

Abstract
Introduction: Mitochondrial diseases are a clinically, biochemically and genetically heterogeneous group of disorders with a variable age of onset and rate of disease progression. It might therefore be expected that this variation be reflected in the age and cause of death. However, to date, little has been reported regarding the ‘end-of-life’ period and causes of death in mitochondrial disease patients. For some specific syndromes, the associated clinical problems might predict the cause of death, but for many patients, it remains difficult to provide an accurate prognosis.
Aims: To describe a retrospective cohort of adult mitochondrial disease patients who had attended the NHS Highly Specialised Services for Rare Mitochondrial Diseases in Newcastle upon Tyne (UK), evaluate life expectancy and causes of death and assess the consequences for daily patient care.
Methods: All deceased adult patients cared for at this centre over a period of 10 years were included in the study. Patient history, data on laboratory findings, biochemical investigations and genetic studies were analysed retrospectively.
Results: A total of 30 adult mitochondrial patients died within the time period of the study. The main mitochondrial disease-related causes of death in this patient cohort were respiratory failure, cardiac failure and acute cerebral incidents such as seizures and strokes. In almost half of the patients, the cause of death remained unknown. Based on our study, we present recommendations regarding the care of patients with mitochondrial disease.
Keywords: Adult patient cohort, Audit, Causes of death, Life expectancy, Mitochondrial disease
Introduction
Mitochondrial Function and Mitochondrial Disease
Mitochondria are cytoplasmic organelles that undertake key metabolic functions, prime of which is the generation of adenosine triphosphate (ATP), the principal energy source of the cell. This process of ATP production, known as oxidative phosphorylation (OXPHOS), takes place via the mitochondrial respiratory chain, a series of four multi-subunit enzymatic complexes (I–IV) that are coupled with the mitochondrial ATP synthase (complex V) and located in the inner mitochondrial membrane. Despite the fact that many metabolic processes take place in mitochondria, the term mitochondrial disease is generally reserved for clinical phenotypes associated with primary impairment of OXPHOS. Mitochondrial diseases can arise from mutations in the small, circular, double-stranded, maternally inherited mitochondrial DNA (mtDNA) or as a result of mutations in nuclear genes intimately linked to mitochondrial function (McFarland et al. 2010). One consequence of this complex genetic arrangement is that the phenotypic diversity of mitochondrial disease is enormous (DiMauro and Schon 2003). Generally, tissues with high energy demands such as the brain, liver and muscle are most likely to be affected (Smeitink et al. 2006), but clinical manifestations vary from single organ (e.g. optic neuropathy) to multisystem involvement. However, central nervous system disease with, for example, ataxia, dementia, spasticity, behaviour abnormalities, seizures or movement disorders is common (Morava et al. 2006a, 2010).
Given the heterogeneous disease manifestations, the clinical course and long-term outcomes of affected patients vary widely. Onset of mitochondrial disease in early childhood is in most cases multisystem and often rapidly progressive (McFarland and Turnbull 2009), whereas in the adult population, ‘classic mitochondrial syndromes’ such as Leber’s hereditary optic neuropathy (LHON); myoclonic epilepsy with ragged red fibres (MERRF); mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS); maternally inherited diabetes and deafness (MIDD); and chronic progressive external ophthalmoplegia (CPEO) are more often encountered and associated with variable clinical courses. In Kearns–Sayre Syndrome (KSS), caused by either a large-scale deletion or complex mtDNA rearrangements, and MELAS, a clinical phenotype associated with the m.3243A>G mutation (MTTL1 (MIM 590050)), life expectancy is often markedly reduced due to cardiac conduction block (Kearns–Sayre Syndrome) and progressive cardiomyopathy (MELAS) (Ashizawa and Subramony 2001; McFarland and Turnbull 2009). In contrast, in some ‘milder’ genotypes, such as those leading to CPEO, reduced life expectancy has not been described.
Although mitochondrial disease is one of the commonest metabolic and neuromuscular disorders, estimating the exact prevalence has been complicated by the diversity of phenotypes, genotypes and their loose association. Studies in North East England found that in an adult population, 9.2 per 100,000 people have clinically manifest mtDNA disease and a further 16.5 per 100,000 children and adults are at increased risk of developing mtDNA disease (Schaefer et al. 2008). One particular mutation, m.3243 A>G, was found to have an overall point prevalence of 16.3/100,000 of the adult population in Northern Finland (Majamaa et al. 1998).
Mortality in Mitochondrial Disease
The mortality of patients with mitochondrial disease is likely to be influenced by the age of the patient and the underlying genotype, but the extensive phenotypic diversity complicates the study of this aspect of disease. As described above, several mitochondrial syndromes present with skeletal muscle weakness, affecting especially the proximal muscles of the hip and shoulder girdle. This myopathy may eventually also affect respiratory muscles causing respiratory failure, a need for ventilatory support and a markedly reduced life expectancy (McFarland et al. 2010). Respiratory failure appears to be a relatively common cause of death in mitochondrial disease (Arpa et al. 2003).
Smaller cohorts of mitochondrial patients have been reported with cardiopulmonary failure, status epilepticus, aspiration pneumonia and pulmonary embolism as the underlying cause of death (Klopstock et al. 1999). Other papers focus on the cause of death in specific patient groups (Coenen et al. 1999; Wortmann et al. 2007; Morava et al. 2006b; de Vries et al. 2007; Majamaa-Voltti et al. 2008). Currently, there is little data available on mortality in larger cohorts of mitochondrial patients and especially the adult patient group. We have attempted to address this shortfall by performing a retrospective review of the adult population of mitochondrial disease patients who attended the NHS Highly Specialised Services for Rare Mitochondrial Diseases at the Royal Victoria Infirmary in Newcastle upon Tyne, UK. We assessed mean survival rates, causes of death, genotypes, phenotypes and, where available, diagnostic information from skeletal muscle biopsy. In particular, we wanted to identify any potentially avoidable deaths and learn how we might better individualise patient care.
Methods
We performed a retrospective study in all deceased adult and mitochondrial disease patients diagnosed by the Mitochondrial Disease Clinical and Diagnostic Service in Newcastle upon Tyne, UK, who died between 1 Jan 2000 and 31 Dec 2009. In this 10-year period from a total adult patient population of 380 patients, with histochemical and/or biochemical evidence of a respiratory chain deficiency (with or without a known underlying genetic defect), we evaluated 30 deceased patients above the age of 18 years.
All medical files, including correspondence with all involved medical specialists and family doctors, death certificates, postmortems and other reports related to the patients’ death, of these 30 patients, were gathered and analysed retrospectively. Relevant information regarding onset of disease, symptoms, clinical investigations (including DNA analysis, muscle biopsy and mutation load) and all available information referring to the death of the patient was systematically recorded in a database. Inclusion criteria were death in the presence of an ascertained diagnosis of mitochondrial disease, established by muscle biopsy or DNA analysis. Figure 1 provides an overview of this cohort of mitochondrial disease patients including data on the common mutations.
Fig. 1.
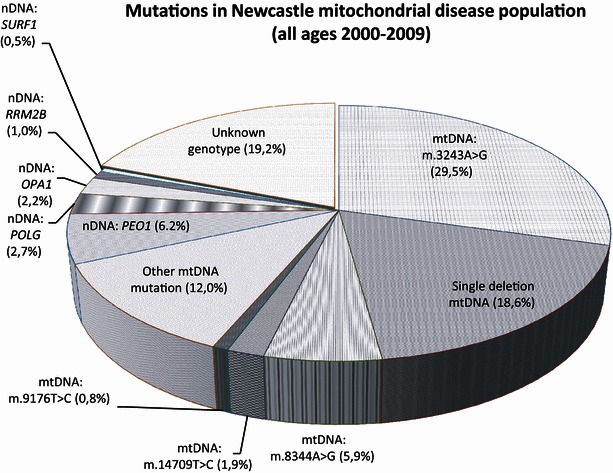
Mutations in the Newcastle mitochondrial disease population. This figure shows cumulative epidemiological data regarding common mutations diagnosed in a 10-year time period in the Newcastle mitochondrial patient cohort
Results
In the time period studied, our centre has largely been treating adult patients. The deceased adult patient group consisted of a heterogeneous mix of 30 patients with mitochondrial disease (see Table 1).
Table 1.
Epidemiologic, genetic, clinical and biochemical characteristics of adult mitochondrial disease patients from the Newcastle cohort, deceased from 2000 to 2009
| No. | Sex | Year of birth | Diagnosis | Age at onset of symptoms | DNA analysis | Muscle biopsy specimen | Level of mutant DNA (%) | Age at death (yr) | Cause of death | |
|---|---|---|---|---|---|---|---|---|---|---|
| Hist | Complex def | |||||||||
| 1 | M | 1985 | MELAS | 9–10 | MTTL1 | NK | NK | BL: 59 | 22 | Drowning incident |
| m.3243A>G | UEC: 89 | |||||||||
| 2 | F | 1977 | MELAS | 16–19 | MTTL1 | COX- | NK | BL: 27 | 28 | NK |
| m.3243A>G | RRF | UEC: 77 | ||||||||
| 3 | M | 1973 | MELAS | 5 | MTTL1 | COX- | I, IV | BL: NRD | 34 | Ventricle fibrillation possibly due to hyperglycaemia/ketoacidosis after RTI |
| m.3243A>G | UEC: 37 | |||||||||
| H: high (PM) | ||||||||||
| 4 | F | 1967 | MELAS | 18 | MTTL1 | COX- | NK | BL: 33 | 36 | Cardiac arrest after seizure |
| m.3243A>G | RRF | UEC: 21 | ||||||||
| 5 | F | 1959 | MELAS | 21 | MTTL1 | COX- | NK | BL: 20 | 41/42 | NK |
| m.3243A>G | RRF | MU: 80 | ||||||||
| 6 | F | 1960 | MELAS | 21 | MTTL1 | NK | NK | BL: 23 (PM) | 42 | NK |
| m.3243A>G | H: 72 (PM) | |||||||||
| MU: 87 (PM) | ||||||||||
| 7 | F | 1945 | MELAS | 26 | MTTL1 | COX- | NK | BL: 5 | 59 | Multi-organ failure |
| m.3243A>G | RRF | UEC:72 | ||||||||
| H: 85 (PM) | ||||||||||
| MU: 75 (PM) | ||||||||||
| 8 | M | 1930 | MELAS | NK | MTTL1 | COX- | I | UEC: 30 | 78 | NK; possibly cardiac failure after MI |
| m.3243A>G | RRF | MU: 29 | ||||||||
| 9 | M | 1978 | CPEO+ | 14 | MTTL1 | COX- | NK | UEC: 96 | 30 | NK; possibly cardiac failure |
| CM | m.3243A>G | RRF | MU: 71 | |||||||
| MM | ||||||||||
| DM | ||||||||||
| Deafness | ||||||||||
| 10 | M | 1961 | CPEO+ | 28 | MTTL1 | COX- | NK | BL: 14 | 43 | Inhalation of gastric contents |
| MIDD | m.3243A>G | UEC: 60 | ||||||||
| MM | MU: 69 | |||||||||
| CNSI | ||||||||||
| Dysphagia | ||||||||||
| Dementia | ||||||||||
| 11 | M | 1963 | Kearns–Sayre | 15 | MTdel5Kb | COX- | None | CB: 27 (PM) | 40 | Respiratory failure due to RTI after surgery for hip fracture (ataxia) |
| RRF | ||||||||||
| 12 | M | 1952 | Kearns–Sayre | 15 | MTdel5Kb | COX- | I, IV | NP | 56 | Cardiopulmonary and renal failure, deterioration after RTI |
| RRF | ||||||||||
| 13 | M | 1934 | Kearns–Sayre | NK | MTdel5Kb | COX- | NK | NP | 66 | Respiratory failure |
| 14 | F | 1934 | CPEO+ | NK | Mult. del. | COX- | NK | NP | 73 | Sepsis due to pneumonia after MI |
| MM | RRF | |||||||||
| CNSI | ||||||||||
| Dysphagia | ||||||||||
| Neuropathy | ||||||||||
| Deafness | ||||||||||
| 15 | F | 1935 | DOA+ | <18 | OPA1 | COX- | I | NP | 71/72 | NK |
| CPEO | c.1635C>G | RRF | ||||||||
| MM | ||||||||||
| Ataxia | ||||||||||
| Dysphagia | ||||||||||
| Deafness | ||||||||||
| 16 | M | 1949 | CPEO+ | NK | POLG | COX- | None | NP | 50 | NK |
| MM | c.1399G>A | RRF | ||||||||
| CNSI | ||||||||||
| Ataxia | ||||||||||
| Neuropathy | ||||||||||
| Deafness | ||||||||||
| OA | ||||||||||
| 17 | F | 1954 | CPEO+ | 17 | MTdel5Kb | COX- | I, IV | BL: low | 53 | NK |
| CNSI | RRF | |||||||||
| MM | ||||||||||
| Ataxia | ||||||||||
| Neuropathy | ||||||||||
| Dysphagia | ||||||||||
| 18 | M | 1936 | CPEO | 5 | Mult. del. | NP | NP | NP | 63 | NK |
| Dysphagia | ||||||||||
| 19 | M | 1937 | CPEO+ | NK | MTdel5Kb | COX- | IV | NP | 65 | Upper GI bleed due to varices (alcoholic chronic liver disease) |
| CNSI | RRF | |||||||||
| Ataxia | ||||||||||
| Neuropathy | ||||||||||
| Dementia | ||||||||||
| MM | ||||||||||
| 20 | M | 1946 | CPEO+ | NK | POLG | COX-(PM) | NK | NP | 59 | NK |
| CNSI | c.2542G>A c.3311C>G (compound heterozygous) | RRF (PM) | ||||||||
| Parkinsonism | ||||||||||
| Dementia | ||||||||||
| MM | ||||||||||
| Neuropathy | ||||||||||
| Dysphagia | ||||||||||
| 21 | F | 1942 | CPEO+ | NK | Mult. del. | COX- | NK | NP | 66 | Probably due to lung cancer |
| CNSI | RRF | |||||||||
| MM | ||||||||||
| Ataxia | ||||||||||
| Deafness | ||||||||||
| Dysphagia | ||||||||||
| 22 | F | 1964 | MERRF | 18 | MT-TK | NP | NP | BL: 92 | 41 | NK; possible respiratory failure due to aspiration or epileptic seizure |
| m.8344A>G | ||||||||||
| 23 | F | 1969 | Strokes | NK | MT-ND5 (MIM 516005) | NK | I, II (PM) | NP | 34 | Cardiac arrest after stroke or seizure |
| CNSI | m.13094T>C | |||||||||
| Ataxia | ||||||||||
| (PM Leigh) | ||||||||||
| 24 | F | 1976 | CNSI | 20 | POLG | None | NK | NP | 24 | Respiratory failure due to RTI in terminal phase with reduced consciousness |
| Ataxia | c.1399G>A | |||||||||
| c.2243G>C (compound heterozygous) | ||||||||||
| 25 | M | 1947 | DM | NK | MT-TE (MIM 590025) | NK | I,II,IV | BL: 100 | 55 | Cardiac arrest due to MI after above knee amputation (thrombosed aortic bypass graft) |
| MM | ||||||||||
| CNSI | m.14709T>C | |||||||||
| Ataxia | ||||||||||
| IHD | ||||||||||
| CM | ||||||||||
| 26 | F | 1931 | MM | <40 | MT-TE | NP | NP | NP | 70 | Respiratory failure due to RTI in bedridden patient |
| Neuropathy | m.14709T>C | |||||||||
| DM | ||||||||||
| IHD | ||||||||||
| 27 | F | 1951 | MM | NK | MT-TC | COX- | None | MU: 100 | 51 | NK |
| CNSI | (MIM 590020) | |||||||||
| Dystonia | m.5816A>G | |||||||||
| Ataxia | ||||||||||
| Dysphagia | ||||||||||
| 28 | M | 1952 | MM | 12 | MT-ND6 (MIM 590020) | COX- | I,II,IV | NP | 54 | Respiratory failure due to RTI after period of increased muscle weakness |
| CM | m.14484T>C | RRF | ||||||||
| 29 | M | 1935 | Neuropathy | <12 | NK | COX- | None | NP | 36 | NK |
| Ataxia | RRF | |||||||||
| Dysphagia | ||||||||||
| MM | ||||||||||
| CNSI (cognitive decline) | ||||||||||
| 30 | F | 1936 | DM | 33 | MT-TS2 (MIM 590085) | NP | NP | NP | 72 | NK |
| CNSI | m.12258G>A | |||||||||
| Ataxia | ||||||||||
| OA + RP | ||||||||||
| Deafness | ||||||||||
| Dementia | ||||||||||
BL blood, CB cerebellum, CM cardiomyopathy, CNSI central nervous system involvement, Complex def complex deficiency, COX cytochrome c oxidase-negative fibre, CPEO chronic progressive external ophthalmoplegia, DM diabetes mellitus, DOA dominant optic atrophy, F female, GI gastrointestinal, H heart, Hist histology, IHD ischemic heart disease, M male, MELAS mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes, MERRF myoclonic epilepsy with ragged red fibres, MI myocardial infarct, MIDD maternally inherited diabetes and deafness, MM mitochondrial myopathy, MU muscle, Mult. del. multiple deletions, NK not known, No. patient number, NP not performed, NRD not reliably detectable, OA optic atrophy, PM postmortem, RRF ragged red fibres, RP retinitis pigmentosa, RTI respiratory tract infection, UEC urinary epithelial cells
MELAS
Eight patients were diagnosed as having a MELAS phenotype (patients 1–8). The onset of disease was in late childhood or adolescence, and the first symptoms were in most cases muscle weakness, headaches or epileptic seizures. All eight patients had the MTTL1 (m.3243A>G) mutation. Tissue-specific mutation levels varied widely in this group. In most of the muscle biopsies, COX-deficient and red ragged fibres (RRF) were seen. The mean and median ages of death were, respectively, 42.5 and 38.75 years (age range 22 to 78 years). Patient 1 died because of accidental drowning. Suicide was considered but was difficult to confirm. He had been stable for a long time following his good recovery from a number of severe stroke-like episodes. In 3 of the MELAS (m.3243A>G) patients, death due to a cardiac cause was described (patients 3, 4 and possibly 8). Patient 3 was known to have hypertrophic cardiomyopathy and suffered from recurrent chest infections and respiratory muscle weakness. Since he failed to attend clinic, evaluation data from the year prior to his death were not available, but his clinical condition seemed stable before then. He died from cardiac arrest after hyperglycaemia and ketoacidosis after an initial respiratory tract infection. Patient 4 had asymptomatic hypertrophic cardiomyopathy from the age of 30 years. Severe biventricular failure, developing 9 months before death, was treated with cardiac medication (including a beta blocker, diuretic and angiotensin-converting enzyme (ACE) inhibitor). Regular cardiac evaluation was performed, with no deterioration noticed two weeks prior to death. Defibrillator placement had been considered but was rejected given his associated comorbidity and the risk of sudden cardiac death being considered low. The patient died of cardiac arrest, most likely tachyarrhythmic, after a seizure. Postmortem examination revealed a mildly enlarged heart with mild dilatation of both ventricles. Patient 7 was severely affected by mitochondrial disease. During the last months of her life, she deteriorated quickly, both physically and mentally, and eventually died of multi-organ failure. In 4 of the MELAS patients, the precise cause of death was not known (patients 2, 5, 6 and 8). Some 5 months prior to the death of patient 8, an echocardiogram identified severely reduced left ventricular function, but as he remained asymptomatic, follow-up was planned for 6 months. Prior to further follow-up, he had a myocardial infarction with subsequent pulmonary oedema. Since his age at death was 78 years, the myocardial infarction may be, in part at least, age related.
Chronic Progressive External Ophthalmoplegia (CPEO)
In nine patients, a CPEO or ‘CPEO-plus’ syndrome was diagnosed (patients 9, 10, 14, 16–21). The underlying genetic causes were point mutations in POLG (MIM 174763) or MTTL1, large-scale single deletion of mtDNA or multiple deletions of mtDNA where the nuclear gene responsible remained unidentified. The majority of these patients presented with one or more of ptosis, ophthalmoplegia, myopathy or ataxia. The mean age of death was 55.8 years and the median 59 (age range 30 to 73 years). Patient 10, known to suffer from dementia and muscle weakness, died after inhalation of gastric contents though dysphagia had not been clinically identified prior to death. Patient 14 died of sepsis after pneumonia following a myocardial infarction at the age of 73 years. She did have cardiac medication and regular cardiac checkups. Patient 19 died of complications of chronic alcoholic liver disease and patient 21 died of lung cancer. In five patients, the cause of death was unknown. One of them, patient 20, showed symptoms of Parkinsonism.
Kearns–Sayre Syndrome
In our cohort, three male adult patients were diagnosed with Kearns–Sayre Syndrome (patients 11–13), caused by large-scale single deletions of mtDNA. Initial presentation in all three cases was with muscle weakness and ophthalmoplegia. All died of cardiopulmonary complications following respiratory tract infection at an age varying from 40 to 66 years.
MERRF
One patient (patient 22) was diagnosed with MERRF, MTTK (MIM 590060), and died suddenly at the age of 41 years from possible respiratory failure due to a seizure or aspiration. She had severe muscle involvement and was care dependent.
Heterogeneous Phenotypes
The other nine patients (patient 15, 23–30) were phenotypically heterogeneous and died between the ages of 24 and 72 years. The cause of death was unknown in 4 patients (patient 15, 27, 29, 30). The other five patients (patient 23–26, 28) died of cardiac and/or respiratory failure during a respiratory tract infection, myocardial infarction, stroke or seizure.
Overall, the mean and median ages of death in the total adult patient group (n = 30) were, respectively, 50.4 and 52 (interquartile range 29.25) years, and in 50% of patients, no cause of death was registered. The respiratory chain deficiencies identified in the adult patient group differed in all phenotype and genotype groups.
Discussion
In this study we aimed to review the causes of death in a large, well-characterised population of patients with confirmed mitochondrial disease attending the NHS Highly Specialised Services for Rare Mitochondrial Diseases in Newcastle (UK). In this cohort, the three adult patients with KSS died of respiratory failure. Although perhaps not a typical cause of death for KSS patients, respiratory infections often have fatal consequences in debilitated, immobile patients with severe respiratory muscle weakness and proximal myopathy irrespective of their aetiology (Benditt and Boitano 2013; Palmio et al. 2014). However, the risk of respiratory compromise might be anticipated to be higher in patients with mitochondrial disease where dysphagia and associated aspiration pneumonia are not an uncommon occurrence. Impaired cardiac function, identified in all three patients, may also have contributed to their death. Approximately 57% of KSS patients exhibit cardiac symptoms (Berenberg et al. 1977), and multiple cases of sudden complete heart block have been described in both adults and children (Welzing et al. 2009; Katsanos et al. 2002; Chawla et al. 2008).
In our MELAS patient group (patients 1–8), we predominantly found cardiovascular causes of death. This finding concords well with the published literature where cardiovascular or neurological diseases were found to account for 30% of immediate causes of death and 32% of underlying causes in m.3243A>G carriers (Majamaa-Voltti et al. 2008). One MELAS patient died of a drowning incident, possibly suicide. Chronic illness is a risk factor for developing mood disorders (Greden 2001), and several studies have indicated a biologically plausible relationship between mitochondrial disease and depression (Morava et al. 2010; Mancuso et al. 2013a). However, this was the only potential suicide recorded in the cohort, and it is therefore difficult to conclude that mitochondrial disease was responsible.
Next to cardiopulmonary failure and pulmonary embolism, respiratory muscle weakness and dysphagia have been reported as a cause of death in CPEO patients (Klopstock et al. 1999; Caballero et al. 2007; Smits et al. 2011), and this was the case in 2 of the CPEO patients reported here. Interestingly one of the CPEO patients showed symptoms of Parkinsonism, a rare and late-onset symptom in CPEO-plus syndromes (Hudson et al. 2007; Mancuso et al. 2004) and an independent risk factor for developing dysphagia (Pfeiffer 2011); unfortunately the cause of death in this patient remains unknown.
The patient with MERRF syndrome died suddenly of possible respiratory failure due to a seizure or aspiration. Epilepsy is perhaps one of the most predictable causes of death in MERRF syndrome although overwhelming lactic acidosis and cerebral haemorrhage are also reported (Sanger and Jain 1996; Huang et al. 2002; Kato et al. 2006; Herrero-Martín et al. 2010; Mancuso et al. 2013b).
Cardiopulmonary failure appears to be one of the most frequent causes of death in mitochondrial disease patients. Cardiac involvement in adult mitochondrial disease patients, such as cardiomyopathy, has been reported with a prevalence up to 25% (Limongelli et al. 2010; Wahbi et al. 2010; Yilmaz et al. 2012). Early recognition of cardiac involvement is important to initiate appropriate therapeutic measures and may possibly prevent complications such as heart failure, arrhythmias and embolism (Finsterer and Kothari 2014). Subclinical myocardial abnormalities can already be seen in mitochondrial disease patients with two-dimensional strain echocardiography, while conventional echocardiographic findings did not show any signs of ventricular systolic dysfunction at all (Marcus et al. 2011). Likewise cardiac MRI is detecting abnormalities of cardiac function in mitochondrial disease patients that are not picked up by conventional echocardiography (Bates et al. 2013).
A few patients in this cohort died of causes not obviously related to mitochondrial disease (patients 19, 21), namely, alcoholic chronic liver disease and lung cancer. The impaired physical condition of many mitochondrial disease patients compared with the general population may be a contributing factor to the poor outcome of other, apparently unrelated diseases that mitochondrial disease patients encounter, but further work in this field is required.
Our patients were born between 1930 and 1985. National life expectancy data show a life expectancy of 58.7 years for males born in 1930 to 71.7 years for males born in 1985 and for females 63.0 years when born in 1930 to 77.4 years in 1985 (Sweet 2011). The age of death in this cohort varies widely, ranging from 22 to 78 years, with a mean age of death of 50.4 years, considerably less than expected on national statistics. Notably those with the CPEO phenotype and large-scale rearrangements/deletions of mtDNA died at younger age than patients with CPEO phenotype caused by other (point) mutations in mtDNA. The patient with MERRF syndrome died at the age of 41. The MERRF phenotype shows high clinical heterogeneity with generalized epilepsy being a negative prognostic factor (Mancuso et al. 2013a). This patient had very severe muscle involvement and was entirely dependent on others for her care. Given the small number of deceased patients in our clinic in the mentioned period and the additional high percentage of unknown causes of death, statements regarding causes of death in specific mitochondrial syndromes cannot be formulated.
In this cohort of patients with mitochondrial disease, a correlation between the degree of heteroplasmy in several tissues and the age of death could not be found, though the precise level of heteroplasmy at, or near, the time of death was known in only a small number of patients. The different complex deficiencies and the presence of COX negative and ragged red fibres also did not seem to relate to the age of death. Postmortem examination of patients 3 and 7, who suffered from hypertrophic cardiomyopathy and died of multi-organ failure, respectively, showed high levels of mutated mtDNA in cardiac tissue from both individuals.
Conclusion and Recommendations
Despite the relatively limited number of patients described, we can carefully draw some conclusions and propose recommendations for the daily care of mitochondrial disease patients. One of the striking findings in this study is the limited information reported about the time period preceding the death of our patients and even about the cause of death in a substantial part of our cohort. Some of the patients died in their home environment, receiving palliative care coordinated by their general practitioner without awareness of the involved tertiary specialists. Better communication between caregivers would aid the study of death in mitochondrial disease patients and probably subsequently improve the care for mitochondrial disease patients.
As a consequence of specific care required, most patients are seen periodically in tertiary-level specialist centres. Optimization of the information exchange between the multidisciplinary team members (specialist clinicians, nurses, paramedical therapists, primary care physicians) involved and the patient might also contribute to early recognition in changes in the medical condition of the patient. For example, the primary care physician should coordinate the patient care and the specialist centre should provide a point of contact for the patient. This communication is equally important in relation to end-of-life issues where effective management may avoid unnecessary intervention in the terminal phase of an illness and allow early institution of palliative care. It may also permit appropriate postmortem samples to be obtained, with minimal distress to relatives, and address the issue of the relatively low number of postmortems performed in our cohort. The timing of when to have ‘end-of-life discussions’ with patients and family will depend on a number of factors including the particular form of mitochondrial disease, the patient’s willingness to engage and the rate of disease progression. The latter will be obvious in rapidly progressive disease, but much more difficult to determine in patients who experience gradual deterioration. A possible approach to objectify disease progression might be by regular review using functional quality scales, such as the adult (NMDAS) forms of the Newcastle Mitochondrial Disease Rating Scale (Schaefer et al. 2006). Objective evidence of deterioration might aid both the physician and patient in discussing future prognosis and planning.
The majority of causes of death in our cohort were cardiac related. This study endorses that early detection of cardiac involvement is important, as it is associated with a poor prognosis, and timely therapeutic intervention may improve both quality and quantity of life. New investigative modalities, such as cardiac magnetic resonance spectroscopy, to detect cardiomyopathy in the initial stages look promising and may permit an even earlier institution of therapy. In this light, we underscore the importance of an interdisciplinary approach and communication between the primary care physician and the cardiology team. Our study highlights that a common cause of death in mitochondrial disease is cardiopulmonary failure following respiratory tract infection. Groups at higher risk of respiratory infection, including those patients with dysphagia or immobility, may therefore benefit from antibiotic prophylaxis, earlier institution of gastrostomy feeding or regular chest physiotherapy.
Finally, in the cohort of mitochondrial patients described in this study, 8% died in a time period of ten years. In almost half of these patients, all of whom were attending a specialist mitochondrial centre, the cause of death remains unknown. If we hope to influence the progression and outcome of mitochondrial disease for these patients, then we need to take careful note of the cause of death and any contributing factors. Good communication between primary care physician and specialist clinicians following the death of patients is essential in reaching this goal.
Take-Home Messages
Mitochondrial disease can cause significant morbidity and mortality.
Mitochondrial disease is incurable and ‘end-of-life’ planning should be discussed with patients appropriately.
Compliance with Ethics Guideline
Conflict of Interest
Marlieke Barends and Lotte Verschuren declare that they have no conflicts of interest. Victoria Nesbitt is a Clinical Research Associate for the Medical Research Council Mitochondrial Diseases Patient Cohort Study, UK. Eva Morava is a guest editor for the Journal of Inherited Metabolic Disease. Doug Turnbull and Robert McFarland are both PIs on the same study.
Contributions of Individual Authors
Marlieke Barends and Lotte Verschuren conducted the collection, analysis and interpretation of data and drafted the article. Eva Morava contributed to the revision of the article. Victoria Nesbitt contributed to data collection and revision of the article. Doug Turnbull contributed to the conception and design of the study and to the revision of the article. Robert McFarland was responsible for the conception and design of the audit, interpretation of data and to the revision of the article; he is the guarantor for the article.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Footnotes
Competing interests: None declared
Contributor Information
Robert McFarland, Email: Robert.mcfarland@ncl.ac.uk.
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Arpa J, Cruz-Martínez A, Campos Y, et al. Prevalence and progression of mitochondrial diseases: a study of 50 patients. Muscle Nerve. 2003;28:690–695. doi: 10.1002/mus.10507. [DOI] [PubMed] [Google Scholar]
- Ashizawa T, Subramony SH. What is Kearns–Sayre syndrome after all? Arch Neurol. 2001;58:1053–1054. doi: 10.1001/archneur.58.7.1053. [DOI] [PubMed] [Google Scholar]
- Bates M, Hollingsworth KG, Newman JH, et al. Concentric hypertrophic remodelling and subendocardial dysfunction in mitochondrial DNA point mutation carriers. Eur Heart J Cardiovasc Imaging. 2013;14:650–658. doi: 10.1093/ehjci/jes226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benditt JO, Boitano LJ. Pulmonary issues in patients with chronic neuromuscular disease. Am J Respir Crit Care Med. 2013;187:1046–1055. doi: 10.1164/rccm.201210-1804CI. [DOI] [PubMed] [Google Scholar]
- Berenberg RA, Pellock JM, DiMauro S, et al. Lumping or splitting? “Ophthalmoplegia-plus” or Kearns–Sayre syndrome? Ann Neurol. 1977;1:37–54. doi: 10.1002/ana.410010104. [DOI] [PubMed] [Google Scholar]
- Caballero PE, Candela MS, Alvarez CI, et al. Chronic progressive external ophthalmoplegia: a report of 6 cases and a review of the literature. Neurologist. 2007;13:33–36. doi: 10.1097/01.nrl.0000252953.49721.f5. [DOI] [PubMed] [Google Scholar]
- Chawla S, Coku J, Forbes T, et al. Kearns–Sayre syndrome presenting as complete heart block. Pediatr Cardiol. 2008;29:659–662. doi: 10.1007/s00246-007-9040-z. [DOI] [PubMed] [Google Scholar]
- Coenen MJ, van den Heuvel LP, Nijtmans LG, et al. SURFEIT-1 gene analysis and two-dimensional blue native gel electrophoresis in cytochrome c oxidase deficiency. Biochem Biophys Res Commun. 1999;265:339–344. doi: 10.1006/bbrc.1999.1662. [DOI] [PubMed] [Google Scholar]
- de Vries MC, Rodenburg RJ, Morava E, et al. Multiple oxidative phosphorylation deficiencies in severe childhood multi-system disorders due to polymerase gamma (POLG1) mutations. Eur J Pediatr. 2007;166:229–234. doi: 10.1007/s00431-006-0234-9. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- Finsterer J, Kothari S. Cardiac manifestations of primary mitochondrial disorders. Int J Cardiol. 2014;177:754–763. doi: 10.1016/j.ijcard.2014.11.014. [DOI] [PubMed] [Google Scholar]
- Greden JF. The burden of disease for treatment-resistant depression. J Clin Psychiatry. 2001;62:26–31. [PubMed] [Google Scholar]
- Herrero-Martín MD, Avuso T, Tuñón MT, et al. A MELAS/MERRF phenotype associated with the mitochondrial DNA 5521G>A mutation. J Neurol Neurosurg Psychiatry. 2010;81:471–472. doi: 10.1136/jnnp.2009.173831. [DOI] [PubMed] [Google Scholar]
- Huang CC, Kuo HC, Chu CC, et al. Clinical phenotype, prognosis and mitochondrial DNA mutation load in mitochondrial encephalomyopathies. J Biomed Sci. 2002;9:527–533. doi: 10.1007/BF02254979. [DOI] [PubMed] [Google Scholar]
- Hudson G, Schaefer AM, Taylor RW, et al. Mutation of the linker region of the Polymerase γ-1 (POLG1) gene associated with progressive external Ophthalmoplegia and Parkinsonism. Arch Neurol. 2007;64:553–557. doi: 10.1001/archneur.64.4.553. [DOI] [PubMed] [Google Scholar]
- Kato H, Uchigata M, Iijima M, et al. Fatal cerebral hemorrhage in mitochondrial encephalomyopathy. Clinical and pathological data of a case. J Neurol. 2006;253:529–530. doi: 10.1007/s00415-005-0010-1. [DOI] [PubMed] [Google Scholar]
- Katsanos KH, Pappas CJ, Patsouras D, et al. Alarming atrioventricular block and mitral valve prolapse in the Kearns–Sayre syndrome. Int J Cardiol. 2002;83:179–181. doi: 10.1016/S0167-5273(02)00040-2. [DOI] [PubMed] [Google Scholar]
- Klopstock T, Jaksch M, Gasser T. Age and cause of death in mitochondrial diseases. Neurology. 1999;53:855–857. doi: 10.1212/WNL.53.4.855. [DOI] [PubMed] [Google Scholar]
- Limongelli G, Tome-Esteban M, Dejthevaporn C, et al. Prevalence and natural history of heart disease in adults with primary mitochondrial respiratory chain disease. Eur J Heart Fail. 2010;12:114–121. doi: 10.1093/eurjhf/hfp186. [DOI] [PubMed] [Google Scholar]
- Majamaa K, Moilanen JS, Uimonen S, et al. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: prevalence of the mutation in an adult population. Am J Hum Genet. 1998;63:447–454. doi: 10.1086/301959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majamaa-Voltti K, Turkka J, Kortelainen ML, et al. Causes of death in pedigrees with the 3243A>G mutation in mitochondrial DNA. J Neurol Neurosurg Psychiatry. 2008;79:209–211. doi: 10.1136/jnnp.2007.122648. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Filosto M, Oh SJ, et al. A novel polymerase c mutation in a family with ophthalmoplegia, neuropathy, and Parkinsonism. Arch Neurol. 2004;61:1777–1779. doi: 10.1001/archneur.61.11.1777. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Orsucci D, Ienco EC, et al. Psychiatric involvement in adult patients with mitochondrial disease. Neurol Sci. 2013;34:71–74. doi: 10.1007/s10072-011-0901-0. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Orsucci D, Angelini C, et al. Phenotypic heterogeneity of the 8344>G mtDNA “MERRF” mutation. Neurology. 2013;80:2049–2054. doi: 10.1212/WNL.0b013e318294b44c. [DOI] [PubMed] [Google Scholar]
- Marcus KA, Barends M, Morava-Kozicz E, et al. Early detection of myocardial dysfunction in children with mitochondrial disease: An ultrasound and two-dimensional strain echocardiography study. Mitochondrion. 2011;11:405–412. doi: 10.1016/j.mito.2010.12.005. [DOI] [PubMed] [Google Scholar]
- McFarland R, Turnbull DM. Batteries not included: diagnosis and management of mitochondrial disease. J Intern Med. 2009;265:210–228. doi: 10.1111/j.1365-2796.2008.02066.x. [DOI] [PubMed] [Google Scholar]
- McFarland R, Taylor RW, Turnbull DM. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010;9:829–840. doi: 10.1016/S1474-4422(10)70116-2. [DOI] [PubMed] [Google Scholar]
- Morava E, van den Heuvel L, Hol F, et al. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006;67:1823–1826. doi: 10.1212/01.wnl.0000244435.27645.54. [DOI] [PubMed] [Google Scholar]
- Morava E, Rodenburg RJ, Hol F, et al. Clinical and biochemical characteristics in patients with a high mutant load of the mitochondrial T8993G/C mutations. Am J Med Genet A. 2006;140:863–868. doi: 10.1002/ajmg.a.31194. [DOI] [PubMed] [Google Scholar]
- Morava E, Gardeitchik T, Kozicz T, et al. Depressive behaviour in children diagnosed with a mitochondrial disorder. Mitochondrion. 2010;10:528–533. doi: 10.1016/j.mito.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Palmio J, Evilä A, Chapon F, et al. Hereditary myopathy with early respiratory failure: occurrence in various populations. J Neurol Neurosurg Psychiatry. 2014;85:345–353. doi: 10.1136/jnnp-2013-304965. [DOI] [PubMed] [Google Scholar]
- Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Parkinsonism Relat Disord. 2011;17:10–15. doi: 10.1016/j.parkreldis.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Sanger TD, Jain KD. MERRF syndrome with overwhelming lactic acidosis. Pediatr Neurol. 1996;14:57–61. doi: 10.1016/0887-8994(95)00226-X. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Phoenix C, Elson JL, et al. Mitochondrial disease in adults: a scale to monitor progression and treatment. Neurology. 2006;66:1932–1934. doi: 10.1212/01.wnl.0000219759.72195.41. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, McFarland R, Blakely EL, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–39. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- Smeitink JA, Zeviani M, Turnbull DM, et al. Mitochondrial medicine: a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab. 2006;3:9–13. doi: 10.1016/j.cmet.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Smits BW, Heijdra YF, Cuppen FW, et al. Nature and frequency of respiratory involvement in chronic progressive external ophthalmoplegia. J Neurol. 2011;258:2020–2025. doi: 10.1007/s00415-011-6060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet D (2011) Office for National Statistics. National Records of Scotland; Northern Ireland Statistics and Research Agency. Social Trends 41 – Health data. ‘Life expectancy at birth and age 65 and infant and neonatal mortality rate United Kingdom’. http://www.ons.gov.uk/ons/index.htm
- Wahbi K, Larue S, Jardel C, et al. Cardiac involvement is frequent in patients with the m.8344A N G mutation of mitochondrial DNA. Neurology. 2010;74:674–677. doi: 10.1212/WNL.0b013e3181d0ccf4. [DOI] [PubMed] [Google Scholar]
- Welzing L, von Kleist-Retzow JC, Kribs A, et al. Rapid development of life-threatening complete atrioventricular block in Kearns–Sayre syndrome. Eur J Pediatr. 2009;168:757–775. doi: 10.1007/s00431-008-0831-x. [DOI] [PubMed] [Google Scholar]
- Wortmann SB, Rodenburg RJ, Backx AP, et al. Early cardiac involvement in children carrying the A3243G mtDNA mutation. Acta Paediatr. 2007;96:450–451. doi: 10.1111/j.1651-2227.2006.00158.x. [DOI] [PubMed] [Google Scholar]
- Yilmaz A, Gdynia HJ, Ponfick M, et al. Cardiovascular magnetic resonance imaging (CMR) reveals characteristic pattern of myocardial damage in patients with mitochondrial myopathy. Clin Res Cardiol. 2012;101:255–261. doi: 10.1007/s00392-011-0387-z. [DOI] [PubMed] [Google Scholar]