

Abstract
Bipolar disorder (BD) and schizophrenia (SZ) are known to share common genetic and psychosocial risk factors. A recent epigenome-wide association study performed on blood samples from SZ patients found significant hypomethylation of FAM63B in exon 9. Here, we used iPLEX-based methylation analysis to investigate two CpG sites in FAM63B in blood samples from 459 BD cases and 268 controls. Both sites were significantly hypomethylated in BD cases (lowest p value = 3.94 × 10−8). The methylation levels at the two sites were correlated, and no strong correlation was found with nearby single nucleotide polymorphisms (SNPs), suggesting that methylation differences at these sites are not readably picked up by genome-wide association studies. Overall, FAM63B hypomethylation was found in BD patients, thus replicating the initial finding in SZ patients. This study suggests that FAM63B is a shared epigenetic risk gene for the two disorders.
Electronic supplementary material
The online version of this article (doi:10.1186/s13148-016-0221-6) contains supplementary material, which is available to authorized users.
Keywords: Epigenetics, DNA methylation, Candidate gene, Mental disorder, iPLEX, FAM63B, Bipolar disorder
Introduction
Bipolar disorder (BD) and schizophrenia (SZ) are severe complex mental disorders known to have common genetic [1] and psychosocial risk factors [2], as well as overlap in some of their symptoms [3]. Together they are classified as major psychosis and contribute significantly to the global burden of disease [4]. Several studies have investigated differences in DNA methylation between individuals with major psychosis and healthy controls in various tissues and associated this epigenetic modification with psychiatric phenotype [5–8]. A recent sequencing-based epigenome-wide association study (EWAS) performed on blood samples from 759 SZ cases and 738 control individuals identified differential DNA methylation in family with sequence similarity 63, member B (FAM63B) gene in a region of exon 9 as their top EWAS finding (p value = 6.3 × 10−11), thereby becoming the first to implicate this gene in a psychiatric disorder [9]. In the same publication, the authors then used targeted bisulfite pyrosequencing of three specific CpG sites in an 18 bp region of FAM63B to follow up the top hit in an independent replication sample of more than a thousands individuals. For all three CpG sites, they obtained independent replication with p values ranging from 2.76 × 10−12 (first CpG site in 18 bp region) to 2.31 × 10−10 (last site in the 18 bp region) [9]. The FAM63B gene spans 86 kb on chromosome 15q22.1, contains nine exons in total and one CpG island that overlaps with exon 1 (Fig. 1). The role of FAM63B in the pathophysiology of SZ is unclear. Due to the high degree of comorbidity between SZ and BD, as well as the established evidence for shared genetic risk factors for the two disorders [1, 10], it is highly relevant to investigate if the CpG sites in SZ risk locus in FAM63B demonstrates the same methylation pattern in BD. The aim of the study was to determine if methylation FAM63B is associated with BD. We used the Sequenom MassARRAY assays targeting specifically the two CpG sites identified as the sites of most interest in SZ [9] to analyze DNA derived from whole blood from 459 BD patients and 268 control individuals.
Fig. 1.
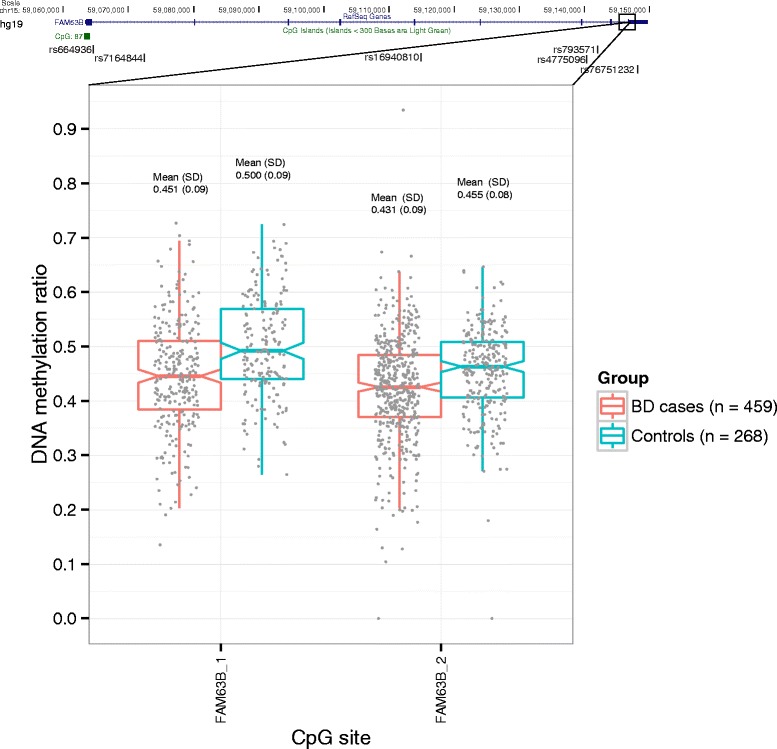
DNA methylation levels in cases and controls of two CpG sites in FAM63B exon 9 (lower panel). Gene structure of FAM63B and position of six relevant SNPs (four genotyped from Affymetrix 500K array, a significant cis-mQTL (rs76751232), and one SNP associated with SZ in a genome-wide association analysis (rs793571) [21]. The upper and lower hinges correspond to the 25th and 75th percentiles, while the whiskers extend from hinges to the highest and lowest values within 1.5 * IQR (inter-quartile range)
Methods
Study population
Bipolar disorder patients and control individuals were collected at UCL and collaborating clinical centers. Cases (n = 459) were Caucasian individuals who received clinical diagnoses of BD according to UK National Health Service (NHS) psychiatrists at interview using the categories of the International Classification of Disease version 10 (ICD10). Control individuals (n = 268) were recruited from London branches of the National Blood Service, from local NHS family doctor clinics, and from university student volunteers. All control individuals were interviewed with the Schizophrenia and Affective Disorders Schedule-Life Time version to exclude all psychiatric disorders [11]. The sample is partly overlapping with the UCL sample described in Sklar et al. [12]. The overlap is defined as all individuals from the UCL sample where DNA was still available and where the DNA was extracted from whole blood (n = 587). Additional cases and controls were included using similar inclusion criteria (n = 140), reaching a sample size of 727 participants. For a description of the sample see Additional file 1: Table S1.
Ethics, consent, and permissions
The project was approved by the Metropolitan Multicenter Research Ethics Committee, and all participants provided written informed consent.
Methylation methods
Genomic DNA was bisulfite converted with the use of EZ DNA Methylation Kit (Zymo Research, Freiburg, Germany). The MassARRAY Designer tool in Typer 4.0 (Sequenom, San Diego, CA, USA) was used to design independent iPLEX assays for the same CpG sites as investigated in the Swedish EWAS study [9]. The FAM63B_1 assay targeted CpG at position chr15:5,9146,738 (hg19) (corresponding to the first site in 18 bp region in Aberg et al.), and FAM63B_2 targeted CpG site at position chr15:59,146,756 (hg19) (corresponding to the last site in 18 bp region in Aberg et al. [9]). The two CpG sites are positioned 18 bp apart in exon 9 of FAM63B and are not part of a CpG island (Fig. 1). Primers for iPLEX assays can be found in Additional file 2: Table S2. Bisulfite converted DNA samples were prepared according to the iPLEX manufacturer’s protocol. The converted DNA was dispensed onto a SpectroCHIPII and analyzed on a MassARRAY workstation in the Allelotype mode (Sequenom). Eighteen samples were independently bisulfite converted and analyzed in duplicates for the FAM63B_2 assay. This analysis demonstrated a good overall consistency between duplicate samples (Additional file 3: Figure S1). Additionally six samples were subjected to pyrosequencing, demonstrating that the variation in methylation levels for FAM63B_2 between individuals was also evident with a different technology (Additional file 3: Figure S1). A regression analysis including BD group status and gender was initially performed on logit-transformed methylation. As the age distribution was different between cases (mean = 47.7, SD = 12.4) and controls (mean = 35.7, SD = 13.5), we tested for the effect of age using regression analysis on logit-transformed methylation in the control group and case group separately. In addition, a comparison of DNA methylation between cases and controls was performed using a nonparametric Mann-Whitney test. Each CpG site was analyzed independently, as well as in combination as a differentially methylated region (DMR) by taking the average of the methylation level of the two sites for each individual. Finally, patients with bipolar I and II disorders were compared using a nonparametric Mann-Whitney test.
Genotypes, PCA, imputing, and mQTL analysis
Genotypes from the Affymetrix Gene Chip Human Mapping 500K Array were available for 587 of the individuals, as they were part of a published genome-wide association study [12]. Principle component analysis (PCA) was performed using SmartPCA [13] with an LD-pruned set of single nucleotide polymorphisms (SNPs) (R2 < 0.1, 39918 independent SNPs).
For cis methylation Quantitative Trait Loci (mQTL) analysis, SNPs in FAM63B and an additional 5 kb up- and downstream were imputed using Markov Chain Haplotyping software (MACH) [14]. Imputation was performed on data from four genotyped SNPs in FAM63B (rs664936, rs7164844, rs16940810 and rs4775096, all indicated at the top of Fig. 1) and the 1000Genome phase 1 reference panel. Genotype counts for the four genotyped SNPs can be found in Additional file 4: Table S3. Imputed SNPs were filtered by removing monomorphic markers and those with an RSQ score ≤0.3. This resulted in a final dataset of 173 imputed SNPs. To test for trans-mQTL we performed an additional analysis that included 9071 genotyped SNPs on chromosome 15. For the trans-mQTL analysis we filtered the results using an unadjusted p value <0.05 and Δβ ≥ 0.1. All mQTL analyses were performed using linear regression in PLINK [15].
Results
A regression analysis of methylation levels found significant effect of affection status (p value = 7.92 × 10−8 for FAM63B_1 and p value = 3.06°×°10−3 for FAM63B_2), and no effect of gender (p value = 0.41 for FAM63B_1 and p value = 0.57 for FAM63B_2). No significant effect of age on methylation was found in a regression analysis among the controls (lowest p value = 0.48) or cases (lowest p value = 0.30).
In a direct comparison of cases and controls with Mann-Whitney test we found significant DNA hypomethylation in BD cases compared to controls (FAM63B_1 p value = 3.94 × 10−8, Δβ = −0.05 and FAM63B_2 p value = 8.59 × 10−7, Δβ = −0.02) (Fig. 1). Consistent with these CpG sites being located 18 bp apart, the methylation levels at the two sites were significantly correlated (Spearman correlation rho = 0.64, p value <2.2 × 10−16). When analyzing the two sites combined as a DMR, the region also displayed significant hypomethylation in BD (p value = 6.85 × 10−6, Δβ = −0.04). Overall, our BD data replicates the FAM63B hypomethylation findings that had been reported in SZ.
The PCA found no evidence of population stratification among the genotyped individuals (Additional file 5: Figure S2), suggesting the differences were not driven by ancestry stratification. No methylation differences were found between bipolar I and bipolar II disorder patients, but because of the small number of individuals with a bipolar II diagnose, this may not be generalizable.
To investigate whether methylation levels were correlated with nearby genotypes, we carried out mQTL analysis. In the cis-mQTL analysis, using only SNPs in the near neighborhood, we found no significant mQTLs with strong effects (β threshold ≥0.1). The most significant mQTL was found in exon 9 (imputed SNP rs76751232) for FAM63B_2 (unadjusted p value = 0.011 and Δβ = −0.025 for the G allele). The full cis-mQTL analysis can be found in Additional file 6: Table S4. In order to assess the effect of trans-mQTLs, which are less frequent but can be notable [16], an mQTL analysis was performed including 9071 genotyped SNPs on chromosome 15. Again using the thresholds (p value <0.05 and Δβ ≥ 0.1) analysis revealed one nominally significant mQTL for FAM63B_1 (rs4774353 in FOXB1, chr15:60309674 (hg19), with p value = 0.006 and Δβ = −0.11 for the A allele). Overall, this suggests that the FAM63B methylation differences seen in whole blood from BD and controls are not strongly driven by SNPs in FAM63B, at least those that are on the Affymetrix 500K array. Of note, a potential mQTL may exist a few genes upstream, in FOXB1, which needs further validation, as it can also be a false positive resulting from multiple testing.
Due to the relative large variation in methylation levels observed in both groups, confirmed by technical replicates and pyrosequencing, we speculated if differences in blood cell composition could influence FAM63B methylation. In the absence of methylation array data to estimate cell composition in our sample, we instead downloaded a publicly available Illumina 450K array methylation dataset for nine different cell types from human whole blood [17]. The nearest probe cg21149266 (chr15:59,146,882 (hg19)) was positioned 126 bp downstream (3’) to FAM63B_2. Although not ideal, the methylation level of cg21149266 may for this purpose serve as a reasonable proxy for our two CpG sites, as methylation status of closely positioned sites is often correlated. In the cell-specific dataset, the methylation levels for cg21149266 were found to vary markedly between cell types with highest level in B cells, T cells, and NK cells, and lowest in monocytes, granulocytes and neutrophils (Additional file 7: Figure S3). It is thus possible that variation in cell composition contributes to the variation in FAM63B methylation observed in the entire dataset. If cell type heterogeneity is a systematic confounder between cases and controls, the effect should be shared across BD and SZ.
Discussion
We report FAM63B methylation to be significantly associated with BD, thus replicating the same finding recently published for SZ [9], with consistency in genomic position (same CpG sites) and direction (lower methylation in cases) across both studies. This is of interest in the light of phenotypic overlap between BD and SZ [18], with relatives to BD patients having increased risk for developing SZ, schizoaffective disorder or major depression [19, 20]. It has also been shown that the two disorders share common genetic risk variants, with a SNP co-heritability estimate of 0.15 [1]. The discovery of shared epigenetic risk loci across the two diseases, with differential methylation at the same sites and in the same direction in the same tissue, as in the case of FAM63B, may help progress the understanding of the shared etiology.
Interestingly, a large recent genome-wide association study (GWAS) meta-analysis of SZ identified association with FAM63B SNP, rs793571, as the most associated finding in this gene (p value = 4.54 × 10−6) [21]. Rs793571 is positioned in intron 7 of FAM63B (~5 kb from exon 9) and could potentially affect methylation levels of the 18 bp region of interest. In our data, rs793571 was however not an mQTL for the two CpG sites and thus unlikely to be involved in the methylation differences in the 18 bp region of interest in blood, at least in our sample.
In general, we do not find strong local mQTLs for the two investigated CpG sites in blood. In human fetal brain, a recent study characterized systematically mQTLs and found no Bonferroni-corrected mQTLs for FAM63B [16]; however, larger samples sizes across different tissues may reveal robust mQTLs for FAM63B. The present literature and data does in our opinion not imply that methylation of this gene is strongly driven by genotypes of known common SNPs, suggesting that methylation differences at these sites are not readably picked up by GWAS. In the near future, dense genotyping coupled with DNA methylation and expression levels across different tissue and cell types from projects like PsychENCODE [22] will help to resolve whether the differences are driven by genetic variation or if they represent independent epigenetic signal.
The strength of the study was the use of the same tissue and analysis of the exact same CpG sites, as identified as the top differentially methylated sites in the large EWAS of SZ [9]. Also, we used a relatively large sample of bipolar cases and controls, with genotypes available for the majority of participants.
A limitation of this study is the possible confounder of age. Because the BD cases in our study were slightly older than controls, we analyzed the controls and cases separately and found no effect of age on methylation at this locus. Also, because the FAM63B hypomethylation was initially identified in a large EWAS study of SZ [9], and replicated in an independent sample, while adjusting for age, we find it unlikely that the hypomethylation in our study is driven by this covariate.
A second limitation is the possible confounder of blood cell composition on methylation that we were not able to correct for because of the candidate-gene approach in this study. This limitation is not specific to our study. While we acknowledge that cell heterogeneity may explain some of the variation in the entire dataset (also among controls), it becomes a confounder only if cell composition is systematically different in cases. If cell composition is a confounder, it must be shared between BD and SZ.
Thirdly, medication could influence the methylation levels of FAM63B. Interestingly, the use of mood-stabilizing medication has been shown to influence the DNA methylation patterns in blood in a recent study by Houtepen and co-workers [23], using primarily the Infinium HumanMethylation27 to measure methylation levels. This array does not cover sites in FAM63B, so the effect of medication on FAM63B methylation in BD (and SZ) remains to be determined. It would be of great interest in the future to collect medication information (drug type, dosage and patient response). This not only would permit correction for possible medication effects in methylation data but also would also allow separation of the BD patients into pharmacological subphenotypes that could provide a link between methylation signatures and medication response.
The biological function of FAM63B is currently not well described. The gene is expressed across most tissues, with highest expression in the cerebellar hemisphere and cerebellum according to the genotype-tissue expression (GTEx) portal [24] (Additional file 8: Figure S4). In the BioGPS reference panel of normal human tissues [25], high FAM63B expression was specifically apparent in the pineal body (Additional file 9: Figure S5), a small endocrine gland positioned in the center of the brain. Interestingly, the pineal body is known to be involved in producing and releasing melatonin which is involved in the circadian clock [26]. Disruption of the circadian clock has been linked to the etiology of multiple psychiatric disorders [27]. In the future, inclusion of information on diurnal mood variation and sleeping patterns of investigated individuals could allow for investigation of possible correlations between DNA methylation levels of FAM63B and the circadian clock.
In conclusion, we have identified FAM63B hypomethylation in BD. Our data supports that FAM63B is a shared epigenetic risk gene for BD and SZ.
Acknowledgements
We acknowledge Anne Hedemand for excellent technical assistance in the laboratory.
Funding
This study was funded by the Agnes og Poul Friis Fond, The Toyota-Foundation, and the Lundbeck Foundation, Denmark. Genetic analysis of the UCL cohort has been supported by UK Medical Research Council project grants G9623693N,G0500791, G0701007, and G1000708.
Disclaimer
These funders had no involvement in any aspect of the study. All authors declare no competing financial interests in relation to the work described.
Abbreviations
- BD
bipolar disorder
- DMR
differentially methylated region
- DNA
deoxyribonucleic acid
- EWAS
epigenome-wide association study
- FAM63B
family with sequence similarity 63, member B
- GTEx
genotype-tissue expression (GTEx)
- GWAS
genome-wide association study
- MACH
Markov Chain Haplotyping software
- mQTL
methylation quantitative trait loci
- PCA
principal component analysis
- SNP
single nucleotide polymorphism
- SZ
schizophrenia
- UCL
University College London
Additional files
Overview of participants in the study. (DOCX 42 kb)
Primer sequences for the two FAM63B iPLEX assays. (DOCX 43 kb)
Technical assessment of the iPLEX method. Samples analyzed with iPLEX and pyrosequencing for FAM63B_2 site (panel A). Technical replicates for the FAM63B_2 assay run with iPLEX (panel B). (PDF 150 kb)
Overview for genotype counts for SNPs genotyped in FAM63B. (DOCX 39 kb)
Scatterplot of PC1 and PC2 obtained from PCA of genome-wide independent SNPs (n = 39918, R 2 < 0.1) in UCL sample. (EPS 801 kb)
Overview of all genotyped and imputed SNPs in FAM63B (plus 5 kb up- and downstream region) used in the cis-mQTL analysis of the two investigated FAM63B CpG sites. (DOCX 178 kb)
{kind=link}
DNA methylation levels for cg21149266, an Illumina 450 K methylation array probe in the region of interest in FAM63B across different blood cell types [17]. (PNG 242 kb)
Expression of FAM63B across different tissues according to the GTEx portal (http://www.gtexportal.org/home/). (PDF 185 kb)
{kind=link}
Expression of FAM63B across tissues according to BioGPS portal (http://biogps.org/). (PNG 137 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors meet the four authorship criteria of the International Committee of Medical Journal Editors. MN and AS provided the study conception and design. AS performed the experimental work and analyzed the data. DD supervised the imputing and mQTL analyses. AM, NLO, ADB, and OM contributed with samples. MN, AS, NS, and ALN interpreted the data. MN, AS, and AM drafted the manuscript. All authors performed critical revision of the manuscript and approved the final version.
References
- 1.Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013;45:984–94. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lichtenstein P, Yip BH, Björk C, Pawitan Y, Cannon TD, Sullivan PF, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373:234–9. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bambole V, Johnston M, Shah N, Sonavane S, Desouza A, Shrivastava A. Symptom overlap between schizophrenia and bipolar mood disorder: diagnostic issues. Open J. Psychiatry. 2013;03:8–15. doi: 10.4236/ojpsych.2013.34A002. [DOI] [Google Scholar]
- 4.Patel V, Prince M. Global mental health: a new global health field comes of age. JAMA. 2010;303:1976–7. doi: 10.1001/jama.2010.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum. Mol. Genet. 2011;20:4786–96. doi: 10.1093/hmg/ddr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen C, Zhang C, Cheng L, Reilly JL, Bishop JR, Sweeney JA, et al. Correlation between DNA methylation and gene expression in the brains of patients with bipolar disorder and schizophrenia. Bipolar Disord. 2014;16:790–9. doi: 10.1111/bdi.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruzicka WB, Subburaju S, Benes FM. Circuit- and diagnosis-specific DNA methylation changes at γ-aminobutyric acid-related genes in postmortem human hippocampus in schizophrenia and bipolar disorder. JAMA Psychiatry. 2015;72:541–51. doi: 10.1001/jamapsychiatry.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pidsley R, Mill J. Epigenetic studies of psychosis: current findings, methodological approaches, and implications for postmortem research. Biol. Psychiatry. 2011;69:146–56. doi: 10.1016/j.biopsych.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 9.Aberg KA, McClay JL, Nerella S, Clark S, Kumar G, Chen W, et al. Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiatry. 2014;71:255–64. doi: 10.1001/jamapsychiatry.2013.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Endicott J, Spitzer RL. A diagnostic interview: the schedule for affective disorders and schizophrenia. Arch Gen Psychiatry. 1978;35:837–44. doi: 10.1001/archpsyc.1978.01770310043002. [DOI] [PubMed] [Google Scholar]
- 12.Sklar P, Smoller JW, Fan J, Ferreira MAR, Perlis RH, Chambert K, et al. Whole-genome association study of bipolar disorder. Mol. Psychiatry. 2008;13:558–69. doi: 10.1038/sj.mp.4002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 14.Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–5. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hannon E, Spiers H, Viana J, Pidsley R, Burrage J, Murphy TM, et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat. Neurosci. 2016;19:48–54. doi: 10.1038/nn.4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén S-E, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laursen TM, Agerbo E, Pedersen CB. Bipolar disorder, schizoaffective disorder, and schizophrenia overlap. J. Clin. Psychiatry. 2009;70:1432–8. doi: 10.4088/JCP.08m04807. [DOI] [PubMed] [Google Scholar]
- 19.Craddock N, Jones I. Genetics of bipolar disorder. J. Med. Genet. 1999;36:585–94. doi: 10.1136/jmg.36.8.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Craddock N, O’Donovan MC, Owen MJ. The genetics of schizophrenia and bipolar disorder: dissecting psychosis. J. Med. Genet. 2005;42:193–204. doi: 10.1136/jmg.2005.030718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, Crawford GE, et al. The PsychENCODE project. Nat. Neurosci. 2015;18:1707–12. doi: 10.1038/nn.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Houtepen LC, van Bergen AH, Vinkers CH, Boks MP. DNA methylation signatures of mood stabilizers and antipsychotics in bipolar disorder. Epigenomics. 2016;8:197–208. doi: 10.2217/epi.15.98. [DOI] [PubMed] [Google Scholar]
- 24.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013;45:580–5. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu C, Macleod I, Su AI. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 2013;41:D561–5. doi: 10.1093/nar/gks1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macchi MM, Bruce JN. Human pineal physiology and functional significance of melatonin. Front. Neuroendocrinol. 2004;25:177–95. doi: 10.1016/j.yfrne.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 27.Karatsoreos IN. Links between circadian rhythms and psychiatric disease. Front. Behav. Neurosci. 2014;8:162. doi: 10.3389/fnbeh.2014.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]