

Abstract
Energy homeostasis in eukaryotic cells is a complex and fundamental process that is misregulated in several human diseases. A key component of energy regulation is a process called autophagy that involves the recycling of cellular components. There has been much recent interest in studying the mechanism of autophagy to understand an important cellular process and to evaluate therapeutic potential in targeting autophagy. Activation of a kinase called ULK1 initiates autophagy by driving downstream pathways that lead to the formation of double membrane bound vesicles that surround the cellular contents that are to be degraded. Here, we report the discovery of an inhibitor of ULK1 with improved selectivity and a high-resolution crystal structure of the compound bound to the kinase, which will be useful tools for studying autophagy in cells.
Graphical Abstract

1. Introduction
Autophagy is a conserved process in eukaryotic cells for breaking down cellular components from proteins to entire organelles for energy, building blocks, and quality control[1]. There has been increasing interest in the pathway as a fundamental cellular mechanism and as a complex component of cancer development and survival. Through its ability to provide energy by recycling cellular components, it is thought that cancer cells can survive energy shortage during rapid growth when nutrients are limiting, for example prior to angiogenesis[2]. However autophagy is also thought to protect cells against tumorigenesis by removing damaged components that might harm cellular integrity[3, 4]. Despite its important cellular role and its ability to serve as a potential target in several diseases, including cancer and neurodegeneration[5-7], it is still poorly understood. What is known is that upon activation by upstream nutrient deprivation signals, including mTOR and AMPK[8-10], the autophagy pathway is initiated by a kinase called ULK1[11, 12] (Figure 1A). A closely related kinase, ULK2, has also been implicated in this process, but it has not been as well characterized and has a less clear role in autophagy. Activation of these kinases, through a series of autophagy (atg) protein dependent steps that are only recently becoming clearer[13-17], ULK1 and its complex partners drive the formation of double membrane bound vesicles called autophagophores, which form around cellular components that are to be degraded. These vesicles then fuse with lysosomes to degrade the contents of the autophagophores. There is broad interest in developing small molecule tools to study autophagy and evaluate it as a potential therapeutic target. However, few selective tools are available to specifically target autophagy. Compounds used in the field include rapamycin and chloroquine, two molecules that have broad cellular effects in addition to modulating autophagy[18-21]. Previously we developed a series of non-selective ULK1 inhibitors and solved the structure of ULK1[22]. Here, we report new inhibitors that show improved selectivity for ULK1 and a high-resolution crystal structure of ULK1 bound to an aminopyrimidine scaffold. The compounds should be useful tools for modulating autophagy in cells and studying its role in several pathologies, and the structure will help guide further improvements in ULK1 inhibitors.
Figure 1. Autophagy activation through ULK1 and ULK2.

(a) Schematic of ULK1 and ULK2 activation and role in the autophagy activation pathway. (b) Bacterial purification of ULK2 for in vitro assays. Coumassie gel shows purified SUMO-tagged ULK2 next to molecular weight ladder on the left. The SUMO-tagged ULK2 runs around 47 kDa, with a small degradation band visible around 25 kDa.
2. Results
Recently we reported the first structure of human ULK1 and a series of highly potent active site inhibitors, with compound 1 (see Table 1 below) being the most potent[22]. These compounds were not selective against ULK1 and are therefore not useful in cells for studying autophagy. Attempts to improve selectivity for ULK1 on the aminoquinazoline core were unsuccessful. Here, we explore another lead compound class from our original screen that contained an aminopyrimidine scaffold: BX-795[23] (Table 1). This compound is a potent and relatively selective inhibitor of PDK1[23], which also inhibits TBK1 and IKKε[24]. We anticipated that this compound would be a good starting point for a more selective ULK1 inhibitor that could be used in cells, since it already showed good selectivity for a few kinases and had good potency against ULK1.
Table 1. Potencies of ULK1 inhibitors.
Compounds were tested against ULK1, ULK2 and PDK1 in an in vitro kinase assay. Assay was performed in duplicate.
| Compound | Structure | ULK1 IC50 (nM) | ULK2 IC50 (nM) | PDK1 IC50 (nM) |
|---|---|---|---|---|
| Compound 1 |
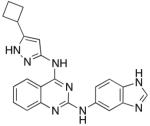
|
5.3 ± 0.91 | 13 ± 3.3 | 420 ± 43 |
| BX-795 |

|
87 ± 7.4 | 310 ± 88 | 65 ± 3.9 |
| Compound 2 |
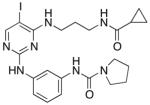
|
67 ± 15 | 200 ± 49 | 56 ± 5.5 |
| Compound 3 |
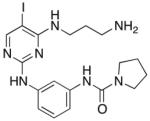
|
120 ± 1.7 | 360 ± 79 | 710 ± 270 |
| MRT67307 |
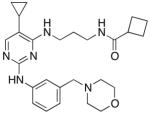
|
170 ± 11 | 230 ± 21 | 421±19 |
We wanted to examine the inhibition of ULK2 with our new compounds as well, since little is known about the biological role of this kinase and since ULK2 had not been tested with either the aminoquinazoline or aminopyrimidine class of compounds. Compounds that inhibit both ULK1 and ULK2 might be more potent inhibitors of autophagy, whereas selective compounds could help tease apart the roles of the two kinases. We developed a bacterial expression system for ULK2, similar to ULK1, to enable the rapid production of protein, the introduction of mutations, and eventual structural studies. Using a sumo-tagged kinase domain construct, we were able to obtain milligram quantities of the kinase similar to ULK1 (Figure 1B). We would then be able to screen both enzymes against this new class of inhibitors.
We started chemical optimization by exploring substitutions for the iodine position on the pyrimidine. However, all attempts at bulkier substituents at this position complete abrogated inhibition of ULK1 (data not shown). To understand the selectivity of the BX-795 compound towards ULK1 and its preference for an iodine substituent on the pyrimidine ring, and to rationally improve its potency, we set out to crystallize ULK1 with BX-795. We were unable, however, to obtain crystals with ULK1 bound to BX-795, either in the same conditions as for compound 1 or in a broad screen. We then analyzed the previous crystal structures and hypothesized that the extended diaminopropyl linker of BX-795 compared to our previous scaffold might interfere with the tight crystal packing between ULK1 protomers in the crystal. In addition, BX-795 is an extremely potent inhibitor of PDK1, so we wanted to increase the selectivity of the compounds for ULK1 over PDK1 to develop a selective autophagy inhibitor. We therefore introduced a series of substitutions around the BX-795 scaffold (Figure 2) to explore the structure activity relationships for inhibition of ULK1 and focused on smaller modifications including the scaffold with the free diaminopropane linker.
Figure 2. Synthetic scheme for ULK1 inhibitors.

Reagents and conditions. (a) ACN, TEA; (b) MeOH, conc. HCl; (c) TFA; (d) cyclopropanecarbonyl chloride, DIPEA, DMF.
The smaller compounds showed good potency against ULK1 (Table 1) showing IC50 values of around 100 nM or better, including compound 2 which was more potent than BX-795 against ULK1. The smaller compound 3 was not quite as potent but substantially smaller than BX-795 and would be ideal for structural studies. Furthermore, compound 3 showed dramatically reduced potency towards PDK1 and good selectivity throughout the kinome (Supplementary Figure 1).
Using compound 3 that has a less bulky diaminopropyl substituent we were able to obtain a high-resolution crystal structure of the inhibitor bound to ULK1 (Figure 3, Table 2, and Supplementary Figure 2). The compound binds in the active site of the kinase making traditional hinge contacts to the protein from the aminopyrimidine (Figure 3A). The overall conformation of the kinase is similar to the two previous structures of ULK1 (PDB 4WNO and PDB 4WNP), with an RMSD of 0.2371 and 0.6687 Å, respectively. The main difference in the kinase is a movement of the β sheet in the N-terminal lobe with Gly 23 being displaced 0.9 Å towards the inhibitor, which allows Ile 22 to twist away from the bulky diaminopropyl substituent on the pyrimidine core (Figures 3B and 3C). The other difference is the gatekeeper methionine moves towards the iodine group, presumably to adopt a favorable dipole-dipole interaction.
Figure 3. Structure of ULK1 bound to inhibitors.

(A) Structure of compound 3 bound to ULK1. The key sidechains (cyan) are shown which surround the compound (shown in yellow) in the active site. The backbone that makes hinge contacts to the inhibitor is also shown, with sidechains omitted for clarity. (B) Our previously reported structure of compound 1 (orange) bound to ULK1. (C) Overlay of the two inhibitor complexes, with the kinase and sidechains from the compound 3 complex shown.
Table 2.
X-ray data collection and refinement statistics.
| Compound 3:ULK1 complex | |
|---|---|
| Data collection | |
| Space group | P42212 |
| Cell dimensions | |
| a, b, c (Å) | 66.86, 66.86, 116.61 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 47.28-1.74 (1.83-1.74)* |
| Rsym or Rmerge | 0.116 (0.690) |
| I / σI | 7.5 (2.0) |
| Completeness (%) | 96.4 (94.3) |
| Redundancy | 4.7 (3.7) |
| Refinement | |
| Resolution (Å) | 43.81 - 1.74 |
| No. reflections | 26552 |
| Rwork / Rfree | 0.1688/0.2069 |
| No. atoms | |
| Protein | 2459 |
| Ligand/ion | 45 |
| Water | 156 |
| B-factors | |
| Protein | 18.31 |
| Ligand/ion | 26.37 |
| Water | 25.14 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.094 |
Highest resolution shell shown in parentheses. Data was collected from a single crystal
Compared to the previous lead compound, 1, compound 3 adopts a similar orientation in the active site with the diaminophenyl group occupying a similar space as the quinazoline. However, the pyrrolidine urea moiety projects off of this phenyl group into previously unoccupied space in the pocket. We speculate that this area provides selectivity to this scaffold, in addition to the aminopropyl group that requires a flexible orientation of Ile 22 to pack above it, since this is the part of the molecule that differs most from our previous scaffold. Lastly we note that the iodine on the new pyrimidine scaffold overlaps with the cyclobutyl substituent off the old core, which suggested a path for optimization of the new scaffold. We can compare our structure to those of closely related kinases bound to the BX-795 scaffold. Structures of PDK1[23] and TBK1[25] show similar orientations of the scaffold to our compound 3 bound to ULK1 (Supplementary Figure 3). The smaller free amine on compound 3 allows our compound to adopt a more compact conformation in our crystal packing. The active sites of these kinases show nearly identical contacts around the scaffold, which is consistent with the selectivity of this scaffold towards these kinases. The main differences are the gatekeeper residues, the residues contacting the urea in the inhibitor, and two sidechains near the pyrrolidine moiety, which are different in all three proteins (Supplementary Figure 3 and Supplementary Figure 4). These sites could provide guidance for further selectivity in the compounds. During the preparation of this manuscript, another report showed a similar compound that exhibited strong potency against ULK1 and ULK2 and showed inhibition of autophagy in cells [26], MRT67307 (see Table 1). This compound indeed combines some of the features of our two classes of compounds, with a cyclopropyl group similar to our cyclobutyl from our quinazolines replacing the iodine substituent on pyrimidine ring.
Lastly, we evaluated the compounds to see if they inhibited autophagy in cells. We treated HeLa cells for 24 hours with compounds 2, 3, BX-795 and MRT67307 at 4μM. We then blotted for LC3 levels as a readout for autophagosome formation (Figure 4). The LC3 protein is lipidated during autophagy so it can be incorporated into the autophagosomes, converting it from the LC3-I form to the faster migrating LC3-II form, which makes it a common marker of autophagosome formation[27]. Incubation with the ULK1 inhibitors led to a clear accumulation of the LC3-I form relative to LC3-II, which suggests a blockage of autophagy due to ULK1 inhibition. Therefore, these compounds show promise as tools to selectively inhibit autophagy in cells. Other features of the quinazoline compounds could be incorporated onto the pyrimidine scaffold to further optimize this kinase inhibitor.
Figure 4. Inhibition of ULK1 in cells.

The ULK1 inhibitors were evaluated for inhibition of autophagy in cells, and then evaluated by Western blot. HeLa cells were treated with the indicated compounds for 24 hours at a concentration of 4 μM. The lysates were then immunoblotted for LC3 and actin (as a loading control). The compounds led to an accumulation of the LC3-I precursor, which suggests a blockage of autophagy.
3. Conclusions
We report a new scaffold for inhibiting ULK1 that should be useful as a tool compound and have a structural understanding of its inhibition. We think this a promising class of compounds to modulate autophagy in cells and explore its role in cancer. Similar compounds confirm the utility of this scaffold for inhibiting ULK1, and our structure shows how the compounds bind. Our structure also explains the resistant mutant reported for MRT67307, Met92Thr, which presents a β-branched amino acid as the gatekeeper residue into the tightly fitting cyclopropyl group. Finally, the structure also suggests the residues that provide selectivity to this class of compounds. Our structures should provide guidance on tuning the compounds to minimize unwanted off-target effects. We also note that care should be taken in the interpretation of cellular experiments using BX-795 given its potent inhibition of ULK1.
4. Experimental
4.1 Expression of ULK1 and ULK2
ULK1 was expressed and purified as previously reported. ULK2 was cloned from cDNA into a sumo-tagged vector using the following primers: MBL319N (5’ CACAGAGAACAGATTGGTGGTG GTTCTATGGAGGTGGTGGGTGACTTCG 3’) and MBL319C (5’ CGTGGCACCAGAGCGAGCTCTT TATTTTACTGGACCTTGCTCAAGAAAAG 3’). The plasmid was then transformed into KRX cells along with lambda phosphatase and the protein was expressed and purified as was previously reported for ULK1.
4.2 Synthesis of inhibitors
General Methods: Reactions were performed in sealed vials with magnetic stirring. 1H and 13C NMR spectra were recorded on a Bruker Avance DRX500 spectrometer, Chemical shifts are reported in δ (ppm) relative to solvent as either s (singlet), d (doublet), t (triplet), or m (multiplet). Low resolution mass spectra (LC/ESI-MS) were recorded in positive mode on a Waters TQ detector with an Acquity UPLC equipped with a BEH C18 column. All commercial reagents were used without further purification. All RP-HPLC runs were performed with a Waters 2545 binary gradient module equipped with an XBridge prep C18 column using H2O + 0.1% formic acid and CH3CN + 0.1% formic acid (5-95% gradient) while monitoring at 254 nm.
N-(3-((4-((3-aminopropyl)amino)-5-iodopyrimidin-2-yl)amino)phenyl)pyrrolidine-1-carboxamide 3
The mixture of 2,4-dichloro-5-iodopyrimidine (400 mg) and N-(3-aminopropyl)carbamic acid tert-butyl ester (0.24 ml) in 3 ml of ACN with 0.23 ml od TEA was stirred overnight at rt. The product was then precipitated by the addition of water and collected and dried as an opaque yellow oil. 20 mg of this product was then reacted with N-(3-aminophenyl)pyrrolidine-1-carboxamide (14.9 mg, prepared previously[28]) overnight at 115°C in 1 ml of MeOH in a sealed vial with 1.5 μl conc. HCl. Then 1 ml of TFA was added overnight at rt to deprotect. The product was then purified by RP HPLC to give the final product, which was then lyophilized to give a white powder. Overall yield of 16%. LC-MS m/z 482.62 [M+H+]. 1H NMR (400 MHz, DMSO-d6) δ PPM 1.81 (m, 2H), 1.85 (m, 4H), 2.76 (t, 2H, J= 7.24 Hz ), 3.36 (m, 4H), 3.50 (m, 2H), 6.77 (s, 2H), 6.96 (d, 1H, J = 8.01 Hz), 7.07 (t, 1H, J = 8.01 Hz), 7.24 (d, 1H, J= 8.36 Hz), 7.91 (s, 1H), 8.01 (s, 1H), 8.10 (s, 1H), 8.35 (s, 1H), 9.10 (s, 1H)13C NMR (400 MHz, DMSO-d6) δ ppm 25.044, 27.517, 36.890, 37.822, 45.678, 64.777, 111.044, 112.928, 113.249, 127.887, 140.557, 140.619, 153.995, 158.981, 160.066, 161.744
N-(3-((4-((3-(cyclopropanecarboxamido)propyl)amino)-5-iodopyrimidin-2-yl)amino)phenyl)pyrrolidine-1-carboxamide 2
Cyclopropanecarbonyl chloride (3 μl) was added to compound 2 in 1 ml DMF with 20 μl DIPEA on ice. After five minutes the reaction was moved to rt for 1 hour. The final compound was then purified by RP HPLC and lyophilized to give the final product as a white powder with 80% yield. LC-MS m/z 550.72 [M+H+]. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.65 (m, 4H), 1.49 (m, 1H), 1.67 (m, 2H), 1.85 (m, 4H), 3.11 (m, 2H), 3.35 (m, 4H), 3.44 (m, 2H), 6.64 (t, 1H, J= 5.84 Hz ), 6.97 (d, 1H, J = 8.08 Hz ), 7.08 (t, 1H, J = 8.03 Hz), 7.27 (d, 1H, J = 8.03 Hz), 7.88 (s, 1H), 7.99 (s, 1H), 8.07 (t, 1H, J = 5.67 Hz ), 8.10 (s, 1H), 9.07 (s, 1H) 13C NMR (400 MHz, DMSO-d6) δ ppm 6.129, 13.641, 25.027. 29.088, 36.188, 38.393, 45.650, 64.861, 111.228, 112.901. 113.323, 127.822. 140.499, 140.674, 153.981, 159.030, 159.923, 161.603, 172.610
4.3 Inhibition assay
For ULK1 and ULK2 in vitro kinase assays, the enzymes were tested at 5 nM with 50 μM cold ATP, 0.05 μCi/μl 32P-γ-ATP and 20 μM myelin basic protein (Millipore) as the substrate, as previously reported. For PDK1 assays, recombinant PDK1 and synthetic PDK1tide (sequence: KTFCGTPEYLAPEVRREPRILSEEEQ-EMFRDFDYIADWC) were purchased from Promega. The PDK1 assay used 5 nM enzyme, 50 μM peptide in a buffer containing 50 mM HEPES pH 7.5, 10 mM MgCl2, 1 mg/ml BSA, 0.02 % Triton-X, and 50 μM DTT. The reactions were terminated after 7 minutes by spotting onto nitrocellulose paper, which was then washed in 1% phosphoric acid three times and then dried and imaged on a Typhoon imager. IC50 values were calculated with GraphPad Prism. Compound 3 was profiled against 50 kinases by the International Centre for Protein Kinase Profiling in duplicate using a radioactive filter-binding assay.
4.4 Crystallization and structure determination
Untagged ULK1 was purified as previously reported. ULK1 at 7 mg/ml was incubated with 300 μM compound 3. Crystals were obtained by seeding the compound 3:ULK1 complex with crystals of compound 1:ULK1 using the seed bead kit (Hampton) grown in 1.55 M sodium malonate pH 7 with 0.35 M sodium malonate pH 5. The compound 3:ULK1 crystals were iteratively seeded 3 times before being cryoprotected in 1.8 M sodium malonate pH 7, 0.4 M sodium malonate pH 5 and 17 % (v/v) glycerol and flash frozen in liquid nitrogen. Data was collected at ALS beamline 8.2.1. The data was processed with iMosflm and scaled using Scala in the CCP4 suite to 1.74 Å. The structure was then solved by molecular replacement using the ULK1 structure (pdb 4WNO) as a search model. The ligand was optimized using Phenix ELBOW[29] and the structure was refined in Phenix[30] using coordinate, ADP, and TLS[31, 32] refinement, with intermittent manual adjustments in Coot[33] to a final Rwork/Rfree of 0.1688/0.2069. All structural figures were made with PyMOL[34].
The coordinates of the ULK1:Compound 3 complex have been deposited in the Protein Data Bank with accession code XXXX
4.5 Cellular assays and immunoblotting
To evaluate the cellular efficacy of the compounds, HeLa cells were grown in Dulbecco modified Eagle medium (DMEM) with 10% fetal bovine serum to approximately 50% confluency in 6 cm dishes and treated for 24 hours with the indicated compounds or DMSO. Cells were then washed with cold PBS and lysed in buffer containing TBS pH 7.6, 1% Triton-X, 0.2 % SDS, and protease and phosphatase inhibitor tablets (Roche). After pelleting the cell debris, the lysate was quantified using a protein assay (Bio-Rad) and immunoblotted. The LC3 antibody was from Cell Signaling (#12741), the actin antibody was from Sigma (A1978) and the blots were developed using a LI-COR imaging system.
Supplementary Material
Acknowledgements
This work was supported by the Howard Hughes Medical Institute and NIH U19AI109622 and R01AI099245 to K.M.S. M.B.L. is a Merck fellow of the Helen Hay Whitney Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 2.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, Mautner J, Tonon G, Haigis M, Shirihai OS, Doglioni C, Bardeesy N, Kimmelman AC. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat. Rev. Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang X, Overholtzer M, Thompson CB. Autophagy in cellular metabolism and cancer. J. Clin. Invest. 2015;125:47–54. doi: 10.1172/JCI73942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nature reviews. Drug discovery. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karantza-Wadsworth V, White E. Role of autophagy in breast cancer. Autophagy. 2007;3:610–613. doi: 10.4161/auto.4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007;282:25464–25474. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- 12.Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy. 2013;9:124–137. doi: 10.4161/auto.23323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 14.Fujioka Y, Suzuki SW, Yamamoto H, Kondo-Kakuta C, Kimura Y, Hirano H, Akada R, Inagaki F, Ohsumi Y, Noda NN. Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat. Struct. Mol. Biol. 2014;21:513–521. doi: 10.1038/nsmb.2822. [DOI] [PubMed] [Google Scholar]
- 15.Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J. Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kofinger J, Ragusa MJ, Lee IH, Hummer G, Hurley JH. Solution structure of the atg1 complex: implications for the architecture of the phagophore assembly site. Structure. 2015;23:809–818. doi: 10.1016/j.str.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stjepanovic G, Davies CW, Stanley RE, Ragusa MJ, Kim do J, Hurley JH. Assembly and dynamics of the autophagy-initiating Atg1 complex. Proc. Natl. Acad. Sci. U. S. A. 2014;111:12793–12798. doi: 10.1073/pnas.1407214111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J, Vinckier S, Vankelecom H, Garmyn M, Vion AC, Radtke F, Boulanger C, Gerhardt H, Dejana E, Dewerchin M, Ghesquiere B, Annaert W, Agostinis P, Carmeliet P. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell. 2014;26:190–206. doi: 10.1016/j.ccr.2014.06.025. [DOI] [PubMed] [Google Scholar]
- 19.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 20.Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA, Debnath J, Shokat KM, Weiss WA. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Science signaling. 2010;3:ra81. doi: 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu L, Gu C, Zhong D, Shi L, Kong Y, Zhou Z, Liu S. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett. 2014;355:34–45. doi: 10.1016/j.canlet.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 22.Lazarus MB, Novotny CJ, Shokat KM. Structure of the Human Autophagy Initiating Kinase ULK1 in Complex with Potent Inhibitors. ACS Chem. Biol. 2015;10:257–261. doi: 10.1021/cb500835z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feldman RI, Wu JM, Polokoff MA, Kochanny MJ, Dinter H, Zhu D, Biroc SL, Alicke B, Bryant J, Yuan S, Buckman BO, Lentz D, Ferrer M, Whitlow M, Adler M, Finster S, Chang Z, Arnaiz DO. Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J. Biol. Chem. 2005;280:19867–19874. doi: 10.1074/jbc.M501367200. [DOI] [PubMed] [Google Scholar]
- 24.Clark K, Plater L, Peggie M, Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J. Biol. Chem. 2009;284:14136–14146. doi: 10.1074/jbc.M109.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, Bowman KK, Starovasnik MA, Dueber EC. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc. Natl. Acad. Sci. U. S. A. 2012;109:9378–9383. doi: 10.1073/pnas.1121552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petherick KJ, Conway OJ, Mpamhanga C, Osborne SA, Kamal A, Saxty B, Ganley IG. Pharmacological Inhibition of ULK1 Kinase Blocks Mammalian Target of Rapamycin (mTOR)-dependent Autophagy. J. Biol. Chem. 2015;290:11376–11383. doi: 10.1074/jbc.C114.627778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamguney T, Zhang C, Fiedler D, Shokat K, Stokoe D. Analysis of 3-phosphoinositide-dependent kinase-1 signaling and function in ES cells. Exp. Cell Res. 2008;314:2299–2312. doi: 10.1016/j.yexcr.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moriarty NW, Grosse-Kunstleve RW, Adams PD. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta crystallographica. Section D, Biological crystallography. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta crystallographica. Section D, Biological crystallography. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Painter J, Merritt EA. A molecular viewer for the analysis of TLS rigid-body motion in macromolecules. Acta crystallographica. Section D, Biological crystallography. 2005;61:465–471. doi: 10.1107/S0907444905001897. [DOI] [PubMed] [Google Scholar]
- 32.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta crystallographica. Section D, Biological crystallography. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 33.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica. Section D, Biological crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schrodinger LLC. The PyMOL Molecular Graphics System. 2010 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.