

Abstract
We have demonstrated that glucagon like peptide-1 (GLP-1) protects the heart against ischemic injury. However, the physiological mechanism by which GLP-1 receptor (GLP-1R) initiates cardioprotection remains to be determined. The objective of this study is to elucidate the functional roles of MAPK kinase 3 (MKK3) and Akt-1 in mediating exendin-4-elicited protection in the infarcted hearts. Adult mouse myocardial infarction (MI) was created by ligation of the left descending artery. Wild-type, MKK3−/−, Akt-1−/−, and Akt-1−/−;MKK3−/− mice were divided into one of several groups: 1) sham: animals underwent thoracotomy without ligation; 2) MI: animals underwent MI and received a daily dose of intraperitoneal injection of vehicle (saline); 3) MI + exendin-4: infarcted mice received daily injections of exendin-4, a GLP-1R agonist (0.1 mg/kg, ip). Echocardiographic measurements indicate that exendin-4 treatment resulted in the preservation of ventricular function and increases in the survival rate, but these effects were diminished in MKK3−/−, Akt-1−/−, and Akt-1−/−;MKK3−/− mice. Exendin-4 treatments suppressed cardiac hypotrophy and reduced scar size and cardiac interstitial fibrosis, respectively, but these beneficial effects were lost in genetic elimination of MKK3, Akt-1, or Akt-1−/−;MKK3−/− mice. GLP-1R stimulation stimulated angiogenic responses, which were also mitigated by deletion of MKK3 and Akt-1. Exendin-4 treatment increased phosphorylation of MKK3, p38, and Akt-1 at Ser129 but decreased levels of active caspase-3 and cleaved poly (ADP-ribose) polymerase; these proteins were diminished in MKK3−/−, Akt-1−/−, and Akt-1−/−;MKK3−/− mice. These results reveal that exendin-4 treatment improves cardiac function, attenuates cardiac remodeling, and promotes angiogenesis in the infarcted myocardium through MKK3 and Akt-1 pathway.
Keywords: glucagon-like peptide-1 receptor, MAPK kinase 3, Akt-1, myocardial infarction, function
glucagon-like peptide-1 (GLP-1) is a naturally occurring incretin that is implicated in the control of appetite and satiety (23). GLP-1 has been studied extensively in type 2 diabetes as a novel insulinotropic peptide whose actions are dependent on the ambient glucose concentration. GLP-1 acts through the GLP-1 receptor (GLP-1R), a 463-amino acid member of the G protein-coupled receptor superfamily (23). GLP-1 is rapidly cleaved by dipeptidyl-peptidase-4 (DPP4), which results in the generation of largely inactive molecular GLP-19–36 amide and GLP-19–37 forms (14). The majority of GLP-1 leaving the intestinal venous circulation has already been cleaved by DPP4 expressed in capillary surrounding gut L cells, which provides an estimated half-life of 1–2 min for intact GLP-1 in vivo (12). The GLP-1 receptor is widely distributed in tissues, including brain, pancreas, intestine, lung, stomach, and kidney. Recently, multiple GLP-1 receptor agonists with longer duration of effect in vivo have been explored, among which is exendin-4, a 39-amino acid peptide that shares 53% sequence homology with GLP-1, making it a more favorable target for GLP-1 for a therapeutic approach in treating diabetes (16, 22, 26, 40). More importantly, patients with myocardial infarction (MI), metabolic syndrome, and diabetes exhibited a higher prevalence in association with higher risks of deaths and major cardiovascular events. Emerging evidence indicates that the use of GLP-1-based therapies as metabolic modulators in heart disease represents a new therapeutic approach in this important cardiovascular domain (1).
Experimental studies and clinical data demonstrate that infusion of GLP-1 promotes regional and global left ventricular (LV) function recovery in patients with acute MI (23, 25). Recent observation suggests a possible link between the GLP-1R agonist and mitogen-activated protein kinase pathway in initiating cellular signalings (13). Our previous studies showed that activation of p38 protected the heart against ischemia/reperfusion injury (43). We have demonstrated that GLP-1 protected the heart against ischemia/reperfusion injury, which is related to p38 phosphorylation. Noticeably, activation of p38 has been demonstrated to play an essential role in GLP-1-induced protective effects in heart failure (8). Therefore, direct inhibition of p38 by genetic ablation of MAPK kinase 3 (MKK3) will provide evidence for the role of p38 in GLP-1R-mediated cardiac protection. Akt-1 pathway is essential in protecting against myocardial loss in ischemic hearts (20). Inhibitors of phosphoinositide-3-kinase (PI3K) abolished the GLP-1-induced infarct size limitation in rat hearts (9, 10). Exendin-4 treatment was found to induce phosphorylation of Akt-1 in mice with dilated cardiomyopathy (36). However, the functional role of Akt-1 and MKK3 in direct linkage with GLP-1R in the protection of ischemic heart remains to be determined.
In the present study, we employed MKK3−/−, Akt-1−/−, and Akt-1−/−;MKK3−/− mice to define the effect of exendin-4 treatment on MKK3 and Akt-1 and to further elucidate the functional roles of MKK3 and Akt-1 in GLP-1R stimulation by restoring cardiac function, attenuating interstitial fibrosis, suppressing cardiac hypertrophy, and promoting angiogenesis in the infarcted heart. Our results reveal that exendin-4 treatment elicits a protective effect through MKK3 and Akt-1 in the infarcted myocardium.
MATERIALS AND METHODS
Animals.
Adult C57BL/6 mice were purchased from Charles River Laboratory (Bar Harbor, ME); Akt-1−/−, MKK3−/−, and Akt-1−/−;MKK3−/− mice were bred and maintained. Akt-1−/−;MKK3−/− mice double-transgenic mice were obtained from breeding between Akt-1−/− and MKK3−/− mice. All animal experiments were conducted under a protocol approved by the Institutional Animal Care and Use Committee, which conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
MI and study protocol.
The mouse MI model was created as described previously (44). MI was created by ligation of the left anterior descending coronary artery. Mice in the sham group were anesthetized and underwent thoracotomy without coronary ligation. Following MI, exendin-4 at a dose of (0.1 mg/kg) or vehicle (saline, 0.1 ml) was immediately intraperitoneally injected to infarcted mice after recovery from the surgical operation on a daily basis for the following consecutive 9 wk, and this included mice from MKK3−/−, Akt-1−/−, Akt-1−/−;MKK3−/−, and wild-type control backgrounds. All sham, MI + vehicle, and MI + exendin-4 treatment animals in all the above backgrounds were euthanized at the end of 9 wk after MI. Echocardiographic measurements were carried out every 3 wk. Mice were anesthetized with 1.5% isoflurane, and temperature was maintained at 37°C. Nair lotion (Church & Dwight Canada, Mississauga, Ontario, Canada) was applied on the precordial region for 3 min to cleanly remove the hair, and the region was covered with prewarmed ultrasound transmission gel (Aquasonic; Parker Laboratory, Fairfield, NJ). Transthoracic echocardiography was performed using an Acuson Sequoia C512 system with a 15L8 linear array probe. All images were acquired at a depth setting of 25 mm. Two-dimensional B-mode and M-mode echocardiographic images were obtained at the level of the papillary muscles from the parasternal short-axis view. Wall thickness and chamber dimension were determined from M-mode tracings using cardiac calcs software. All LV dimensions are presented as the average of measurements using six to nine consecutive selected beats.
Histological analysis.
Sections (10 μm) were prepared from paraffin-embedded tissues. Myocyte cross-sectional area was measured from images captured from the sections obtained mid-distance from the base to the apex. Wheat germ agglutinin (WGA) staining was carried out using immunofluorescent staining to measure cell size. Suitable cross sections were defined as having nearly circular to oval myocyte sections. The outline of myocytes was traced using NIH Image J software to determine myocyte cross-sectional area. A value from each heart was calculated by the measurements of ∼400–600 cells in a remote area from infarction of an individual heart. To evaluate cardiac fibrosis, LV sections were stained with picrosirius red, and collagen content was quantitated in images taken under a microscope coupled to a computerized morphometry system (Olympus BX41). Interstitial collagen density was expressed as a percentage of myocardial area in each image (44). Infarct scar area and the total area of LV myocardium were traced manually in the digital images and measured automatically by the computer. A score for scar size was assigned as a percentage of overall LV circumference.
Immunohistology.
Tissue sections were deparaffinized as described previously (44). Microvessels were identified by smooth muscle cells by anti-α-smooth muscle actin (α-SMA) monoclonal antibody (Sigma, St. Louis, MO) and endothelial cells by CD31 (anti- platelet endothelial cell adhesion molecule-1)(Millipore, Billerica, MA). The total number of vessels from each group was calculated and normalized to the tissue area. The stained numbers of each section were counted in ∼10 randomized fields of the tissue sections, which were taken in the middle plane of each heart and contained infarct and border regions.
Western blot analysis for molecular signaling.
We prepared samples from the myocardium and performed immunoblotting analysis. Proteins (50 μg/lane) were separated by SDS-PAGE and then transferred onto a nitrocellulose membrane. The blots were incubated with their respective antibodies: phosphorylated and nonphosphorylated rabbit-MKK3 polyclonal, phosphorylated and nonphosphorylated rabbit-Akt-1 polyclonal, phosphorylated and nonphosphorylated p38 polyclonal, p85 subunit of PI3K (Cell Signaling, Danvers, MA), rabbit monoclonal phosphorylated Akt-1, active caspase-3, cleaved poly (ADP-ribose) polymerase (PARP) monoclonal rabbit (E51) (Abcam, Cambridge, MA), β-actin polyclonal antibodies (1:1,000) (Cell Signaling), respectively, for 2 h and visualized by incubation with anti-rabbit horseradish peroxidase-conjugated secondary antibody (1:5,000) for 1 h and developed with enhanced chemiluminescence detection reagent (Amersham Pharmacia Biotech, Piscataway, NJ). The myocardium was separated into noninfarcted remote and border-zone areas for analysis of apoptosis in MI hearts.
The measurement of hydroxyproline in myocardium.
All chemicals used in this assay were purchased from Sigma-Aldrich or Thermo-Fisher (Waltham, MA) unless otherwise noted. Hydroxyproline content in myocardial tissues was determined according to a previous method with modifications (29). In brief, LV myocardial tissues were homogenized and hydrolyzed at 120°C for 20 min. 4-Hydroxy-l-proline standards, ranging from 2–20 μg, were prepared from a 1 mg/ml hydroxyproline stock solution. Detected samples and hydroxyproline standards were first mixed with a solution of 0.056 M chloramine T and then processed at room temperature for 25 min. Enrich's reagent (1 M P-dimethylamino-benzaldehyde dissolved in N-propanol/perchloric acid) was added to each sample followed by incubation at 60°C for 20 min. Absorbances of each sample and standards were read at 550 nm using an Eon High Performance Microplate Spectrophotometer (BioTek, Winooski, VT). Hydroxyproline content was determined using the standard curve generated by 4-hydroxy-l-proline standards.
Statistics.
All measurements are expressed as means ± SE. Differences among the groups were analyzed by two-way ANOVA, followed by post hoc Bonferroni correction. Statistical differences were considered significant with a value of P < 0.05.
RESULTS
Stimulation of Akt-1 and MKK3 following exendin-4 treatment.
As shown in Fig. 1, A and B, MI induced a slight decrease in Akt-1 and MKK3 phosphorylation. Exendin-4 treatment resulted in marked increases in both Akt-1 and MKK3 phosphorylation (Fig. 1, C and D). Both total and phosphorylated Akt-1 were not detected in Akt-1−/− mice (Fig. 1A). Likewise, neither the total nor phosphorylated MKK3 were detectable in MKK3−/− mice (Fig. 1B).
Fig. 1.
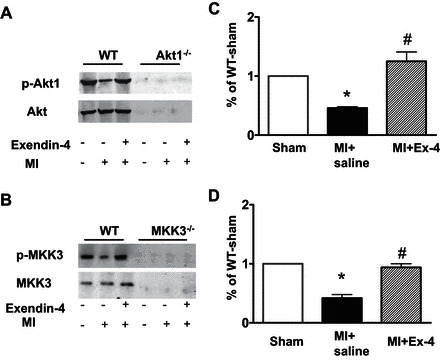
Exendin-4 (Ex-4) treatment resulted in increases in phosphorylated Akt-1 (A and C) and phosphorylated MAPK kinase 3 (MKK3) (B and D) in the myocardial infarction (MI). C and D show densitometric analyses of protein levels in each group, and values represent mean ± SE (n = 3/per group), *P < 0.05 vs. sham; #P < 0.05 vs. MI. WT, wild-type.
Post-MI animal survivals.
The survival rates were evaluated using the Kaplan-Meier survival curve for up to 9 wk in wild-type, MKK3−/−, Akt-1−/−, and Akt-1−/−;MKK3−/− infarcted mice. As shown in Fig. 2, A–D, all MI mice that received vehicle treatment displayed a gradual decline in survival rate, but wild-type mice receiving exendin-4 treatments demonstrated a significant improvement throughout in survival rate from 3 wk to the end of experiment following MI (Fig. 2A). However, this improvement in survival rate by exendin-4 treatment was not observed in Akt-1−/−, MKK3−/−, and Akt-1−/−;MKK3−/− mice, indicating that the increase in the survival rate by exendin-4 relies on MKK3 and Akt-1 (Fig. 2, B–D).
Fig. 2.
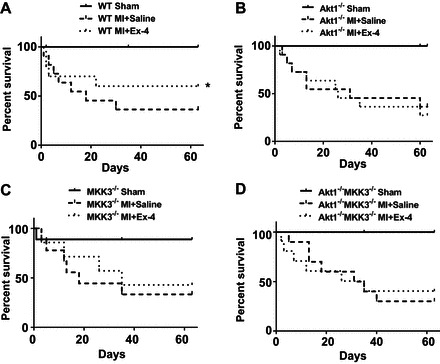
Exendin-4 treatment increases Kaplan-Meier survival rate after MI. Percentage survival of sham-operated mice and post-MI mice injected with saline or Exendin-4. A: WT (n = 10–12/per group). B: Akt-1−/− mice (n = 10–11/per group). C: MKK3−/− (n = 10–11/per group). D: Akt-1−/−;MKK3−/− mice that underwent sham, vehicle-treated MI, and exendin-4 treatment (n = 9–11/per group). Values represent means ± SE, *P < 0.05 vs. vehicle-treated-MI.
Scar size in the post-MI myocardium.
We measured the scar size to determine whether treatment of animals with exendin-4 could mitigate scar formation. Treatment of animals with exendin-4 resulted in a marked reduction in scar size compared with the vehicle treatments in wild-type mice (Fig. 3A). However, genetic elimination of Akt-1 or MKK3 abolished the effect of exendin-4 on decreasing scar size (Fig. 3, B and C). Likewise, simultaneous deletion of both MKK3 and Akt-1 was also unable to cause a reduction in scar formation as demonstrated by exendin-4 treatment (Fig. 3D), suggesting that exendin-4 treatment attenuates scar formation, which is dependent on MKK3 or Akt-1.
Fig. 3.
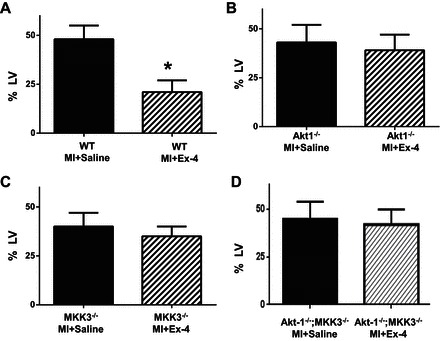
Infarct scar area in postinfarcted myocardium. Infarct scar area and total area of left ventricle (LV) were measured from picrosirius red staining sections. The scar size was expressed as the percentage of area of LV. Values represent means ± SE (n = 3/per group). *P < 0.05 vs. MI + saline.
Ventricular functions.
Serial echocardiographic measurements were performed every 3 wk in sham and infarcted mice. There was no difference in baseline among wild-type and genetic knockout mice (Table 1). As shown in Fig. 4, exendin-4 treatment prevented progressive systolic dysfunction in wild-type mice, as indicated by increases in ejection fraction (Fig. 4, A and E) and fractional shortening compared with vehicle-treated group (Table 2). However, there were no significant differences observed in ejection fraction and fractional shortening in MKK3−/−, Akt-1−/−, and Akt-1−/−;MKK3−/− mice between vehicle-treated groups and exendin-4-treated groups (Fig. 4 and Table 2). An increase in LV internal dimension (LVID) and significant decrease in contractility were observed in all infarcted mice compared with sham-operated mice. Exendin-4 treatment resulted in improvement in both LVID;d and LVID;s in wild-type mice compared with wild-type vehicle-treated MI mice (Table 2). However, the improvements in LVID;d and LVID;s following exendin-4 treatment were lost in MKK3−/− and Akt-1−/− mice. On the other hand, a progressive LV dilation was demonstrated in MKK3−/− and Akt-1−/− mice, which became indistinguishable between vehicle- and exendin-4-treated groups (Fig. 4, Table 2). There is no difference in heart rates among groups (data not included). Furthermore, treatment of Akt-1−/−;MKK3−/− mice with exendin-4 did not result in functional improvements, as shown in wild-type mice (Fig. 4, Table 2). Genetic deletion of both MKK3 and Akt-1 did not cause additional ventricular function depression, suggesting that the synergistic effects of MKK3 and Akt-1 may not play an important role in exendin-4 treatment-induced myocardial protection in MI hearts.
Table 1.
Echocardiographic parameters at baseline between wild-type and transgenic mice
| WT | Akt-1−/− | MKK3−/− | Akt-1−/−MKK3−/− | |
|---|---|---|---|---|
| LVID;d, mm | 2.8 ± 0.3 | 2.9 ± 0.3 | 2.9 ± 0.4 | 3.0 ± 0.3 |
| LVID;s, mm | 1.7 ± 0.3 | 1.9 ± 0.2 | 2.0 ± 0.2 | 1.9 ± 0.3 |
| EF, % | 73.1 ± 4.6 | 67.1 ± 6.3 | 62.4 ± 4.2 | 69.9 ± 6.2 |
| FS, % | 39.3 ± 3.5 | 34.5 ± 3.3 | 36.2 ± 4.1 | 36.7 ± 4.0 |
Values are means ± SE (n = 12 for WT, n = 11 for Akt-1−/−, n = 11 for MKK3−/−, n = 9 for Akt-1−/−MKK3−/−).
WT, wild-type; LVID;d left ventricular internal dimension in diastole; LVID;s, left ventricular dimension in systole; EF, ejection fraction; FS, fractional shortening.
Fig. 4.
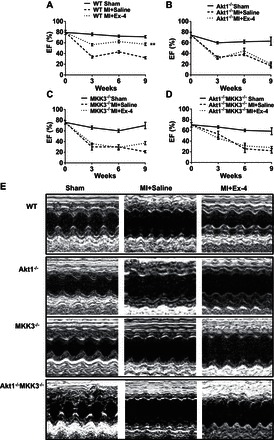
Echocardiographic measurements of ventricular function. Echographic measurements of ventricular functional parameter: ejection fraction (EF) (A–D). Representative images are shown as the M-mode short-axis ultrasound among the groups (E). Values represent means ± SE (n = 4–7/per group). **P < 0.01 vs. saline-treated MI in WT mice.
Table 2.
FS, LVID;d, and LVID;s in sham and treated mice
| Mouse Type | Sham | MI + Saline | MI + Ex-4 |
|---|---|---|---|
| Fractional Shortening, % | |||
| WT | 39.0 ± 3.0 | 12.0 ± 2.1 | 27.0 ± 1.5* |
| Akt-1−/− | 25.0 ± 2.0 | 11.4 ± 0.8 | 15.1 ± 3.3 |
| MKK3−/− | 30.0 ± 1.8 | 9.0 ± 4.0 | 14.0 ± 3.0 |
| Akt-1−/−MKK3−/− | 28.5 ± 3.9 | 9.0 ± 5.5 | 9.8 ± 4.2 |
| LVID;d, mm | |||
| WT | 3.1 ± 0.3 | 5.5 ± 0.2 | 4.1 ± 0.2* |
| Akt-1−/− | 3.1 ± 0.1 | 4.9 ± 0.1 | 5.2 ± 0.3 |
| MKK3−/− | 3.5 ± 0.3 | 5.5 ± 0.2 | 4.9 ± 0.3 |
| Akt-1−/−MKK3−/− | 3.4 ± 0.3 | 5.7 ± 0.4 | 5.5 ± 0.3 |
| LVID;s, mm | |||
| WT | 1.8 ± 0.3 | 4.8 ± 0.2 | 2.3 ± 0.6* |
| Akt-1−/− | 2.1 ± 0.2 | 4.3 ± 0.3 | 3.8 ± 0.1 |
| MKK3−/− | 2.3 ± 0.3 | 5.0 ± 0.2 | 4.0 ± 0.4 |
| Akt-1−/−MKK3−/− | 2.8 ± 0.1 | 4.6 ± 0.1 | 4.2 ± 0.6 |
Values are means ± SE (n = 5–8/per group).
LVID;d and LVID;s were taken at 9 week of post- myocardial infarction (MI) from all groups.
P < 0.01 vs. MI + saline. Ex-4; exendin-4.
Cardiac hypertrophy and interstitial fibrosis.
We measured the heart weight/tibia length and heart weight/body weight ratio to evaluate the hypertrophic response at the organ level. As shown in Table 3, MI resulted in an increase in the heart weight/body weight ratio and heart/tibia ratio compared with sham control animals. Treatment of wild-type infarcted animals with exendin-4 dramatically reduced the heart/tibia ratio and heart weight/body weight ratio. However, deletion of Akt-1 and MKK3 abolished the effects of exendin-4 on the reduction of heart/tibia ratio and heart weight/body weight ratio (Table 3). As shown in Fig. 5A, WGA staining showed that MI led to an increase in the cross-sectional area of cardiomyocytes in all vehicle-treated mice compared with sham-operated groups. Compared with vehicle-treated wild-type MI mice, exendin-4-treated wild-type mice displayed a reduction in cross-sectional area of cardiomyocyte compared with wild-type mice treated with vehicle. However, the anti-hypertrophy effect of exendin-4 was totally lost in Akt-1−/−, MKK3−/−, and/or Akt-1−/−;MKK3−/− mice (Fig. 5, B–E).
Table 3.
Heart weight, tibia length, and body weight in WT and treated mice
| Heart Weight/Tibia Length, mg/mm |
Heart Weight/Body Weight, mg/g |
|||||
|---|---|---|---|---|---|---|
| Sham | MI + Saline | MI + Ex-4 | Sham | MI + Saline | MI + Ex-4 | |
| WT | 9.58 ± 0.24 | 13.75 ± 1.0* | 10.14 ± 0.9† | 5.31 ± 0.28 | 9.01 ± 1.05* | 6.53 ± 0.61† |
| Akt-1−/− | 8.50 ± 0.60 | 15.90 ± 1.50* | 17.70 ± 2.70 | 4.60 ± 0.60 | 7.30 ± 0.60* | 7.40 ± 0.48 |
| MKK3−/− | 9.70 ± 0.30 | 15.70 ± 0.30* | 14.10 ± 2.00 | 5.30 ± 0.20 | 7.50 ± 0.20* | 8.00 ± 0.60 |
| Akt-1−/−MKK3−/− | 10.46 ± 2.20 | 19.44 ± 2.00* | 21.25 ± 1.75 | 3.75 ± 0.62 | 6.56 ± 0.45* | 7.24 ± 0.52 |
Values are means ± SE (n = 3–7/per group).
P < 0.05 vs. sham;
P < 0.05 vs. MI + saline.
Fig. 5.
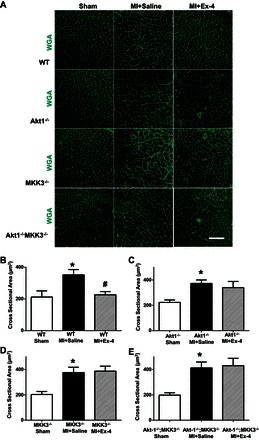
Exendin-4 treatment suppresses cellular hypertrophic response in the infarcted myocardium. A: representative images of wheat germ agglutinin (WGA) staining showing sham, vehicle-treated MI, and exendin-4-treated MI in WT, Akt-1−/−, MKK3−/−, and Akt-1−/−;MKK3−/− mice. Scale bar = 80 μm. B–D: Quantitative analyses of myocyte cross-sectional area; values represent means ± SE (n = 3–5/per group), *P < 0.05 vs. sham. #P < 0.05 vs. MI + saline.
The collagen content was significantly increased after MI in all MI mice receiving vehicle treatment compared with sham-operated group (Fig. 6). Compared with vehicle-treated wild-type mice, exendin-4 treatment resulted in a significant reduction of interstitial collagen following MI (Fig. 6, A and B). However, exendin-4 treatment did not show obvious antifibrotic effects in Akt-1−/−, MKK3−/−, and/or Akt-1−/−;MKK3−/− mice following MI (Fig. 6, C–E). As shown in Fig. 6, F–I, exendin-4 treatment resulted in the reduction of hydroxyproline in the infarcted myocardium in wild-type mice, which was abolished in Akt-1−/−, MKK3−/−, and/or Akt-1−/−;MKK3−/− mice. In addition, exendin-4 treatment reduced the contents of collagen 1a1 (Fig. 7, A–F) and fibronectin (Fig. 7, A–C, and G–I) in the myocardium of wild-type mice (Fig. 7, A–I), but the deletion of Akt-1 and MKK3 abrogated the effect of exendin-4 on reducing collagen 1a1 and fibronectin (Fig. 7, A–I).
Fig. 6.
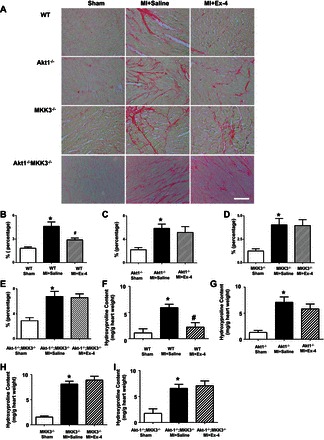
Interstitial fibrosis and hydroxyproline contents in MI hearts. A: representative images showing picrosirius red staining in sham, vehicle-treated MI, and exendin-4 treatments in WT, Akt-1−/−, MKK3−/−, and Akt-1−/−;MKK3−/− mice, respectively. B–E: quantitative analysis of myocyte interstitial collagen deposition. F–I: myocardial hydroxyproline from myocardium in sham, vehicle-treated MI, and exendin-4 treatment in WT, Akt-1−/−, MKK3−/−, and Akt-1−/−;MKK3−/− mice, respectively; values represent mean ± SE (n = 3/per group). *P < 0.05 vs. sham; #P < 0.05 vs. MI + saline. Scale bar = 50 μm.
Fig. 7.
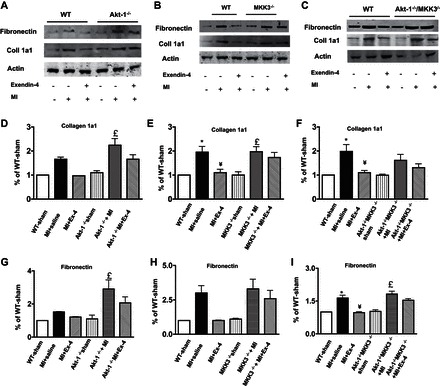
Effect of exendin-4 treatment on collagen 1a1 and fibronectin in myocardium. A–C: representative blots showing that exendin-4 treatment reduced collagen 1a1 and fibronectin in WT, Akt-1−/−, MKK3−/−, and Akt-1−/−/MKK3−/− mice. D–F: densitometric analysis of collagen 1a1 content in each group. G–I: densitometric analysis of fibronectin contents in each group. Values represent means ± SE (n = 3–4/per group), *P < 0.05 vs. sham; ¥P < 0.05 vs. MI + Saline; £P < 0.05 vs. sham in genetic knockout mice.
Signaling proteins.
As shown in Fig. 8, A and B, ablation of Akt-1-1 did not result in a noticeable change in phosphorylated MKK3 following exendin-4 treatment. However, exendin-4 treatment was unable to increase phosphorylated Akt-1 in MKK3−/− mice (Fig. 8, C and D). Additionally, exendin-4 treatment mitigated the magnitude of reduction of the phosphorylated p38, which was absent in MKK3 mice (Fig. 8, E and F). Exendin-4 treatment caused increases in the phosphorylation of p85, but the ablation of Akt-1 did not affect phosphorylated p-85 (Fig. 8, G–J).
Fig. 8.
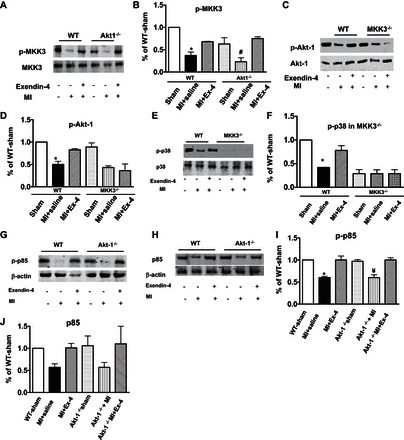
A–D: phosphorylated MKK3 and Akt-1 following exendin-4 treatment in Akt-1−/− mice and MKK3−/− mice. A and B: phosphorylated MKK3 in Akt-1−/− mice. C and D: phosphorylated Akt-1 in MKK3−/− mice. B and D: densitometric analyses of protein levels in each group, and values represent means ± SE (n = 3/per group), *P < 0.05 vs. sham; #P < 0.05 vs. MI + exendin-4. E and F: deletion of MKK3 attenuated p38 phosphorylation in exendin-4-treated myocardium. E: representative blots showing decreased p38 phosphorylation in MKK3−/− myocardium that received exendin-4 treatment. F: densitometric analysis of phosphorylated p38, and values represent means ± SE (n = 3/per group); *P < 0.05 vs. sham and MI + exendin-4. The preparation of samples and protocol for detecting proteins is described in detail in materials and methods. G–J: effect of exendin-4 treatment on phosphoinositide-3-kinase (PI3K)-p85 in Akt-1−/− mice. G and H: representative blots showing that exendin-4 treatment increased phosphorylated p85 and p85 and was not affected by ablation of Akt-1, downstream of PI3K. I and J: densitometric analyses of p-p85 and p85 in each group, and values represent means ± SE (n = 3/per group), *P < 0.05 vs. WT sham and MI + exendin-4; ¥P < 0.05 vs. Akt-1−/− sham and Akt-1−/− MI + exendin-4.
Apoptosis in the infarcted myocardium.
To estimate whether exendin-4 treatment would mitigate the occurrence of apoptosis and death in the infarcted mice, we detected the protein levels of cleaved PARP and active caspase-3 in both noninfarcted remote and border zones of an infarcted myocardium. As shown in Fig. 9, A–D, both cleaved PARP and active caspase-3 increased in all vehicle-treated MI mice compared with sham-operated animals in both noninfarcted remote and border zone infarcted myocardium. The densitometry analyses indicated that exendin-4 treatment dramatically reduced the levels of cleaved PARP and active caspase-3 in the infarcted wild-type mice compared with vehicle-treated mice (Fig. 9, E–L). However, deletion of Akt-1 (Fig. 9, A, B, E, F, I, and J), MKK3 (Fig. 9, C, D, G, H, K, and L), and/or both MKK3 and Akt-1 (Fig. 10, A–F) abrogated the effects of exendin-4 treatment on the suppression of cleaved PARP and active caspase-3. These results suggest that the antiapoptotic effect of exendin-4 treatment is dependent on Akt-1 and MKK3 in the MI heart.
Fig. 9.
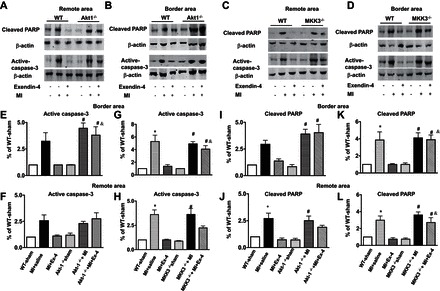
Exendin-4 treatment reduced active caspase-3 and cleaved poly(ADP-ribose)polymerase (PARP) in MI hearts. Representative blots of cleaved PARP and active caspase-3 in remote and border areas in sham, vehicle-treated MI, and exendin-4 treatment in WT, Akt-1−/− (A and B), and MKK3−/− mice (C and D), respectively. E–H: densitometric analysis of active caspase-3 in Akt-1−/− mice (E and F) and MKK3−/− mice (G and H). Cleaved PARP in sham, vehicle-treated MI, and exendin-4 treatment in WT, Akt-1−/− mice (I and J), and MKK3−/− mice (K and L), respectively. Values represent means ± SE (n = 3/per group), *P < 0.05 vs. WT sham; WT MI + exendin-4; #P < 0.05 vs. Akt-1−/− or MKK3−/− sham; &P < 0.05 vs. WT MI + exendin-4.
Fig. 10.
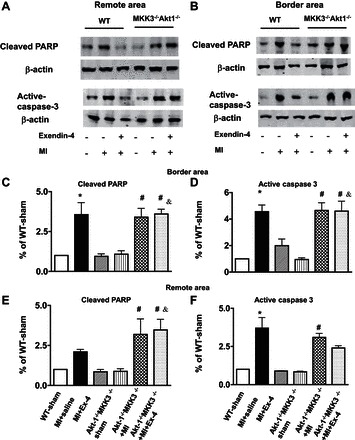
Cleaved PARP and active caspase-3 in Akt-1−/−;MKK3−/− sham, MI, and MI + exendin-4 treatment. A: remote. B: border area. Representative blots showing cleaved PARP and active caspase-3 in Akt-1−/−;MKK3−/− mice that underwent different treatments. C and E: densitometric analysis of cleaved PARP. D and F: analysis of active caspase-3 from remote and border area in Akt-1−/−;MKK3−/− mice. Values represent means ± SE (n = 3/per group); *P < 0.05 vs. WT sham and MI + exendin-4; #P < 0.05 vs. Akt-1−/−;MKK3−/− sham; &P < 0.05 vs. WT MI + exendin-4. The preparation of samples and protocol for detecting proteins is described in detail in materials and methods.
Angiogenesis in post-MI myocardium.
Vascular endothelial cell (CD31) and vascular α-SMA were utilized to examine microvessels in the infarcted myocardium. Exendin-4 treatment resulted in a robust increase in CD31 (Fig. 11A) and α-SMA positive capillary density (Fig. 11E) in the border area of infarct heart compared with vehicle-treated infarcted wild-type mice. However, the increase in CD31-stained angiogenesis following exendin-4 treatment was not observed in mice deficient of MKK3 and Akt-1 (Fig. 11, B–D). Likewise, there were no significant differences in α-SMA-positive microvessels in MKK3−/−, Akt-1−/−, and Akt-1−/−;MKK3−/− mice following the administration of exendin-4 (Fig. 11, F–H).
Fig. 11.
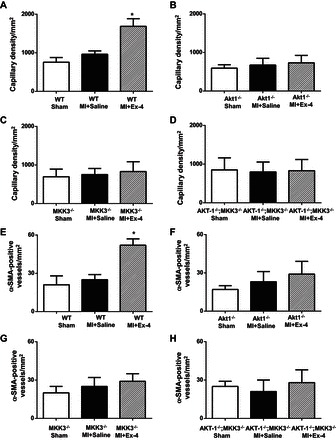
Exendin-4 treatment promotes angiogenesis in the infarcted myocardium. A–H: quantitative analyses in CD-31- and α-smooth muscle actin (α-SMA)-positive capillary density in WT (A and E), Akt-1−/− (B and F), MKK3−/− (C and G), and Akt-1−/−;MKK3−/− (D and H) mice that underwent different treatments, as described in materials and methods. Total number of vessels was calculated and normalized to the tissue area. Values are shown as means ± SE (n = 3/per group). *P < 0.05 vs. MI + saline.
DISCUSSION
Salient findings.
Our study is the first to use genetic and physiological approaches to document that exendin-4 treatment preserves myocardial function and prevents cardiac remodeling through MKK3/Akt-1. The improvement in functional recovery and the decreased remodeling observed by exendin-4 were abolished by genetic elimination of MKK3 and/or Akt-1. Exendin-4 treatment mitigated cardiac hypertrophy and interstitial fibrosis and attenuated scar size, but these effects were abrogated in mice deficient of MKK3 and Akt-1. Exendin-4 treatment decreased active-caspase 3 and cleaved PARP levels, but the antiapoptotic effects of exendin-4 treatment were inhibited by elimination of MKK3 and Akt-1. Furthermore, exendin-4 treatment induced an angiogenic response, but deletion of MKK3 and Akt-1 diminished the angiogenic response induced by exendin-4.
A plethora of experimental data has been generated concerning the role of GLP-1 in diabetes, but very limited evidence has focused on its cardiovascular effects. Both GLP-1(7–36) amide and the GLP-1 receptor agonist, exendin-4, are shown to increase heart rate and blood pressure in both anesthetized and conscious restrained rats. Although the mechanisms are controversial (6, 38), initial observation suggests that GLP-1 has the potential to modulate the cardiovascular system other than lowering glucose level (30). Promising clinical data showed that GLP-1 infusion improved regional and global function in patients with acute myocardial ischemia and severe systolic dysfunction after successful primary angioplasty (33). Sokos et al. (34) reported that a long-term infusion of GLP-1 improves both LV ejection fraction and functional capacity in human patients with advanced heart failure (34). This is in agreement with another pilot study showing that treatment with GLP-1 (7–36) improved LV function in patients with coronary artery disease (28).
The stimulation of GLP signaling with GLP-1 has also been demonstrated to improve cardiac performance in conscious dogs with dilated cardiomyopathy and rat ischemia/reperfusion injury (24, 42). Evidence from Yellon's observations suggests that GLP-1 added before ischemia caused a significant reduction of infarct size (10). This is consistent with our observation that exendin-4 treatment remarkably preserved myocardial function improvement in post-MI.
Our previous studies have suggested that activation of p38 is associated with the protection induced by adenosine receptor, histone deacetylase inhibition, and GLP-1 (41, 42). Deletion of MKK3 suppressed the preconditioning effect induced by histone deacetylase inhibition (41). MKK3 overexpression protected the heart against ischemia/reperfusion injury although MKK3 was reported to be involved in the early cardio-depressant action of tumor necrosis factor (7, 19). Moreover, it was reported that GLP-1 induced phosphorylation of MKK3/MKK6 in CHO/GLPR and RIN 1046-38 cells (21). In the present study, our data indicated that exendin-4 treatment resulted in a marked increase in phosphorylation of both MKK3 and p38, but the exendin-4-induced improvement in cardiac function was rendered completely absent by deletion of MKK3. These findings further address an involvement of p38 signaling in exendin-4-produced beneficial effect.
Exenatide was demonstrated to reduce myocardial infarction in association with an increase of Akt-1 phosphorylation (35). However, in dilated cardiomyopathy independent of an insulinotropic effect, GLP-1 did not show an increase in the phosphorylation of Akt-1 at Ser473 (8). GLP-1-induced cardioprotection against acute ischemia/reperfusion in the rat was not in association with Akt-1 Ser473 (42). However, in this observation, exendin-4 treatment induced a profound increase in Akt-1 phosphorylation at Ser129; this is a phosphorylation of Akt-1 at Ser129 that was not examined previously in response to GLP-1R stimulation. These studies are well characterized by the observation in which the functional role of Akt-1 depends on the extent to which an activated Akt-1 is induced (32). The discrepancy of GLP-1 on Akt-1 is likely to be associated with the differences in animal models used as well as pharmacological approaches (GLP-1 vs. GLP-1R agonist). GLP-1R agonist caused an increase in the phosphorylation of Akt-1 and induced cardioprotection in MI, which extends our knowledge of the relationship between GLP-1R and Akt-1 in modulating myocardial remodeling. This is consistent with a recent report demonstrating an improved cardiac function in Akt-1 transgenic mice (2). Inhibition of DPP4 increased angiogenic responses in diabetic cardiomyopathy (31). Human mesenchymal stem cells engineered with GLP-1 also activate a proangiogenic signaling pathway in limb ischemia (15). This is in line with a report showing that GLP-1 promotes angiogenesis in human endothelial cells through the Akt pathway (3). In our study, exendin-4 treatment significantly promoted capillary density, but this effect was abolished by the elimination of MKK3 and Akt-1, suggesting that Akt-1 and MKK3 mediate the GLP-1R mediated-proangiogenic effect. The increased MKK3 phosphorylation following exendin-4 treatments was not attenuated in Akt-1−/− mice. However, exendin-4-induced Akt-1 phosphorylation was lost in MKK3−/− myocardium, indicating that Akt-1 phosphorylation was secondary to MKK3 following exendin-4 treatment. Noticeably, the synergistic effect of MKK3 and Akt-1 was not demonstrated in MI hearts following exendin-4 treatment, which supports a cascade sequence of MKK3 and Akt-1.
Antiapoptosis is regarded as one of the most important cytoprotections among the pleiotropic actions reported for GLP-1 (17, 27). Exendin-4 treatment resulted in a decrease in cleaved PARP and active caspase-3; these decreases were abrogated by deletion of MKK3 and Akt-1. LV ablation-induced heart failure in rats treated with the DPP4 inhibitor, sitagliptin, attenuated cardiac remodeling and cardiomyocyte apoptosis (11). Delivery of GLP-1 encapsulating genetically modified mesenchymal stem cells in a post-MI pig improved LV function and reduced epicardial infarct size, which was associated with increased angiogenesis and an altered remodeling response (37). Exendin-4 and DPP4 attenuated cardiac remodeling in rat diabetic cardiomyopathy by reducing cardiac fibrosis (5). It was reported that GLP-1 and the exendin-4 analog AC3174 prevented cardiac remodeling in post-MI rat heart (18), which is supported by a recent clinical investigation showing that circulating dipeptidyl peptidase-4 activity is well correlated with cardiac dysfunction in human heart failure (11). The present study demonstrated that exendin-4 mitigates the hypertrophic response and prevented the deposition of collagen content, which was abrogated by depletion of MKK3 and Akt-1. However, in a long-term post-MI cardiac remodeling model, vildagliptin, an inhibitor of DPP4, produced no substantial protective effects on cardiac function or preservations in cardiac remodeling (39). This is likely due to the effects of different pharmacological agents and a different animal model. A previous report indicated that a large dose of exendin-4 could still produce protective effects in an isolated GLP-1R−/− mouse heart, implying that a GLP-1R-independent pathway mediates the cardioprotective effects of exendin-4 (4).
Conclusion.
The present study is the first to demonstrate that exendin-4 preserves cardiac functional recovery, reduces scar size, and attenuates myocardial remodeling in postinfarcted hearts. The improvement in cardiac function, prevention of myocardial remodeling, and reduction in scar size following exendin-4 treatment are mitigated by depletion of MKK3 and Akt-1. Exendin-4 treatment induces a robust neovascularization in the postinfarcted myocardium, which is dependent on MKK3 and Akt-1. Furthermore, exendin-4 treatment remarkably attenuated apoptotic protein levels, which were also diminished by the elimination of MKK3 and Akt-1. Taken together, our study reveals that exendin-4 treatment-induced cardioprotective effects in the infarcted hearts are dependent on both MKK3 and Akt-1.
GRANTS
The work is supported by the National Heart, Lung, and Blood Institute Grant (R01 HL089405 and R01 HL115265).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.D., L.Z., Z.W., N.Y., and Y.T.Z. performed experiments; J.D., L.Z., Z.W., and N.Y. analyzed data; J.D., L.Z., Z.W., and N.Y. prepared figures; Z.W., P.D.-S., S.Z., G.Q., and T.C.Z. interpreted results of experiments; L.W., P.Y.L., G.Q., and T.C.Z. conception and design of research; L.W. and T.C.Z. edited and revised manuscript; T.C.Z. drafted manuscript; T.C.Z. approved final version of manuscript.
REFERENCES
- 1.Angeli FS, Shannon RP. Incretin-based therapies: can we achieve glycemic control and cardioprotection? J Endocrinol 221: T17–T30, 2014. [DOI] [PubMed] [Google Scholar]
- 2.Araki S, Izumiya Y, Hanatani S, Rokutanda T, Usuku H, Akasaki Y, Takeo T, Nakagata N, Walsh K, Ogawa H. Akt-1-mediated skeletal muscle growth attenuates cardiac dysfunction and remodeling after experimental myocardial infarction. Circ Heart Fail 5: 116–125, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aronis KN, Chamberland JP, Mantzoros CS. GLP-1 promotes angiogenesis in human endothelial cells in a dose-dependent manner, through the Akt, Src and PKC pathways. Metabolism 62: 1279–1286, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 117: 2340–2350, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Barakat GM, Nuwayri-Salti N, Kadi LN, Bitar KM, Al-Jaroudi WA, Bikhazi AB. Role of glucagon-like peptide-1 and its agonists on early prevention of cardiac remodeling in type 1 diabetic rat hearts. Gen Physiol Biophys 30: 34–44, 2011. [DOI] [PubMed] [Google Scholar]
- 6.Barragán JM, Rodríguez RE, Eng J, Blázquez E. Interactions of exendin-(9–39) with the effects of glucagon-like peptide-1-(7–36) amide and of exendin-4 on arterial blood pressure and heart rate in rats. Regul Pept 67: 63–68, 1996. [DOI] [PubMed] [Google Scholar]
- 7.Bellahcene M, Jacquet S, Cao XB, Tanno M, Haworth RS, Layland J, Kabir AM, Gaestel M, Davis RJ, Flavell RA, Shah AM, Avkiran M, Marber MS. Activation of p38 mitogen-activated protein kinase contributes to the early cardiodepressant action of tumor necrosis factor. J Am Coll Cardiol 48: 545–555, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Bhashyam S, Fields AV, Patterson B, Testani JM, Chen L, Shen YT, Shannon RP. Glucagon-like peptide-1 increases myocardial glucose uptake via p38alpha MAP kinase-mediated, nitric oxide-dependent mechanisms in conscious dogs with dilated cardiomyopathy. Circ Heart Fail 3: 512–521, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bose AK, Mocanu MM, Carr RD, Yellon DM. Myocardial ischaemia-reperfusion injury is attenuated by intact glucagon like peptide-1 (GLP-1) in the in vitro rat heart and may involve the p70s6K pathway. Cardiovasc Drugs Ther 21: 253–256, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Bose AK, Mocanu MM, Carr RD, Brand CL, Yellon DM. Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes 54: 146–151, 2005. [DOI] [PubMed] [Google Scholar]
- 11.dos Santos L, Salles TA, Arruda-Junior DF, Campos LC, Pereira AC, Barreto AL, Antonio EL, Mansur AJ, Tucci PJ, Krieger JE, Girardi AC. Circulating dipeptidyl peptidase IV activity correlates with cardiac dysfunction in human and experimental heart failure. Circ Heart Fail 6: 1029–1038, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Drucker DJ. Biological actions and therapeutic potential of the glucagon-like peptides. Gastroenterology 122: 531–544, 2002. [DOI] [PubMed] [Google Scholar]
- 13.González N, Acitores A, Sancho V, Valverde I, Villanueva-Peñacarrillo ML. Effect of GLP-1 on glucose transport and its cell signalling in human myocytes. Regul Pept 126: 203–211, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Heo KS, Fujiwara K, Abe J. Glucagon-like peptide-1 and its cardiovascular effects. Curr Atheroscler Rep 14: 422–428, 2012. [DOI] [PubMed] [Google Scholar]
- 15.Katare R, Riu F, Rowlinson J, Lewis A, Holden R, Meloni M, Reni C, Wallrapp C, Emanueli C, Madeddu P. Perivascular delivery of encapsulated mesenchymal stem cells improves postischemic angiogenesis via paracrine activation of VEGF-A. Arterioscler Thromb Vasc Biol 33: 1872–1880, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Kolterman OG, Buse JB, Fineman MS, Gaines E, Heintz S, Bicsak TA, Taylor K, Kim D, Aisporna M, Wang Y, Baron AD. Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 88: 3082–3089, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drucker DJ. Glucagon-like peptide-1 receptor signaling modulates cell apoptosis. J Biol Chem 278: 471–478, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Liu Q, Anderson C, Broyde A, Polizzi C, Fernandez R, Baron A, Parkes DG. Glucagon-like peptide-1 and the exenatide analogue AC3174 improve cardiac function, cardiac remodeling, and survival in rats with chronic heart failure. Cardiovasc Diabetol 9: 76, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martindale JJ, Wall JA, Martinez-Longoria DM, Aryal P, Rockman HA, Guo Y, Bolli R, Glembotski CC. Overexpression of mitogen-activated protein kinase kinase 6 in the heart improves functional recovery from ischemia in vitro and protects against myocardial infarction in vivo. J Biol Chem 280: 669–676, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao W, Luo Z, Kitsis RN, Walsh K. Intracoronary, adenovirus-mediated Akt gene transfer in heart limits infarct size following ischemia-reperfusion injury in vivo. J Mol Cell Cardiol 32: 2397–402, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Montrose-Rafizadeh C, Avdonin P, Garant MJ, Rodgers BD, Kole S, Yang H, Levine MA, Schwindinger W, Bernier M. Pancreatic glucagon-like peptide-1 receptor couples to multiple G proteins and activates mitogen-activated protein kinase pathways in Chinese hamster ovary cells. Endocrinology 140: 1132–1140, 1999. [DOI] [PubMed] [Google Scholar]
- 22.Nielsen LL, Baron AD. Pharmacology of exenatide (synthetic exendin-4) for the treatment of type 2 diabetes. Curr Opin Investig Drugs 4: 401–405, 2003. [PubMed] [Google Scholar]
- 23.Nikolaidis LA, Elahi D, Hentosz T, Doverspike A, Huerbin R, Zourelias L, Stolarski C, Shen YT, Shannon RP. GLP-1 increased myocardial glucose uptake and improved LV performance in conscious dogs with pacing induced dilated cardiomyopathy. Circulation 110: 955–961, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Nikolaidis L, Elahi D, Shen Y, Shannon RP. Active metabolite of GLP-1 mediates myocardial glucose uptake and improves ventricular performance in conscious dogs with dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 289: H2401–H2408, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Nikolaidis LA, Mankad S, Sokos GG, Miske G, Shah A, Elahi D, Shannon RP. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 109: 962–965, 2004. [DOI] [PubMed] [Google Scholar]
- 26.Parkes DG, Pittner R, Jodka C, Smith P, Young A. Insulinotropic actions of exendin-4 and glucagon-like peptide-1 in vivo and in vitro. Metabolism 50: 583–589, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Ravassa S, Zudaire A, Díez J. GLP-1 and cardioprotection: from bench to bedside. Cardiovasc Res 94: 316–323, 2012. [DOI] [PubMed] [Google Scholar]
- 28.Read PA, Khan FZ, Dutka DP. Cardioprotection against ischaemia induced by dobutamine stress using glucagon-like peptide-1 in patients with coronary artery disease. Heart 98: 408–413, 2012. [DOI] [PubMed] [Google Scholar]
- 29.Reddy GK, Enwemeka CS. A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem 29: 225–229, 1996. [DOI] [PubMed] [Google Scholar]
- 30.Richter G, Feddersen O, Wagner U, Barth P, Goke R, Goke B. GLP-1 stimulates secretion of macromolecules from airways and relaxes pulmonary artery. Am J Physiol Lung Cell Mol Physiol 265: L374–L381, 1993. [DOI] [PubMed] [Google Scholar]
- 31.Shigeta T, Aoyama M, Bando YK, Monji A, Mitsui T, Takatsu M, Cheng XW, Okumura T, Hirashiki A, Nagata K, Murohara T. Dipeptidyl peptidase-4 modulates left ventricular dysfunction in chronic heart failure via angiogenesis-dependent and -independent actions. Circulation 126: 1838–1851, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 115: 2108–2118, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sokos GG, Bolukoglu H, German J, Hentosz T, Magovern GJ Jr, Maher TD, Dean DA, Bailey SH, Marrone G, Benckart DH, Elahi D, Shannon RP. Effect of glucagon-like peptide-1 (GLP-1) on glycemic control and left ventricular function in patients undergoing coronary artery bypass grafting. Am J Cardiol 100: 824–829, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Sokos GG, Nikolaidis LA, Mankad S, Elahi D, Shannon RP. Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. J Card Fail 12: 694–699, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Timmers L, Henriques JP, de Kleijn DP, Devries JH, Kemperman H, Steendijk P, Verlaan CW, Kerver M, Piek JJ, Doevendans PA, Pasterkamp G, Hoefer IE. Exenatide reduces infarct size and improves cardiac function in a porcine model of ischemia and reperfusion injury. J Am Coll Cardiol 53: 501–510, 2009. [DOI] [PubMed] [Google Scholar]
- 36.Vyas AK, Yang KC, Woo D, Tzekov A, Kovacs A, Jay PY, Hruz PW. Exenatide improves glucose homeostasis and prolongs survival in a murine model of dilated cardiomyopathy. PLoS One 6: e17178, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wright EJ, Farrell KA, Malik N, Kassem M, Lewis AL, Wallrapp C, Holt CM. Encapsulated glucagon-like peptide-1-producing mesenchymal stem cells have a beneficial effect on failing pig hearts. Stem Cells Transl Med 1: 759–769, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto H, Lee CE, Marcus JN, Williams TD, Overton JM, Lopez ME, Hollenberg AN, Baggio L, Saper CB, Drucker DJ, Elmquist JK. Glucagon-like peptide-1 receptor stimulation increases blood pressure and heart rate and activates autonomic regulatory neurons. J Clin Invest 110: 43–52, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin M, Silljé HH, Meissner M, van Gilst WH, de Boer RA. Early and late effects of the DPP-4 inhibitor vildagliptin in a rat model of post myocardial infarction heart failure. Cardiovasc Diabetol 10: 85, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young AA, Gedulin BR, Bhavsar S, Bodkin N, Jodka C, Hansen B, Denaro M. Glucose-lowering and insulin-sensitizing actions of exendin-4: studies in obese diabetic (ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus monkeys (Macaca mulatta). Diabetes 48: 1026–1034, 1999. [DOI] [PubMed] [Google Scholar]
- 41.Zhao TC, Du J, Zhuang S, Liu P, Zhang LX. HDAC inhibition elicits myocardial protective effect through modulation of MKK3/Akt-1. PLoS One 8: e65474, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao T, Parikh P, Bhashyam S, Bolukoglu H, Poornima I, Shen YT, Shannon RP. Direct effects of glucagon-like peptide-1 on myocardial contractility and glucose uptake in normal and postischemic isolated rat hearts. J Pharmacol Exp Ther 317: 1106–1113, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Zhao TC, Taher MM, Valerie KC, Kukreja RC. p38 Triggers late preconditioning elicited by anisomycin in heart: involvement of NF-kappaB and iNOS. Circ Res 89: 915–922, 2001. [DOI] [PubMed] [Google Scholar]
- 44.Zhang L, Chen B, Zhao Y, Dubielecka PM, Wei L, Qin GJ, Chin YE, Wang Y, Zhao TC. Inhibition of histone deacetylase-induced myocardial repair is mediated by c-kit in infarcted hearts. J Biol Chem 287: 39338–39348, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]