

Abstract
Metabolism research has made tremendous progress over the last several decades in establishing the adipocyte as a central rheostat in the regulation of systemic nutrient and energy homeostasis. Operating at multiple levels of control, the adipocyte communicates with organ systems to adjust gene expression, glucoregulatory hormone exocytosis, enzymatic reactions and nutrient flux to equilibrate the metabolic demands of a positive or negative energy balance. The identification of these mechanisms has great potential to identify novel targets for the treatment of diabetes and related metabolic disorders. Herein, we review the central role of the adipocyte in the maintenance of metabolic homeostasis highlighting three critical mediators: adiponectin, leptin, and fatty acids.
Keywords: adipokines, adiponectin, leptin, lipid signaling
Graphical abstract
INTRODUCTION
With the increasing incidence of obesity and Type 2 Diabetes Mellitus in the U.S. and abroad, the lipotoxic effects of overnutrition have become a stark reality. Consequently, the field has focused on novel signaling molecules and pathways that regulate metabolic homeostasis with the hope of identifying a pharmacological target to limit obesity and/or its pathophysiological consequences. Within the scope of these efforts, it has become evident that adipose tissue-derived signaling molecules are amongst the most influential. In fact, adipose tissue has gained respect as a bona fide endocrine organ that regulates systemic metabolic homeostasis, responding to nutrient flux to equally match the metabolic demands of positive or negative energy balance. In turn, the secretion of adiponectin, leptin, and fatty acids (among other adipokines) are regulated by fasting and feeding. Fasting increases adipose tissue lipolysis, and thus non-esterified fatty acid release. In contrast, fasting acutely decreases circulating leptin (Boden et al., 1996), while adiponectin tends to increase with fasting (Halberg et al., 2005).
Prompted by the discovery of leptin more than 20 years ago (Zhang et al., 1994), followed by that of adiponectin (Scherer et al., 1995), and potentiated by the identification of 600+ adipocyte-derived secretory products since (Halberg et al., 2008) (Lehr et al., 2012b) (Lehr et al., 2012a), our appreciation for white adipose tissue (WAT) as a highly influential driver in the regulation of systemic metabolic homeostasis continues to expand. As such, polypeptides secreted by adipocytes, termed adipokines, have increasingly gained respect for their potent effects on systemic energy homeostasis. With the recent developments in our understanding of adipose tissue function as an endocrine organ, we are expanding our appreciation of the role that adipocytes take in the regulation of whole body homeostasis. The downstream effects of adipose tissue signaling range from central regulation of energy expenditure and satiety to altering the secretion of glucoregulatory hormones in the endocrine pancreas. Furthermore, we are beginning to appreciate the reciprocal signals from other tissues that communicate with adipose and alter adipokine production and release, hence the term “crosstalk”.
Over the course of the past 20 years, our laboratory has focused on understanding the mechanisms mediating the “healthy” expansion of adipose tissue during positive energy balance. In fact, we have shown that a fully functional, insulin-sensitive fat pad has a positive systemic impact, rescuing other peripheral organs from the lipotoxic effects of dyslipidemia during times of excess caloric intake (Kim et al., 2007; Kusminski et al., 2012). These effects are, in part, mediated by the adipocyte communicating with target tissues to affect metabolic homeostasis. Importantly, these findings are robustly supported by human studies demonstrating that, independent of total body fat mass, adipose tissue dysfunction, inflammation, and an imbalance of circulating adipokines are associated with insulin resistance in obese individuals (Hardy et al., 2011; Kloting et al., 2010).
Indeed, numerous of adipocyte-derived secretory factors have been identified to play a role in the maintenance of glucose, lipid, and energy homeostasis, each contributing to the communication between the adipocyte and tissues involved in the maintenance of glucose, lipid, and energy homeostasis. Here, we are focusing on the inter-cellular and inter-organ communication axis that has the adipocyte as a central nexus highlighting three critical mediators: adiponectin, leptin, and fatty acids. We do not wish to distract from the importance of many additional adipocyte-derived products that are physiologically important. However, we believe that these three factors serve as excellent paradigms for adipocyte-derived products that exert a profound influence on systemic metabolic homeostasis.
ADIPOCYTE COMMUNICATION REGULATES LIPID METABOLISM: EFFECTS ON ECTOPIC LIPID ACCUMULATION
Adipose Tissue Lipid Metabolism
Adiponectin
Although most research efforts aimed at understanding the effects of adipokines have focused on endocrine functions, these adipocyte-derived signals also have profound autocrine and paracrine functions. In fact, adiponectin appears to encourage the “healthy” expansion of adipose tissue. Adiponectin has repeatedly been shown to enhance insulin sensitivity (Berg et al., 2001) and maintains healthy adipose tissue expansion while rescuing ectopic lipid accumulation in animal models (Xu et al., 2003; Yamauchi et al., 2001). Accordingly, several human studies have associated an increased prevalence of diabetes with polymorphisms in the gene encoding adiponectin that lead decreased levels of adiponectin (Hara et al., 2002; Stumvoll et al., 2002). In leptin deficient (ob/ob) mice, maintaining adiponectin at lean levels as opposed to the widely observed drop of adiponectin in the obese state, increases fat mass, yet improves insulin sensitivity (Kim et al., 2007). In fact, adiponectin enhances adipocyte lipid storage, thereby preventing ectopic lipid accumulation. Adiponectin overexpression in 3T3-L1 cells increases lipid storage and adipogenesis (Fu et al., 2005).
Leptin
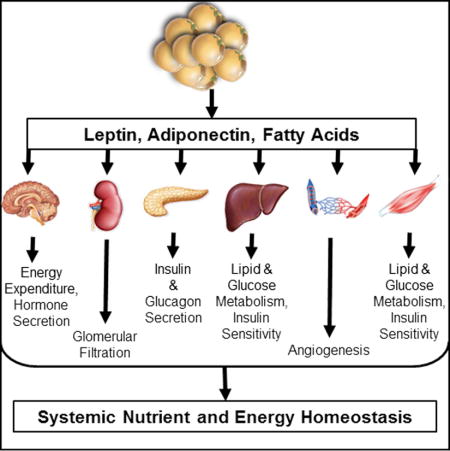
Leptin was first recognized for its prominent action on the hypothalamus to control food intake, energy expenditure, and, hence, body weight (Friedman and Halaas, 1998; Halaas et al., 1997; Halaas et al., 1995; Montez et al., 2005; Zhang et al., 1994). The discoveries of leptin and adiponectin were the first indications that adipose tissue was an endocrine organ with widespread control over systemic energy homeostasis. Although the precise mechanisms by which leptin and adiponectin are secreted from the adipocyte remain elusive, their metabolic effects on target tissues are robust. While the adipocyte lacks a classical triggered secretory pathway for adipokines, it is clear that the release of both leptin and adiponectin can be acutely enhanced through a variety of factors. Circulating levels of leptin are proportional to fat mass and leptin receptors are abundantly expressed in many tissues, including on the adipocyte, suggesting leptin may regulate adipose tissue metabolism through autocrine signaling. Fasting acutely decreases leptin (Boden et al., 1996), suggesting that factors aside from fat mass regulate its secretion (ie: fasting-induced changes in circulating insulin (Barr et al., 1997)). In vitro, leptin decreases lipogenesis, increases triglyceride hydrolysis, and increases fatty acid oxidation (William et al., 2002). However, the predominant effects of leptin on adipose tissue may be mediated through the peripheral nervous system. Leptin increases the sympathetic efferent signal to both brown adipose tissue (Rezai-Zadeh and Munzberg, 2013; Scarpace and Matheny, 1998) and white adipose tissue (WAT) to increase lipolysis (Bartness et al., 2005; Geerling et al., 2014). To fully identify the role of leptin, Zeng and colleagues (2015) recently explored the role of leptin in the neuro-adipose junction (Zeng et al., 2015). Utilizing optogenetic stimulation of sympathetic innervation in white fat, the authors visualized and identified in vivo the neuro-adipose junctions that mediate the lipolytic effect of leptin, establishing that the leptin induced lipolysis is mediated by sympathetic neurons that innervate adipocytes in white adipose tissue (Zeng et al., 2015). Thus, although leptin is affecting adipose tissue, this effect does not appear to be entirely autocrine in nature, but instead depends on an intact sympathetic nervous system (Figure 1).
Figure 1. Autocrine signaling in the adipocyte.
Adiponectin, which increases as fat mass decreases, acts locally on the adipocyte to increase GLUT4- mediated glucose uptake, while enhancing adipogenesis and adipocyte lipid storage. Fasting and leptin independently increase adipose tissue lipolysis, while fasting decreases leptin secretion. The predominant lipolytic action of leptin may be mediated through the peripheral nervous system. A rise in some fatty acids (see text for details) can increase adipocyte GLUT4-mediated glucose uptake.
Hepatic Lipid Metabolism
As the master regulator of systemic lipid and glucose homeostasis, the liver is the organ most affected by ectopic lipid accumulation. Accordingly, non-alcoholic fatty liver disease (NAFLD), and non-alcoholic steatohepatitis (NASH) are now considered the most common forms of liver disease in the U.S. NAFLD and NASH are common in obesity, with up to 90% prevalence of NAFLD in the obese population (Bellentani et al., 2010). NAFLD and NASH can be associated with insulin resistance, though not in all cases, and a subgroup of individuals amongst this group has a higher risk of progressing to cirrhosis and, eventually, hepatocellular carcinoma (Cohen et al., 2011; Starley et al., 2010). Hence, excess lipid deposition in the liver can have severe physiological consequences in some individuals.
The canonical lipid transport mechanisms mediated by albumin and lipoproteins regulate the flux of lipids between tissues. As such, lipids are continuously transported either from adipose to the liver and skeletal muscle or from the liver to adipose and skeletal muscle. Studies demonstrating that a 16 hour fast in mice induces nearly a ten-fold increase in hepatic lipid accumulation illustrate the powerful impact that adipose tissue lipolysis has on lipid accumulation in the liver (Renquist et al., 2012). Adding to our classical understanding of the role of lipid transport, synthesis, and utilization in hepatic lipid metabolism and deposition, we (Holland et al., 2013; Xia et al., 2015) and others (Awazawa et al., 2009; Yamauchi et al., 2002) have identified key mechanistic links that provide the basis for an adipose tissue-liver communication axis that regulates systemic metabolic homeostasis.
Adiponectin
Both subtypes of adiponectin receptors (adipoR1 and adipoR2) are expressed in the liver (Yamauchi et al., 2003). Serum adiponectin is decreased in obese, insulin resistant, type 2 diabetic rodents and humans (Scherer, 2006). Suggesting a possible causative role of this depressed adiponectin in insulin resistance of obesity, adiponectin overexpression prevents high fat diet induced hepatic lipid accumulation in rodent models of obesity (Kim et al., 2007). Furthermore, genetically eliminating adiponectin in leptin deficient (ob/ob) obese mice intensifies hepatosteatosis (Holland et al., 2013).
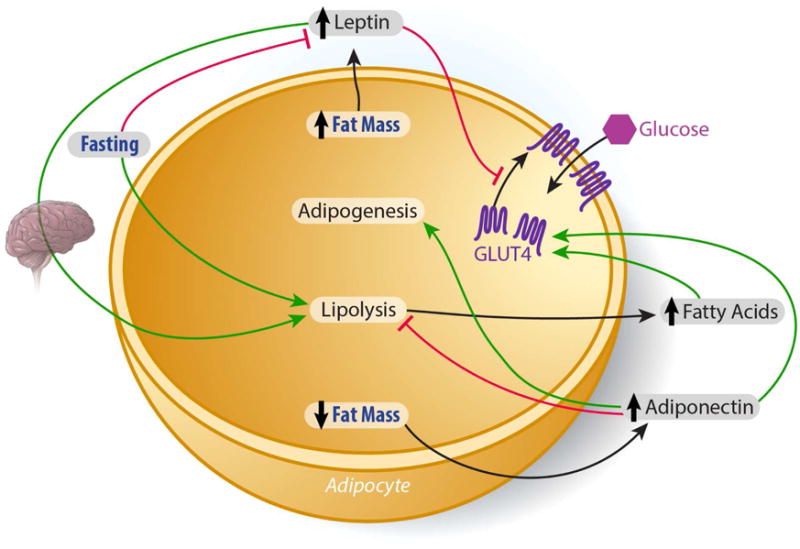
Adiponectin decreases hepatic lipogenesis and increases β-oxidation through adipoR1 mediated activation of AMP protein kinase (AMPK) and peroxisome proliferator-activated receptor (PPARα) (Awazawa et al., 2009; Miyamoto et al., 2012; Yamauchi et al., 2002; Yamauchi et al., 2007). As a cellular energy sensor, AMPK inhibits lipogenesis by phosphorylating the rate-limiting enzyme of de novo lipogenesis, acetyl CoA carboxylase-1 (ACC-1). This decreases in ACC-1 activity, decreases malonyl CoA production, thereby relieving inhibition of carnitine palmitoyl transferase-1 (CPT-1) activity and enhancing fatty acid transport into the mitochondria to undergo β-oxidation. These observations are in contrast to the effects on hepatic glucose output. Birnbaum and colleagues reported that the hepatic loss of the upstream activator of AMPK, LKB1 partially impaired the ability of adiponectin to lower serum glucose, though other actions of the hormone were preserved, including reduction of gluconeogenic gene expression and hepatic glucose production (Miller et al., 2011). Others report that at the transcriptional level, adiponectin signaling through adipoR1 induces the liver kinase B (LKB)-AMPK pathway which decreases the expression of genes involved in hepatic lipogenesis and cholesterol synthesis by suppressing sterol response element binding protein-1C (SREBP1c) expression (Awazawa et al., 2009) (Figure 2). SREBP1c, a transcription factor, acts as a master regulator of lipogenic gene expression in the liver. Activated by downstream insulin signaling, coupled with liver X receptor (LXR) nuclear receptors in the liver, SREBP1c decreases the expression of lipogenic enzymes, namely ACC-1 (Chen et al., 2004). Accordingly, genetic ablation of SREBP1c prevents hepatic lipid accumulation in high fat diet fed and leptin deficient mice (Yahagi et al., 2002). The Kadowaki laboratory recently reported a mechanism which links SREBP1c to the anti-lipogenic effects of adiponectin. Through an AMPK mediated inhibition of SREBP1c activity, the authors demonstrated that adiponectin inhibits the expression of SREBP1c through the adioR1 receptor which mediates the liver kinase B (LKB)-AMPK pathway (Awazawa et al., 2009). Intraperitoneal administration of adiponectin suppressed hepatic SREBP1c within 4 hours and 8 hours post injection, the mRNA expression of ACC-1 was decreased significantly. Importantly, the authors observed that during this time course, plasma insulin and glucose remained unchanged. Thus, neither insulin signaling, nor changes in glycemia could account for this adiponectin-induced decrease in lipogenic gene expression (Awazawa et al., 2009).
Figure 2. Adipocyte/liver crosstalk to maintain systemic lipid and glucose homeostasis.
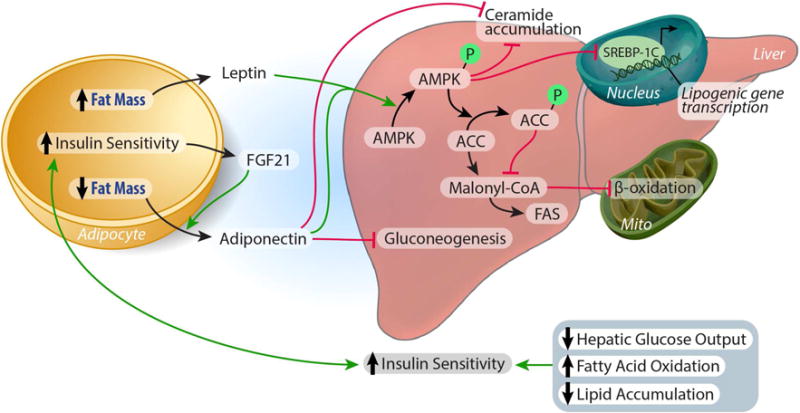
Both Leptin and Adiponectin decrease hepatic lipogenesis and increase β-oxidation through activation of AMP protein kinase. Phospo-AMPK inhibits lipogenesis by 1) suppressing SREBP1c expression, and 2) by phosphorylating acetyl CoA carboxylase-1 (ACC-1), the rate-limiting enzyme of de novo lipogenesis. This decrease in ACC-1 activity, limits malonyl CoA production, relieving the inhibition of carnitine palmitoyl transferase-1 (CPT-1) activity and enhancing fatty acid transport into the mitochondria to undergo β-oxidation. Adiponectin lowers hepatic ceramide accumulation, independent of AMPK activity, by enhancing ceramidase activity in the liver. FGF21 stimulates adiponectin secretion. Adiponectin also inhibits hepatic gluconeogenesis, independent of AMPK, decreasing glucose output and improving glycemia. Consequently, enhanced hepatic insulin sensitivity feeds back to the adipocyte to promote adipose tissue insulin sensitivity, resulting in enhanced systemic lipid and glucose homeostasis (see text for details).
Adiponectin can ameliorate the hyperlipidemia and the resultant excess hepatic lipid accumulation associated with metabolic dysfunction through a mechanism upstream of AMPK signaling (Holland et al., 2011). When hepatocellular ceramide levels are high, these lipids can decrease insulin sensitivity and increase apoptosis (Bikman and Summers, 2011). We found that adiponectin lowers hepatic ceramide accumulation by enhancing ceramidase activity associated with its two receptors, AdipoR1 and AdipoR2 to enhance ceramide catabolism. Importantly, these effects were independent of AMPK activity (Holland et al., 2011) (Figure 2). Most recently, we have extended our studies that originally identified adiponectin’s receptor-associated ceramidase activity and consequent hepatic ceramide lowering properties. With an appreciation that the communication between adipose tissue and liver is crucial to the maintenance of systemic energy homeostasis, we sought to understand this “cross-talk” from a lipid-centric perspective. Through the development of transgenic mouse models that inducibly overexpress acid ceramidase in either adipose tissue or liver, we found that ceramides readily exchange between adipose tissue and the liver. Overexpression of acid ceramidase in either adipose tissue or the liver prevented hepatic lipid accumulation and improved insulin sensitivity in both the liver and adipose tissue of mice challenged with high fat diet (Xia et al., 2015). Thus, the enhanced ceramidase activity induced by adiponectin signaling may be key to its potential therapeutic efficacy.
Adiponectin facilitates the metabolic effects of FGF21
Fibroblast growth factor 21 (FGF21), a member of the FGF superfamily is secreted by adipose tissue, liver, (Coskun et al., 2008; Kharitonenkov and Larsen, 2011; Kharitonenkov et al., 2005) and skeletal muscle. Originally cloned by Nobuyuki Itoh’s group (Nishimura et al., 2000) and later identified in 2005 at Lilly Pharmaceuticals as a novel regulator of energy metabolism (Kharitonenkov et al., 2005), it has since been studied as a potential treatment for TDM and related metabolic disease. FGF21 restores euglycemia, ameliorates hyperlipidemia, and decreases fat mass in animal models of obesity and T2DM (Coskun et al., 2008; Kharitonenkov and Larsen, 2011; Kharitonenkov et al., 2005). Of particular interest to our group was the observation that chronic FGF21 administration increased circulating adiponectin levels (Adams et al., 2012). Thus, we sought to elucidate the relationship between the metabolic actions of FGF21 and adiponectin. In so doing, we demonstrated that FGF21 decreases the accumulation of ceramides in the liver, while stimulating adiponectin secretion in mice (Figure 2). In fact, the concentration of adiponectin rose within 5 minutes of FGF21 exposure and was significantly elevated within 15 minutes. By genetically ablating the gene encoding adiponectin in both Leptin deficient (ob/ob) and diet-induced obese (DIO) mice, we further demonstrated that FGF21 relies on adiponectin to exert its blood-glucose and lipid- lowering effects in obese mice (Holland et al., 2013).
Leptin
Similar to its catabolic action in the adipocyte, leptin prevents lipogenesis, while activating β-oxidation of fatty acids in the liver. Leptin receptors are abundantly expressed in the liver and their expression is increased in response to leptin administration and short term fasting (Cohen et al., 2005), demonstrating that the liver is a direct target for leptin as well. In addition to presenting as hyperphagic and obese, leptin deficient ob/ob mice and leptin receptor-deficient db/db mice have hypertriglyceridemia, hypercholesterolemia (Nishina et al., 1994), and impaired lipid tolerance (Haluzik et al., 2004), resulting in hepatic steatosis (Trak-Smayra et al., 2011). To test the liver specific-effects of leptin signaling, Huynh and colleagues recently generated a liver-specific leptin receptor knock out mouse which displays decreased circulating levels of apolipoprotein-B, increased levels of low density lipoprotein (VLDL) triglycerides, coupled with an increase in hepatic lipoprotein lipase activity (Huynh et al., 2013). These results advocate for a role of leptin in the incorporation of triglyceride into VLDL, allowing for the export of lipid to target tissues. Similar to adiponectin, leptin increases hepatic fatty acid oxidation and decreases de novo lipogenesis through the phosphorylation of ACC-1 (Huang et al., 2006) (Figure 2).
In addition to the aforementioned anti-lipotoxic effects, adiponectin and leptin both exert beneficial anti-inflammatory actions on the liver to prevent the transition from NAFLD to NASH. Adiponectin’s anti-fibrotic and anti-inflammatory roles have been shown to decrease liver fibrosis in multiple models of steatohepatitis (Kamada et al., 2003; Xu et al., 2003). Similarly, recombinant leptin and the leptin analog, metreleptin, are currently employed as a treatment for lipodystrophy and congenital leptin deficiency. These therapeutic approaches reverse hepatic lipid accumulation and decrease the severity of NASH in lipodystrophic patients (Javor et al., 2005). Clinical trials are currently underway to assess the efficacy of metreleptin as a treatment for NAFLD (ClinicalTrials.gov Identifier: NCT00596934), providing a promising future for adipose derived secretory products as therapeutics for the treatment of obesity-associated liver disease. Such a confirmation of leptin action in the clinical setting is of great importance to the success of these therapeutics, as there are some important discrepancies with respect to the effect of leptin and leptin deficiency between rodents and humans (Wang et al., 2014).
Adipokines and Vascular Health
Obesity and Type 2 diabetes increase the risk for the development of atherosclerosis, a disease process beginning with endothelial dysfunction, leading to increased vasoconstriction, and ultimately coagulation and inflammation. The adipocyte secretes numerous factors that play a role in angiogenesis (Cao, 2007). While pro-inflammatory cytokines released from the adipocyte have been associated with systemic inflammation and endothelial dysfunction, other adipokines have direct beneficial effects on the vasculature.
Adiponectin
The cardioprotective role of adiponectin has been demonstrated in numerous rodent and cell culture models, and human studies demonstrating decreased circulating adiponectin in patients with cardiovascular disease corroborate these findings (Baker et al., 2006; Hotta et al., 2000). Adiponectin stimulates angiogenesis through activation of AMP Kinase and AKT in vivo and in vitro, thereby promoting endothelial nitric oxide synthesis within the endothelial cell (Ouchi et al., 2004). Additional studies have shown that the high molecular weight adiponectin form in particular inhibits endothelial apoptosis in vitro (Kobayashi et al., 2004). While the pro-angiogenic role of adiponectin is beneficial to cardiovascular health, these actions may be detrimental in the context of some cancers. For example, our lab has shown that deletion of the adiponectin gene in a mouse model of aggressive mammary tumor development leads to a reduction in angiogenic profile, promoting tumor-associated cell death (Landskroner-Eiger et al., 2009). Additional studies support our findings, demonstrating that inactivation of the T-cadherin gene, a binding partner for high molecular weight adiponectin, attenuates mammary tumor growth by restricting tumor vascularization (Hebbard et al., 2008).
Leptin
The relationship between leptin and cardiovascular disease remains somewhat controversial, particularly in human studies in which the severity of disease can vary greatly (Sweeney, 2010). Rodent models of obesity and hyperleptinemia demonstrate that leptin’s effects on vasodilation are concentration dependent. Within normal physiological concentrations, leptin stimulates the release of nitric oxide, thus promoting vasodilation. In contrast, hyperleptinemia leads to the opposite effect. Knutson and colleagues showed that leptin prevents the acetylcholine induced vasodilation mediated by nitric oxide both in vivo and in vitro under obese concentrations of leptin, but not by normal physiological concentrations of leptin (Knudson et al., 2005). Studies in leptin deficient mice provide conflicting findings as to whether leptin is protective or detrimental to vascular health. While leptin deficient mice on the background of low density lipoprotein receptor deficiency developed severe hyperlipidemia (Hasty et al., 2001), leptin deficient mice on an apolipoprotein-E deficient background fed an atherogenic diet were protected against the development of atherosclerotic lesions in the form of fibrous plaques (Chiba et al., 2008).
The relationship that both adiponectin and leptin have with vascular function is an example of the dichotomous role that adipocyte-derived secretory products have in maintaining metabolic homeostasis. Depending on the physiological state, adipokine actions can be extraordinarily beneficial or detrimental.
Skeletal Muscle Lipid Metabolism
Adiponectin
Adiponectin improves the systemic metabolic milieu by potentially enhancing skeletal muscle fatty acid oxidation (Fruebis et al., 2001). These early results were obtained with the truncated globular form of adiponectin. This form of the protein has been widely used for pharmacological studies, even though it is still controversial whether such a form of the protein exists in vivo as a bioactive protein or whether it is merely an unspecific degradation product generated during clotting. Also, it is not clear whether the pharmacological effects attributed to globular adiponectin in any way reflect the effects of the full-length version of adiponectin in vivo. As such, globular adiponectin activates AMPK in skeletal muscle, stimulating fatty acid oxidation via activation of p38 mitogen-activated kinase and PPARα (Yoon et al., 2006). By promoting fatty acid oxidation in skeletal muscle, globular adiponectin decreases intramuscular lipid accumulation, thereby preserving insulin sensitivity (Ceddia et al., 2005; Civitarese et al., 2006; Fruebis et al., 2001; Yamauchi et al., 2002). However, whether skeletal muscle is a bona fide target for adiponectin in vivo under normal physiological conditions has yet to be fully elucidated.
Leptin
Leptin receptors are abundantly expressed in human (Guerra et al., 2007) and rodent (Liu et al., 1997) skeletal muscle and are upregulated in response to atrophy (Chen et al., 2007), indicating that leptin stimulates skeletal muscle growth. Leptin increases skeletal muscle fatty acid oxidation and decreases triglyceride accumulation via activation of AMPK (Minokoshi et al., 2002). To test the effects of leptin on skeletal muscle lipid oxidation, Muoio et al measured [14C]oleate incorporation into CO2 and triacylglycerol in explants of mouse soleus muscle with insulin, leptin, or insulin plus leptin. While insulin increased decreased fatty acid oxidation and increased incorporation into TAG, incubation with leptin increased fatty acid oxidation by 42%, and decreased fatty acid incorporation in TAG by 35% (Muoio et al., 1997). When tissues were treated with both, leptin blunted the lipogenic effects of insulin by 50%. Thus, leptin promotes catabolic regulation of skeletal muscle lipid metabolism and the utilization of fatty acids to generate energy and decrease intramuscular lipid accumulation.
Ectopic Lipid Accumulation in the Pancreatic β-cell
Adiponectin
Subsequent to the cloning of the adiponectin receptors adipoR1 and adipoR2, (Yamauchi et al., 2003), Kharroubi and colleagues showed that both human and rodent pancreatic β-cells express mRNAs for the adiponectin receptors, adipoR1 and adipoR2, at high levels (Kharroubi et al., 2003). Further, the authors noted the expression levels were comparable to liver. Just as the liver is sensitive to lipid overload, pancreatic β-cells are susceptible to the lipotoxic effects of overnutrition (Holland et al., 2011; Park et al., 2011; Rakatzi et al., 2004) (Figure 3). We (Holland et al., 2011) and others (Park et al., 2011; Rakatzi et al., 2004) have shown that adiponectin is capable of protecting the β-cell against this toxicity. In fact, adiponectin prevents obesity and type I diabetes-induced decreases in β-cell mass (Holland et al., 2011; Kim et al., 2007) and prevents the attenuation of glucose stimulated insulin secretion in β-cells challenged with lipid overload (Rakatzi et al., 2004). By generating a mouse model of inducible β-cell apoptosis in combination with transgenic overproduction of adiponectin, we have demonstrated that adiponectin protects β-cells against caspase-8-mediated apoptosis (Holland et al., 2011). Additionally, adiponectin prevents lipid and cytokine-induced apoptosis in INS-1 β-cells (Holland et al., 2011; Rakatzi et al., 2004). Interestingly, Adiponectin receptor expression is increased by β-cell exposure to oleate (Kharroubi et al., 2003), suggesting that lipids facilitate the anti-lipotoxic response of adiponectin. We recently demonstrated that adiponectin is essential to maintain lipid homeostasis under insulinopenic conditions (Ye et al., 2014). Upon near complete elimination of insulin, adiponectin is critical for insulin signaling, endocytosis, and lipid uptake in subcutaneous white adipose tissue. In the absence of both insulin and adiponectin, severe lipoatrophy and hyperlipidemia ensue and lead to lethality. Transgenic elevation of adiponectin rescues systemic lipid metabolism, even in the near absence of insulin. Adiponectin can also reduce local ceramide levels, thereby mitigating lipotoxicity in pancreatic islets, thereby promoting reconstitution of β-cell mass, eventually reinstating glycemic control. The key pathways activated by adiponectin that lead to the reconstitution of β cell mass are governed by HNF4α and PPARα, suggesting that transcriptional programs that lead to a local improvement in lipid metabolism are key pathways activated by adiponectin to achieve recovery.
Figure 3. A working model of adipocyte/islet communication.
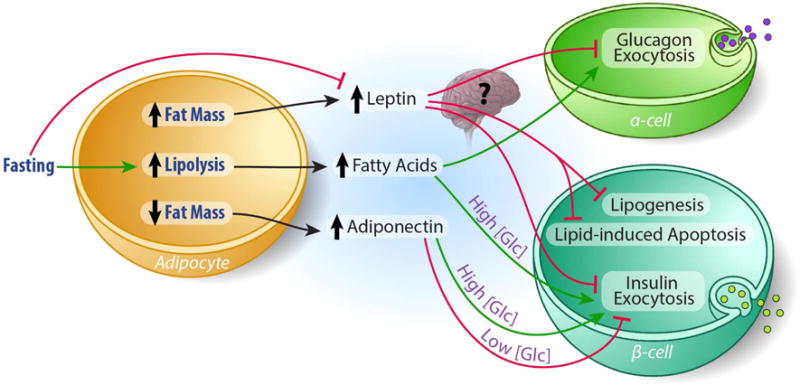
Leptin inhibits insulin and glucagon secretion from β- and α-cells, respectively. Whether this is a direct action of leptin or centrally mediated has not been entirely elucidated. The effects of adiponectin on β -cell insulin secretion may depend on circulating glucose concentration, with adiponectin inhibiting insulin secretion at low glucose concentrations and stimulating insulin secretion at high glucose concentrations. Fatty acids enhance glucose stimulated insulin secretion at high glucose conditions. Under fasting conditions, fatty acids may increase glucagon secretion indirectly by limiting somatostatin’s inhibitory effect on α-cell exocytosis and fasting-induced decreases in leptin secretion may act to relieve leptin’s inhibitory effect on glucagon secretion under low blood glucose concentrations.
Leptin
Similar to the protective role of adiponectin, leptin has also been shown to protect β-cells from lipotoxicity in several rodent and in vitro models. Adenovirus-mediated hyperleptinemia in prevents streptozotocin-induced islet cell apoptosis and preserves β-cell mass by blocking lipogenesis in diabetic ZDF rats (Lee et al., 2007). Additionally, the Unger group has shown that leptin reduces blood glucose and improves hyperlipidemia in insulin-deficient, lean mice by inhibiting glucagon production by the α-cell (Wang et al., 2010).
Lipid Metabolism in the Kidney
Renal lipotoxicity is detrimental to kidney health and function (Moorhead et al., 1982). As a result of altered expression of genes that regulate fatty acid oxidation, diabetic nephropathy and chronic kidney disease result in renal lipid accumulation (Bobulescu, 2010; Kang et al., 2015; Simon and Hertig, 2015). Increasing fatty acid oxidation, genetically or pharmacologically, through local overexpression of PGC1α, reduces folate-induced fibrosis and tissue lipid accumulation (Kang et al., 2015). The overall substrate utilization switch to lipid oxidation in diabetes leads to increased mitochondrial generation of reactive oxygen species (ROS) (Rosca et al., 2012). As a result, tubule sodium, ammonia, and albumin transport are reduced, leading to decreased pH buffering capacity in blood and urine (Bobulescu et al., 2009; Erkan et al., 2007; Liu et al., 2012). Lipid metabolism in the kidney is thus a delicate balance, which is, at least in part, regulated via adipocyte-secreted factors.
Adiponectin
Adiponectin is renoprotective in multiple mouse models and clinical studies suggest that adiponectin is beneficial to kidney disease outcomes (Sweiss and Sharma, 2014). Adiponectin receptors are ubiquitously expressed in the kidney and activate AMPK upon stimulation (Tanabe et al., 2015). In fact, AMPK activation limits the effects of high fat diet-induced renal dysfunction (Decleves et al., 2011) and chronic inactivation of AMPK leads to CKD (Ix and Sharma, 2010; Sweiss and Sharma, 2014). Accordingly, adiponectin knockout mice demonstrate elements of CKD (Sharma et al., 2008) Interestingly, Fenofibrate, a PPARα agonist, improves kidney function partially through adiponectin signaling (Christou and Kiortsis, 2014).
Leptin
Leptin appears to play a role in renal triglyceride accumulation. In a mouse model of lipoatrophic diabetes, wherein leptin is low due to highly limited fat mass, restoration of circulating leptin normalized systemic glucose levels and reduced lipid accumulation and proteinuria (Suganami et al., 2005). Leptin receptor deficient db/db mice exhibit diabetic nephropathy with increased renal triglyceride accumulation through an SREBP-1 and -2 dependent mechanism (Wang et al., 2005). Leptin-deficient ob/ob mice also present with triglyceride accumulation in the kidney. Hyperleptinemic mice, however, have decreased renal triglycerides compared to ob/ob mice, suffer however from increased ROS and endoplasmic reticulum (ER) stress.
Fatty Acids
The podocyte is the first cell-type that is damaged in the progression of diabetic nephropathy. When treated with palmitate, podocytes accumulate lipid droplets, increase ER stress and ROS, and show reduced insulin-mediated Akt phosphorylation. Fatty acid accumulation in the cortex reduces the expression and activity of sodium–hydrogen exchanger 3 (NHE3), which promotes proximal tubule sodium absorption (Bobulescu et al., 2009). Interestingly, while renal cortex triglyceride content was positively correlated with BMI, total ceramide content was inversely proportional, suggesting an obesity-induced shift in tubule fatty acid utilization (Bobulescu et al., 2014). In cultured podocytes, however, palmitate induced ceramide accumulation and insulin resistance is reversed by inhibiting ceramide synthesis (Lennon et al., 2009).
ADIPOCYTE COMMUNICATION MAINTAINS SYSTEMIC GLUCOSE HOMEOSTASIS
The dysregulation of glucose homeostasis in insulin resistance results from reduced glucose clearance coupled with increased hepatic glucose production. Treatments that enhance systemic insulin sensitivity have been shown to enhance glucose uptake by peripheral tissues, while decreasing hepatic glucose output (Pernicova and Korbonits, 2014). Healthy, fully functional adipose tissue is required for the maintenance of systemic insulin sensitivity. This is exemplified in individuals with lipodystrophy and in mice with loss of specific adipose tissue depots or complete ablation of adipose tissue. In the absence of adipose tissue, severe insulin resistance ensues (Gorden and Gavrilova, 2003; Moitra et al., 1998; Pajvani et al., 2005). Yet, adipose tissue excess can also lead to severe insulin resistance, resulting in a decrease in adipose tissue glucose clearance. This depression in adipose glucose clearance is a result of adipose tissue hypoxia, fibrosis, and the resultant inflammatory, lipotoxic response (Kim et al., 2007; Kusminski et al., 2012). The initial discovery of adiponectin (Scherer et al., 1995) and subsequent recognition of its potent actions on numerous tissues and cell types as an insulin sensitizing, anti-apoptotic, and anti-inflammatory peptide secreted by adipocytes (Berg et al., 2001; Combs et al., 2001; Combs et al., 2004; Holland et al., 2011), coupled with the discovery of leptin just one year prior (Zhang et al., 1994) and its insulin sensitizing actions (Chinookoswong et al., 1999; Pelleymounter et al., 1995; Shimomura et al., 1999), provided the diabetes and obesity fields with promising therapeutic targets for obesity-related hyperglycemia. (Okada-Iwabu et al., 2013; Ziemke and Mantzoros, 2010).
Adipocyte Glucose Metabolism
Adiponectin
The insulin sensitizing action of adiponectin is impressively conserved among mammals, extending from mice and humans (Turer and Scherer, 2012) to the yellow-bellied marmot and dolphin (Florant et al., 2004; Venn-Watson et al., 2013). While the insulin sensitizing effects are a combination of the vast metabolic effects exerted on multiple tissues, adiponectin also acts locally on the adipocyte to increase glucose uptake, contributing to euglycemia and promotion of adipose tissue expansion. Accordingly, lentiviral overexpression of adiponectin in cultured 3T3-L1 cells increases glucose uptake (Fu et al., 2005). Adiponectin increases glucose uptake at submaximal insulin concentrations independent of insulin signaling in primary rat adipocytes (Wu et al., 2003). While tyrosine phosphorylation of insulin receptor and AKT phosphorylation was unaffected, adiponectin treatment increased phosphorylation and catalytic activity of AMPK. Pharmacological inhibition of AMPK abolished the increase in glucose uptake, demonstrating that AMPK mediates adiponectin stimulated glucose update in the adipocyte (Wu et al., 2003).
We have recently shown that increasing insulin signaling in the mature adipocyte enhances systemic insulin sensitivity (Morley et al., 2015) which may be mediated by an increase in adiponectin secretion. We assessed the role of enhanced insulin signaling in the adipocyte, using an inducible model of adipocyte-specific deletion of the PTEN (phosphatase and tensin homologue) gene, an inhibitor of the insulin signal transduction cascade. Although these transgenic mice gained more weight in response to high fat diet feeding, they had reduced adipose tissue inflammation and elevated circulating adiponectin levels. As such, these improvements lead to reduced hepatic steatosis and enhanced hepatic insulin sensitivity (Morley et al., 2015). The results of this study initially appear to contradict the observation that mice with adipose-tissue specific deletion of the insulin receptor (FIRKO) are leaner and are protected against age-related and diet induced glucose intolerance (Bluher et al., 2002). However, combined, these studies suggest that insulin signaling is required for the development of excess adiposity, which can lead to insulin insensitivity and glucose intolerance. However, if insulin signaling can be maintained, so can glucose tolerance.
The rise in circulating adiponectin in our mouse model of enhanced insulin signaling, suggests that adiponectin secretion from the adipocyte is regulated by local insulin sensitivity. These observations offer us an opportunity to appreciate a more nuanced view of insulin signaling within the adipocyte. While our study suggests that selective enhancement in insulin sensitivity at the level of the adipocyte can improve whole-body glucose homeostasis when induced in the mature adipocyte, it is possible that specific components of the insulin signaling cascade in the adipocyte, rather than the entire receptor mediated signal itself have the potential to modulate adiponectin secretion and systemic insulin sensitivity.
Leptin
Leptin receptors are abundantly expressed on the adipocyte. Early studies demonstrated that leptin suppresses insulin-stimulated glucose uptake in cultured primary rat adipocytes (Muller et al., 1997). Prolonged exposure of isolated rat adipocytes to leptin prevents insulin binding and 20 minutes of exposure to high (80–800 ng/mL) concentrations of leptin increases the expression of SOCS3 (an inhibitor of insulin receptor auto-phosphorylation), suggesting that leptin inhibits insulin receptor activation (Perez et al., 2004). Because adipocytes receive sympathetic innervation and respond to insulin, it has been hypothesized that leptin may also regulate adipocyte glucose metabolism indirectly through neuronal signaling.
Fatty Acids
Lipids secreted from the adipocyte serve as signaling molecules that regulate energy metabolism. As such, the variations in the composition of fatty acids released from the adipocyte are vast, and so are their distinct actions on target tissues. We previously reviewed the role of ceramides in the control of insulin sensitivity. Yet, additional classes of lipids appear to also control metabolic flux. Using Glut4 overexpressing mice that present with elevated lipogenesis and increased glucose tolerance, despite being obese and hyperlipidemic, Kahn and colleagues lab has identified a class of endogenous lipids, fatty acid esters of hydroxy fatty acids (termed ‘FAHFAs’), derived from adipose tissue that potently increase adipose tissue insulin sensitivity (Yore et al., 2014). In humans, serum, palmitic-acid-9-hydroxy-stearic-acid (PAHSA), a specific FAHFA isomer, concentration is positively correlated with insulin sensitivity and, in mice, fasting induces an increase in PAHSA accumulation in WAT. PAHSAs exert their action on adipocytes by binding to the G-Protein coupled receptor GPR120, increasing GLUT4 translocation, and thus, glucose uptake into the adipocyte. Treatment of diabetic mice with these lipids also stimulated GLP-1 and insulin secretion, leading to improved glucose tolerance and a reduction of adipose tissue inflammation (Yore et al., 2014). The observed enhancement in insulin secretion indicates that PAHSAs may also directly regulate β-cell insulin exocytosis. As suspected, PAHSAs enhanced glucose stimulated insulin secretion in isolated human islets at high (20mM) but not low (2.5mM) glucose conditions (Yore et al., 2014). The stimulatory, yet conditional, effect this new lipid class has on β-cell insulin exocytosis reflects the elegant precision with which adipocyte secreted factors exert their actions.
Secretion of Glucoregulatory Hormones from the Pancreatic Islet
Glucose homeostasis is tightly regulated by insulin and glucagon, secreted by pancreatic β-cells and α-cells, respectively. Pancreatic α-cells and β-cells are sensitive to glucose induced changes in the ATP:ADP ratio that directly manipulate membrane polarity to differentially regulate the exocytosis of glucagon and insulin, respectively. Glucose stimulated increases in cellular ATP inhibit the release of glucagon, while stimulating the release of insulin. In both the α- and β-cell, the increase in ATP:ADP ratio blocks KATP channels leading to cell depolarization. This depolarization differentially regulates Ca2+ flux into the α- and β- cells. In α-cells, depolarization resulting from closure of the KATP channels reduces Ca2+ entry through T-type calcium channels, while glucose induced increases in ATP activate Ca2+ sequestration in the sarcoplasmic reticulum through Ca2+ATPase (Barg et al., 2000; Gopel et al., 2000). In contrast, β-cells respond to depolarization with an increase in Ca++ influx, through voltage dependent Ca++ channels, that stimulates insulin secretion. Although the direct role of glucose in stimulating β-cell insulin secretion while inhibiting α-cell glucagon secretion is fundamental to the opposing secretory function these two cell-types, recent studies have demonstrated that some adipokines also regulate the secretion of these glucoregulatory hormones.
β-cell Insulin Secretion
Leptin
Whether β-cells express leptin receptors is still in question and currently debated. While previous studies show that the β-cell expresses leptin receptors (Seufert et al., 1999), Soedling and colleagues recently reported that leptin receptor mRNA expression in isolated β-cells was at or below the level of detection and, using a newly developed Cre to specifically target Leptin receptor deletion to the β-cell, the authors reported no metabolic abnormalities (Soedling et al., 2015). Contrary to these findings, previous studies in isolated pancreatic islets from rodents and humans (Emilsson et al., 1997; Fehmann et al., 1997; Kieffer et al., 1997) showed that the pancreatic β-cell is a major target of leptin. Further, studies in mice with targeted β-cell ablation of the Leptin receptor (ObR) gene have confirmed this in vivo (Covey et al., 2006; Morioka et al., 2007). Leptin inhibits insulin gene expression and insulin secretion in human pancreatic islets (Seufert et al., 1999) and the immortalized INS-1 β-cell line (Laubner et al., 2005). The inhibition of insulin secretion is in response to the leptin induced increase in cell surface expression and K+ conductance through KATP channels, which is achieved through leptin induced activation of AMPK and subsequent PKA activity (Chen et al., 2013; Park et al., 2013). This increased K+ leak from the cell decreases Ca++ flux into the cell and insulin secretion. Given the more recent findings of Soedling and colleagues, it is possible that the effects of leptin on insulin secretion are mediated by leptin signaling in the brain (Fujikawa et al., 2013) (Figure 3).
Adiponectin
Similar to leptin, the insulin secretory response to adiponectin is not entirely elucidated. Adiponectin treatment of isolated mouse islets exposed to 5.6mM glucose stimulated insulin secretion. In vivo experiments support this stimulatory role of adiponectin, as intravenous injection of adiponectin increased insulin secretion in mice (Okamoto et al., 2008). In contrast, Winzel et al found that adiponectin administration on islets from normal mice had no effect on insulin secretion. Yet, in islets from obese mice, adiponectin depressed insulin release at basal glucose concentrations and potentiated insulin release at stimulatory glucose (16.7mM) concentrations (Winzell et al., 2004). Given the systemic insulin sensitizing effects of adiponectin, this dichotomous action of adiponectin on pancreatic β-cells further touts this adipokine for its plasticity in actions on a single cell-type, based on the glycemic environment. Although, additional studies are required to fully understand the role of adiponectin in the control of insulin release, we can hypothesize that the increased responsiveness of islets from obese mice may result from the obesity induced differences in in vivo adiponectin exposure.
Fatty Acids
Non-esterified fatty acid (NEFA) induced β-cell insulin secretion has been well recognized for decades (Malaisse and Malaisse-Lagae, 1968; Opara et al., 1992; Vara et al., 1988; Warnotte et al., 1994), providing a link between the hyperlipidemia and hyperinsulinemia seen in T2DM. Itoh and colleagues showed that NEFAs stimulate insulin secretion from the β-cell through activation of the G-protein-coupled receptor, GPR40, inducing a rise in cytosolic Ca2+ concentrations, thereby stimulating insulin exocytosis (Itoh et al., 2003).
α-cell Glucagon Secretion
Similar to dysregulated insulin secretion in the diabetic β-cell, diabetic α-cells are hypersecretory and do not appropriately respond to elevated blood glucose levels with a decrease in glucagon secretion (Muller et al., 1970). Because glucagon potentiates hepatic glucose production, while inhibiting adipose tissue glucose clearance, it is increasingly appreciated as an important target to treat T2DM (Dimitriadis et al., 1985; Pernicova and Korbonits, 2014).
Leptin
Glucagon-secreting α-cells express leptin receptors and in vitro studies show that leptin inhibits glucagon secretion (Soedling et al., 2015) (Tuduri et al., 2009). Tudori and colleagues have shown that leptin hyperpolarizes α-cells and prevents the hypoglycemia induced glucagon secretion (Tuduri et al., 2009). Leptin also decreases expression of the gene encoding glucagon in immortalized α-cells (αTC1-9) (Marroqui et al., 2011), demonstrating that leptin regulates pancreatic islet function at the level of both gene transcription and secretory function. Mouse models that are deficient in leptin signaling develop hyperglucagonemia (Dunbar and Walsh, 1980; Stearns and Benzo, 1978), suggesting that leptin chronically suppresses glucagon secretion. However, the impact of leptin on glucagon secretion in vivo has yet to be fully understood. Employing their newly developed Cre to specifically target Leptin receptor deletion to the α-cell, Soedling and colleagues showed that leptin receptor signaling in the α-cell has minimal effect on glucagon secretion under hypoglycemic conditions. Thus, similar to the β-cell, the direct role of leptin on α-cell function in vivo requires further investigation, and leptin may exert its glucagon lowering effects indirectly through a centrally mediated mechanism (Figure 3).
Adiponectin
The impact of adiponectin signaling in the α-cell has yet to be explored. Given the insulin sensitizing actions of adiponectin on multiple cell types, along with its protective capacity in the β-cell, the role of adiponectin in the α-cell provides for an exciting potential target to treat the hyperglycemia associated with type 1 and type 2 diabetes.
Fatty Acids
Recent studies suggest that short-term exposure to fatty acids stimulates glucagon exocytosis (Bollheimer et al., 2004; Hong et al., 2007; Olofsson et al., 2004). Similar to the stimulatory effect of low glucose concentrations, palmitate has been shown to increase Ca2+ mediated glucagon release by opening L-type Ca2+ channels. Simultaneously, NEFA may act to increase glucagon secretion by limiting somatostatin’s inhibitory effect on α-cell exocytosis (Olofsson et al., 2004). Interestingly, this NEFA induced glucagon release provides a mechanism by which fasting- induced adipose tissue lipolysis can encourage glucagon release.
Hepatic Glucose Metabolism
The hyperglycemia associated with insulin resistance results from reduced glucose clearance coupled with increased hepatic glucose production (gluconeogenesis). Hence, inhibition of hepatic gluconeogenesis appears to provide a promising intervention to treat insulin resistance and related metabolic disorders (Lee et al., 2011; Wang et al., 2015). Accordingly, there are a number of studies evaluating the role of adipokines in hepatic glucose output in type 1 and type 2 diabetics.
Adiponectin
Adiponectin decreases hepatic gluconeogenesis (Combs et al., 2001). However, the mechanism remains in question. Recombinant adiponectin treatment decreases mRNA expression of the rate-limiting gluconeogenic enzymes phosphoenolpyruvate kinase (PEPCK) and glucose-6-phosphotase (G6Pase) through the AMPK signaling pathway (Yamauchi et al., 2002). We also hypothesized that our initial observation that the blood glucose lowering actions of adiponectin administration was mechanistically linked to AMPK mediated inhibition of gluconeogenesis (Combs et al., 2001). However, recent studies have revealed that adiponectin activation of AMPK is much lower in hepatocytes than in other tissues (Miller et al., 2011).
Moreover, ablation of LKB1 and AMPK in primary mouse hepatocytes did not affect adiponectin induced reduction of gluconeogenic gene expression or glucose output (Miller et al., 2011). In vivo deletion of LKB1, an upstream activator or AMPK, partially prevented the adiponectin induced decrease in serum glucose (Miller et al., 2011). Yet, in line with the in vitro studies, adiponectin decreased gluconeogenic gene expression and hepatic glucose production (Miller et al., 2011). Together these studies suggest that adiponectin depresses hepatic glucose production independent of AMPK signaling (Figure 2).
Skeletal Muscle Glucose Metabolism
As the chief site of insulin-stimulated glucose uptake, skeletal muscle plays an essential role in maintaining systemic glucose homeostasis. Adipokines and fatty acids released from adipose tissue both affect skeletal muscle insulin sensitivity. Both leptin (Miyamoto et al., 2012; Shimomura et al., 1999) and adiponectin (Yamauchi et al., 2001), attenuate skeletal muscle insulin resistance and hyperglycemia. Ectopic accumulation of lipid in skeletal muscle, resulting from elevated adipose tissue lipolysis or depressed adipose tissue lipid clearance from the circulation, result in skeletal muscle insulin resistance and hyperglycemia. Thus, adipose tissue plays an essential role in the maintenance of skeletal muscle glucose clearance.
Leptin
Leptin administration in ob/ob mice improves insulin sensitivity in peripheral tissues independent of effects on food intake or body weight (Levin et al., 1996; Mistry et al., 2004). In the absence of insulin, Leptin acutely increases glucose uptake in cultured L6 muscle cells, but has no long term effect. (Bates et al., 2002). In streptozotocin (STZ) induced type 1 diabetic mice hyperleptinemia, induced via leptin encoding adenovirus, ameliorates hyperglycemia, ketosis, and weight loss (Yu et al., 2008). Leptin overexpression also increased skeletal muscle PI3K, IRS-1, and IGF-1 receptor phosphorylation, suggesting that leptin acts as an insulin-mimetic in skeletal muscle (Yu et al., 2008). Further supporting the insulin mimetic hypothesis, leptin activates the insulin receptor substrate-2 (IRS-2) and janus kinase-2 (JAK-2) signaling molecules to initiate signaling through the phosphatidylinositol 3-kinase (PI3K) signaling pathway which induces translocation of Glut4 to the cell surface and stimulates glucose uptake in C2C12 myotubes (Kellerer et al., 1997).
Adiponectin
While the translocation of GLUT4 vesicles to the cell surface of the myocyte is dependent on insulin signaling, the expression of the GLUT4 gene is regulated by multiple factors, two of which are the phosphorylation/activation of AMPK and increased cytosolic Ca2+. AMPK increases the expression of GLUT4 by phosphorylating its transcriptional repressor and allowing for its nuclear export (McGee et al., 2008). Elevated cytosolic Ca2+ activates Ca2+/calmodulin-dependent protein kinase II in skeletal muscle which also increases the expression of GLUT4 (Chin, 2005). Accordingly, adiponectin stimulates GLUT4 mediated glucose uptake in isolated skeletal muscle and cultured myocytes (Tomas et al., 2002; Yamauchi et al., 2002) via phosphorylation/activation of AMPK and by increasing cytosolic Ca2+. Furthermore, adipoR1 in skeletal muscle stimulates extracellular Ca2+ influx, increasing cytosolic Ca2+ (Iwabu et al., 2010). Ceddia and colleagues (2005) showed that adiponectin increases glucose uptake in L6 myotubes through enhanced GLUT4 translocation without affecting the affinity of the transporter. As expected, AMPK signaling phosphorylates glycogen synthase, inactivating this limiting enzyme in glycogenesis and decreasing the rate of glycogen synthesis (source). In line with a decrease in glucose flux toward glycogen, no change in glucose oxidation, and enhanced glucose uptake through GLUT4, adiponectin treatment enhanced insulin stimulated lactate production (Ceddia et al., 2005).
Glucose Metabolism in the Kidney
Diabetic nephropathy and obesity-associated albuminuria suggest that the kidney is a primary target of hyperglycemia. However, the kidney as a whole is a net producer of glucose, which by some estimates, accounts for 20% of endogenous glucose release under basal conditions (Cano, 2002). Renal gluconeogenesis is specific to tubular epithelial cells of the proximal tubule and its regulation is insulin dependent. An imbalance of PEPCK-mediated glucose production and glycolysis in the distal nephron can thus influence systemic glucose levels (Cano, 2002; Gerich et al., 2001). An increase in gluconeogenesis, and hence, PEPCK activity, generates additional HCO3− exported by the proximal tubule to raise blood pH during metabolic acidosis (Curthoys and Moe, 2014). Deletion of the insulin receptor in tubule epithelium induces systemic hyperglycemia in the mouse (Tiwari et al., 2013). Increased glucose uptake under high glucose and high insulin conditions result in increased glycogen deposition (Gerich et al., 2001) and a reduction in fatty acid oxidation in the cortex (Meyer et al., 1997). A similar outcome occurs in glomerular podocytes, wherein hyperglycemia incudes ROS-mediated apoptosis, a first step in the progression of diabetic nephropathy (Susztak et al., 2006). High fatty acid concentrations also cause insulin resistance in cultured podocytes (Lennon et al., 2009). As in other tissues, adipokines that impact insulin sensitivity and glucose uptake likely impact renal health and function. Adiponectin, leptin, and fatty acids have all demonstrated relationships with glomerular filtration commiserate with the metabolic syndrome, but whether these adipokines act directly to regulate glucose metabolism in the kidney is currently being explored (Briffa et al., 2013).
ADIPOCYTE COMMUNICATION REGULATES ENERGY METABOLISM
In his 1932 publication, Max Kleiber provided the energetics field with the basis for the notion that an animal’s metabolic rate is independent of body size (Kleiber, 1932). Since Kleiber’s initial hypothesis, countless genes, transcription factors, and tissue-derived signaling molecules which regulate energy homeostasis have been identified. Nearly a century later, with obesity and the related metabolic disorders rising at an alarming rate, we continue to discover new pathways that regulate energy homeostasis, of which adipokines are at the forefront. In fact, more than 60 years ago, before the term adipokine was coined, it was proposed that that signals secreted in proportion to adipose tissue stores regulate energy intake and energy expenditure (Kennedy, 1953). Two years later, Mayer (Mayer, 1955) asserted that energy homeostasis was regulated by ‘glucostatic’ and ‘lipostatic’ signals, the latter of which mirrored long-term energy stores. We now embrace a feedback model, originally hypothesized by Bray (Bray, 1991) in which adipose tissue, the gastrointestinal tract, and other peripheral tissues provide endocrine and neural signals to the brain to regulate of energy balance. Building upon Kleiber’s and Kennedy’s assertions, Bray’s model is constantly evolving as we gain new insights on how the adipocyte communicates its nutritional status to the central nervous system and peripheral tissues to regulate energy homeostasis.
Leptin
While multiple components comprise the energy balance equation (ie: adaptive thermogenesis, excretory losses, activity, etc.), it can be simplified as: Total body energy = energy in-energy out. Leptin signals the energetic status of adipocyte lipid stores to the hypothalamus to regulate both components of this equation: food intake and energy expenditure (Friedman and Halaas, 1998) (Halaas et al., 1997; Zhang et al., 1994). Leptin increases sympathetic nervous system (SNS) tone (Satoh et al., 1999). Through its interaction with the melanocortin system, leptin increases gene expression (Harris et al., 2001) and secretion of (Ortiga-Carvalho et al., 2002; Seoane et al., 2000) thyrotropin-releasing hormone to increase thyroid hormone signaling and, thus, energy expenditure. This role of leptin to increase energy expenditure is so dominant that Leptin-deficient ob/ob mice quickly become obese, despite pair feeding with control littermates (Halaas et al., 1995). In fact, leptin prevents the decrease in basal metabolic rate induced by calorie restriction (Ahima et al., 1996; Doring et al., 1998), while low circulating leptin, during times of negative energy balance, serves as a signal to encourage a state of torpor (Gavrilova et al., 1999; Trayhurn et al., 1977) as a means of conserving energy. As such, leptin signals the energetic status of adipocyte lipid stores to the brain and the intensity of this signal mirrors that of circulating leptin concentrations (Friedman and Halaas, 1998).
In obesity, leptin no longer adequately regulates energy expenditure. Obese individuals with low basal metabolic rates, despite high circulating leptin concentrations are commonly referred to as “leptin resistant”. And with weight loss leptin drops, yet leptin sensitivity is low. Leptin treatment in clinical trials partially restores the reduction in basal metabolic rate and circulating thyroid hormones associated with fat- loss in patients maintaining a reduction in body weight following acute weight loss (Rosenbaum et al., 2005; Rosenbaum et al., 2002).
Adiponectin
Adiponectin exerts potent effects on the brain to regulate energy expenditure, independent of AMPK signaling (Park et al., 2011; Qi et al., 2004). Qi and colleagues showed that intravenous administration of adiponectin to leptin deficient ob/ob mice increases thermogenesis and weight loss (Qi et al., 2004). Further, when co-administered, adiponectin enhanced the thermogenic effects of leptin (Qi et al., 2004). Intracerebroventricular infusion of adiponectin, increases energy expenditure by activating hypothalamic leptin and insulin signaling pathways, independent of AMPK signaling (Park et al., 2011). Similarly, our studies linking adiponectin signaling to the energetic effects of FGF21 administration demonstrate that adiponectin is necessary for FGF21-induced enhancement in energy expenditure (Holland et al., 2013). Together, these studies shed light on the collaborative nature of adipokine signaling. Not only do they provide communication between tissues, we now have evidence to suggest that these signals interact with each other to regulate energy homeostasis.
CONCLUDING REMARKS
Metabolism research has made tremendous progress over the last several decades in establishing the adipocyte as a central rheostat in the regulation of systemic nutrient and energy homeostasis. Reflecting upon this progress, it is astounding to imagine that adipocyte function was once thought to be merely that of lipid storage with no other physiologically meaningful utility. A mouse completely devoid of adipose tissue or a human with lipodystrophy are equally insulin resistant and vulnerable to the ectopic lipid deposition as seen in obesity. Yet, in both extremes, treatment with adipokines (i.e.: leptin or adiponectin) can ameliorate these maladies. Through the sophisticated precision with which adipocyte secreted factors exert their actions, the adipocyte reaches far beyond a simple on/off switch in maintaining systemic metabolic homeostasis. Operating at multiple levels of control, the adipocyte communicates with organ systems to precisely adjust gene expression, glucoregulatory hormone exocytosis, enzymatic action, and nutrient flux to equally match the metabolic demands of positive or negative energy balance. The identification of these mechanisms has great potential to identify novel targets for the treatment of diabetes and related metabolic disorders.
Acknowledgments
The authors were supported by the National Institutes of Health (grants R01-DK55758, R01-DK099110 and P01-DK088761 to P.E.S.) as well as a grant from the Cancer Prevention and Research Institute of Texas (CPRIT RP140412). J.H.S was supported by the National Institutes of Health (grants T32-DK007307 and F32-DK107058). J.M.R. was supported by the American Heart Association (12SDG12050287). Figures were illustrated by Richard Howdy, certified medical illustrator, at Visually Medical (VisuallyMedical.com).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
J.H.S., J.M.R., and P.E.S. wrote the manuscript. J.H.S. and P.E.S edited the manuscript.
Stern et al. review the central role of the adipocyte in the maintenance of metabolic homeostasis, highlighting the role of adiponectin, leptin, and fatty acids in meeting the metabolic demands of a positive or negative energy balance to regulate systemic nutrient and energy utilization.
References
- Adams AC, Yang C, Coskun T, Cheng CC, Gimeno RE, Luo Y, Kharitonenkov A. The breadth of FGF21’s metabolic actions are governed by FGFR1 in adipose tissue. Molecular metabolism. 2012;2:31–37. doi: 10.1016/j.molmet.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- Awazawa M, Ueki K, Inabe K, Yamauchi T, Kaneko K, Okazaki Y, Bardeesy N, Ohnishi S, Nagai R, Kadowaki T. Adiponectin suppresses hepatic SREBP1c expression in an AdipoR1/LKB1/AMPK dependent pathway. Biochemical and biophysical research communications. 2009;382:51–56. doi: 10.1016/j.bbrc.2009.02.131. [DOI] [PubMed] [Google Scholar]
- Baker AR, Silva NF, Quinn DW, Harte AL, Pagano D, Bonser RS, Kumar S, McTernan PG. Human epicardial adipose tissue expresses a pathogenic profile of adipocytokines in patients with cardiovascular disease. Cardiovasc Diabetol. 2006;5:1. doi: 10.1186/1475-2840-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg S, Galvanovskis J, Gopel SO, Rorsman P, Eliasson L. Tight coupling between electrical activity and exocytosis in mouse glucagon-secreting alpha-cells. Diabetes. 2000;49:1500–1510. doi: 10.2337/diabetes.49.9.1500. [DOI] [PubMed] [Google Scholar]
- Barr VA, Malide D, Zarnowski MJ, Taylor SI, Cushman SW. Insulin stimulates both leptin secretion and production by rat white adipose tissue. Endocrinology. 1997;138:4463–4472. doi: 10.1210/endo.138.10.5451. [DOI] [PubMed] [Google Scholar]
- Bartness TJ, Kay Song C, Shi H, Bowers RR, Foster MT. Brain-adipose tissue cross talk. The Proceedings of the Nutrition Society. 2005;64:53–64. doi: 10.1079/pns2004409. [DOI] [PubMed] [Google Scholar]
- Bates SH, Gardiner JV, Jones RB, Bloom SR, Bailey CJ. Acute stimulation of glucose uptake by leptin in l6 muscle cells. Hormone and metabolic research = Hormon-und Stoffwechselforschung = Hormones et metabolisme. 2002;34:111–115. doi: 10.1055/s-2002-23192. [DOI] [PubMed] [Google Scholar]
- Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Digestive diseases. 2010;28:155–161. doi: 10.1159/000282080. [DOI] [PubMed] [Google Scholar]
- Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nature medicine. 2001;7:947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- Bikman BT, Summers SA. Ceramides as modulators of cellular and whole-body metabolism. The Journal of clinical investigation. 2011;121:4222–4230. doi: 10.1172/JCI57144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluher M, Michael MD, Peroni OD, Ueki K, Carter N, Kahn BB, Kahn CR. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Developmental cell. 2002;3:25–38. doi: 10.1016/s1534-5807(02)00199-5. [DOI] [PubMed] [Google Scholar]
- Bobulescu IA. Renal lipid metabolism and lipotoxicity. Current opinion in nephrology and hypertension. 2010;19:393–402. doi: 10.1097/MNH.0b013e32833aa4ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobulescu IA, Dubree M, Zhang J, McLeroy P, Moe OW. Reduction of renal triglyceride accumulation: effects on proximal tubule Na+/H+ exchange and urinary acidification. American journal of physiology. Renal physiology. 2009;297:F1419–1426. doi: 10.1152/ajprenal.00177.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobulescu IA, Lotan Y, Zhang J, Rosenthal TR, Rogers JT, Adams-Huet B, Sakhaee K, Moe OW. Triglycerides in the human kidney cortex: relationship with body size. PloS one. 2014;9:e101285. doi: 10.1371/journal.pone.0101285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden G, Chen X, Mozzoli M, Ryan I. Effect of fasting on serum leptin in normal human subjects. The Journal of clinical endocrinology and metabolism. 1996;81:3419–3423. doi: 10.1210/jcem.81.9.8784108. [DOI] [PubMed] [Google Scholar]
- Bollheimer LC, Landauer HC, Troll S, Schweimer J, Wrede CE, Scholmerich J, Buettner R. Stimulatory short-term effects of free fatty acids on glucagon secretion at low to normal glucose concentrations. Metabolism: clinical and experimental. 2004;53:1443–1448. doi: 10.1016/j.metabol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Bray GA. Obesity, a disorder of nutrient partitioning: the MONA LISA hypothesis. The Journal of nutrition. 1991;121:1146–1162. doi: 10.1093/jn/121.8.1146. [DOI] [PubMed] [Google Scholar]
- Briffa JF, McAinch AJ, Poronnik P, Hryciw DH. Adipokines as a link between obesity and chronic kidney disease. American journal of physiology. Renal physiology. 2013;305:F1629–1636. doi: 10.1152/ajprenal.00263.2013. [DOI] [PubMed] [Google Scholar]
- Cano N. Bench-to-bedside review: glucose production from the kidney. Critical care. 2002;6:317–321. doi: 10.1186/cc1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y. Angiogenesis modulates adipogenesis and obesity. The Journal of clinical investigation. 2007;117:2362–2368. doi: 10.1172/JCI32239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceddia RB, Somwar R, Maida A, Fang X, Bikopoulos G, Sweeney G. Globular adiponectin increases GLUT4 translocation and glucose uptake but reduces glycogen synthesis in rat skeletal muscle cells. Diabetologia. 2005;48:132–139. doi: 10.1007/s00125-004-1609-y. [DOI] [PubMed] [Google Scholar]
- Chen G, Liang G, Ou J, Goldstein JL, Brown MS. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11245–11250. doi: 10.1073/pnas.0404297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PC, Kryukova YN, Shyng SL. Leptin regulates KATP channel trafficking in pancreatic beta-cells by a signaling mechanism involving AMP-activated protein kinase (AMPK) and cAMP-dependent protein kinase (PKA) The Journal of biological chemistry. 2013;288:34098–34109. doi: 10.1074/jbc.M113.516880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YW, Gregory CM, Scarborough MT, Shi R, Walter GA, Vandenborne K. Transcriptional pathways associated with skeletal muscle disuse atrophy in humans. Physiological genomics. 2007;31:510–520. doi: 10.1152/physiolgenomics.00115.2006. [DOI] [PubMed] [Google Scholar]
- Chiba T, Shinozaki S, Nakazawa T, Kawakami A, Ai M, Kaneko E, Kitagawa M, Kondo K, Chait A, Shimokado K. Leptin deficiency suppresses progression of atherosclerosis in apoE-deficient mice. Atherosclerosis. 2008;196:68–75. doi: 10.1016/j.atherosclerosis.2007.01.040. [DOI] [PubMed] [Google Scholar]
- Chin ER. Role of Ca2+/calmodulin-dependent kinases in skeletal muscle plasticity. Journal of applied physiology. 2005;99:414–423. doi: 10.1152/japplphysiol.00015.2005. [DOI] [PubMed] [Google Scholar]
- Chinookoswong N, Wang JL, Shi ZQ. Leptin restores euglycemia and normalizes glucose turnover in insulin-deficient diabetes in the rat. Diabetes. 1999;48:1487–1492. doi: 10.2337/diabetes.48.7.1487. [DOI] [PubMed] [Google Scholar]
- Christou GA, Kiortsis DN. The role of adiponectin in renal physiology and development of albuminuria. The Journal of endocrinology. 2014;221:R49–61. doi: 10.1530/JOE-13-0578. [DOI] [PubMed] [Google Scholar]
- Civitarese AE, Ukropcova B, Carling S, Hulver M, DeFronzo RA, Mandarino L, Ravussin E, Smith SR. Role of adiponectin in human skeletal muscle bioenergetics. Cell metabolism. 2006;4:75–87. doi: 10.1016/j.cmet.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Yang G, Yu X, Soukas AA, Wolfish CS, Friedman JM, Li C. Induction of leptin receptor expression in the liver by leptin and food deprivation. The Journal of biological chemistry. 2005;280:10034–10039. doi: 10.1074/jbc.M413684200. [DOI] [PubMed] [Google Scholar]
- Combs TP, Berg AH, Obici S, Scherer PE, Rossetti L. Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. The Journal of clinical investigation. 2001;108:1875–1881. doi: 10.1172/JCI14120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs TP, Pajvani UB, Berg AH, Lin Y, Jelicks LA, Laplante M, Nawrocki AR, Rajala MW, Parlow AF, Cheeseboro L, et al. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology. 2004;145:367–383. doi: 10.1210/en.2003-1068. [DOI] [PubMed] [Google Scholar]
- Coskun T, Bina HA, Schneider MA, Dunbar JD, Hu CC, Chen Y, Moller DE, Kharitonenkov A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018–6027. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- Covey SD, Wideman RD, McDonald C, Unniappan S, Huynh F, Asadi A, Speck M, Webber T, Chua SC, Kieffer TJ. The pancreatic beta cell is a key site for mediating the effects of leptin on glucose homeostasis. Cell metabolism. 2006;4:291–302. doi: 10.1016/j.cmet.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Curthoys NP, Moe OW. Proximal tubule function and response to acidosis. Clinical journal of the American Society of Nephrology : CJASN. 2014;9:1627–1638. doi: 10.2215/CJN.10391012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decleves AE, Mathew AV, Cunard R, Sharma K. AMPK mediates the initiation of kidney disease induced by a high-fat diet. Journal of the American Society of Nephrology : JASN. 2011;22:1846–1855. doi: 10.1681/ASN.2011010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitriadis GD, Pehling GB, Gerich JE. Abnormal glucose modulation of islet A- and B-cell responses to arginine in non-insulin-dependent diabetes mellitus. Diabetes. 1985;34:541–547. doi: 10.2337/diab.34.6.541. [DOI] [PubMed] [Google Scholar]
- Doring H, Schwarzer K, Nuesslein-Hildesheim B, Schmidt I. Leptin selectively increases energy expenditure of food-restricted lean mice. International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity. 1998;22:83–88. doi: 10.1038/sj.ijo.0800547. [DOI] [PubMed] [Google Scholar]
- Dunbar JC, Walsh MF. Glucagon and insulin secretion by islets of lean and obese (ob/ob) mice. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 1980;12:39–40. doi: 10.1055/s-2007-996193. [DOI] [PubMed] [Google Scholar]
- Emilsson V, Liu YL, Cawthorne MA, Morton NM, Davenport M. Expression of the functional leptin receptor mRNA in pancreatic islets and direct inhibitory action of leptin on insulin secretion. Diabetes. 1997;46:313–316. doi: 10.2337/diab.46.2.313. [DOI] [PubMed] [Google Scholar]
- Erkan E, Devarajan P, Schwartz GJ. Mitochondria are the major targets in albumin-induced apoptosis in proximal tubule cells. Journal of the American Society of Nephrology : JASN. 2007;18:1199–1208. doi: 10.1681/ASN.2006040407. [DOI] [PubMed] [Google Scholar]
- Fehmann HC, Berghofer P, Brandhorst D, Brandhorst H, Hering B, Bretzel RG, Goke B. Leptin inhibition of insulin secretion from isolated human islets. Acta diabetologica. 1997;34:249–252. doi: 10.1007/s005920050083. [DOI] [PubMed] [Google Scholar]
- Florant GL, Porst H, Peiffer A, Hudachek SF, Pittman C, Summers SA, Rajala MW, Scherer PE. Fat-cell mass, serum leptin and adiponectin changes during weight gain and loss in yellow-bellied marmots (Marmota flaviventris) Journal of comparative physiology. B, Biochemical, systemic, and environmental physiology. 2004;174:633–639. doi: 10.1007/s00360-004-0454-0. [DOI] [PubMed] [Google Scholar]
- Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, Bihain BE, Lodish HF. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Luo N, Klein RL, Garvey WT. Adiponectin promotes adipocyte differentiation, insulin sensitivity, and lipid accumulation. Journal of lipid research. 2005;46:1369–1379. doi: 10.1194/jlr.M400373-JLR200. [DOI] [PubMed] [Google Scholar]
- Fujikawa T, Berglund ED, Patel VR, Ramadori G, Vianna CR, Vong L, Thorel F, Chera S, Herrera PL, Lowell BB, et al. Leptin engages a hypothalamic neurocircuitry to permit survival in the absence of insulin. Cell metabolism. 2013;18:431–444. doi: 10.1016/j.cmet.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilova O, Leon LR, Marcus-Samuels B, Mason MM, Castle AL, Refetoff S, Vinson C, Reitman ML. Torpor in mice is induced by both leptin-dependent and - independent mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:14623–14628. doi: 10.1073/pnas.96.25.14623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerling JJ, Boon MR, Kooijman S, Parlevliet ET, Havekes LM, Romijn JA, Meurs IM, Rensen PC. Sympathetic nervous system control of triglyceride metabolism: novel concepts derived from recent studies. Journal of lipid research. 2014;55:180–189. doi: 10.1194/jlr.R045013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerich JE, Meyer C, Woerle HJ, Stumvoll M. Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes care. 2001;24:382–391. doi: 10.2337/diacare.24.2.382. [DOI] [PubMed] [Google Scholar]
- Gopel SO, Kanno T, Barg S, Weng XG, Gromada J, Rorsman P. Regulation of glucagon release in mouse -cells by KATP channels and inactivation of TTX-sensitive Na+ channels. The Journal of physiology. 2000;528:509–520. doi: 10.1111/j.1469-7793.2000.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorden P, Gavrilova O. The clinical uses of leptin. Current opinion in pharmacology. 2003;3:655–659. doi: 10.1016/j.coph.2003.06.006. [DOI] [PubMed] [Google Scholar]
- Guerra B, Santana A, Fuentes T, Delgado-Guerra S, Cabrera-Socorro A, Dorado C, Calbet JA. Leptin receptors in human skeletal muscle. Journal of applied physiology. 2007;102:1786–1792. doi: 10.1152/japplphysiol.01313.2006. [DOI] [PubMed] [Google Scholar]
- Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8878–8883. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Halberg N, Henriksen M, Soderhamn N, Stallknecht B, Ploug T, Schjerling P, Dela F. Effect of intermittent fasting and refeeding on insulin action in healthy men. Journal of applied physiology. 2005;99:2128–2136. doi: 10.1152/japplphysiol.00683.2005. [DOI] [PubMed] [Google Scholar]
- Halberg N, Wernstedt-Asterholm I, Scherer PE. The adipocyte as an endocrine cell. Endocrinol Metab Clin North Am. 2008;37:753–768. x–xi. doi: 10.1016/j.ecl.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haluzik M, Colombo C, Gavrilova O, Chua S, Wolf N, Chen M, Stannard B, Dietz KR, Le Roith D, Reitman ML. Genetic background (C57BL/6J versus FVB/N) strongly influences the severity of diabetes and insulin resistance in ob/ob mice. Endocrinology. 2004;145:3258–3264. doi: 10.1210/en.2004-0219. [DOI] [PubMed] [Google Scholar]
- Hara K, Boutin P, Mori Y, Tobe K, Dina C, Yasuda K, Yamauchi T, Otabe S, Okada T, Eto K, et al. Genetic variation in the gene encoding adiponectin is associated with an increased risk of type 2 diabetes in the Japanese population. Diabetes. 2002;51:536–540. doi: 10.2337/diabetes.51.2.536. [DOI] [PubMed] [Google Scholar]
- Hardy OT, Perugini RA, Nicoloro SM, Gallagher-Dorval K, Puri V, Straubhaar J, Czech MP. Body mass index-independent inflammation in omental adipose tissue associated with insulin resistance in morbid obesity. Surg Obes Relat Dis. 2011;7:60–67. doi: 10.1016/j.soard.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M, Aschkenasi C, Elias CF, Chandrankunnel A, Nillni EA, Bjoorbaek C, Elmquist JK, Flier JS, Hollenberg AN. Transcriptional regulation of the thyrotropin-releasing hormone gene by leptin and melanocortin signaling. The Journal of clinical investigation. 2001;107:111–120. doi: 10.1172/JCI10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty AH, Shimano H, Osuga J, Namatame I, Takahashi A, Yahagi N, Perrey S, Iizuka Y, Tamura Y, Amemiya-Kudo M, et al. Severe hypercholesterolemia, hypertriglyceridemia, and atherosclerosis in mice lacking both leptin and the low density lipoprotein receptor. The Journal of biological chemistry. 2001;276:37402–37408. doi: 10.1074/jbc.M010176200. [DOI] [PubMed] [Google Scholar]
- Hebbard LW, Garlatti M, Young LJ, Cardiff RD, Oshima RG, Ranscht B. T-cadherin supports angiogenesis and adiponectin association with the vasculature in a mouse mammary tumor model. Cancer Res. 2008;68:1407–1416. doi: 10.1158/0008-5472.CAN-07-2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, Bauer SM, Wade M, Singhal E, Cheng CC, et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell metabolism. 2013;17:790–797. doi: 10.1016/j.cmet.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nature medicine. 2011;17:55–63. doi: 10.1038/nm.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Jeppesen PB, Nordentoft I, Hermansen K. Fatty acid-induced effect on glucagon secretion is mediated via fatty acid oxidation. Diabetes/metabolism research and reviews. 2007;23:202–210. doi: 10.1002/dmrr.663. [DOI] [PubMed] [Google Scholar]
- Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, Iwahashi H, Kuriyama H, Ouchi N, Maeda K, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000;20:1595–1599. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- Huang W, Dedousis N, Bandi A, Lopaschuk GD, O’Doherty RM. Liver triglyceride secretion and lipid oxidative metabolism are rapidly altered by leptin in vivo. Endocrinology. 2006;147:1480–1487. doi: 10.1210/en.2005-0731. [DOI] [PubMed] [Google Scholar]
- Huynh FK, Neumann UH, Wang Y, Rodrigues B, Kieffer TJ, Covey SD. A role for hepatic leptin signaling in lipid metabolism via altered very low density lipoprotein composition and liver lipase activity in mice. Hepatology. 2013;57:543–554. doi: 10.1002/hep.26043. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422:173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature. 2010;464:1313–1319. doi: 10.1038/nature08991. [DOI] [PubMed] [Google Scholar]
- Ix JH, Sharma K. Mechanisms linking obesity, chronic kidney disease, and fatty liver disease: the roles of fetuin-A, adiponectin, and AMPK. Journal of the American Society of Nephrology : JASN. 2010;21:406–412. doi: 10.1681/ASN.2009080820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javor ED, Ghany MG, Cochran EK, Oral EA, DePaoli AM, Premkumar A, Kleiner DE, Gorden P. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology. 2005;41:753–760. doi: 10.1002/hep.20672. [DOI] [PubMed] [Google Scholar]
- Kamada Y, Tamura S, Kiso S, Matsumoto H, Saji Y, Yoshida Y, Fukui K, Maeda N, Nishizawa H, Nagaretani H, et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology. 2003;125:1796–1807. doi: 10.1053/j.gastro.2003.08.029. [DOI] [PubMed] [Google Scholar]
- Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nature medicine. 2015;21:37–46. doi: 10.1038/nm.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellerer M, Koch M, Metzinger E, Mushack J, Capp E, Haring HU. Leptin activates PI-3 kinase in C2C12 myotubes via janus kinase-2 (JAK-2) and insulin receptor substrate-2 (IRS-2) dependent pathways. Diabetologia. 1997;40:1358–1362. doi: 10.1007/s001250050832. [DOI] [PubMed] [Google Scholar]
- Kennedy GC. The role of depot fat in the hypothalamic control of food intake in the rat. Proceedings of the Royal Society of London. Series B, Biological sciences. 1953;140:578–596. doi: 10.1098/rspb.1953.0009. [DOI] [PubMed] [Google Scholar]
- Kharitonenkov A, Larsen P. FGF21 reloaded: challenges of a rapidly growing field. Trends in endocrinology and metabolism: TEM. 2011;22:81–86. doi: 10.1016/j.tem.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, et al. FGF-21 as a novel metabolic regulator. The Journal of clinical investigation. 2005;115:1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharroubi I, Rasschaert J, Eizirik DL, Cnop M. Expression of adiponectin receptors in pancreatic beta cells. Biochemical and biophysical research communications. 2003;312:1118–1122. doi: 10.1016/j.bbrc.2003.11.042. [DOI] [PubMed] [Google Scholar]
- Kieffer TJ, Heller RS, Leech CA, Holz GG, Habener JF. Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic beta-cells. Diabetes. 1997;46:1087–1093. doi: 10.2337/diab.46.6.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. The Journal of clinical investigation. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiber M. Body size and metabolism. Hilgardia. 1932;6:315–353. [Google Scholar]
- Kloting N, Fasshauer M, Dietrich A, Kovacs P, Schon MR, Kern M, Stumvoll M, Bluher M. Insulin-sensitive obesity. American journal of physiology. Endocrinology and metabolism. 2010;299:E506–515. doi: 10.1152/ajpendo.00586.2009. [DOI] [PubMed] [Google Scholar]
- Knudson JD, Dincer UD, Zhang C, Swafford AN, Jr, Koshida R, Picchi A, Focardi M, Dick GM, Tune JD. Leptin receptors are expressed in coronary arteries, and hyperleptinemia causes significant coronary endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2005;289:H48–56. doi: 10.1152/ajpheart.01159.2004. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Ouchi N, Kihara S, Walsh K, Kumada M, Abe Y, Funahashi T, Matsuzawa Y. Selective suppression of endothelial cell apoptosis by the high molecular weight form of adiponectin. Circ Res. 2004;94:e27–31. doi: 10.1161/01.RES.0000119921.86460.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, Askew GR, Simcox JA, McClain DA, Li C, et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nature medicine. 2012;18:1539–1549. doi: 10.1038/nm.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landskroner-Eiger S, Qian B, Muise ES, Nawrocki AR, Berger JP, Fine EJ, Koba W, Deng Y, Pollard JW, Scherer PE. Proangiogenic contribution of adiponectin toward mammary tumor growth in vivo. Clin Cancer Res. 2009;15:3265–3276. doi: 10.1158/1078-0432.CCR-08-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laubner K, Kieffer TJ, Lam NT, Niu X, Jakob F, Seufert J. Inhibition of preproinsulin gene expression by leptin induction of suppressor of cytokine signaling 3 in pancreatic beta-cells. Diabetes. 2005;54:3410–3417. doi: 10.2337/diabetes.54.12.3410. [DOI] [PubMed] [Google Scholar]
- Lee Y, Ravazzola M, Park BH, Bashmakov YK, Orci L, Unger RH. Metabolic mechanisms of failure of intraportally transplanted pancreatic beta-cells in rats: role of lipotoxicity and prevention by leptin. Diabetes. 2007;56:2295–2301. doi: 10.2337/db07-0460. [DOI] [PubMed] [Google Scholar]
- Lee Y, Wang MY, Du XQ, Charron MJ, Unger RH. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes. 2011;60:391–397. doi: 10.2337/db10-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehr S, Hartwig S, Lamers D, Famulla S, Muller S, Hanisch FG, Cuvelier C, Ruige J, Eckardt K, Ouwens DM, et al. Identification and validation of novel adipokines released from primary human adipocytes. Molecular & cellular proteomics : MCP. 2012a;11:M111010504. doi: 10.1074/mcp.M111.010504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehr S, Hartwig S, Sell H. Adipokines: a treasure trove for the discovery of biomarkers for metabolic disorders. Proteomics. Clinical applications. 2012b;6:91–101. doi: 10.1002/prca.201100052. [DOI] [PubMed] [Google Scholar]
- Lennon R, Pons D, Sabin MA, Wei C, Shield JP, Coward RJ, Tavare JM, Mathieson PW, Saleem MA, Welsh GI. Saturated fatty acids induce insulin resistance in human podocytes: implications for diabetic nephropathy. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association – European Renal Association. 2009;24:3288–3296. doi: 10.1093/ndt/gfp302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin N, Nelson C, Gurney A, Vandlen R, de Sauvage F. Decreased food intake does not completely account for adiposity reduction after ob protein infusion. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:1726–1730. doi: 10.1073/pnas.93.4.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Kennedy DJ, Yan Y, Shapiro JI. Reactive Oxygen Species Modulation of Na/K-ATPase Regulates Fibrosis and Renal Proximal Tubular Sodium Handling. International journal of nephrology. 2012;2012:381320. doi: 10.1155/2012/381320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YL, Emilsson V, Cawthorne MA. Leptin inhibits glycogen synthesis in the isolated soleus muscle of obese (ob/ob) mice. FEBS letters. 1997;411:351–355. doi: 10.1016/s0014-5793(97)00732-1. [DOI] [PubMed] [Google Scholar]
- Malaisse WJ, Malaisse-Lagae F. Stimulation of insulin secretion by noncarbohydrate metabolites. The Journal of laboratory and clinical medicine. 1968;72:438–448. [PubMed] [Google Scholar]
- Marroqui L, Vieira E, Gonzalez A, Nadal A, Quesada I. Leptin downregulates expression of the gene encoding glucagon in alphaTC1-9 cells and mouse islets. Diabetologia. 2011;54:843–851. doi: 10.1007/s00125-010-2024-1. [DOI] [PubMed] [Google Scholar]
- Mayer J. Regulation of energy intake and the body weight: the glucostatic theory and the lipostatic hypothesis. Annals of the New York Academy of Sciences. 1955;63:15–43. doi: 10.1111/j.1749-6632.1955.tb36543.x. [DOI] [PubMed] [Google Scholar]
- McGee SL, van Denderen BJ, Howlett KF, Mollica J, Schertzer JD, Kemp BE, Hargreaves M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes. 2008;57:860–867. doi: 10.2337/db07-0843. [DOI] [PubMed] [Google Scholar]
- Meyer C, Nadkarni V, Stumvoll M, Gerich J. Human kidney free fatty acid and glucose uptake: evidence for a renal glucose-fatty acid cycle. The American journal of physiology. 1997;273:E650–654. doi: 10.1152/ajpendo.1997.273.3.E650. [DOI] [PubMed] [Google Scholar]
- Miller RA, Chu Q, Le Lay J, Scherer PE, Ahima RS, Kaestner KH, Foretz M, Viollet B, Birnbaum MJ. Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. The Journal of clinical investigation. 2011;121:2518–2528. doi: 10.1172/JCI45942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- Mistry AM, Swick AG, Romsos DR. Leptin acts peripherally to limit meal-induced increases in plasma insulin concentrations in mice: a brief communication. Experimental biology and medicine. 2004;229:1033–1037. doi: 10.1177/153537020422901007. [DOI] [PubMed] [Google Scholar]
- Miyamoto L, Ebihara K, Kusakabe T, Aotani D, Yamamoto-Kataoka S, Sakai T, Aizawa-Abe M, Yamamoto Y, Fujikura J, Hayashi T, et al. Leptin activates hepatic 5′-AMP-activated protein kinase through sympathetic nervous system and alpha1-adrenergic receptor: a potential mechanism for improvement of fatty liver in lipodystrophy by leptin. The Journal of biological chemistry. 2012;287:40441–40447. doi: 10.1074/jbc.M112.384545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B, Feigenbaum L, Lee E, Aoyama T, Eckhaus M, et al. Life without white fat: a transgenic mouse. Genes & development. 1998;12:3168–3181. doi: 10.1101/gad.12.20.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montez JM, Soukas A, Asilmaz E, Fayzikhodjaeva G, Fantuzzi G, Friedman JM. Acute leptin deficiency, leptin resistance, and the physiologic response to leptin withdrawal. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2537–2542. doi: 10.1073/pnas.0409530102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead JF, Chan MK, El-Nahas M, Varghese Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet. 1982;2:1309–1311. doi: 10.1016/s0140-6736(82)91513-6. [DOI] [PubMed] [Google Scholar]
- Morioka T, Asilmaz E, Hu J, Dishinger JF, Kurpad AJ, Elias CF, Li H, Elmquist JK, Kennedy RT, Kulkarni RN. Disruption of leptin receptor expression in the pancreas directly affects beta cell growth and function in mice. The Journal of clinical investigation. 2007;117:2860–2868. doi: 10.1172/JCI30910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley TS, Xia JY, Scherer PE. Selective enhancement of insulin sensitivity in the mature adipocyte is sufficient for systemic metabolic improvements. Nature communications. 2015;6:7906. doi: 10.1038/ncomms8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller G, Ertl J, Gerl M, Preibisch G. Leptin impairs metabolic actions of insulin in isolated rat adipocytes. The Journal of biological chemistry. 1997;272:10585–10593. doi: 10.1074/jbc.272.16.10585. [DOI] [PubMed] [Google Scholar]
- Muller WA, Faloona GR, Aguilar-Parada E, Unger RH. Abnormal alpha-cell function in diabetes. Response to carbohydrate and protein ingestion. The New England journal of medicine. 1970;283:109–115. doi: 10.1056/NEJM197007162830301. [DOI] [PubMed] [Google Scholar]
- Muoio DM, Dohm GL, Fiedorek FT, Jr, Tapscott EB, Coleman RA. Leptin directly alters lipid partitioning in skeletal muscle. Diabetes. 1997;46:1360–1363. doi: 10.2337/diab.46.8.1360. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Nakatake Y, Konishi M, Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochimica et biophysica acta. 2000;1492:203–206. doi: 10.1016/s0167-4781(00)00067-1. [DOI] [PubMed] [Google Scholar]
- Nishina PM, Lowe S, Wang J, Paigen B. Characterization of plasma lipids in genetically obese mice: the mutants obese, diabetes, fat, tubby, and lethal yellow. Metabolism: clinical and experimental. 1994;43:549–553. doi: 10.1016/0026-0495(94)90194-5. [DOI] [PubMed] [Google Scholar]
- Okada-Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami K, Matsuda K, Yamaguchi M, Tanabe H, Kimura-Someya T, Shirouzu M, et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature. 2013;503:493–499. doi: 10.1038/nature12656. [DOI] [PubMed] [Google Scholar]
- Okamoto M, Ohara-Imaizumi M, Kubota N, Hashimoto S, Eto K, Kanno T, Kubota T, Wakui M, Nagai R, Noda M, et al. Adiponectin induces insulin secretion in vitro and in vivo at a low glucose concentration. Diabetologia. 2008;51:827–835. doi: 10.1007/s00125-008-0944-9. [DOI] [PubMed] [Google Scholar]
- Olofsson CS, Salehi A, Gopel SO, Holm C, Rorsman P. Palmitate stimulation of glucagon secretion in mouse pancreatic alpha-cells results from activation of L-type calcium channels and elevation of cytoplasmic calcium. Diabetes. 2004;53:2836–2843. doi: 10.2337/diabetes.53.11.2836. [DOI] [PubMed] [Google Scholar]
- Opara EC, Hubbard VS, Burch WM, Akwari OE. Characterization of the insulinotropic potency of polyunsaturated fatty acids. Endocrinology. 1992;130:657–662. doi: 10.1210/endo.130.2.1733714. [DOI] [PubMed] [Google Scholar]
- Ortiga-Carvalho TM, Oliveira KJ, Soares BA, Pazos-Moura CC. The role of leptin in the regulation of TSH secretion in the fed state: in vivo and in vitro studies. The Journal of endocrinology. 2002;174:121–125. doi: 10.1677/joe.0.1740121. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, Funahashi T, Walsh K. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. The Journal of biological chemistry. 2004;279:1304–1309. doi: 10.1074/jbc.M310389200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, Kitsis RN, Scherer PE. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nature medicine. 2005;11:797–803. doi: 10.1038/nm1262. [DOI] [PubMed] [Google Scholar]
- Park S, Kim DS, Kwon DY, Yang HJ. Long-term central infusion of adiponectin improves energy and glucose homeostasis by decreasing fat storage and suppressing hepatic gluconeogenesis without changing food intake. Journal of neuroendocrinology. 2011;23:687–698. doi: 10.1111/j.1365-2826.2011.02165.x. [DOI] [PubMed] [Google Scholar]
- Park SH, Ryu SY, Yu WJ, Han YE, Ji YS, Oh K, Sohn JW, Lim A, Jeon JP, Lee H, et al. Leptin promotes K(ATP) channel trafficking by AMPK signaling in pancreatic beta-cells. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:12673–12678. doi: 10.1073/pnas.1216351110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- Perez C, Fernandez-Galaz C, Fernandez-Agullo T, Arribas C, Andres A, Ros M, Carrascosa JM. Leptin impairs insulin signaling in rat adipocytes. Diabetes. 2004;53:347–353. doi: 10.2337/diabetes.53.2.347. [DOI] [PubMed] [Google Scholar]
- Pernicova I, Korbonits M. Metformin–mode of action and clinical implications for diabetes and cancer. Nature reviews. Endocrinology. 2014;10:143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- Qi Y, Takahashi N, Hileman SM, Patel HR, Berg AH, Pajvani UB, Scherer PE, Ahima RS. Adiponectin acts in the brain to decrease body weight. Nature medicine. 2004;10:524–529. doi: 10.1038/nm1029. [DOI] [PubMed] [Google Scholar]
- Rakatzi I, Mueller H, Ritzeler O, Tennagels N, Eckel J. Adiponectin counteracts cytokine- and fatty acid-induced apoptosis in the pancreatic beta-cell line INS-1. Diabetologia. 2004;47:249–258. doi: 10.1007/s00125-003-1293-3. [DOI] [PubMed] [Google Scholar]
- Renquist BJ, Murphy JG, Larson EA, Olsen D, Klein RF, Ellacott KL, Cone RD. Melanocortin-3 receptor regulates the normal fasting response. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E1489–1498. doi: 10.1073/pnas.1201994109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezai-Zadeh K, Munzberg H. Integration of sensory information via central thermoregulatory leptin targets. Physiology & behavior. 2013;121:49–55. doi: 10.1016/j.physbeh.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca MG, Vazquez EJ, Chen Q, Kerner J, Kern TS, Hoppel CL. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes. 2012;61:2074–2083. doi: 10.2337/db11-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum M, Goldsmith R, Bloomfield D, Magnano A, Weimer L, Heymsfield S, Gallagher D, Mayer L, Murphy E, Leibel RL. Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. The Journal of clinical investigation. 2005;115:3579–3586. doi: 10.1172/JCI25977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum M, Murphy EM, Heymsfield SB, Matthews DE, Leibel RL. Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulating concentrations of thyroid hormones. The Journal of clinical endocrinology and metabolism. 2002;87:2391–2394. doi: 10.1210/jcem.87.5.8628. [DOI] [PubMed] [Google Scholar]
- Satoh N, Ogawa Y, Katsuura G, Numata Y, Tsuji T, Hayase M, Ebihara K, Masuzaki H, Hosoda K, Yoshimasa Y, et al. Sympathetic activation of leptin via the ventromedial hypothalamus: leptin-induced increase in catecholamine secretion. Diabetes. 1999;48:1787–1793. doi: 10.2337/diabetes.48.9.1787. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M. Leptin induction of UCP1 gene expression is dependent on sympathetic innervation. The American journal of physiology. 1998;275:E259–264. doi: 10.1152/ajpendo.1998.275.2.E259. [DOI] [PubMed] [Google Scholar]
- Scherer PE. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes. 2006;55:1537–1545. doi: 10.2337/db06-0263. [DOI] [PubMed] [Google Scholar]
- Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. The Journal of biological chemistry. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- Seoane LM, Carro E, Tovar S, Casanueva FF, Dieguez C. Regulation of in vivo TSH secretion by leptin. Regulatory peptides. 2000;92:25–29. doi: 10.1016/s0167-0115(00)00145-2. [DOI] [PubMed] [Google Scholar]
- Seufert J, Kieffer TJ, Leech CA, Holz GG, Moritz W, Ricordi C, Habener JF. Leptin suppression of insulin secretion and gene expression in human pancreatic islets: implications for the development of adipogenic diabetes mellitus. The Journal of clinical endocrinology and metabolism. 1999;84:670–676. doi: 10.1210/jcem.84.2.5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Ramachandrarao S, Qiu G, Usui HK, Zhu Y, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, et al. Adiponectin regulates albuminuria and podocyte function in mice. The Journal of clinical investigation. 2008;118:1645–1656. doi: 10.1172/JCI32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401:73–76. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- Simon N, Hertig A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Frontiers in medicine. 2015;2:52. doi: 10.3389/fmed.2015.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soedling H, Hodson DJ, Adrianssens AE, Gribble FM, Reimann F, Trapp S, Rutter GA. Limited impact on glucose homeostasis of leptin receptor deletion from insulin- or proglucagon-expressing cells. Molecular metabolism. 2015;4:619–630. doi: 10.1016/j.molmet.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–1832. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- Stearns SB, Benzo CA. Glucagon and insulin relationships in genetically diabetic (db/db) and in streptozotocin-induced diabetic mice. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 1978;10:20–23. doi: 10.1055/s-0028-1093473. [DOI] [PubMed] [Google Scholar]
- Stumvoll M, Tschritter O, Fritsche A, Staiger H, Renn W, Weisser M, Machicao F, Haring H. Association of the T-G polymorphism in adiponectin (exon 2) with obesity and insulin sensitivity: interaction with family history of type 2 diabetes. Diabetes. 2002;51:37–41. doi: 10.2337/diabetes.51.1.37. [DOI] [PubMed] [Google Scholar]
- Suganami T, Mukoyama M, Mori K, Yokoi H, Koshikawa M, Sawai K, Hidaka S, Ebihara K, Tanaka T, Sugawara A, et al. Prevention and reversal of renal injury by leptin in a new mouse model of diabetic nephropathy. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005;19:127–129. doi: 10.1096/fj.04-2183fje. [DOI] [PubMed] [Google Scholar]
- Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- Sweeney G. Cardiovascular effects of leptin. Nat Rev Cardiol. 2010;7:22–29. doi: 10.1038/nrcardio.2009.224. [DOI] [PubMed] [Google Scholar]
- Sweiss N, Sharma K. Adiponectin effects on the kidney. Best practice & research. Clinical endocrinology & metabolism. 2014;28:71–79. doi: 10.1016/j.beem.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe H, Fujii Y, Okada-Iwabu M, Iwabu M, Nakamura Y, Hosaka T, Motoyama K, Ikeda M, Wakiyama M, Terada T, et al. Crystal structures of the human adiponectin receptors. Nature. 2015;520:312–316. doi: 10.1038/nature14301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari S, Singh RS, Li L, Tsukerman S, Godbole M, Pandey G, Ecelbarger CM. Deletion of the insulin receptor in the proximal tubule promotes hyperglycemia. Journal of the American Society of Nephrology : JASN. 2013;24:1209–1214. doi: 10.1681/ASN.2012060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas E, Tsao TS, Saha AK, Murrey HE, Zhang Cc C, Itani SI, Lodish HF, Ruderman NB. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:16309–16313. doi: 10.1073/pnas.222657499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trak-Smayra V, Paradis V, Massart J, Nasser S, Jebara V, Fromenty B. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. International journal of experimental pathology. 2011;92:413–421. doi: 10.1111/j.1365-2613.2011.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trayhurn P, Thurlby PL, James WP. Thermogenic defect in pre-obese ob/ob mice. Nature. 1977;266:60–62. doi: 10.1038/266060a0. [DOI] [PubMed] [Google Scholar]
- Tuduri E, Marroqui L, Soriano S, Ropero AB, Batista TM, Piquer S, Lopez-Boado MA, Carneiro EM, Gomis R, Nadal A, et al. Inhibitory effects of leptin on pancreatic alpha-cell function. Diabetes. 2009;58:1616–1624. doi: 10.2337/db08-1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turer AT, Scherer PE. Adiponectin: mechanistic insights and clinical implications. Diabetologia. 2012;55:2319–2326. doi: 10.1007/s00125-012-2598-x. [DOI] [PubMed] [Google Scholar]
- Vara E, Fernandez-Martin O, Garcia C, Tamarit-Rodriguez J. Palmitate dependence of insulin secretion, “de novo” phospholipid synthesis and 45Ca2+-turnover in glucose stimulated rat islets. Diabetologia. 1988;31:687–693. doi: 10.1007/BF00278753. [DOI] [PubMed] [Google Scholar]
- Venn-Watson S, Smith CR, Stevenson S, Parry C, Daniels R, Jensen E, Cendejas V, Balmer B, Janech M, Neely BA, et al. Blood-Based Indicators of Insulin Resistance and Metabolic Syndrome in Bottlenose Dolphins (Tursiops truncatus) Frontiers in endocrinology. 2013;4:136. doi: 10.3389/fendo.2013.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Chandrasekera PC, Pippin JJ. Leptin- and leptin receptor-deficient rodent models: relevance for human type 2 diabetes. Curr Diabetes Rev. 2014;10:131–145. doi: 10.2174/1573399810666140508121012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MY, Chen L, Clark GO, Lee Y, Stevens RD, Ilkayeva OR, Wenner BR, Bain JR, Charron MJ, Newgard CB, et al. Leptin therapy in insulin-deficient type I diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4813–4819. doi: 10.1073/pnas.0909422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MY, Yan H, Shi Z, Evans MR, Yu X, Lee Y, Chen S, Williams A, Philippe J, Roth MG, et al. Glucagon receptor antibody completely suppresses type 1 diabetes phenotype without insulin by disrupting a novel diabetogenic pathway. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:2503–2508. doi: 10.1073/pnas.1424934112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiang T, Li J, Proctor G, McManaman JL, Lucia S, Chua S, Levi M. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes. 2005;54:2328–2335. doi: 10.2337/diabetes.54.8.2328. [DOI] [PubMed] [Google Scholar]
- Warnotte C, Gilon P, Nenquin M, Henquin JC. Mechanisms of the stimulation of insulin release by saturated fatty acids. A study of palmitate effects in mouse beta-cells. Diabetes. 1994;43:703–711. doi: 10.2337/diab.43.5.703. [DOI] [PubMed] [Google Scholar]
- William WN, Jr, Ceddia RB, Curi R. Leptin controls the fate of fatty acids in isolated rat white adipocytes. The Journal of endocrinology. 2002;175:735–744. doi: 10.1677/joe.0.1750735. [DOI] [PubMed] [Google Scholar]
- Winzell MS, Nogueiras R, Dieguez C, Ahren B. Dual action of adiponectin on insulin secretion in insulin-resistant mice. Biochemical and biophysical research communications. 2004;321:154–160. doi: 10.1016/j.bbrc.2004.06.130. [DOI] [PubMed] [Google Scholar]
- Wu X, Motoshima H, Mahadev K, Stalker TJ, Scalia R, Goldstein BJ. Involvement of AMP-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes. 2003;52:1355–1363. doi: 10.2337/diabetes.52.6.1355. [DOI] [PubMed] [Google Scholar]
- Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, Sifuentes AJ, McDonald JG, Gordillo R, Scherer PE. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell metabolism. 2015;22:266–278. doi: 10.1016/j.cmet.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. The Journal of clinical investigation. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahagi N, Shimano H, Hasty AH, Matsuzaka T, Ide T, Yoshikawa T, Amemiya-Kudo M, Tomita S, Okazaki H, Tamura Y, et al. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. The Journal of biological chemistry. 2002;277:19353–19357. doi: 10.1074/jbc.M201584200. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nature medicine. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nature medicine. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, Okada-Iwabu M, Kawamoto S, Kubota N, Kubota T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nature medicine. 2007;13:332–339. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- Ye R, Holland WL, Gordillo R, Wang M, Wang QA, Shao M, Morley TS, Gupta RK, Stahl A, Scherer PE. Adiponectin is essential for lipid homeostasis and survival under insulin deficiency and promotes beta-cell regeneration. eLife. 2014;3 doi: 10.7554/eLife.03851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MJ, Lee GY, Chung JJ, Ahn YH, Hong SH, Kim JB. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes. 2006;55:2562–2570. doi: 10.2337/db05-1322. [DOI] [PubMed] [Google Scholar]
- Yore MM, Syed I, Moraes-Vieira PM, Zhang T, Herman MA, Homan EA, Patel RT, Lee J, Chen S, Peroni OD, et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell. 2014;159:318–332. doi: 10.1016/j.cell.2014.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Park BH, Wang MY, Wang ZV, Unger RH. Making insulin-deficient type 1 diabetic rodents thrive without insulin. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14070–14075. doi: 10.1073/pnas.0806993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Pirzgalska RM, Pereira MM, Kubasova N, Barateiro A, Seixas E, Lu YH, Kozlova A, Voss H, Martins GG, et al. Sympathetic Neuro-adipose Connections Mediate Leptin-Driven Lipolysis. Cell. 2015;163:84–94. doi: 10.1016/j.cell.2015.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- Ziemke F, Mantzoros CS. Adiponectin in insulin resistance: lessons from translational research. The American journal of clinical nutrition. 2010;91:258S–261S. doi: 10.3945/ajcn.2009.28449C. [DOI] [PMC free article] [PubMed] [Google Scholar]