

Abstract
Background
Plants have the remarkable property to elaborate entire body plan from any tissue part. The conversion of lateral root primordium (LRP) to shoot is an ideal method for plant propagation and for plant researchers to understand the mechanism underlying trans-differentiation. Until now, however, a robust method that allows the efficient conversion of LRP to shoot is lacking. This has limited our ability to study the dynamic phases of reprogramming at cellular and molecular levels.
Results
Here we present an efficient protocol for the direct conversion of LRP to a complete fertile shoot system. This protocol can be readily applied to the various ecotypes of Arabidopsis. We show that, the conversion process is highly responsive to developmental stages of LRP and changes in external environmental stimuli such as temperature. The entire conversion process can be adequately analyzed by histological and imaging techniques. As a demonstration, using a battery of cell fate specific markers, we show that confocal time-lapse imaging can be employed to uncover the early molecular events, intermediate developmental phases and relative abundance of stem cell regulators during the conversion of LRP to shoot.
Conclusion
Our method is highly efficient, independent of genotypes tested and suitable to study the reprogramming of LRP to shoot in intact plants as well as in excised roots.
Electronic supplementary material
The online version of this article (doi:10.1186/s13007-016-0127-5) contains supplementary material, which is available to authorized users.
Keywords: Arabidopsis, Auxin, Cytokinin, Lateral root, Shoot, Regeneration, Trans-differentiation
Background
The profound capacity of somatic cells to reproduce an entire organism has been widely exploited in in vitro plant regeneration [1, 2]. This spectacular property of plant cells has great impact on both, basic plant research and biotechnological application. On the one hand, in vitro plant regeneration can be exploited to understand many fundamental questions pertaining to subjects such as stem cell specification, cell fate determination, cell differentiation, meristem formation and organogenesis in the absence of positional information laid down during embryogenesis. On the other hand, the relatively simple and robust approach of this system attracts biotechnology sector for in vitro micropropagation, haploid production and genetic engineering.
Regeneration can be induced from the explant either directly or indirectly and are defined based on the presence or absence of an intermediate phase that leads to the formation of callus, a regenerative mass of cells. Indirect regeneration demands pre-treatment of the explant with high concentration of auxin which induces callus formation. Callus formation involves the activation of pericycle or pericycle-like cells and largely follows lateral root developmental pathway regardless of the origin of explants [3–5]. Root or shoot can be regenerated from callus when it is cultivated on media containing high auxin relative to cytokinin or high cytokinin relative to auxin, respectively. On the other hand, organs can also be regenerated directly from the explants without passing through a callus phase. Direct conversion of lateral root primordia (LRP) to shoot upon cytokinin treatment is one such example [4, 6, 7]. LRP is formed from xylem pole pericycle cells and it is induced by auxin [8, 9]. Application of cytokinin can convert LRP to shoot in a concentration-dependent manner and this process provides an elegant example of trans-differentiation [4, 6, 7]. Cytokinin rapidly induces the expression of a shoot stem cell regulator, WUSCHEL (WUS) in the LRP, and facilitates the conversion process [6, 7]. Consistent with the rapid upregulation of WUS during cytokinin-induced LRP-to-shoot conversion, inducible ectopic overexpression of WUS can convert LRP to shoot in the absence of external inductive cues and can induce shoot formation from Arabidopsis primary root meristem [6, 10].
Direct conversion of LRP to shoot can be exploited in sterile plants and in those where normal propagation by seed is not possible. This approach can substitute callus-mediated shoot regeneration that is more time consuming, laborious and difficult to achieve in some plant species. Moreover, explants that undergo prolonged sub-culture might encounter somaclonal variation associated with callus growth, and thus, such long-term callus-mediated culturing may not be desirable for the regeneration of elite crop species [11]. Therefore, direct conversion of LRP to shoot offers an attractive alternative for plant breeding industry.
Conversion of LRP to shoot must involve massive reprogramming in response to shoot inductive cues. Study of early cellular and molecular events regulating the reprogramming is essential to probe the underlying mechanisms. The model dicot species, Arabidopsis, can be used to study the in depth mechanism and it can serve as a reference for studies in other plant species [12–14]. Recent studies in Arabidopsis have begun to address this process [4, 6, 7]. Although previous studies in Arabidopsis have attempted to understand the molecular players involved in LRP-to-shoot conversion, they do not provide a robust method that can monitor the transient intermediate phases of conversion in real time [4, 7]. Dissection of the intermediate phases in which the converting structures are likely to remain morphologically identical to untreated LRP is instrumental to understand the mechanisms. Here, we describe a simple and highly reproducible method that can be used for the synchronized and efficient conversion of LRP to complete fertile shoot system in Arabidopsis which allows us to dissect the transient intermediate developmental phases of conversion (Fig. 1). We reveal the unrecognized role of external environmental stimulus, temperature, in the conversion of LRP to shoot. Using a battery of cell fate specific fluorescent markers, we show that time-lapse imaging is a useful tool to gain deeper insights into these processes. Furthermore, we illustrate the use of classical histological analysis to understand the anatomical changes during the conversion. Our methods can be used to study the process in both, the excised root as well as in growing plants.
Fig. 1.
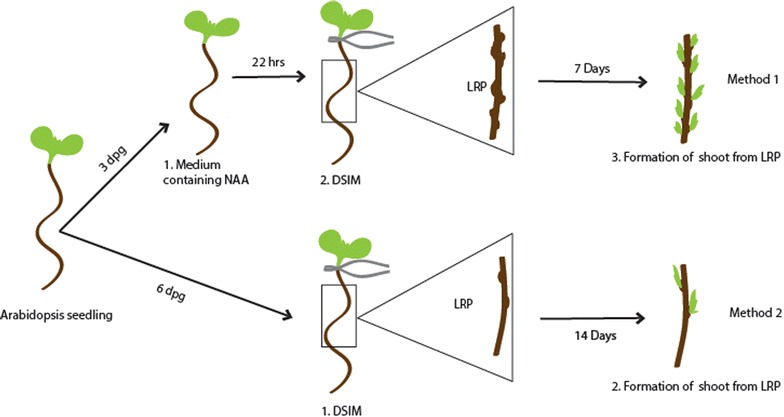
Schematic representation of direct conversion of LRP to shoot in Arabidopsis thaliana
Materials
Reagents
-
Reagents for seed sterilization
Dried seeds of Arabidopsis thaliana, 20 % sodium hypochlorite, 70 % ethanol and autoclaved distilled water.
-
Murashige and skoog (MS) medium
Use de-ionized Milli Q water to prepare growth media. All the hormones and antibiotics should be filter-sterilized and should be added to autoclaved, cool medium (45–50 °C). Preparation of hormone stocks is described in Table 1.
To prepare 1L MS medium, add 2.165 g (for half strength) or 4.3 g (for full strength) of MS salt (Sigma, M5524) and 10 g of sucrose (Sigma, S0389). Adjust pH to 5.8 with 1 N KOH and add 8 g of plant Agar (Sigma, A7921). Autoclave the medium (121 °C for 20 min) and cool it to 45–50 °C. Add 1 ml of 100 mg/ml filter-sterilized Ampicillin (final conc. 100 µg/ml) to 1L medium and pour 50 ml each into square Petri dishes within the LAF.
Note Addition of Ampicillin to the medium can prevent bacterial contamination.
-
NAA pretreatment medium
Add 50 µl of 1.86 mg/ml NAA (naphthaleneacetic acid) (final concentration 10 µM) to 50 ml full strength MS agar medium and pour into square Petri dishes in the LAF.
-
Direct shoot inducing medium (DSIM)
Add 4.3 g of MS salt (Sigma, M5524) and 10 g of Sucrose (Sigma, S0389), 0.5 g of MES (2-(N-Morpholino) ethane sulfonic acid) (Sigma, M2933) and 1 ml of Gamborg’s vitamin solution (Sigma, G1019) for 1 l of 1× DSIM. Adjust pH to 5.7 with 1 N KOH and add 8 g of plant Agar (Sigma, A7921). After autoclaving, cool the medium to 45–50 °C and add 1 ml of 100 mg/ml filter-sterilized Ampicillin (final conc. 100 µg/ml) and specific hormones to 1 l medium as mentioned below. Pour 50 ml medium into each square Petri dish in the LAF. Two hormonal combinations for DSIM are described here.
DSIM-1: 10 µM 2iP (6-(γ,γ-dimethylallylamino) purine)
DSIM-2: 10 µM trans-zeatin
-
Root inducing medium (RIM)
Add 500 µl of 1 mg/ml indol-3-butyric acid (IBA) into the basal composition of DSIM (without cytokinin) to make 1 l RIM. Pour into square Petri dishes in the LAF.
-
Soilrite–vermiculite mixture
Mix soilrite and exfoliated vermiculite (1:1) with 1× MS solution (without sucrose and agar) for ex vitro establishment of seedlings. It is desirable to autoclave both soilrite and vermiculite before mixing them with MS solution to destroy germs and weeds.
-
Hormone/antibiotic stocks
The hormone stocks should be prepared in DMSO and filter sterilized by 0.22 µm filter. All the stocks should be stored at −20 °C.
-
Staining reagents for confocal microscopy
10 µg/ml propidium iodide (filter sterilized) (Sigma).
10 µg/ml FM4-64 (filter sterilized) (molecular probes).
Both propidium iodide and FM4-64 should be dissolved in sterile distilled water followed by filter sterilization. Store in amber coloured tubes at 4 °C.
Note Caution! Propidium iodide is a potential carcinogen. Wear protective gloves to avoid contact with skin.
-
Fixing solution preparation for histological analysis
401 ml ddH2O
25 ml 1 M Na3PO4 with pH 7.2
54 ml 37 % formaldehyde
20 ml 25 % glutaraldehyde
Note Formaldehyde and glutaraldehyde are toxic. The preparation should be performed in fume hood.
-
Solution A preparation
Leica historesin embedding kit is used for embedding. Dissolve 0.5 g historesin activator into 50 ml basic resin, mix thoroughly. Solution A can be stored at 4 °C, dark, for up to 2 weeks.
-
Embedding solution preparation
Add hardener into solution A with the volume ratio 1:14. Mix thoroughly by vortexing.
Note Embedding solution should be made freshly before use as it cannot be stored. It hardens within 10 min.
-
Plant materials and constructs
Arabidopsis ecotypes Col-0, Ws and Ler are used in this study. pPLT2::PLT2-vYFP [6, 15] pSCR::H2B-vYFP, and pWUS::erCFP [6] have been described previously. pARF10::ARF10-YFP construct is generated by incorporating genomic sequence of ARF10 between its 4.984 kb upstream regulatory sequence at the 5′ end and the coding region of vYFP at the 3′ end. The genomic sequence of STM is cloned with its 5.752 kb upstream regulatory sequence fused in translational frame with the vYFP to generate pSTM::STM-YFP. All the DNA fragments mentioned above are amplified from Col-0 genomic DNA using the primers listed in Table 2. Multisite Gateway recombination (Invitrogen) based cloning system is used to clone all components into pCAMBIA 1300 destination vector. Floral-dip method is used to transform Col-0 plants and transgenics are selected based on hygromycin resistance.
Table 1.
Details of hormone/antibiotic stock preparation
| Antibiotic/hormone | Stock concentration | Solvent | Concentration in media | Amount to be added for 50 ml medium (µl) | Amount to be added for 1 l medium (ml) |
|---|---|---|---|---|---|
| Ampicillin | 100 mg/ml | 40 % ethanol | 100 µg/ml | 50 | 1 |
| NAA | 1.86 mg/ml | DMSO | 10 µM | 50 | 1 |
| 2iP | 10 mM | DMSO | 10 µM | 50 | 1 |
| Trans zeatin | 10 mM | DMSO | 10 µM | 50 | 1 |
Table 2.
Details of oligonucleotide primers (gateway compatible) used in this study (5′–>3′)
| Primer name | Forward primer | Reverse primer |
|---|---|---|
| ARF10 promoter | ggggacaactttgtatagaaaagttgttgtcacgattagaatgcacatgcagttcgt | ggggactgcttttttgtacaaacttgtctagacgaagttgtgtaacccccaaattct |
| ARF10 gene | ggggacaagtttgtacaaaaaagcaggctgtatggagcaagagaaaagcttggatccacaac | Ggggaccactttgtacaagaaagctgggttagcgaagatgctgagcggaccagtcttcgtag |
| STM gene | ggggacaagtttgtacaaaaaagcaggctgtatggagagtggttccaacagcacttcttgtc | Ggggaccactttgtacaagaaagctgggttaagcatggtggaggagatgtgatccattgg |
Equipment
Equipment for in vitro culture
Laminar air flow chamber (LAF)
Microscissors-Vannas scissor, straight (Ted Pella, 1346) (flame sterilized)
Forceps (flame sterilized)
Scalpel and scalpel blades (flame sterilized)
Micro pipettes
Sterile pipette-tips (200 µl and 1 ml)
1.5 ml micro-centrifuge tubes
Spirit lamp
Sterile disposable square Petri plates, size: 120 mm × 120 mm (Himedia PW050-1)
Parafilm
Gloves
Plant growth chamber (Percival AR-100L3)
Equipment for microscopy
Stereo microscope (Leica M205FA) for bright field imaging
Confocal laser scanning microscope (Leica TCS SP5 II) for time-lapse fluorescent imaging
Microscope slides (Himedia CG029)
Microscope cover glass 22 × 22 mm (Corning 2850-22)
Equipment for resin cross section
Micro pipettes
Sterile pipette tips (200 µl and 1 ml)
1.5 ml micro centrifuge tubes
13 × 19 × 5 mm Polyethylene mold cups
Flat bottom 6 well cell culture plates
20 × 15 × 10 mm wooden block
Microscope slides
Microtome (Leica, Jung RM2055)
Microtome knife
Water bath (Kunz instruments, HIS-2)
Flattening table (Kunz instruments, HP-3)
Copier films (Folex, A4 size)
Double side adhesive tape
Scissors and forceps
Equipment setup
-
Embedding chamber making
Cut the copier film into size of 3 × 4 cm, cover it with double side adhesive tape, then dig a hole in the middle with the size 1 × 2 cm, stick it to another piece of intact copier film, the chamber left can be used to embed material. The chamber depth equals to the film thickness. If the material is thick, just stick two or more layers of film together to make the chamber deeper.
Note This method is quite flexible, this can be modified by using another material instead of copier film, as long as the material is not organic solution permeable and if a chamber to hold the samples can be made. The chamber size can be adjusted according to the sample size. This kind of chamber is recyclable.
Methods
Seed sterilization and inoculation
A sterile working environment should be ensured during all steps of plant tissue culture, from seed sterilization to shoot regeneration. Clean the working area of LAF and the surfaces of necessary materials/tools (petri plates, pipettes, tip box, forceps, scissors, spirit lamp and bottles containing ethanol, bleach and sterile water) with 70 % ethanol. Flame-sterilize forceps and scissors before contact with each explant. Sterilize the LAF and the materials/tools by UV irradiation for 20 min. Hands should be wiped with 70 % ethanol prior to the commencement of the work.
A liquid surface sterilization method for seeds is introduced here. The vapour phase method can also be used as described in Ref. [16].
-
Add 1 ml of 70 % ethanol to the seeds in 1.5 ml sterile micro-centrifuge tube.
Note The maximum amount of seeds that can be effectively sterilized in a 1.5 ml microfuge tube is until 50 μl mark.
Gently mix it for 5 min by inverting the tubes followed by a brief spin at 6000 rpm.
-
Discard the ethanol and add 1 ml of 20 % bleach (sodium hypochlorite) and mix well for 5 min followed by a brief spin at 6000 rpm.
Note It should be ensured that bleach treatment does not exceed maximum 10 min as it adversely affects seed germination.
Wash the seeds with 1 ml of sterile deionized water 5–7 times.
-
Store the sterilized seeds in a micro-centrifuge tube containing 1 ml water at 4 °C for 2–5 days for stratification.
Note Stratification is important for synchronized seed germination. Stratification treatment can be provided even in plate after seed inoculation.
Pipette the sterilized seeds and inoculate over ½ MS agar plate in 2 rows. Spread the seeds distinctly in a linear manner using 1 ml sterile pipette tip. One row of square petri plate (120 mm × 120 mm) can accommodate approximately 30 seeds. Seal the plates with parafilm.
Incubate the plates in plant growth chamber maintained at 22 °C, 45 µmol/m2 s and 16 h light/8 h dark. The plates can be placed slanting in racks made into plastic trays.
-
Follow the seed germination and collect the explants for shoot regeneration after 3rd or 6th day post germination (dpg) depending upon which method is followed.
Note Plants should not be subjected to any kind of stress like nutrient stress, water stress or contamination before and after the explant collection. This is because stress can affect the shoot conversion efficiency. LRP-to-shoot conversion is hyper sensitive to temperature. Any deviation in temperature affects root-to-shoot conversion. Therefore, it is necessary to ensure that the water, which accumulates at the bottom of the plates during incubation in growth chamber should be removed on periodic basis as it can lead to bacterial contamination.
Explant excision and LRP-to-shoot conversion
In order to convert LRP directly into shoot, two methods based on Refs. [4, 6, 7] with modifications are introduced here. First method utilizes 3 dpg old seedlings and it requires a short auxin pre-treatment before transferring into cytokinin rich DSIM. The second method exploits 6 dpg old seedlings and the explants are directly transferred into DSIM (Fig. 2).
Fig. 2.
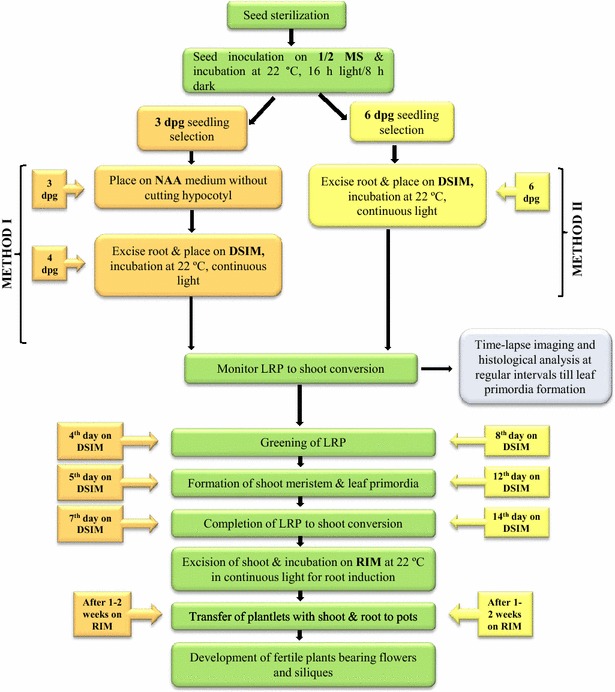
Schematic representation of the steps and time involved in direct conversion of LRP to shoot in Arabidopsis thaliana
Method-I The two major advantages of the first method over the second method are its speed and efficiency. Method-1 is speedy as it utilizes very young (3 dpg) explants and the LRP-to-shoot conversion occurs within 6–7 days. This method is more efficient in converting maximum number of LRP into shoot. One of the disadvantages of this method is that it requires an additional step of NAA pre-treatment.
-
Transfer 3 dpg old seedlings from MS plate onto NAA (10 µM) containing medium. Seal the plate with parafilm and incubate for 20–22 h in plant growth chamber under the same conditions mentioned above. NAA pretreatment can ensure LRP formation throughout the root. LRP stages at II and III are more responsive towards cytokinin treatments. NAA treatment should not exceed 24 h as it promotes lateral root outgrowth which is less sensitive to cytokinin. LRP can be observed adjacent to one another in all directions throughout the root using DIC/confocal microscope. More number of LRP can be noticed at the basal meristem (transition zone) where auxin sensitivity is high.
Note Pay attention to hold the seedlings at the cotyledon/leaf with flame-sterilized and cooled forceps during the transfer. Holding the seedlings at hypocotyl or root may damage the seedlings.
-
After 20–22 h of treatment on NAA-containing medium, transfer the seedlings onto DSIM using flame-sterilized and cooled forceps. Excise the root from the seedling by cutting at hypocotyl junction with flame-sterilized and cooled Vannas scissor. Root excision can be done on the medium. The wound scratches made on the medium while the excision of root will not affect conversion. Gently press the root explant on the medium using forceps so that the explant is in direct contact with the medium. Discard the shoot part along with hypocotyl.
Note Use separate scissors and forceps for each seedling to avoid cross contamination. It is critical to ensure that the explants do not dry during these processes. The plate should not be kept open for a long time in LAF and the spirit lamp should be kept away from the plates to avoid the explants from drying.
Seal the plates with parafilm and incubate the same for 5–7 days under continuous light at 22 °C. The LRP will turn green in 4 days and shoot meristem and leaf primordia will be formed in 5 days. These can be viewed under high resolution microscope. LRP-to-shoot conversion will be completed by 7 days of incubation on DSIM. The developmental changes associated with LRP-to-shoot conversion can be monitored by time-lapse imaging under confocal microscope (see below). Newly formed leaves appear similar to the rosette leaves formed in seedlings with regular phyllotaxy and these exhibit trichomes on adaxial surface (Fig. 3). Narrow or filamentous leaves can also be observed occasionally.
-
Excise the converted shoot from the explant with flame-sterilized and cooled scalpel and place it on ½ MS medium and incubate for 4–5 more days to induce more number of leaves on the shoot. Transfer the plantlets further to RIM plate to make roots. Seal the RIM plate with parafilm and transfer to the growth chamber under continuous light at 22 °C.
Note If the shoots are left undetached from root explant for long time, inflorescence stem will be formed with cauline leaves and flowers. But we suggest the excision of the shoot from the explant at 4–6 leaves stage and transfer to RIM plate because the shoots will be clustered otherwise.
Rooting can be noticed after 1 week of treatment on RIM. Transfer the plantlets having both shoot and root to soilrite–vermiculite mixture for ex vitro establishment. (Soilrite–vermiculite mixture is made wet with 1× MS solution). Cover the pot with saran wrap, to maintain humidity, and shift the pot in a tray into the growth chamber.
Gradually expose the plantlets to low humidity by progressive removal of the saran wrap after a few days. Add water to the tray containing the pots at regular interval and add ½ MS solution at every fifth round. The regenerated plants will be fertile with flowers and siliques. No watering is required once the siliques have matured.
Fig. 3.

Conversion of LRP to complete fertile shoot system. a–c Stages of direct conversion of LRP to shoot by Method I after transfer from NAA to DSIM. d LRP-to-shoot conversion in growing plant after shoot inductive cues. Arrows mark converted shoots. e Root induction from regenerated shoot after incubation on RIM. f–i Direct LRP-to-shoot conversion by Method II after incubation on DSIM. j Root formation from the bulges developed in LRP at high temperature. k LRP converted shoot bearing rosette leaves and inflorescence. l Ex vitro established complete fertile shoot system bearing flowers and siliques. Scale bar 1 mm
Method II This method employs direct transfer of 6 dpg old root explants into DSIM for the conversion of LRP into shoot.
Pick 6 dpg old seedlings from ½ MS plate by holding the leaves with flame sterilized and cooled forceps and transfer onto DSIM. Gently press the root on the medium so that the entire explant is in contact with DSIM.
Excise the root from the seedling by cutting at hypocotyl junction using flame sterilized and cooled Vannas scissor. Discard the shoot part along with the hypocotyl.
Seal the plate with parafilm and incubate the same for 2 weeks under continuous light at 22 °C. The LRP will turn green by 8 days and shoot meristem and leaf primordia will be formed by 12 days. The developmental changes during LRP-to-shoot conversion can be monitored by time-lapse imaging. LRP-to-shoot conversion will be completed by 14 days of incubation on DSIM. Shoots will be arranged acropetally on root in an alternating fashion similar to lateral root formation.
Excise the shoot from the explant with flame-sterilized and cooled scalpel and place on ½ MS medium and incubate for 7 more days to induce more number of leaves on the shoot. Transfer the plantlets further to RIM plate to make roots. Seal the RIM plate with parafilm and transfer to growth chamber under continuous light at 22 °C.
Follow steps 5 and 6 of Method I for rooting and ex vitro establishment.
Histological analysis
For histological sectioning seedlings are fixed according to Ref. [17] and embedded using Leica Historesin kit according to Ref. [18]. In this method, samples are embedded in ‘sandwich’ chambers, which is particularly useful when the samples are small. This is a fast and versatile method, and many samples can be analysed in one section.
Add proper contents of fixing solution to the plate’s well, gently remove and soak the explants into the fixing solution. Seal the plates using parafilm and leave it overnight.
Discard the fixing solution, replace by ethanol gradients, starting from 10 %, then proceed to 30, 50, 70, 95, 100 %, and again 100 %, 30 min for each incubation. The material can be stored in 70 % ethanol.
Add equal volume of pure ethanol to solution A to get the half solution A. Immerse the explants in half solution A for at least 3 h. Lastly change to solution A for another 3 h. For older material, a longer incubation is recommended.
Gently place the explants in embedding chamber, use forceps to align them properly, make sure all explants are positioned in the same direction, add some drops of embedding solution, then carefully cover the chamber with another piece of film to insulate it from air. Make sure no air bubbles are left in the chamber. The sample-containing embedding solution will completely harden within 3 h.
Remove the cover film from the chamber, and take the sample containing solidified embedding piece out. Trim the embedding piece properly. Cut it into smaller pieces every 3 mm, and put them on top of each other to make a stack. All these pieces should be placed in the same direction and order relative to original explants. Add some drops of embedding solution to make them stick to each other. Their relative position should not be changed. In this way, you will get the results of more than one sample and more than one position in one section.
3 h later, when the stack hardens, transfer the stack into the polyethylene mold cup (place the root containing stacks vertically for cross section and horizontally for longitudinal section). Add about 2 ml embedding solution to soak the stack and then cover a lid to insulate the air. Keep it over night before microtome.
Take the plastic block out from the mold cup. Trim it and stick it to a wooden block using super glue. Set the microtome machine, set the water bath temperature at 50 °C and the flattening table at 60 °C. Then make normal cross sections. Transfer each section piece to water bath to make it expand, then fish it out using a slide and dry the slide on the flattening table.
Immerse the slides in toluidine blue for 3–6 s and gently wash it using tap water. Add some drops of water on the slide and mount the slide. Observe under the microscope.
Confocal time-lapse imaging
Developmental changes during the conversion of LRP into shoot can be monitored by time-lapse imaging/live imaging of fluorescent protein tagged cell fate specific markers using confocal microscope. We are introducing a method to follow the time lapse imaging of the explant from untreated 0-day sample till complete shoot formation without contaminating the same. Live imaging can also be done using different explants to follow the developmental changes. The confocal settings we describe are based on Leica TCS SP5 II laser scanning microscope. The excitation/emission wavelengths will be invariable in all microscopes, but slight modifications may be required for laser power and intensity in different microscopes even if it is the same model. Imaging of the explants should commence before transferring to DSIM (method I: LRP of 20 h NAA treated explant. Method II: LRP of 6 dpg explant). Make sure that all materials used for time-lapse imaging are sterile. Microscope slides, cover glasses and pipette tips used during time-lapse imaging should be autoclaved. To avoid sticking together of cover glasses due to water vapour and to avoid the formation of water film during autoclaving, cover them individually in tissue paper and then wrap in an autoclavable, sealed polythene cover. Propidium iodide and FM4-64 should be filter-sterilized. (Dye concentration is given under materials section above).
Pipette 60–80 µl propidium iodide/FM4-64 onto clean microscope slide. Use propidium iodide for LRP imaging at 0-day and early days on DSIM. FM4-64 stain should be used for shoot meristem and shoot primordia imaging. Propidium iodide stains the cell wall while FM4-64 stains cell membrane to mark the cell boundaries. (Caution!: propidium iodide is a potential carcinogen, wear protective gloves during handling).
Aseptically transfer the explants from the respective medium into propidium iodide/FM4-64 on the slide using sterilized forceps.
Immerse the explants in propidium iodide/FM4-64 and stain for 2 min. This step is crucial to stain all cell files adequately.
Place the cover glass carefully over the sample without introducing air bubbles. The entire explant can be covered with 22 × 22 mm cover glass if the explant is kept in S shape. Hypocotyl and shoot part of 0-day explant should be kept out of the cover glass so that the root part is in single plane. However, ensure that the shoot part does not dehydrate during imaging. Cover glass with 24 × 60 mm which cover entire slide can also be used, but 0-day explants may not remain stable in single plane, therefore, it may interfere with single section imaging.
-
Keep the slide under the confocal microscope and focus the LRP/shoot meristem. Use 10× air or 20× oil objective for imaging. The confocal settings described here is based on our recent paper [6]. The settings for commonly used fluorescent signals are listed in Table 3.
The laser power and intensity may vary based on the nature of the marker tagged with fluorescent protein and the extent of staining. Use 20–30 % power for argon laser. Laser intensity should be minimal in order to reduce bleaching of the fluorescent signal and to avoid over saturation. Adjust the intensity from 5 to 30 % and also adjust the same with gain. Capture single middle section images for LRP at 0 day and for early days on DSIM. For imaging the shoot meristem and the leaf primordia, capture the Z-stack of all sections.
Once imaging is completed, carefully remove the coverglass from the slide. To remove the coverglass without damaging the explants, hold the slide slanting and add surplus of sterile water from one side, so that the coverglass will be washed off.
Carefully transfer the explant into a fresh petri plate containing sterile water to remove the propidium iodide/FM4-64 content. Gently swirl the plate for some time to ensure the proper removal of the stain. Pay attention to avoid any external source of contamination during these processes.
Transfer the explants to its respective medium and incubate in plant growth chamber until next imaging. Follow the imaging process at regular interval till the leaf primordia forms.
Table 3.
Details of the settings for commonly used fluorescent signals
| Signal | Excitation wavelength (nm) | Emission wavelength range (nm) |
|---|---|---|
| CFP | 458 | 465–515 |
| GFP | 488 | 495–530 |
| YFP/VENUS | 514 | 520–545 |
| Propidium iodide | 514/561 | 585–630 |
| FM4-64 | 514/561 | 620–750 |
| Chlorophyll autofluorescence | 488/514/561 | 620–750 |
Results and discussion
Here we provide detailed methods which can be readily used to study the conversion of LRP-to-shoot in the model organism, Arabidopsis. Both methods described here were effective in converting LRP into complete shoot system (Fig. 3a–l). We examined if shoot conversion can be achieved only in excised root or if this method can also be implemented to growing plants where complete root system is in the context of growing shoot. We found that our method can also be used to trigger shoot formation from LRP in intact plant (Fig. 3d). The ability to convert the LRP to shoot by external inductive cues in both intact plant and excised root is consistent with our previous study where inducible ectopic overexpression of shoot stem cell regulator, WUS, triggered shoot from LRP of both intact plant as well as excised root [6].
Though previous studies [4, 6, 7] have shown that LRP can be converted to shoot, it was not known if the developing shoot can be used to generate complete fertile shoot system. We show that the converted shoots bearing flowers and siliques can be successfully established ex vitro (Fig. 3k, l). Method I, which demanded auxin pre-treatment, displayed more number of LRP-to-shoot conversion. This is due to the presence of more number of LRP in the explants of method I than those of method II. As previously shown, NAA pre-treatment induced more number of LRP along the entire length of the root at both xylem pole pericycle [7, 19]. Upon DSIM treatment, these LRP converted into shoot. But it was not known if LRP at all stages can convert into shoot. We have tested the capacity of different stages of LRP to convert into shoot. We found that LRP stages II and III were more responsive to convert into shoot (Fig. 5a, e, i, m). LRP at the very late stage of development (lateral root emergence stage) was recalcitrant to convert into shoot (Additional file 1). The degree of shoot conversion ability of LRP at different developmental stages might be due to the varying sensitivity of LRP to cytokinin. It has been reported that stage V/VI LRP is least sensitive to cytokinin [20]. Thus, the developmental stage of LRP plays a significant role in determining the conversion process.
Fig. 5.
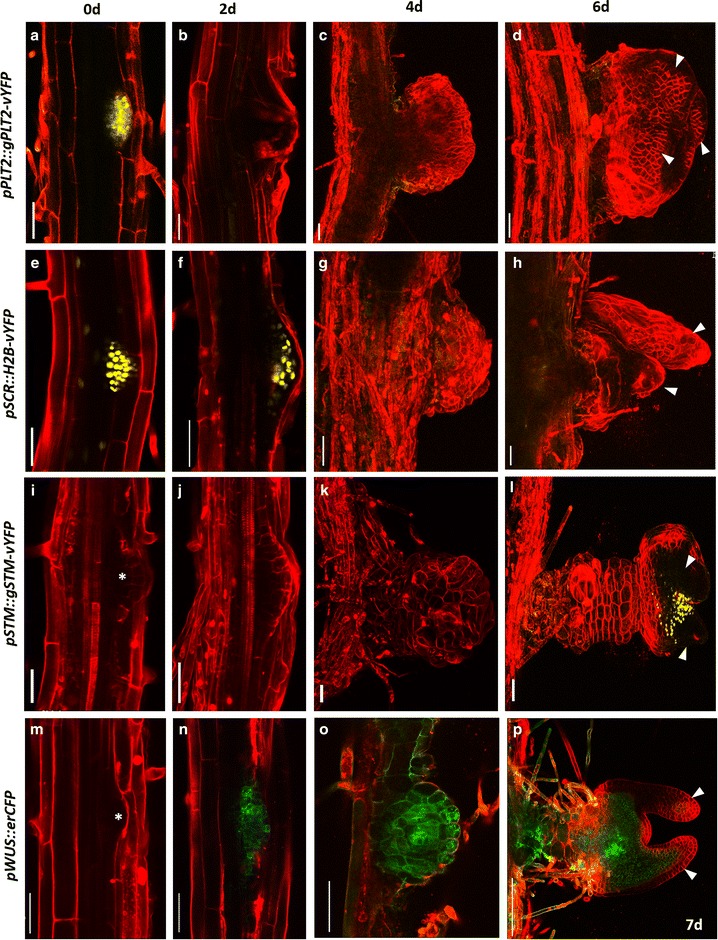
Confocal time lapse imaging during LRP to shoot conversion. a–d Time-lapse imaging showing PLT2-vYFP expression during the conversion of LRP to shoot. Note the down-regulation of PLT2-vYFP during the development of shoot meristem. e–h Dynamic expression pattern of pSCR::H2B-vYFP during the conversion of root to shoot. i–l Live imaging showing STM-vYFP expression during the shoot conversion. l Note the appearance of STM-vYFP in the nascent shoot meristem. m–p Spatiotemporal expression pattern of pWUS::erCFP. p Note the rapid upregulation of pWUS::erCFP upon cytokinin treatment. Arrowhead in (d, h, l, p) marks leaf primordium. Red signal is propidium iodide stain in (a, b, e, f, i, j, m, n) and FM4-64 stain in the remaining. Scale bar 50 µm
Besides the developmental stage of LRP, the concentration of cytokinin is also a vital factor to accomplish LRP-to-shoot conversion. At lower concentration, cytokinin perturbs LRP organization and thereby suppresses lateral root outgrowth [20, 21]. At higher concentration, cytokinin triggers trans-differentiation leading to the conversion of LRP to shoot. We have tested various concentrations of three cytokinins such as 6-(γ,γ-dimethylallylamino) purine (2iP), trans-zeatin (TZ) and kinetin (KT) to identify the most effective concentration of cytokinin that can convert maximum number of LRP to shoot. We found that both 2iP and TZ (10 and 20 µM) were more efficient to convert LRP into shoot than KT (Method-I: 2iP = 8 ± 0.57, TZ = 7.8 ± 0.87, KT = 4.1 ± 0.85). While 10 µM 2iP or TZ was sufficient to convert LRP to shoot in method I, 20 µM of 2iP or TZ was required in method II.
Different ecotypes can respond differently to the conversion of LRP to shoot. We have tested our methods in three different ecotypes of Arabidopsis, namely, Columbia (Col-0), Wassilewskija (Ws) and Landsberg erecta (Ler). Both methods described here for LRP-to-shoot conversion were effective in all the three ecotypes with variations in shoot formation efficiency (Table 4). Ecotype Col-0 displayed the conversion of maximum number of LRP to shoot followed by Ler and Ws.
Table 4.
Frequency of LRP-to-shoot conversion that occurred in different Arabidopsis ecotypes (based on method 1, DSIM-1)
| Ecotypes | Frequency of shoot regeneration (per explant) |
|---|---|
| Col-0 | 8.62 ± 0.83 |
| Ler | 6.5 ± 1.17 |
| Ws | 5.5 ± 0.38 |
In addition to the developmental stages of root and hormonal concentrations, external environmental stimuli may play a crucial role in LRP-to-shoot conversion. The culture temperature can be a potential factor that influences the conversion. To test this, we cultured the explants at both normal temperature (22 °C) and high temperature (29 °C). Many of the LRP cultured at high temperature arrested (42 %) during the conversion process despite the abundance of cytokinin (Fig. 3j). These arrested LRP made green bulges. Interestingly, some of these bulges eventually lead to the formation of lateral roots (Fig. 3j). Consistent with our observation, a recent study shows that seedlings grown at high temperature make more lateral roots [22]. Incubation at lower temperature (18 °C/20 °C cycle) marginally reduced the efficiency of LRP-to-shoot conversion (Additional file 2). Our data suggests that LRP-to-shoot conversion is hypersensitive to temperature and the LRP choose default root developmental pathway at high temperature even in the presence of high cytokinin. We have also tested the effect of continuous light and diurnal (16 h light/8 h dark) conditions on LRP to shoot conversion. We did not notice any significant difference in conversion between these conditions (Additional file 3).
To study the anatomical changes during the LRP conversion to shoot meristem on DSIM, histological sections were generated (Fig. 4a–d). Our study shows that LRP-to-shoot conversion is accompanied by organized cell proliferation in LRP. These divisions, further, lead to the formation of shoot meristem and leaf primordium. Histological analysis can help to ascertain the cells in LRP that are competent to convert into shoot fate. Coupling these cellular changes with molecular changes can lead to the deeper insights into the trans-differentiation during LRP-to-shoot conversion.
Fig. 4.
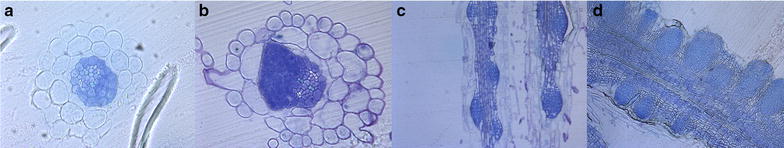
Anatomical changes during the conversion of LRP. a–d Histological section depicting the anatomical changes during the conversion of LRP to shoot meristem. a Cross section of a 3 dpg root treated with NAA for 20 h (i.e. first, lateral root induction step in method I). b Cross section of a 3 dpg root treated with NAA for 20 h followed by 4 day incubation on DSIM medium (i.e. second, shoot conversion step in method I). c Longitudinal section after lateral root induction step (3 days + 20 h NAA). d Longitudinal section after shoot conversion step (3 days + 20 h NAA + 4 days DSIM)
Cytokinin triggers massive reprogramming associated with trans-differentiation. Studying the spatio-temporal expression patterns of cell fate specific regulators using confocal time-lapse imaging would enable to understand the early molecular events during trans-differentiation. Our method illustrates how time-lapse imaging could be revealing to study the process of conversion of LRP to shoot. We have used a set of cell fate specific fluorescent markers to analyze their expression patterns. We observed significant changes in the expression patterns of these cell fate specific markers even before morphological changes were visible in LRP. First we monitored the expression pattern of root specific stem cell regulator, PLETHORA2 (PLT2) [23] tagged with yellow fluorescent protein (YFP) during the conversion of LRP into shoot. The translational fusion pPLT2::PLT2-vYFP was expressed in stem cell niche of primary root meristem and in LRP from stage II onwards as previously reported [13, 15]. Time-lapse imaging showed the drastic reduction in PLT2-vYFP expression in LRP soon after transferring to DSIM and it was undetectable after 24 h, suggesting the rapid cell fate changes (Figs. 5b, 6a–f). As expected, PLT2-vYFP expression remained absent in the nascent shoot meristem and leaf primordia (Fig. 5c, d). Next we examined the expression pattern of QC and endodermis specific SCARECROW (SCR) gene [24, 25] during LRP-to-shoot conversion. Before transferring to DSIM, pSCR::H2B-vYFP was expressed in LRP (Figs. 5e, 6g). pSCR::H2B-vYFP expression was detectable even after 63 h of DSIM treatment (Figs. 5f, 6h–k) but its expression was down regulated afterwards (Figs. 5g, 6l). pSCR::H2B-vYFP expression was reinstated in regenerating leaves at petiole and mid-vein. Next, we analyzed the spatio-temporal expression pattern of shoot meristem specific marker, SHOOT MERISTEMLESS (STM) [26] during the LRP-to-shoot conversion. Expression of pSTM::STM-vYFP was neither detectable in untreated LRP nor in LRP at early days on DSIM (Fig. 5i–k). The earliest pSTM-vYFP expression was noticed when the shoot meristem developed from LRP (Fig. 5l). We next examined the expression pattern of shoot stem cell regulator, WUS [27] during the conversion. pWUS::erCFP was expressed in the organizing center of shoot apical meristem and not in LRP as previously reported (Fig. 5m) [6]. Upon DSIM treatment, pWUS::erCFP expression was rapidly induced in LRP (Figs. 5n, o, 6o–r). Once the shoot meristem is formed, expression of pWUS::erCFP got confined to a smaller domain of shoot meristem as reported in our previous study (Fig. 5p) [6]. We noted the pWUS::erCFP expression in treated LRP (at 24 h) much prior to the disappearance of pSCR::H2B-vYFP (at 66 h) suggesting the intermediate developmental phases during LRP-to-shoot conversion (Figs. 5f, n, 6l, o). It is important to note that the onset of pSTM:STM-vYFP expression is much delayed as compared to pWUS::erCFP during this process (Figs. 5l, n, 6o). Together, the expression pattern of tissue specific cell fate markers for root and shoot clearly demonstrate dynamic changes in the relative abundance of stem cell regulators and rapid cell fate changes during the conversion.
Fig. 6.
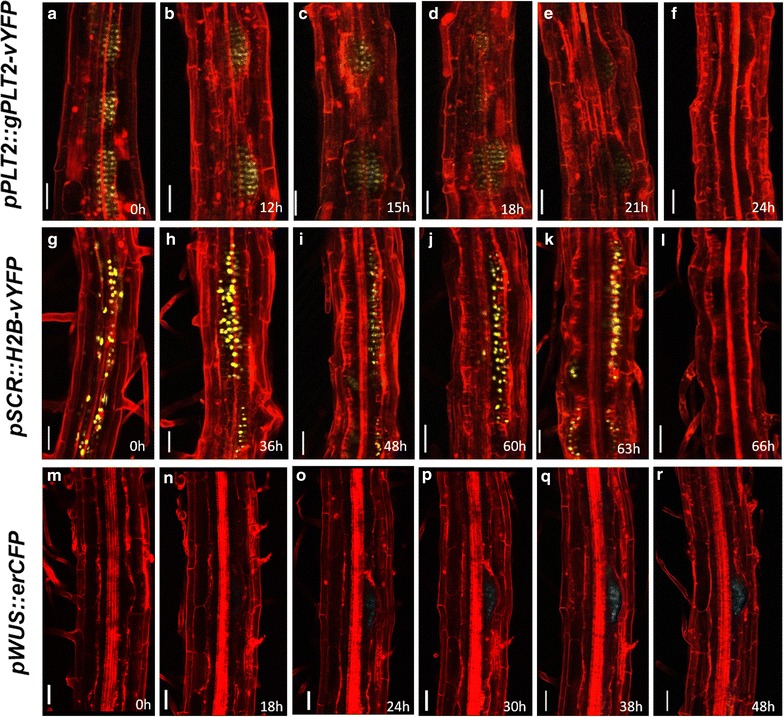
Confocal time lapse imaging of hourly resolution during LRP to shoot transition. a–f Time-lapse imaging showing PLT2-vYFP expression during the early hours of LRP to shoot conversion. f PLT2-vYFP expression is undetectable at 24 h on DSIM. g–l Gradual disappearance of pSCR::H2B-vYFP expression upon DSIM treatment. l 66 h of DSIM treatment leads to the loss of pSCR::H2B-vYFP expression from LRP. m–r Induction of pWUS::erCFP on 24 h of DSIM treatment. Red signal is propidium iodide stain. Scale bar 50 µm
Further we analyzed the spatiotemporal expression pattern of the factors involved in auxin signaling pathway. AUXIN RESPONSE FACTOR10 (ARF10) regulates the auxin mediated signal transduction pathway [28]. pARF10::ARF10-vYFP was expressed in untreated primary root meristem and LRP (Fig. 7a). In LRP its expression was predominantly detected in epidermis. Upon cytokinin treatment, ARF10-vYFP expression persisted in LRP undergoing fate changes, although its level was reduced (Fig. 7b, c). High level of ARF10-vYFP expression was observed in shoot meristem and leaf primordia (Fig. 7d).
Fig. 7.
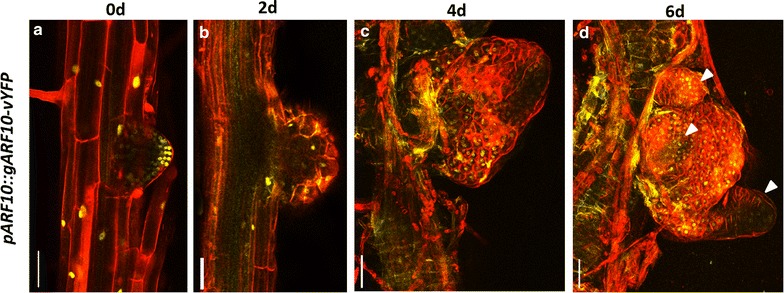
Time lapse imaging of auxin response factor during conversion. a–d Confocal time-lapse imaging showing ARF10-YFP expression during the conversion of LRP to shoot. Note the persistence of ARF10-YFP expression in both LRP and leaf primordia. Red signal is propidium iodide stain in (a, b) and FM4-64 stain in (c, d). Scale bar 50 µm
In different plant species, the ideal culture conditions such as culture temperature, developmental stages of LRP and hormonal concentration required for the transition can vary. Based on our results in Arabidopsis, we recommend to standardize (1) temperature, (2) developmental stages of lateral root primordia (preferably early stages of LRP), (3) concentration of cytokinin, to adapt our protocol for other plant species.
Collectively, our method describes the use of time-lapse imaging technique for the fine dissection of the early developmental changes during trans-differentiation in Arabidopsis. We have also explored the role of external stimuli such as light and temperature in the LRP to shoot conversion. Further, this method may be adopted for other plant species on standardizing the parameters that influence the conversion process.
Conclusion
Studies on the conversion of LRP-to-shoot are important in the light of the fact that many plant species adopt root-to-shoot conversion as one of the natural means of vegetative propagation. Many species like murraya, poplars, guava etc. have the natural capacity to make shoots from root when attached to or detached from the mother plant. The newly formed plant is called ‘a sucker’ or ‘a root sprout’ and these eventually grow as an independent plant. Although the frequency of sucker formation is less, some plant species mainly depend upon this mode of reproduction when seed based propagation is rare. Studies on this mode of propagation are highly fragmentary and the mechanisms underlying root-to-shoot conversion remain unknown. Although our method has been developed for Arabidopsis, it can be used as a reference to study the cellular and molecular mechanism underlying trans-differentiation in other plant species. Taken together, our method for the LRP-to-shoot conversion is highly efficient, independent of genotype tested and is amenable for time-lapse imaging to delineate the mechanism underlying this process.
Authors’ contributions
AK, DR, XW, SB and ZBT performed research. AK, DR, XW, KS, JX, APM and KP analyzed data. AK and KP wrote the paper with the help of all co-authors. XW, JX and APM contributed new reagents and tools. All authors read and approved the final manuscript.
Acknowledgements
KP acknowledges the Grant from Department of Biotechnology (DBT), Govt. of India and infrastructures and financial support from IISER-TVM. AK is supported by IISER-TVM fellowship. DR acknowledges UGC fellowship. XW and APM acknowledge the grant from Academy of Finland and University of Helsinki. XW is also supported by a grant from the Chinese Scholarship Council. ZBT acknowledges DST-Inspire fellowship. JX acknowledges National University of Singapore Young Investigator Award and an AcRF Tier 2 Grant (MOE2014-T2-1-128) from the Ministry of Education of Singapore. The authors acknowledge Eswar Reddy and Anzal KS for their generous technical support.
Competing interests
The authors declare that they have no competing interests.
Additional files
10.1186/s13007-016-0127-5 Stage of LRP determines root to shoot conversion. (A) Late stage LRP of wild type plant on DSIM does not convert to shoot (B) Wild type untreated plant on ½ MS with outgrown lateral roots. Arrowhead in A marks outgrown lateral roots. Scale bar: 1 mm.
10.1186/s13007-016-0127-5 Effect of temperature on LRP to shoot conversion. Incubation at lower temperature (18 °C/20 °C cycle) marginally reduced the efficiency of LRP-to-shoot conversion as compared to 22 °C.
10.1186/s13007-016-0127-5 Effect of light on LRP to shoot conversion. Diurnal and continuous light conditions produced no significant difference in LRP to shoot conversion in explants (p > 0.05).
Footnotes
Dhanya Radhakrishnan and Xin Wang contributed equally to this work
Contributor Information
Abdul Kareem, Email: kareemvk@iisertvm.ac.in.
Dhanya Radhakrishnan, Email: dhanyaradhakrishnan15@iisertvm.ac.in.
Xin Wang, Email: xin.wang@helsinki.fi.
Subhikshaa Bagavathiappan, Email: subhikshaab@iisertvm.ac.in.
Zankhana B. Trivedi, Email: zankhanatrivedi2413@iisertvm.ac.in
Kaoru Sugimoto, Email: kaoru_sugimoto@rs.tus.ac.jp.
Jian Xu, Email: dbsxj@nus.edu.sg.
Ari Pekka Mähönen, Email: AriPekka.Mahonen@helsinki.fi.
Kalika Prasad, Email: kalika@iisertvm.ac.in.
References
- 1.Birnbaum KD, Alvarado AS. Slicing across kingdoms: regeneration in plants and animals. Cell. 2008;132(4):697–710. doi: 10.1016/j.cell.2008.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pulianmackal AJ, Kareem AV, Durgaprasad K, Trivedi ZB, Prasad K. Competence and regulatory interactions during regeneration in plants. Front Plant Sci. 2014;5:142. doi: 10.3389/fpls.2014.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sugimoto K, Jiao Y, Meyerowitz EM. Arabidopsis regeneration from multiple tissues occurs via a root development pathway. Dev Cell. 2010;18(3):463–471. doi: 10.1016/j.devcel.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Atta R, Laurens L, Boucheron-Dubuisson E, Guivarc’h A, Carnero E, Giraudat-Pautot V, Rech P, Chriqui D. Pluripotency of Arabidopsis xylem pericycle underlies shoot regeneration from root and hypocotyl explants grown in vitro. Plant J. 2009;57(4):626–644. doi: 10.1111/j.1365-313X.2008.03715.x. [DOI] [PubMed] [Google Scholar]
- 5.Che P, Lall S, Howell SH. Developmental steps in acquiring competence for shoot development in Arabidopsis tissue culture. Planta. 2007;226(5):1183–1194. doi: 10.1007/s00425-007-0565-4. [DOI] [PubMed] [Google Scholar]
- 6.Kareem A, Durgaprasad K, Sugimoto K, Du Y, Pulianmackal AJ, Trivedi ZB, Abhayadev PV, Pinon V, Meyerowitz EM, Scheres B, Prasad K. PLETHORA genes control regeneration by a two-step mechanism. Curr Biol. 2015;25(8):1017–1030. doi: 10.1016/j.cub.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatfield SP, Capron R, Severino A, Penttila PA, Alfred S, Nahal H, Provart NJ. Incipient stem cell niche conversion in tissue culture: using a systems approach to probe early events in WUSCHEL-dependent conversion of lateral root primordia into shoot meristems. Plant J. 2013;73(5):798–813. doi: 10.1111/tpj.12085. [DOI] [PubMed] [Google Scholar]
- 8.Dubrovsky JG, Rost TL, Colon-Carmona A, Doerner P. Early primordium morphogenesis during lateral root initiation in Arabidopsis thaliana. Planta. 2001;214(1):30–36. doi: 10.1007/s004250100598. [DOI] [PubMed] [Google Scholar]
- 9.Celenza JL, Jr, Grisafi PL, Fink GR. A pathway for lateral root formation in Arabidopsis thaliana. Genes Dev. 1995;9(17):2131–2142. doi: 10.1101/gad.9.17.2131. [DOI] [PubMed] [Google Scholar]
- 10.Gallois JL, Nora FR, Mizukami Y, Sablowski R. WUSCHEL induces shoot stem cell activity and developmental plasticity in the root meristem. Genes Dev. 2004;18:375–380. doi: 10.1101/gad.291204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee M, Phillips RL. The chromosomal basis of somaclonal variation. Annu Rev Plant Physiol Plant Mol Biol. 1988;39(1):413–437. doi: 10.1146/annurev.pp.39.060188.002213. [DOI] [Google Scholar]
- 12.Krizek BA, Eaddy M. AINTEGUMENTA-LIKE6 regulates cellular differentiation in flowers. Plant Mol Biol. 2011;78(3):199–209. doi: 10.1007/s11103-011-9844-3. [DOI] [PubMed] [Google Scholar]
- 13.Hofhuis H, Laskowski M, Du Y, Prasad K, Grigg S, Pinon V, Scheres B. Phyllotaxis and rhizotaxis in Arabidopsis are modified by three PLETHORA transcription factors. Curr Biol. 2013;23(11):956–962. doi: 10.1016/j.cub.2013.04.048. [DOI] [PubMed] [Google Scholar]
- 14.Prasad K, Grigg SP, Barkoulas M, Yadav R, Sanchez-Perez GF, Pinon V, Blilou I, Hofhuis H, Dhonukshe P, Galinha C, Mähönen A, Muller WH, Raman S, Verkleij AJ, Snel B, Reddy VG, Tsiantis M, Scheres B. Arabidopsis PLETHORA transcription factors control phyllotaxis. Curr Biol. 2011;21(13):1123–1128. doi: 10.1016/j.cub.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 15.Mähönen A, ten Tusscher K, Siligato R, Smetana O, Díaz-Triviño S, Salojärvi J, Wachsman G, Prasad K, Heidstra R, Scheres B. PLETHORA gradient formation mechanism separates auxin responses. Nature. 2014;515(7525):125–129. doi: 10.1038/nature13663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugimoto K, Meyerowitz EM. Regeneration in Arabidopsis tissue culture. Methods Mol Biol. 2013;959:265–275. doi: 10.1007/978-1-62703-221-6_18. [DOI] [PubMed] [Google Scholar]
- 17.Scheres B, Wolkenfelt H, Willemsen V, Terlouw M, Lawson E, Dean C, Weisbeek P. Embryonic origin of the Arabidopsis primary root and root-meristem initials. Development. 1994;120(9):2475–2487. [Google Scholar]
- 18.Mahonen AP, Bonke M, Kauppinen L, Riikonen M, Benfey PN, Helariutta Y. A novel two-component hybrid molecule regulates vascular morphogenesis of the Arabidopsis root. Genes Dev. 2000;14(23):2938–2943. doi: 10.1101/gad.189200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Himanen K, Boucheron E, Vanneste S, de Almeida Engler J, Inze D, Beeckman T. Auxin-mediated cell cycle activation during early lateral root initiation. Plant Cell. 2002;14(10):2339–2351. doi: 10.1105/tpc.004960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bielach A, Podlesakova K, Marhavy P, Duclercq J, Cuesta C, Muller B, Grunewald W, Tarkowski P, Benkova E. Spatiotemporal regulation of lateral root organogenesis in Arabidopsis by cytokinin. Plant Cell. 2012;24(10):3967–3981. doi: 10.1105/tpc.112.103044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laplaze L, Benkova E, Casimiro I, Maes L, Vanneste S, Swarup R, Weijers D, Calvo V, Parizot B, Herrera-Rodriguez MB, Offringa R, Graham N, Doumas P, Friml J, Bogusz D, Beeckman T, Bennett M. Cytokinins act directly on lateral root founder cells to inhibit root initiation. Plant Cell. 2007;19(12):3889–3900. doi: 10.1105/tpc.107.055863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang R, Zhang Y, Kieffer M, Yu H, Kepinski S, Estelle M. HSP90 regulates temperature-dependent seedling growth in Arabidopsis by stabilizing the auxin co-receptor F-box protein TIR1. Nat Commun. 2016;7:10269. doi: 10.1038/ncomms10269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aida M, Beis D, Heidstra R, Willemsen V, Blilou I, Galinha C, Nussaume L, Noh Y-S, Amasino R, Scheres B. The PLETHORA genes mediate patterning of the Arabidopsis root stem cell niche. Cell. 2004;119(1):109–120. doi: 10.1016/j.cell.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 24.Di Laurenzio L, Wysocka-Diller J, Malamy JE, Pysh L, Helariutta Y, Freshour G, Hahn MG, Feldmann KA, Benfey PN. The SCARECROW gene regulates an asymmetric cell division that is essential for generating the radial organization of the Arabidopsis root. Cell. 1996;86(3):423–433. doi: 10.1016/S0092-8674(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 25.Sabatini S, Heidstra R, Wildwater M, Scheres B. SCARECROW is involved in positioning the stem cell niche in the Arabidopsis root meristem. Genes Dev. 2003;17(3):354–358. doi: 10.1101/gad.252503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barton MK, Poethig RS. Formation of the shoot apical meristem in Arabidopsis thaliana—an analysis of development in the wild-type and in the shoot meristemless mutant. Development. 1993;119(3):823–831. [Google Scholar]
- 27.Mayer KF, Schoof H, Haecker A, Lenhard M, Jurgens G, Laux T. Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell. 1998;95(6):805–815. doi: 10.1016/S0092-8674(00)81703-1. [DOI] [PubMed] [Google Scholar]
- 28.Liu PP, Montgomery TA, Fahlgren N, Kasschau KD, Nonogaki H, Carrington JC. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 2007;52(1):133–146. doi: 10.1111/j.1365-313X.2007.03218.x. [DOI] [PubMed] [Google Scholar]