65 (± SD 0.14) | 0.78 (± SD 0.29) |
| Galactosaemia | 40 | 0.75 (0.15) | 0.95 (0.39) |
| Irish | 32 | 0.73 (0.14) | 0.95 (0.36) |
| Dutch | 8 | 0.75 (0.18) | 0.94 (0.50) |
| p-valuea, b | 0.0004658 | 0.007318 |
aStatistical analysis (MANOVA)
bControls vs. total galactosaemia patients
Figure 2 illustrates box–scatter plots of the distribution of G-ratios between patients and controls. As noted in our previous paediatric study, there may be overlap between outlier control and galactosaemia patients (Coss et al. 2013; Knerr et al. 2015). In this context, as previously noted, the test may be best utilised to monitor intervention changes using the patient acting as their own individual control. As with our earlier studies, the adult galactosaemia patients have higher levels of agalactosylated structures relative to the controls (Coss et al. 2012, 2013).
Fig. 2.
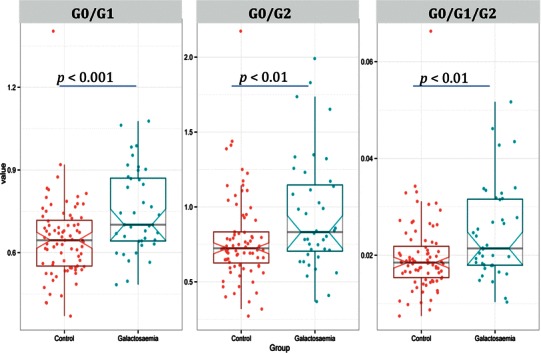
Box–scatter plots of statistically significant G0/G1, G0/G2 and G0/G1/G2 ratios from adult undigested IgG N-glycans between the total galactosaemia group and controls, with respective average G-ratios and SD indicated in Table 2. Data are given in a box–scatter plot to show the spread of the individual values. The diagram is made up of a ‘box’, which lies between the upper and lower quartiles. The median level divides the ‘box’ into two. Values for healthy controls are shown in red and total galactosaemia patients are shown in blue
Discussion
The development of informative and accurate biomarkers which reflect the disease pathogenesis is paramount for the management of galactosaemia, in view of the heterogeneity and disappointing clinical outcomes, identifiable in at least 50% of treated individuals.
In our earlier studies, we have proposed that early stage developmental, and later ongoing, disruptions to glycosylation and related gene expression pathways may have a role in the pathophysiology of classical galactosaemia. Perturbations to glycosylation during prenatal galactose intoxication could lead to systemic defects affecting long-term development in patients (Coss et al. 2013, 2014).
We have previously observed the correction of the gross assembly defects in young galactosaemia children following treatment with galactose restriction (Coss et al. 2013). In addition, we have observed improved glycosylation profiles in a number of adults with short-term moderate dietary galactose liberalisation and now recently in children, suggesting the upregulation of accessory glycosylation pathways (Coss et al. 2012; Knerr et al. 2015).
Beyond the untreated neonatal period, analysis of the whole serum or plasma N-glycome is largely uninformative in identifying subtle differences in glycosylation between adults with galactosaemia (Liu et al. 2012).
Our automated high-throughput HILIC-UPLC method applied to measure N-glycan abnormalities has been applied to the study of IgG N-glycan synthesis abnormalities and to the monitoring of a pilot study of moderate dietary relaxation in children with galactosaemia (Coss et al. 2013; Knerr et al. 2015). We now report the improvement of this method, now applied to a larger cohort of adult patients and controls, with the use of a large independent control dataset. Also of note, the G-ratios observed are similar in the Irish and Dutch classical galactosaemia cohorts (Table 2).
In summary, we have now refined the recently improved high-throughput automated HILIC-UPLC methodology of the IgG N-glycan galactose ratios, (G0/G1) and (G1/G2) as an indicator of processing defects in galactosaemia. In agreement with our earlier studies, in the current study, we have illustrated that treated adult galactosaemia patients generally have increased agalactosylated (G0) and monogalactosylated (G1) structures, with decreases in certain digalactosylated (G2) structures compared to healthy controls. These reductions would seem to indicate a continued lower percentage of galactose incorporation into serum glycoproteins and continuing N-glycan processing defects, in comparison to healthy individuals even after the prenatal, neonatal and childhood periods of intervention.
This further development of the IgG N-glycan G-ratios provides a tool to monitor therapeutic interventions and individualised metabolic/dietary control in affected adults with classical galactosaemia. Further studies will study this application in variant galactosaemia.
Acknowledgements
Funding for these studies was granted by the Irish Medical Research Charities Group (CFFH/TSCUH)/Health Research Board (No 2) was supported by the EU FP7 Research Framework Program ‘HighGlycan’ (Grant Reference No. 278535).
Abbreviations
- G0
Agalactosylated
- G1
Monogalactosylated
- G2
Digalactosylated
- Gal-1-P
Galactose-1-phosphate
- GALT
Galactose-1-phosphate uridyltransferase
- HILIC-UPLC
Hydrophilic interaction ultra-performance liquid chromatography
Synopsis
The use of IgG N-glycosylation galactose incorporation analysis by HILIC-UPLC analysis can be applied for monitoring of optimum metabolic control for individuals with classical galactosaemia.
Compliance with Ethics Guidelines
Conflict of Interest
The authors, Henning Stockmann, Karen P. Coss, M. Estela Rubio-Gozalbo, Ina Knerr, Maria Fitzgibbon, Ashwini Maratha, James Wilson, Pauline Rudd and Eileen P. Treacy, declare that they have no conflict of interest.
All procedures regarding the galactosaemia patient studies and sampling were followed in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
All authors listed above have been involved in the conception or design of this study and in drafting/revising the submitted article. Henning Stockmann and Karen Coss were involved in the direct planning and conduct of the study and experiments. The other authors were involved in interpreting the results, drafting and revising the manuscript.
Footnotes
Competing interests: None declared
Contributor Information
Eileen P. Treacy, Email: etreacy@mater.ie
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Bosch AM, Bakker HD, Wenniger-Prick LJ, Wanders RJ, Wijburg FA. High tolerance for oral galactose in classical galactosaemia: dietary implications. Arch Dis Child. 2004;89:1034–1036. doi: 10.1136/adc.2003.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlwood J, Clayton P, Keir G, Mian N, Winchester B. Defective galactosylation of serum transferrin in galactosemia. Glycobiology. 1998;8:351–357. doi: 10.1093/glycob/8.4.351. [DOI] [PubMed] [Google Scholar]
- Coman DJ, Murray DW, Byrne JC, Rudd PM, Bagaglia PM, Doran PD, Treacy EP. Galactosemia, a single gene disorder with epigenetic consequences. Pediatric Res. 2010;67:286–292. doi: 10.1203/PDR.0b013e3181cbd542. [DOI] [PubMed] [Google Scholar]
- Coss K, Byrne J, Coman D, Adamczyk B et al (2012) IgG N-glycans as potential biomarkers for determining galactose tolerance in Classical Galactosaemia. Mol Genet Metab 105:212–220 [DOI] [PubMed]
- Coss KP, Hawkes CP, Adamczyk B, et al. N-glycan abnormalities in children with galactosemia. J Proteome Res. 2013;13:385–394. doi: 10.1021/pr4008305. [DOI] [PubMed] [Google Scholar]
- Coss K, Treacy E, Cotter E, Knerr I, Murray D, Shin Y, Doran P. Systemic gene dysregulation in classical Galactosaemia: is there a central mechanism? Mol Genet Metab. 2014;113:177–187. doi: 10.1016/j.ymgme.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Dani N, Broadie K. Glycosylated synaptomatrix regulation of trans‐synaptic signalling. Dev Neurobiol. 2012;72(1):2–21. doi: 10.1002/dneu.20891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze HH. Understanding human glycosylation disorders: biochemistry leads the charge. J Biol Chem. 2013;288:6936–6945. doi: 10.1074/jbc.R112.429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey PA. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J. 1996;10:461–470. [PubMed] [Google Scholar]
- Fridovich-Keil JL, Walter JH. Galactosaemia Chapter 72. The online metabolic and molecular bases of inherited disease, OMMBID. New York: McGraw Hill; 2008. [Google Scholar]
- Hughes J, Ryan S, Lambert D, et al. Outcomes of siblings with classical galactosemia. J Pediatr. 2009;154:721–726. doi: 10.1016/j.jpeds.2008.11.052. [DOI] [PubMed] [Google Scholar]
- Hutchesson AC, Murdoch-Davis C, Green A, Preece MA, Allen J, Holton JB, Rylance G. Biochemical monitoring of treatment for galactosaemia: biological variability in metabolite concentrations. J Inherit Metab Dis. 1999;22:139–148. doi: 10.1023/A:1005493701913. [DOI] [PubMed] [Google Scholar]
- Jumbo-Lucioni P, Garber K, Kiel J, et al. Diversity of approaches to classic galactosaemia around the world: a comparison of diagnosis, intervention and outcomes. J Inherit Metab Dis. 2012;35(6):1037–1049. doi: 10.1007/s10545-012-9477-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumbo-Lucioni P, Parkinson W, Broadie K. Altered synaptic architecture and glycosylated synaptomatrix composition in a Drosophila classic galactosemia disease model. Dis Model Mech. 2014;7(12):1365–1378. doi: 10.1242/dmm.017137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knerr I, Coss KP, Kratzsch J, et al. Effects of temporary low-dose galactose supplements in children aged 5–12 years with Classical Galactosaemia: a pilot study. Pediatr Res. 2015 doi: 10.1038/pr.2015.107. [DOI] [PubMed] [Google Scholar]
- Krabbi K, Uudelepp ML, Joost K, Zordania R, Ounap K. Long-term complications in Estonian galactosemia patients with a less strict lactose-free diet and metabolic control. Mol Genet Metab. 2011;103(3):249–253. doi: 10.1016/j.ymgme.2011.03.023. [DOI] [PubMed] [Google Scholar]
- Lai K, Langley SD, Khwaja FW, Schmitt EW, Elsas LJ. GALT deficiency causes UDP-hexose deficit in human galactosemic cells. Glycobiology. 2003;13:285–294. doi: 10.1093/glycob/cwg033. [DOI] [PubMed] [Google Scholar]
- Liu Y, Xia B, Gleason TJ, Castañeda U, He M, Berry GT, Fridovich-Keil JL. N-and O-linked glycosylation of total plasma glycoproteins in galactosemia. Mol Genet Metab. 2012;106:442–454. doi: 10.1016/j.ymgme.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng WG, Xu YK, Kaufman FR, Donnell GN. Deficit of uridine diphosphate galactose in galactosaemia. J Inherit Metab Dis. 1989;12:257–266. doi: 10.1007/BF01799215. [DOI] [PubMed] [Google Scholar]
- Ornstein KS, McGuire EJ, Berry GT, Roth S, Segal S. Abnormal galactosylation of complex carbohydrates in cultured fibroblasts from patients with galactose-1-phosphate uridyltransferase deficiency. Pediatr Res. 1992;31:508–511. doi: 10.1203/00006450-199205000-00020. [DOI] [PubMed] [Google Scholar]
- Petry K, Greinix HT, Nudelman E, Eisen H, Hakomori S, Levy HL, Reichardt JK (1991) Characterization of a novel biochemical abnormality in galactosemia: deficiency of glycolipids containing galactose or N-acetylgalactosamine and accumulation of precursors in brain and lymphocytes. Biochem Med Metab Biol 46:93 [DOI] [PubMed]
- Pučić M, Knežević A, Vidič J et al (2011) High throughput isolation and glycosylation analysis of IgG–variability and heritability of the IgG glycome in three isolated human populations. Mol Cell. Proteomics 10:M111.010090 [DOI] [PMC free article] [PubMed]
- Quintana E, Navarro-Sastre A, Hernández-Pérez JM, et al. Screening for congenital disorders of glycosylation (CDG): transferrin HPLC versus isoelectric focusing (IEF) Clin Biochem. 2009;42:408–415. doi: 10.1016/j.clinbiochem.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Royle L, Campbell MP, Radcliffe CM, White DM, et al. HPLC-based analysis of serum N-glycans on a 96-well plate platform with dedicated database software. Anal Biochem. 2008;376:1–12. doi: 10.1016/j.ab.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Slepak TI, Tang M, Slepak VZ, Lai K. Involvement of endoplasmic reticulum stress in a novel Classic Galactosemia model. Mol Genet Metab. 2007;92:78–87. doi: 10.1016/j.ymgme.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöckmann H, Adamczyk B, Hayes J, Rudd PM. Automated, high- throughput IgG-antibody glycoprofiling platform. Anal Chem. 2013;85:8841–8849. doi: 10.1021/ac402068r. [DOI] [PubMed] [Google Scholar]
- Sturiale L, Barone R, Fiumara A et al (2005) Hypoglycosylation with increased fucosylation and branching of serum transferrin N-glycans in untreated galactosemia. Glycobiology 15:1268–1276 [DOI] [PubMed]