

Duchenne muscular dystrophy is a devastating disease of striated muscle deterioration that is uniformly fatal from respiratory and/or cardiac failure. Here, we show that hypoxia is sufficient to cause significant cardiac lesions in the dystrophic heart, secondary to energetic inefficiency and poor oxygen extraction of the dystrophic myocardium.
Keywords: hypoxia, mitochondria, dystrophic cardiomyopathy, dystrophin, Duchenne muscular dystrophy
Abstract
Duchenne muscular dystrophy (DMD) is a disease of progressive destruction of striated muscle, resulting in muscle weakness with progressive respiratory and cardiac failure. Respiratory and cardiac disease are the leading causes of death in DMD patients. Previous studies have suggested an important link between cardiac dysfunction and hypoxia in the dystrophic heart; these studies aim to understand the mechanism underlying this connection. Here we demonstrate that anesthetized dystrophic mice display significant mortality following acute exposure to hypoxia. This increased mortality is associated with a significant metabolic acidosis, despite having significantly higher levels of arterial Po2. Chronic hypoxia does not result in mortality, but rather is characterized by marked cardiac fibrosis. Studies in isolated hearts reveal that the contractile function of dystrophic hearts is highly susceptible to short bouts of ischemia, but these hearts tolerate prolonged acidosis better than wild-type hearts, indicating an increased sensitivity of the dystrophic heart to hypoxia. Dystrophic hearts display decreased cardiac efficiency and oxygen extraction. Isolated dystrophic cardiomyocytes and hearts have normal levels of FCCP-induced oxygen consumption, and mitochondrial morphology and content are normal in the dystrophic heart. These studies demonstrate reductions in cardiac efficiency and oxygen extraction of the dystrophic heart. The underlying cause of this reduced oxygen extraction is not clear; however, the current studies suggest that large disruptions of mitochondrial respiratory function or coronary flow regulation are not responsible. This finding is significant, as hypoxia is a common and largely preventable component of DMD that may contribute to the progression of the cardiac disease in DMD patients.
NEW & NOTEWORTHY
Duchenne muscular dystrophy is a devastating disease of striated muscle deterioration that is uniformly fatal from respiratory and/or cardiac failure. Here, we show that hypoxia is sufficient to cause significant cardiac lesions in the dystrophic heart, secondary to energetic inefficiency and poor oxygen extraction of the dystrophic myocardium.
duchenne muscular dystrophy (DMD) initially presents as skeletal muscle weakness; muscle dysfunction continues to progress, resulting in the loss of ambulation early in the second decade of life and death in the third or fourth decade (10, 16). Since its first description in the 19th century, respiratory failure has been recognized as the leading cause of mortality in DMD patients (12, 14, 30, 48). Improvements in symptomatic respiratory therapy have delayed mortality associated with respiratory failure in DMD patients (16, 33). The combination of skeletal muscle and respiratory dysfunction often masks the clinically significant cardiac disease that is present in DMD patients. The improved survival of DMD patients with symptomatic respiratory therapy has caused the underlying cardiac disease to become even more clinically significant. Cardiac disease was noted as a component of DMD in many of the earliest clinical reports (23, 24, 58), but understanding the pathophysiology of DMD in the heart has lagged behind that of skeletal muscle. As DMD progresses, respiratory and cardiac dysfunction both become clinically evident sometime after the loss of ambulation. Respiratory failure in DMD patients results from hypoventilation secondary to a weakened diaphragm; this progressive loss of ventilatory function results in transient periods of hypoxia. Bouts of hypoxia initially occur at night, even in patients with normal daytime pulmonary function (2, 35). Furthermore, over 70% of DMD patients display symptoms of respiratory disease before referral for respiratory therapy (2, 19, 36). These results indicate that bouts of hypoxia are routinely experienced by DMD patients, even in patients receiving ideal medical management (8–11). These observations raise the question of what effect bouts of hypoxia might have on the dystrophic heart.
DMD results from the loss of the large cytoskeletal protein dystrophin (28). Dystrophin is expressed in many tissues throughout the body, with the highest levels present in striated muscle. Dystrophin has been implicated in providing mechanical support to the membrane of striated muscle cells by functioning as a molecular shock absorber (54, 68, 73). Dystrophin also has a significant signaling function by acting as a scaffold to localize ion channels and enzymes to specific membrane microdomains (5, 17, 20–22, 63). In skeletal muscle, the absence of dystrophin results in the mislocalization of NOS1 (neuronal nitric oxide synthase), which results in the disruption of an important mechanism mediating contraction-induced vasodilation and results in functional ischemia (5, 60, 63). Furthermore, the loss of dystrophin in the heart results in a significant increase in the diastolic cytosolic Ca2+ concentration within cardiac myocytes (70, 72, 73). Increases in cytosolic Ca2+ are correlated with significant elevation of mitochondrial Ca2+ levels and increased production of reactive oxygen species (70).
The mdx mouse, a genetic model of DMD, presents with skeletal muscle pathology and reduced force generation (7, 45). In addition, mdx mice display significant reductions in cardiac function (44, 49, 56, 73) and significant declines in respiratory function (18, 29, 32). Mild hypoxia results in increased dysfunction (18) and apoptosis (38) in the diaphragm. The cardiac consequences of hypoxic exposure in the dystrophic heart remains an important area of investigation. In the studies presented here, the mdx mouse is used to assess the pathophysiological importance of hypoxia. Recent studies have demonstrated that acute exposure of anesthetized mdx mice to hypoxia is associated with acute myocardial decompensation, characterized by initial diastolic and subsequent systolic dysfunction (65). In the present study, it is shown that this acute hypoxia-induced mortality in anesthetized mice is associated with the development of metabolic acidosis. Interestingly, chronic hypoxia does not cause mortality in dystrophic mice, but does result in widely distributed cardiac fibrosis of the dystrophic heart. These observations have immediate clinical relevance, as they suggest that the short bouts of hypoxia experienced by DMD patients may result in myocardial damage, accelerating the decline in cardiac function. The data presented here implicate hypoxia as a potential inciting factor in the development of dystrophic cardiomyopathy and suggest that earlier initiation of respiratory support could delay the onset of heart disease in DMD patients.
METHODS
Animals.
Mouse colonies were established from control (C57BL/10SnJ) and mdx (C57BL/10 ScSn-Dmdmdx/J) mice obtained from Jackson Laboratories (Bar Harbor, ME). To prevent genetic drift, new breeding stock were obtained every four generations. Animals were fed standard chow diet ad libitum and were kept in a room with 12:12-h light-dark cycles. These studies used mice of both sexes, aged 3–6 mo, well before the onset of significant structural damage to the dystrophic heart. All procedures reported here were reviewed and approved by the University of Minnesota Institutional Animal Care and Use Committee and conform with National Institutes of Health guidelines.
Serial blood-gas and acute hypoxia studies.
Anesthesia in mice was induced with 5% isoflurane, and a surgical plane of anesthesia was maintained with 2% isoflurane in 100% oxygen. Mice were fitted with a pulse oximeter located on their right thigh (Starr Life Sciences, Holliston, MA) and needle electrodes configured to measure lead II ECG. Mice were intubated via a tracheotomy with a 20-gauge angiocath connected to the anesthetic circuit; this allowed tight control of the content of inhaled gases. The left carotid artery and right jugular vein were cannulated for arterial blood sampling and venous volume administration, respectively. Following instrumentation, the isoflurane was set to 1%, a level sufficient to maintain deep sedation. Gas mixtures were generated using a custom-built device consisting of two flow meters: one controlling oxygen and the other controlling nitrogen. The gas mixtures were calibrated using a ProOx P110 oxygen sensor (Biospherix, Lacona, NY) with a correlation coefficient of 0.996. Arterial blood samples (50 μl) were collected from the left carotid artery and quickly analyzed using a Stat Profile pHOx basic blood-gas analyzer (Nova Biomedical, Walthman, MA) providing pH, Po2, and Pco2 levels. The arterial catheter was flushed with heparinized saline, while simultaneously 50 μl of 10% albumin solution were injected into the jugular vein. In pilot studies, this blood sampling method has been demonstrated to have no significant hemodynamic effect. For these studies, death was defined as a heart rate of <200 beats/min, coupled with the loss of pulse measurements by the pulse oximeter. Previous studies have indicated that heart rates of <200 beats/min in the mouse are associated with pulseless electrical activity and occur following hemodynamic collapse (65).
Chronic hypoxia studies.
Wild-type and mdx mice, 3–4 mo of age, were housed within a sealed chamber in which the oxygen concentration was maintained at 10% through regulated injection of 100% nitrogen (Biospherix, Parish, NY). The carbon dioxide concentration was also monitored and averaged 0.11 ± 0.02% (mean ± SD). Mice were given free access to food and water. The chamber was briefly opened daily to assess animal welfare and to change soiled cages. Mice were housed in this hypoxic state for 3 wk before death; during this time there was no mortality observed. Sections of hearts from mdx and wild-type mice were stained with Sirius Red/Fast Green, as previously described (49). One image was collected from a midventricular short-axis cross section from each heart. These images were analyzed using ImageJ in a blinded fashion, briefly following the creation of an image montage (55). Color images were split into individual color channels. Predetermined thresholds were applied to each of these images to determine the total myocardial area and the area of positive Sirius red (i.e., collagen) staining. The resulting images were manually assessed, and areas of false-positive staining were removed. All of these analyses were performed without knowledge of the genotype of the mouse.
Langendorff heart studies.
The heart was excised from a heparinized (150 units) and anesthetized (200 mg/kg pentobarbital) mouse and placed in ice-cold physiological salt solution (in mM: 118 NaCl, 4.7 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.5 CaCl2, 25 NaHCO3, 15 glucose, 0.5 sodium-pyruvate, and 0.5 EDTA). The aorta was cannulated and the heart perfused with oxygenated (95% O2 and 5% CO2) physiological salt solution. A balloon was placed into the left ventricle and inflated to an end-diastolic pressure of ∼10 mmHg, and the heart was stimulated at 7 Hz. All hearts were subjected to an equilibration period of 20 min. Only hearts with left ventricular developed pressures of >90 mmHg and coronary flows between 2 and 5 ml/min were used for experiments. Some hearts were treated with 500 nM dobutamine to assess maximal cardiac function; in these hearts, 50 μM dobutamine were infused at 1/100th the average coronary flow rate. Other hearts were subjected to a metabolic flow protocol consisting of reactive hyperemia, 20 s of no-flow ischemia, a second 20-min recovery period, followed by a hypercapnic challenge. Hypercapnia was created by treating the bicarbonate buffered physiological salt solution with 80% oxygen and 20% carbon dioxide, the challenge consisted of a 10-min perfusion of the hypercapnic solution, followed by a 10-min recovery period.
Isolation of mouse cardiac myocytes.
Isolation of mouse cardiomyocytes was modified from a previously published procedure (34, 62, 69). Briefly, hearts were excised from animals anesthetized with pentobarbital sodium, and their ascending aorta was cannulated and mounted on a perfusion apparatus to allow perfusion of coronary vasculature. Hearts were perfused for 4 min with cell isolation buffer that contained the following (in mM): 113 NaCl, 4.7 KCl, 1.2 MgSO4, 0.6 NaH2PO4, 0.6 KH2PO4 10 2,3-butanedione monoxime, 30 taurine, and 20 glucose. Subsequently, cell isolation buffer containing 620 U/ml collagenase, 0.015 mg/ml DNase 1, and 0.104 mg/ml protease XIV was perfused for 10 min. Digested hearts were triturated in the collagenase-containing buffer to free individual myocytes. Calcium was added back by three separate additions of 1 M CaCl2 (1.5 mM final).
Cellular respiration.
Cardiomyocytes were resuspended in M-199 media (Sigma, St. Louis, MO) containing 0.02% BSA and 5% fetal bovine serum and used to assess cellular respiration. Cardiac myocyte respiration was assessed using the XF24 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA). Cardiac myocytes were plated (10,000 cells/well) on laminin-coated V7 microplates. Following attachment, cells were washed and incubated in warm XF assay media containing 0.2 mM sodium-palmitate/0.034 mM BSA at 37°C for 1 h. Baseline measurements of oxygen consumption rate were obtained, followed by the addition of the protonophore carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 5 μM) to assess FCCP-induced increases in oxygen consumption.
Mitochondrial assessment by electron microscopy.
Hearts were rapidly excised and perfused with cell isolation buffer, followed by fixation with 2.5% glutaraldehyde. A region of the left ventricle was stained with 1% osmium tetraoxide, embedded in resin, and sectioned. From each section, 10–20 non-overlapping images were collected using a JROL 1200 EXII transmission electron microscope. Using ImageJ, the areas of mitochondria were highlighted. The area, intensity relative to background muscle, and homogeneity of the mitochondrial areas were collected from 53 images from 3 mdx hearts containing 3,858 mitochondrial regions and 47 images from 3 C57BL/10 hearts containing 4,733 mitochondrial regions.
Blood lactate measurements.
Blood samples were collected from the cut tip of the tail of anesthetized C57BL/10 or mdx mice. Blood samples were analyzed using a Lactate Pro analyzer (Nova Biomedical, Walthman, MA).
Proteomic assessment of mitochondrial protein levels.
Cardiac muscle samples from dystrophic and wild-type dogs were collected and snap frozen (67). The samples from three dogs of each genotype were homogenized, and crude microsomal membrane fractions were prepared as previously described (52, 66). These membrane fractions were further fractionated and labeled with iTraq isobaric tags for relative quantification; one pair of membranes was double labeled as a technical replicate (71). Peptides were detected using a LTQ Orbitrap Velos (Thermo Scientific, Waltham, MA), and peptides were identified and mapped by ProteinPilot (Sciex, Framingham, MA). From this data set, proteins associated with oxidative phosphorylation were selected using the Kyoto Encyclopedia of Genes and Genomes reference pathways. Corrected iTraq probe areas were computed for each peptide. The probe areas from peptides with >95% confidence match for a given protein were added together to assess the abundance of the protein coming from the various labeled samples; only proteins with more than three peptides were included in subsequent analyses. The level of each protein was normalized to the mean of that present in the samples from wild-type hearts. A two-way ANOVA with a Tukey posttest was used to assess the presence of any statistically significant differences in protein level present in wild-type and dystrophic heart samples.
Statistical analysis.
Statistical analysis was performed using either R (57) or Prism 5 software (GraphPad Software, La Jolla, CA). Where multiple comparisons were performed, one-way ANOVA with Bonferroni posttest was used. Comparisons between two groups used Student's T-test. Kaplan-Meier method was used to create survival curves, followed by a log-rank (Mantel-Cox) test to determine differences in survival. The box-and-whisker plots in Figs. 1–7 show the distribution of the data with the mean displayed as a “+”; points greater than 1.5 times the interquartile distance are displayed separately, but are included in any relevant statistical comparisons.
Fig. 1.

Hypoxia in closed chest anesthetized mice results in mortality preceded by significant metabolic acidosis. A: dystrophic mice with closed chests display a significant degree of mortality following hypoxia exposure. Pulse-ox derived hemoglobin saturation (B) and arterial oxygen levels (C) confirm the presence of hypoxia. Under hypoxic conditions, a significant acidosis develops (D), which is characterized by normal levels of CO2 (E) and a negative base excess (F). Normoxic arterial samples were taken from mice following 10 min of 20% inspired O2 fraction; hypoxic measurements were taken after 10 min of 10% inspired O2 fraction (arrowhead in A). Data are derived from 10–12 mdx and 9 C57BL/10 mice. *P < 0.05 for the indicated comparisons. SpO2, arterial O2 saturation from pulse oximetry.
Fig. 7.

Mitochondrial content is normal in dystrophic hearts. A: oxygen consumption by quiescent isolated adult cardiac myocytes from dystrophic mice is significantly reduced compared with wild-type myocytes. *P < 0.05 for the indicated comparisons. B: uncoupling of the mitochondrial electron transport chain by 5 μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone results in equivalent oxygen consumption between C57BL/10 and mdx myocytes (14–15 wells isolated from 3 animals for each genotype). N.S., nonsignificant. C and D: electron microscopic analysis of mitochondrial content of myocardium reveal equivalent levels of mitochondrial density (data collected from 47–53 non-overlapping images from 3 hearts of each genotype). E: quantitative proteomic data set of cardiac membranes from dystrophic and wild-type dogs was probed for mitochondrial proteins. Results demonstrate that there are no significant differences in the expression of proteins of the electron transport chain in these severely affected dystrophic hearts.
RESULTS
Dystrophic mice have increased mortality and metabolic acidosis following hypoxia.
Previous studies have demonstrated that anesthetized dystrophic mice exposed to acute hypoxia display increased mortality linked to the initial development of diastolic dysfunction followed by cardiac decompensation and death (65). These prior studies used invasive cardiac catheterization featuring a thoracotomy and artificial ventilation, which may have contributed to the increased mortality. To further assess the pathophysiological mechanisms causing this mortality, a less invasive approach was used. Mice were anesthetized and instrumented with both jugular vein and carotid artery catheters. Importantly, these mice were not ventilated with positive pressure, and the chest remained closed so that control of respiration was regulated by the mouse. The anesthetized mice were intubated to ensure control of the inspired gas composition and to limit upper airway obstruction. After an equilibration period at 100% O2, the fraction of oxygen inhaled was reduced to 20% for 10 min and then to 10%. Even in this less invasive protocol, dystrophic mice display significant mortality during the hypoxic period, while wild-type mice tolerate this challenge without incident (Fig. 1A). Pulse oximetry and arterial Po2 (Fig. 1, B and C) assessed 10 min after the initiation of the hypoxic period demonstrate that the degree of hypoxia is not more severe in the dystrophic mice. However, dystrophic mice develop a significant acidosis during this hypoxic period compared with wild-type mice (Fig. 1D). Hypoxic arterial CO2 levels (Fig. 1E) and respiratory rates (data not shown) are not different between wild-type and mdx mice, indicating that respiratory dysfunction is not responsible for the acidosis observed. Thus dystrophic mice display a negative base excess, or an acid excess relative to wild-type mice. This is evident at 20% O2 and exacerbated during hypoxia (Fig. 1F). Resting blood lactates in dystrophic mice are 4.5 ± 0.6 mM, which is significantly higher than the 2.9 ± 0.3 mM lactate observed in wild-type blood. Together these data strongly indicate that dystrophic mice develop a metabolic acidosis in response to hypoxia. This is particularly surprising, given the significantly greater arterial Po2 that is present during hypoxia in the dystrophic mouse (Fig. 1C). The arterial pH present after 10 min of hypoxia is a significant predictor of mortality in mdx mice (P = 0.0149, r2 = 0.54). The magnitude of base excess decline and the hemoglobin saturation levels are also significantly correlated with mortality (P = 0.0043, r2 = 0.66 for base excess and P = 0.0042, r2 = 0.58 for hemoglobin saturation). There is no correlation between survival time and arterial Pco2 or respiratory rate in these studies. Interestingly, the heart rate of dystrophic mice after 10 min of hypoxia is inversely related to mortality (P = 0.0126, r2 = 0.48). It is not possible to confirm causation from these correlations; however, the relatively high metabolic activity of the heart in the face of systemic anaerobic metabolism presents significant challenges to the myocardium. Skeletal muscle is an important source of lactate production and is well suited to tolerate periods of anaerobic metabolism. This is in stark contrast to the heart, which requires aerobic metabolism to function normally, as such periods of hypoxia may be more detrimental to cardiac tissues.
Chronic hypoxia results in significant cardiac fibrosis.
Interestingly, chronic exposure of dystrophic mice to 10% oxygen levels did not result in increased mortality of dystrophic mice. Both dystrophic and wild-type mice survived throughout the 3-wk hypoxic challenge period. However, examination of heart sections collected following this hypoxic period reveal the presence of significant levels of fibrosis in dystrophic hearts (Fig. 2). These observations clearly demonstrate that chronic hypoxia, while not lethal, does induce significant cardiac damage in the dystrophic heart. The Langendorff isolated heart preparation was used to further understand the effects of hypoxia on the cardiac function of the dystrophic heart.
Fig. 2.

Chronic hypoxic exposure causes increased fibrosis in dystrophic hearts. Three-week exposure to hypoxia (10% O2) results in significant levels of fibrosis, as assessed by Sirius red/fast green staining. *P = 0.00075 derived from 8 control hearts and 11 mdx heart sections.
Contractile function of dystrophic hearts is sensitive to ischemia but tolerant of acidosis.
In the Langendorff heart preparation, the dystrophic heart displayed normal contractile function under baseline conditions (Table 1). Wild-type hearts subjected to 20 s of no-flow ischemia respond to reperfusion with a transient increase in systolic function (Fig. 3, B, C, and F–I). This transient increase in contractile function peaked ∼5 s following reperfusion. In contrast, dystrophic hearts show a significant decrease in both systolic and diastolic function in response to reperfusion (Fig. 3, D, E, and F–I). These changes in function are transient in nature, and the function of the dystrophic heart is equivalent to wild-type hearts after a second equilibration period. After this recovery period, hearts were then subjected to a 10-min period of hypercapnic acidosis (pH = 7.2). The onset of acidosis is marked by a dramatic decline in systolic function in both wild-type and dystrophic hearts (Fig. 4A). Following the 10-min period of acidosis, the heart was perfused with normocapnic physiological salt solution for another 10-min recovery period. Wild-type hearts fail to fully recover systolic and diastolic function following this prolonged acidosis. In contrast, dystrophic hearts subjected to the same level of acidosis show significantly greater recovery of both systolic and diastolic function (Fig. 4, B–D). Together, the sensitivity to ischemia and the tolerance of acidosis suggest that hypoxia is particularly injurious to the dystrophic heart.
Table 1.
Summary of the left ventricular contractile parameters of the isolated heart with and without dobutamine stimulation
| C57BL/10 | C57BL/10 + Dobutamine | mdx | mdx + Dobutamine | Dobutamine-induced Δ C57BL/10 | Dobutamine-induced Δ mdx | |
|---|---|---|---|---|---|---|
| LV systolic pressure, mmHg | 120.3 ± 6 | 155 ± 6.5* | 119 ± 3.7 | 133.5 ± 6.2# | 34.6 ± 3.9 | 14.6 ± 3.6‡ |
| LV developed pressure, mmHg | 114.8 ± 5.8 | 151.6 ± 6.5* | 110.6 ± 3.7 | 124.7 ± 6.5#† | 36.8 ± 4.1 | 14.1 ± 4‡ |
| LV dP/dtmax, mmHg/s | 3,802 ± 378 | 5,624 ± 431* | 3,734 ± 256 | 5,023 ± 424# | 1,822 ± 185 | 1,289 ± 237 |
| LV dP/dtmin, mmHg/s | −2,705 ± 171 | −4,457 ± 215* | −2,676 ± 120 | −3,531 ± 232#† | −1,752 ± 136 | −855 ± 153‡ |
| LV rate-pressure product, mmHg/min | 50,168 ± 2,496 | 69,882 ± 3,612* | 50,117 ± 1,608 | 59,858 ± 3,246# | 19,714 ± 1,535 | 9,741 ± 1,916‡ |
Values are means ± SE; n = 11 C57BL/10 and 22 mdx mice. Δ, Change; LV, left ventricle; dP/dtmax and dP/dtmin, maximum and minimum change in pressure over time, respectively. P < 0.05 vs.
C57BL/10 baseline measures, #mdx baseline measures,
C57BL/10 + dobutamine, and
dobutamine-induced Δ C57BL/10.
Fig. 3.
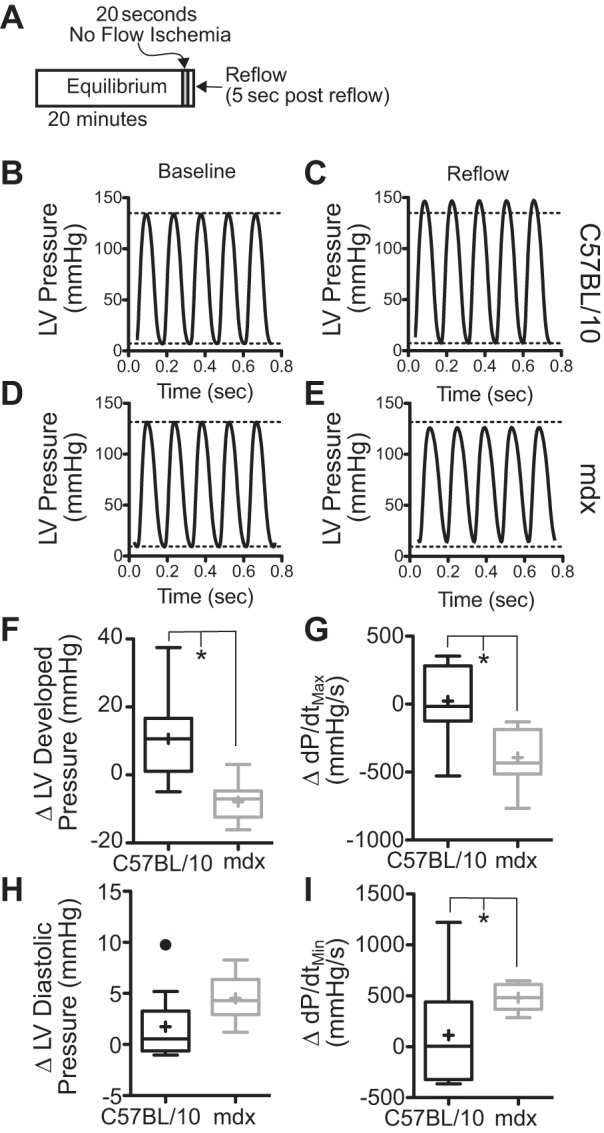
Dystrophic hearts display significant reductions in systolic and diastolic function following brief ischemia. A: following an initial equilibration phase, all coronary flow is blocked for 20 s, and then the heart is reperfused. B and C: in C57BL/10 hearts, shortly (≈5–10 s) following reperfusion, there is a brief peak in systolic function. D and E: in contrast, dystrophic hearts during this period have depressed systolic and diastolic function. There are significant transient reductions in both systolic (F and G) and diastolic (H and I) function in the dystrophic heart. Function is restored 5–10 min following the ischemic period. Data are shown as box plots with mean depicted as a “+”; points greater than 1.5 times the interquartile interval are shown separately. Data are derived from 9 dystrophic and 11 wild-type hearts. *P < 0.05 for the indicated comparisons. LV, left ventricle; Δ, change; dP/dtmin and dP/dtmax, minimum and maximum change in pressure over time, respectively.
Fig. 4.

Dystrophic hearts recover significantly better than controls following prolonged acidosis. A: contractile function is dramatically reduced following the introduction of a hypercapnic (pH ≈ 7.2) perfusate. Values are means ± SE. Following restoration of normocapnic perfusion, dystrophic hearts recover significantly more function than C57BL/10 hearts. This is evident in both systolic (B and C) and diastolic (D) functions. Data are derived from 9 mdx and 7 C57BL/10 hearts. Values are shown as box-and-whisker plots. *P < 0.05 for the indicated comparisons. LVDP, LV developed pressure.
Dystrophic hearts display attenuated response to dobutamine stimulation.
Infusion of dobutamine into the coronary vessels of either dystrophic mdx or wild-type C57BL/10 hearts results in significant increases in systolic function, as observed in significant increases in left ventricular developed pressure, left ventricular maximum change in pressure over time (dP/dt), and the rate pressure product (Table 1). Improved diastolic function is also observed with an increase in the maximal rate of pressure decline (dP/dtmin; Table 1). The magnitude of the response to dobutamine is significantly attenuated in the mdx heart compared with C57BL/10 hearts. The dobutamine-induced changes in both left ventricular developed pressure and rate pressure product are significantly smaller than those observed in C57BL/10 hearts (Table 1). This attenuated response to dobutamine is also evident in diastolic function, as demonstrated by a significantly smaller increase in the rate of relaxation (dP/dtmin; Table 1). Importantly, all of these experiments are preformed in young (4–5 mo) mdx mice, indicating that these functional deficits precede the presence of significant levels of myocardial scarring.
Dystrophic hearts have reduced cardiac efficiency.
Under baseline conditions, the contractile function of the dystrophic heart in the Langendorff preparation is equivalent to that of wild-type hearts (Table 1). However, the oxygen consumed to achieve this level of contractile activity in dystrophic hearts is significantly greater than that of wild-type hearts (Fig. 5A). Together these data indicate that dystrophic hearts have reduced cardiac efficiency, in that they require more oxygen to accomplish the same amount of work (Fig. 5B). Dobutamine increases oxygen consumption in C57BL/10 and mdx hearts to the same degree, 13.2 ± 1.3 and 12.3 ± 1.3 μmol·min−1·g−1, respectively. However, the increases in contractile function are significantly lower in the dystrophic hearts (Table 1). This results in a further decrease in the cardiac efficiency of the dystrophic heart with dobutamine stimulation (Fig. 5B). Despite the higher rate of basal oxygen consumption observed in the dystrophic heart, there is a significant reduction in the percentage of oxygen extracted from the perfusate (Fig. 5C). In wild-type mice, the addition of dobutamine has no significant effect on the percentage of oxygen extracted; in contrast, the addition of dobutamine to the dystrophic heart significantly increases the oxygen extraction. There are several potential causes for this observation, including defects in cardiac perfusion that might result in oxygenated perfusate bypassing metabolically active regions of the heart. To address this possibility, the regulation of coronary flow was assessed in the dystrophic heart.
Fig. 5.
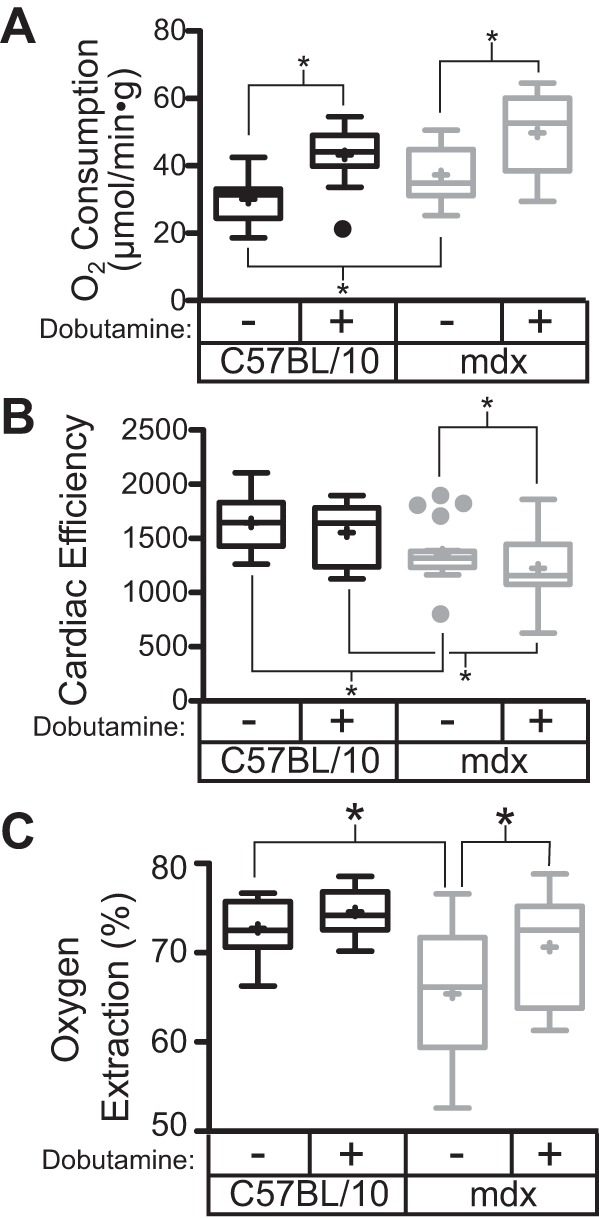
Dystrophic hearts have significant reductions in cardiac efficiency. At baseline in the Langendorff heart preparation, there is no significant contractile dysfunction in the dystrophic heart. However, dystrophic hearts consume more O2 under baseline conditions (A) and thus have significant reductions in cardiac efficiency (B). C: dystrophic hearts demonstrate a significant reduction in oxygen extraction, which is significantly improved with dobutamine treatment. Data are derived from 10 C57BL/10 and 19 mdx hearts. *P < 0.05 based on one-way ANOVA and post hoc tests.
Dystrophic coronary vessels respond normally to metabolic vasodilatory signals.
These studies hypothesized that reductions in oxygen extraction by the Langendorff perfused heart could result from the mismatching of perfusion and oxygen consumption within the dystrophic heart. There is significant literature describing the presence of functional ischemia in the skeletal muscle of mdx mice and DMD and Becker muscular dystrophy patients (47, 60, 63, 64). Other studies have demonstrated defects in flow-mediated dilation in the carotid and mesenteric arteries of dystrophic mice (41–43). Similar defects in the coronary arteries could result in a functional ischemia in regions of the myocardium, that when coupled to hypoxia, may push the heart into a state of unsustainable anaerobic metabolism. To test this hypothesis, the metabolic control of coronary flow in the dystrophic heart was assessed. Using a Langendorff isolated heart preparation, several protocols were used to probe the coronary flow response to metabolic vasodilatory signals in the mdx mouse. Under baseline conditions, dystrophic hearts display a significantly higher coronary flow compared with wild-type hearts (Fig. 6A). This increased flow provides an explanation of the higher total oxygen consumption with a smaller arterial-to-venous oxygen gradient. A brief bout of no-flow ischemia is a very powerful vasodilatory signal that results in significant increases in coronary flow secondary to the accumulation of metabolic waste (3, 31). Dystrophic hearts subjected to 20 s of no-flow ischemia develop an equivalent degree of coronary vasodilation as that seen in wild-type hearts following reperfusion (Fig. 6B). After a 20-min recovery from this short ischemic bout, hearts were subjected to a hypercapnic acidosis (27). During the acidosis challenge, contractile function of the heart is dramatically reduced (Fig. 4A), but coronary flow increases dramatically (Fig. 6C). As with brief ischemia, the coronary flow to the dystrophic heart increases to the same degree as that of wild-type hearts. In other hearts, dobutamine administration was used to assess the coronary flow response to increased contractile activity. Similar to the other challenges of coronary flow, the dystrophic coronary vasculature responds normally to dobutamine infusion (Fig. 2D). Taken together, these data indicate that the dystrophic coronary vessels respond normally to metabolic stimuli of coronary vasodilation, suggesting that any conditions causing regional ischemia would be addressed normally by vasodilation induced by the accumulation of metabolic wastes. This data suggests that the delivery of perfusate in the dystrophic heart is reasonably uniform throughout the myocardium. These results indicating intact oxygen delivery in the dystrophic heart do not support the hypothesis that mismatched perfusion and metabolic demand explain the reduced oxygen consumption.
Fig. 6.

Coronary vasculature responds normally to metabolic vasodilation signals. A: baseline coronary flow is significantly elevated in the dystrophic Langendorff heart. There is no difference in the coronary flow response between mdx and C57BL/10 hearts in response to 20 s of ischemia (B), hypercapnic (acidotic) perfusate (C), or 500 nM dobutamine (D). Baseline data are derived from 22 C57BL/10 and 46 mdx hearts. Ischemia and hypercapnia are derived from 10 C57BL/10 and 9 mdx hearts. Dobutamine data are from 11 C57BL/10 and 22 mdx hearts. Data are shown as box plots with mean depicted as a “+”; points greater than 1.5 times the interquartile interval are shown separately. *P < 0.05 for the indicated comparison.
Mitochondrial respiratory function in dystrophic hearts.
Another potential cause of reduced oxygen extraction is a defect in mitochondrial oxygen utilization. There is general consensus that the mitochondrial content of dystrophic skeletal muscle is reduced relative to wild-type skeletal muscle (15, 25, 39, 59, 61). It remains unclear if these changes are a direct consequence of the absence of dystrophin or secondary to the significant muscle regeneration that is occurring in skeletal muscle. In the heart, there is less agreement in the literature; isolated mitochondrial studies indicate no difference between wild-type and mdx hearts (1, 4). However, other studies indicate energetic deficiencies are present in both the perfused dystrophic heart (26, 74) and more recently in the intact heart (13). The increased levels of oxygen consumption observed in these studies (Fig. 5A) and elsewhere (37) would suggest that mitochondrial oxygen utilization is intact, yet energetic deficiencies remain. The poor oxygen extraction of the dystrophic heart at baseline could be caused by defects within the mitochondrial electron transport chain (ETC), which is responsible for consuming the vast majority of oxygen in the heart. To further assess the mitochondrial respiratory function in the dystrophic heart, the oxygen consumption of acutely isolated noncontracting adult cardiac myocytes was measured. Myocytes from dystrophic mice have a basal level of oxygen consumption that is significantly lower compared with wild-type myocytes (Fig. 7A). Importantly, the dissipation of the proton motive force by FCCP induces an increase in oxygen consumption that is not different between wild-type and dystrophic myocytes, suggesting that the oxygen consumption apparatus is relatively intact in the dystrophic heart (Fig. 7B), consistent with the increased level of oxygen consumption observed in the isolated heart (Fig. 5A). Electron microscopy of sections from intact hearts reveals that the content and morphology of mitochondria in the dystrophic heart are not different from that in wild-type hearts (Fig. 7C). Examination of the mean value and the variation of electron density within mitochondrial areas allows the assessment of mitochondrial morphology. Analysis of these parameters from 3,878 dystrophic and 4,733 normal mitochondrial regions reveals no difference between dystrophic and wild-type mitochondria in either of these measures of morphology (data not shown). Furthermore, a qualitative review of mitochondrial images revealed no evidence of mitochondrial dilation or other morphological indication of mitochondrial dysfunction. Quantitative proteomic studies using heart tissue from the severely affected golden retriever muscular dystrophy model demonstrate normal levels of mitochondrial ETC protein content (Fig. 7E). Together, these results indicate that neither the FCCP-induced oxidative capacity, mitochondrial content, nor morphology are significantly altered in the dystrophic heart. While these measures indicate that there is no dramatic alteration in mitochondrial content, structure, or respiratory function, they do not provide a comprehensive assessment of mitochondrial function. It is possible that alterations in redox potential or calcium handling in dystrophic mitochondria (40, 70) may contribute to the poor oxygen extraction and impaired energetics (13, 26, 74) of the intact dystrophic heart.
DISCUSSION
We demonstrate here that the hypoxia-induced mortality in dystrophic mdx mice is associated with metabolic acidosis, which likely arises from metabolic dysfunction revealed by hypoxia. This observation is particularly important because of the frequency with which patients with DMD are subjected to bouts of hypoxia. Studies in anesthetized mdx mice monitoring hemodynamic function of the dystrophic heart during hypoxia exposure reveal that hypoxia-induced mortality is preceded initially by diastolic dysfunction, which transitions into a gradual decline in systolic function (65). The present studies provide important mechanistic insight into the potential causes of this hypoxia-induced mortality. Here it is demonstrated that hypoxia results in the development of metabolic acidosis, deriving from a systemic shift to anaerobic metabolism. Systemic alterations in blood pH have wide-spread adverse consequences on multiple physiological systems, including the heart where acidosis results in marked declines in contractile function in both wild-type and dystrophic hearts (Fig. 4). Decreases in perfusion following this reduction in cardiac contractile performance would be expected to exacerbate the ongoing metabolic acidosis. The source of the acidosis observed in this study is not entirely clear. The cumulative mass of the skeletal muscle makes it a likely candidate; however, in the anesthetized animals preparation, the expected metabolic activity of skeletal muscle is expected to be very small. DMD patients are at increased risk for malignant hyperthermia (6), which could result in increased oxygen consumption in anesthetized skeletal muscle. However, mdx mice are resistant to developing malignant hyperthermia (46, 53), and no evidence of contracture was present in these preparations. The correlation between cardiac activity and acidosis raises the possibility that the dystrophic heart may be a significant contributor to the net production of lactate during periods of acidosis, although the heart's relatively small mass would require a large level of lactate product for it to be the primary driver of the global acidosis. It is notable that hypoxia-induced mortality is only present in anesthetized dystrophic animals. It is not clear to what degree anesthesia contributes to the development of metabolic acidosis, but it clearly contributes to hypoxia-induced mortality.
During chronic hypoxia exposure, dystrophic hearts develop a significant degree of cardiac fibrosis, underscoring the susceptibility of the dystrophic heart to conditions of limited oxygen. Here we demonstrate that the dystrophic heart both requires more oxygen than wild-type hearts to perform the same level of work and has a reduced ability to utilize oxygen in the coronary perfusate (Fig. 5). This, coupled with the absolute dependence of the heart on aerobic metabolism, makes the dystrophic heart particularly susceptible to hypoxic conditions. This is evident in the studies presented here, as short bouts of no-flow ischemia, which cause both hypoxia and acidosis, have a significant negative impact on both systolic and diastolic function in dystrophic hearts (Fig. 3). This observation that the dystrophic heart is highly sensitive to very short bouts of ischemia are consistent with previous studies demonstrating that the dystrophic heart does very poorly following 20 min of ischemia (1). Importantly, the dystrophic heart is shown to recover better following prolonged global acidosis than wild-type hearts (Fig. 4). The mechanism of this increased tolerance to acidosis is not currently known, although it may be a compensatory response to periods of respiratory acidosis secondary to the diaphragm dysfunction present in the mdx mouse (29, 32, 51). The increased sensitivity to ischemia, coupled with resistance to acidosis, indicates that the dystrophic heart is particularly susceptible to hypoxia.
The significant reduction in cardiac efficiency observed in this study (Fig. 5) and elsewhere (37) likely results from an increased use of ATP for noncontractile functions in the dystrophic heart. The dystrophic heart displays increased sarcolemmal permeability, which dissipates transmembrane ionic gradients and contributes to increases in cytoplasmic Na+ and Ca2+ levels (40, 50, 70, 72, 73), all of which require the hydrolysis of ATP that does not contribute to contraction. Inefficiency in mitochondrial ATP production could also contribute to the overall inefficiency of the dystrophic heart. While the levels of oxygen consumption (Figs. 5 and 7) and measures of mitochondrial mass (Fig. 7) suggest that dystrophic mitochondria are relatively normal, the poor oxygen extraction (Fig. 5) and reductions in cardiac phosophocreatinine-to-ATP ratio in Langendorff perfused hearts (26, 74) and in intact animals (13) all point to potential mitochondrial abnormalities.
There are several potential causes of the reduced oxygen extraction observed in the dystrophic heart. It is possible that shunting of the blood through the coronary arteries could result in a perfusion-consumption mismatching, where some regions are flush with oxygenated perfusate, while others are underperfused. The venous blood from overperfused regions would be expected to have relatively higher levels of oxygen that, when mixed with the perfusate from the rest of the heart, would result in an apparent reduction in oxygen extraction. The normal coronary flow responses to metabolic stresses (Fig. 6) argue against this hypothesis. Another possibility is that the coronary flow in the dystrophic heart is too high to allow for full oxygen extraction. However, this is unlikely, given that dobutamine treatment increases oxygen extraction by the dystrophic heart, while coronary flow rates are increased further (Figs. 5 and 6). This latter observation also suggests that, whatever the defect in oxygen extraction is, it is readily reversed by treatment with dobutamine.
Mitochondria are responsible for the vast majority of oxygen consumption in the heart, thus defects in mitochondrial respiratory function could also result in declines in the oxygen extraction in the dystrophic heart. The studies presented here demonstrate that the ability of the dystrophic hearts or myocytes to consume oxygen is normal, under conditions where oxygen is abundant. As oxygen levels decrease, there is an eventual point at which flux through the ETC stops. It is possible that dystrophic mitochondria are more susceptible to this hypoxia-induced cessation of flux through the ETC, such that dystrophic hearts abandon aerobic metabolism at higher levels of oxygen. The molecular mechanisms underlying such dysfunction in the dystrophic mitochondria are not clear, but alterations to the ETC or changes in cellular redox potential are potential explanations.
In summary, these studies demonstrate that metabolic acidosis induced by acute hypoxia is likely responsible for the mortality of anesthetized dystrophic mice. Furthermore, this metabolic acidosis occurs at significantly higher levels of arterial oxygen compared with wild-type mice exposed to the same hypoxic stress. Additionally, chronic hypoxia does not cause acute mortality, but does result in significant levels of cardiac fibrosis. This hypoxia-induced loss of dystrophic cardiac myocytes likely results from a combination of poor cardiac efficiency and a reduced ability to extract oxygen. Together these deficits contribute to the hypoxia sensitivity observed in dystrophic hearts. These findings have significant implications on the mechanism of the pathogenesis of dystrophic cardiomyopathy, providing a direct mechanism by which hypoxia secondary to respiratory insufficiency could lead to a metabolic crisis within the heart, resulting in myocyte loss and permanent myocardial injury. These findings have particular clinical relevance because currently available therapies can be implemented to limit the occurrence of hypoxia in DMD patients.
GRANTS
This work was supported by Muscular Dystrophy Association Grant 351960 and National Heart, Lung, and Blood Institute Grants K08-HL102066 and R01-HL114832 to D. Townsend.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Z.S., J.S., A.Y., K.F., K.S., and D.T. performed experiments; Z.S., J.S., A.Y., K.S., and D.T. analyzed data; Z.S., J.S., K.S., and D.T. interpreted results of experiments; Z.S., J.S., K.S., and D.T. edited and revised manuscript; J.S., A.Y., and D.T. conception and design of research; D.T. prepared figures; D.T. drafted manuscript; D.T. approved final version of manuscript.
REFERENCES
- 1.Ascah A, Khairallah M, Daussin F, Bourcier-Lucas C, Godin R, Allen BG, Petrof BJ, Rosiers Des C, Burelle Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am J Physiol Heart Circ Physiol 300: H144–H153, 2011. [DOI] [PubMed] [Google Scholar]
- 2.Bersanini C, Khirani S, Ramirez A, Lofaso F, Aubertin G, Beydon N, Mayer M, Maincent K, Boulé M, Fauroux B. Nocturnal hypoxaemia and hypercapnia in children with neuromuscular disorders. Eur Respir J 39: 1206–1212, 2012. [DOI] [PubMed] [Google Scholar]
- 3.Bratkovsky S, Aasum E, Birkeland CH, Riemersma RA, Myhre ESP, Larsen TS. Measurement of coronary flow reserve in isolated hearts from mice. Acta Physiol Scand 181: 167–172, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Braun U, Paju K, Eimre M, Seppet E, Orlova E, Kadaja L, Trumbeckaite S, Gellerich FN, Zierz S, Jockusch H, Seppet EK. Lack of dystrophin is associated with altered integration of the mitochondria and ATPases in slow-twitch muscle cells of MDX mice. Biochim Biophys Acta 1505: 258–270, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 82: 743–752, 1995. [DOI] [PubMed] [Google Scholar]
- 6.Brownell AK, Paasuke RT, Elash A, Fowlow SB, Seagram CG, Diewold RJ, Friesen C. Malignant hyperthermia in Duchenne muscular dystrophy. Anesthesiology 58: 180–182, 1983. [DOI] [PubMed] [Google Scholar]
- 7.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A 81: 1189–1192, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C, DMD Care Considerations Working Group . Diagnosis and management of Duchenne muscular dystrophy. 1. Diagnosis, and pharmacological and psychosocial management. Lancet Neurol 9: 77–93, 2010. [DOI] [PubMed] [Google Scholar]
- 9.Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C, DMD Care Considerations Working Group . Diagnosis and management of Duchenne muscular dystrophy. 2. Implementation of multidisciplinary care. Lancet Neurol 9: 177–189, 2010. [DOI] [PubMed] [Google Scholar]
- 10.Bushby K, Muntoni F, Bourke JP. 107th ENMC international workshop: the management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7th-9th June 2002, Naarden, the Netherlands. Neuromuscul Disord 13: 166–172, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Bushby K, Muntoni F, Urtizberea A, Hughes R, Griggs R. Report on the 124th ENMC International Workshop. Treatment of Duchenne muscular dystrophy; defining the gold standards of management in the use of corticosteroids. 2–4 April 2004, Naarden, The Netherlands. Neuromuscul Disord 14: 526–534, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Clarke JL, Gowers WR. On a case of pseudo-hypertrophic muscular paralysis. Med Chir Trans 57: 247–260, 1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui W, Jang A, Zhang P, Thompson B, Townsend D, Metzger JM, Zhang J. Early detection of myocardial bioenergetic deficits: a 9.4 Tesla complete non invasive 31P MR spectroscopy study in mice with muscular dystrophy. PLoS One 10: e0135000, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duchenne GB. The pathology of paralysis with muscular degeneration (Paralysie Myosclerotique), or paralysis with apparent hypertrophy. Br Med J 2: 541–542, 1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dunn JF, Frostick S, Brown G, Radda GK. Energy status of cells lacking dystrophin: an in vivo/in vitro study of mdx mouse skeletal muscle. Biochim Biophys Acta 1096: 115–120, 1991. [DOI] [PubMed] [Google Scholar]
- 16.Eagle M, Bourke J, Bullock R, Gibson M, Mehta J, Giddings D, Straub V, Bushby K. Managing Duchenne muscular dystrophy–the additive effect of spinal surgery and home nocturnal ventilation in improving survival. Neuromuscul Disord 17: 470–475, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345: 315–319, 1990. [DOI] [PubMed] [Google Scholar]
- 18.Farkas GA, McCormick KM, Gosselin LE. Episodic hypoxia exacerbates respiratory muscle dysfunction in DMD(mdx) mice. Muscle Nerve 36: 708–710, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Finder JD, Birnkrant D, Carl J, Farber HJ, Gozal D, Iannaccone ST, Kovesi T, Kravitz RM, Panitch H, Schramm C, Schroth M, Sharma G, Sievers L, Silvestri JM, Sterni L, American Thoracic Society. Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am J Respir Crit Care Med 170: 456–465, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Fong PY, Turner PR, Denetclaw WF, Steinhardt RA. Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science 250: 673–676, 1990. [DOI] [PubMed] [Google Scholar]
- 21.Franco A, Lansman JB. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature 344: 670–673, 1990. [DOI] [PubMed] [Google Scholar]
- 22.Gavillet B, Rougier JS, Domenighetti AA, Behar R, Boixel C, Ruchat P, Lehr HA, Pedrazzini T, Abriel H. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res 99: 407–414, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Gilroy J, Cahalan JL, Berman R, Newman M. Cardiac and pulmonary complications in Duchenne's progressive muscular dystrophy. Circulation 27: 484–493, 1963. [DOI] [PubMed] [Google Scholar]
- 24.Globus JH. The pathologic findings in the heart muscle in progressive muscular dystrophy. Arch Neurol Psychiatry 9: 59–72, 1923. [Google Scholar]
- 25.Godin R, Daussin F, Matecki S, Li T, Petrof BJ, Burelle Y. Peroxisome proliferator-activated receptor γ coactivator-1 gene α transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post-necrotic mdx mouse skeletal muscle. J Physiol 590: 5487–5502, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graciotti L, Becker J, Granata AL, Procopio AD, Tessarollo L, Fulgenzi G. Dystrophin is required for the normal function of the cardio-protective K(ATP) channel in cardiomyocytes. PLoS One 6: e27034, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heintz A, Damm M, Brand M, Koch T, Deussen A. Coronary flow regulation in mouse heart during hypercapnic acidosis: role of NO and its compensation during eNOS impairment. Cardiovasc Res 77: 188–196, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51: 919–928, 1987. [DOI] [PubMed] [Google Scholar]
- 29.Huang P, Cheng G, Lu H, Aronica M, Ransohoff RM, Zhou L. Impaired respiratory function in mdx and mdx/utrn(+/−) mice. Muscle Nerve 43: 263–267, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inkley SR, Oldenburg FC, Vignos PJ. Pulmonary function in Duchenne muscular dystrophy related to stage of disease. Am J Med 56: 297–306, 1974. [DOI] [PubMed] [Google Scholar]
- 31.Ishizaka H, Gudi SR, Frangos JA, Kuo L. Coronary arteriolar dilation to acidosis: role of ATP-sensitive potassium channels and pertussis toxin-sensitive G proteins. Circulation 99: 558–563, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Ishizaki M, Suga T, Kimura E, Shiota T, Kawano R, Uchida Y, Uchino K, Yamashita S, Maeda Y, Uchino M. Mdx respiratory impairment following fibrosis of the diaphragm. Neuromuscul Disord 18: 342–348, 2008. [DOI] [PubMed] [Google Scholar]
- 33.Jeppesen J, Green A, Steffensen BF, Rahbek J. The Duchenne muscular dystrophy population in Denmark, 1977–2001: prevalence, incidence and survival in relation to the introduction of ventilator use. Neuromuscul Disord 13: 804–812, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Kabaeva Z, Zhao M, Michele DE. Blebbistatin extends culture life of adult mouse cardiac myocytes and allows efficient and stable transgene expression. Am J Physiol Heart Circ Physiol 294: H1667–H1674, 2008. [DOI] [PubMed] [Google Scholar]
- 35.Katz SL, Gaboury I, Keilty K, Banwell B, Vajsar J, Anderson P, Ni A, MacLusky I. Nocturnal hypoventilation: predictors and outcomes in childhood progressive neuromuscular disease. Arch Dis Child 95: 998–1003, 2010. [DOI] [PubMed] [Google Scholar]
- 36.Katz SL, McKim D, Hoey L, Barrowman N, Kherani T, Kovesi T, MacLusky I, Mah JK. Respiratory management strategies for Duchenne muscular dystrophy: practice variation amongst Canadian sub-specialists. Pediatr Pulmonol 48: 59–66, 2013. [DOI] [PubMed] [Google Scholar]
- 37.Khairallah M, Khairallah R, Young ME, Dyck JR, Petrof BJ, Des Rosiers C. Metabolic and signaling alterations in dystrophin-deficient hearts precede overt cardiomyopathy. J Mol Cell Cardiol 43: 119–129, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Kozłowska H, Rowiński J, Karkucińska-Wieckowska A. Effect of acute hypobaric hypoxia on the histological changes of diaphragm in mice. Folia Histochem Cytobiol 37: 129–130, 1999. [PubMed] [Google Scholar]
- 39.Kuznetsov AV, Winkler K, Wiedemann FR, von Bossanyi P, Dietzmann K, Kunz WS. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol Cell Biochem 183: 87–96, 1998. [DOI] [PubMed] [Google Scholar]
- 40.Kyrychenko V, Poláková E, Janíček R, Shirokova N. Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 58: 186–195, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loufrani L, Dubroca C, You D, Li Z, Levy B, Paulin D, Henrion D. Absence of dystrophin in mice reduces NO-dependent vascular function and vascular density: total recovery after a treatment with the aminoglycoside gentamicin. Arterioscler Thromb Vasc Biol 24: 671–676, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loufrani L, Levy BI, Henrion D. Defect in microvascular adaptation to chronic changes in blood flow in mice lacking the gene encoding for dystrophin. Circ Res 91: 1183–1189, 2002. [DOI] [PubMed] [Google Scholar]
- 43.Loufrani L, Matrougui K, Gorny D, Duriez M, Blanc I, Levy BI, Henrion D. Flow (shear stress)-induced endothelium-dependent dilation is altered in mice lacking the gene encoding for dystrophin. Circulation 103: 864–870, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu S, Hoey A. Changes in function of cardiac receptors mediating the effects of the autonomic nervous system in the muscular dystrophy (MDX) mouse. J Mol Cell Cardiol 32: 143–152, 2000. [DOI] [PubMed] [Google Scholar]
- 45.Lynch GS, Hinkle RT, Chamberlain JS, Brooks SV, Faulkner JA. Force and power output of fast and slow skeletal muscles from mdx mice 6–28 months old. J Physiol 535: 591–600, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mader N, Gilly H, Bittner RE. Dystrophin deficient mdx muscle is not prone to MH susceptibility: an in vitro study. Br J Anaesth 79: 125–127, 1997. [DOI] [PubMed] [Google Scholar]
- 47.Martin EA, Barresi R, Byrne BJ, Tsimerinov EI, Scott BL, Walker AE, Gurudevan SV, Anene F, Elashoff RM, Thomas GD, Victor RG. Tadalafil alleviates muscle ischemia in patients with Becker muscular dystrophy. Sci Transl Med 4: 162ra155–162ra155, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCormack WM, Spalter HF. Muscular dystrophy, alveolar hypoventilation, papilledema. JAMA 197: 957–960, 1966. [PubMed] [Google Scholar]
- 49.Meyers TA, Townsend D. Early right ventricular fibrosis and reduction in biventricular cardiac reserve in the dystrophin-deficient mdx heart. Am J Physiol Heart Circ Physiol 308: H303–H315, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mijares A, Altamirano F, Kolster J, Adams JA, López JR. Age-dependent changes in diastolic Ca(2+) and Na(+) concentrations in dystrophic cardiomyopathy: role of Ca(2+) entry and IP3. Biochem Biophys Res Commun 452: 1054–1059, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson CA, Hunter RB, Quigley LA, Girgenrath S, Weber WD, McCullough JA, Dinardo CJ, Keefe KA, Ceci L, Clayton NP, McVie-Wylie A, Cheng SH, Leonard JP, Wentworth BM. Inhibiting TGF-β activity improves respiratory function in mdx mice. Am J Pathol 178: 2611–2621, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohlendieck K, Campbell KP. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol 115: 1685–1694, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patel VK, Dierdorf SF, Krishna G, Bonsett C. Negative halothane-caffeine contracture test in mdx (dystrophin-deficient) mice. Metabolism 40: 883–887, 1991. [DOI] [PubMed] [Google Scholar]
- 54.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A 90: 3710–3714, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25: 1463–1465, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord 14: 491–496, 2004. [DOI] [PubMed] [Google Scholar]
- 57.R Core Team. R: A language and Environment for Statistical Computing (Online) R Foundation for Statistical Computing https://www.r-project.org. [Google Scholar]
- 58.Ross J. On a case of pseudo-hypertrophic paralysis. Br Med J 1: 200–202, 1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rybalka E, Timpani CA, Cooke MB, Williams AD, Hayes A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS One 9: e115763, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, Thomas GD, Victor RG. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 97: 13818–13823, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schuh RA, Jackson KC, Khairallah RJ, Ward CW, Spangenburg EE. Measuring mitochondrial respiration in intact single muscle fibers. Am J Physiol Regul Integr Comp Physiol 302: R712–R719, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Strakova J, Dean JD, Sharpe KM, Meyers TA, Odom GL, Townsend D. Dystrobrevin increases dystrophin's binding to the dystrophin-glycoprotein complex and provides protection during cardiac stress. J Mol Cell Cardiol 76: 106–115, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thomas GD, Sander M, Lau KS, Huang PL, Stull JT, Victor RG. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc Natl Acad Sci U S A 95: 15090–15095, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thomas GD, Ye J, De Nardi C, Monopoli A, Ongini E, Victor RG. Treatment with a nitric oxide-donating NSAID alleviates functional muscle ischemia in the mouse model of Duchenne muscular dystrophy. PLoS One 7: e49350, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Townsend D. Diastolic dysfunction precedes hypoxia-induced mortality in dystrophic mice. Physiol Rep 3: e12513, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Townsend D, Blankinship MJ, Allen JM, Gregorevic P, Chamberlain JS, Metzger JM. Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Mol Ther 15: 1086–1092, 2007. [DOI] [PubMed] [Google Scholar]
- 67.Townsend D, Turner I, Yasuda S, Martindale J, Davis J, Shillingford M, Kornegay JN, Metzger JM. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest 120: 1140–1150, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Townsend D, Yasuda S, Chamberlain J, Metzger JM. Cardiac consequences to skeletal muscle-centric therapeutics for Duchenne muscular dystrophy. Trends Cardiovasc Med 19: 50–55, 2009. [DOI] [PubMed] [Google Scholar]
- 69.Townsend D, Yasuda S, McNally E, Metzger JM. Distinct pathophysiological mechanisms of cardiomyopathy in hearts lacking dystrophin or the sarcoglycan complex. FASEB J 25: 3106–3114, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Viola HM, Davies SMK, Filipovska A, Hool LC. L-type Ca2+ channel contributes to alterations in mitochondrial calcium handling in the mdx ventricular myocyte. Am J Physiol Heart Circ Physiol 304: H767–H775, 2013. [DOI] [PubMed] [Google Scholar]
- 71.Warren CM, Geenen DL, Helseth DL, Xu H, Solaro RJ. Sub-proteomic fractionation, iTRAQ, and OFFGEL-LC-MS/MS approaches to cardiac proteomics. J Proteomics 73: 1551–1561, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Williams IA, Allen DG. Intracellular calcium handling in ventricular myocytes from mdx mice. Am J Physiol Heart Circ Physiol 292: H846–H855, 2007. [DOI] [PubMed] [Google Scholar]
- 73.Yasuda S, Townsend D, Michele DE, Favre EG, Day SM, Metzger JM. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 436: 1025–1029, 2005. [DOI] [PubMed] [Google Scholar]
- 74.Zhang W, Hove ten M, Schneider JE, Stuckey DJ, Sebag-Montefiore L, Bia BL, Radda GK, Davies KE, Neubauer S, Clarke K. Abnormal cardiac morphology, function and energy metabolism in the dystrophic mdx mouse: an MRI and MRS study. J Mol Cell Cardiol 45: 754–760, 2008. [DOI] [PubMed] [Google Scholar]