

Abstract
Objective:
To determine whether GGGGCC (G4C2) repeat expansions at loci other than C9orf72 serve as common causes of amyotrophic lateral sclerosis (ALS).
Methods:
We assessed G4C2 repeat number in 28 genes near known ALS and frontotemporal dementia (FTD) loci by repeat-primed PCR coupled with fluorescent fragment analysis in 199 patients with ALS (17 familial, 182 sporadic) and 136 healthy controls. We also obtained blood from patients with ALS4 for evaluation of repeats surrounding the SETX gene locus. C9orf72 expansions were evaluated in parallel.
Results:
Expansions of G4C2 repeats in C9orf72 explained 8.8% of sporadic and 47% of familial ALS cases analyzed. Repeat variance was observed at one other locus, RGS14, but no large expansions were observed, and repeat sizes were not different between cases and controls. No G4C2 repeat expansions were identified at other ALS or FTD risk loci or in ALS4 cases.
Conclusions:
G4C2 expansions near known ALS and FTD loci other than C9orf72 are not a common cause of ALS.
A GGGGCC (G4C2) hexanucleotide repeat expansion in the first intron of C9orf72 is the most common monogenic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD).1,2 There are normally between 2 and 23 G4C2 repeats at this locus. The repeat expands to hundreds in affected individuals,2 although 30 repeats may be sufficient to elicit G4C2-specific pathology.3
A growing body of evidence suggests that the C9orf72 repeat elicits toxicity primarily through gain-of-function mechanisms that are independent of the genetic locus where the repeat resides. Specifically, Drosophila and mouse models of ALS with expression of the repeat outside its normal genomic context suggest that instability and expansion of G4C2 repeats elsewhere in the genome could also cause ALS or FTD. A precedent for this phenomenon is found in spinocerebellar ataxia, in which CAG repeat expansions in a diverse set of genes elicit overlapping clinical phenotypes.4
We therefore hypothesized that cryptic repeat expansions at loci other than C9orf72 could also contribute to ALS and FTD pathogenesis. Using repeat-primed PCR assays, we evaluated whether G4C2 repeats near known ALS and FTD loci identified by linkage analysis or genome-wide association studies (GWAS) exhibited expansions in a cohort of patients with ALS and controls in the University of Michigan ALS Patient Biorepository. Our results confirm that repeat instability and large expansions at C9orf72 are common in sporadic ALS in the United States, but expansions at other disease-associated loci are rare in this population and are unlikely to be a common cause of ALS.
METHODS
Standard protocol approvals, registrations, and patient consents.
This study was approved by the Institutional Review Board of the University of Michigan. Individual patients and controls who contributed these DNA samples provided written informed consent via representatives from the Coriell Institute, University of Michigan ALS Patient Biorepository, or individually to a member of the research group.
ALS and control patient cohorts and DNA isolation.
DNA samples for repeat-primed PCR and genomic PCR were from the following sources: 1 μg of genomic DNA from 199 patients with ALS and 136 healthy controls from the University of Michigan ALS Patient Biorepository. Patients with ALS met the revised El Escorial criteria5 and were recruited from the University of Michigan ALS Clinic; controls were recruited via the University of Michigan clinical trials Web site (https://umclinicalstudies.org/). Demographic data were analyzed using SAS9.5 software (SAS Institute Inc., Cary, NC) and summarized in table e-1 at Neurology.org/ng. Two hundred fifty nanograms of genomic DNA from 86 patients with sporadic ALS from Coriell Cell Repository panel #NDPT026 (Coriell Institute) was used for determination of C9orf72 repeat status only. One hundred micrograms of genomic DNA from a patient with ALS4 and 1 non-ALS sibling from a previously reported large pedigree6 was extracted from 4 mL of whole blood using a commercial DNA isolation kit (DNeasy Blood & Tissue kit; Qiagen, Netherlands). Genomic DNA from a second published ALS4 case with a family history7 was extracted from patient fibroblast cells obtained from a collaborator's laboratory using the same kit.
Candidate gene selection.
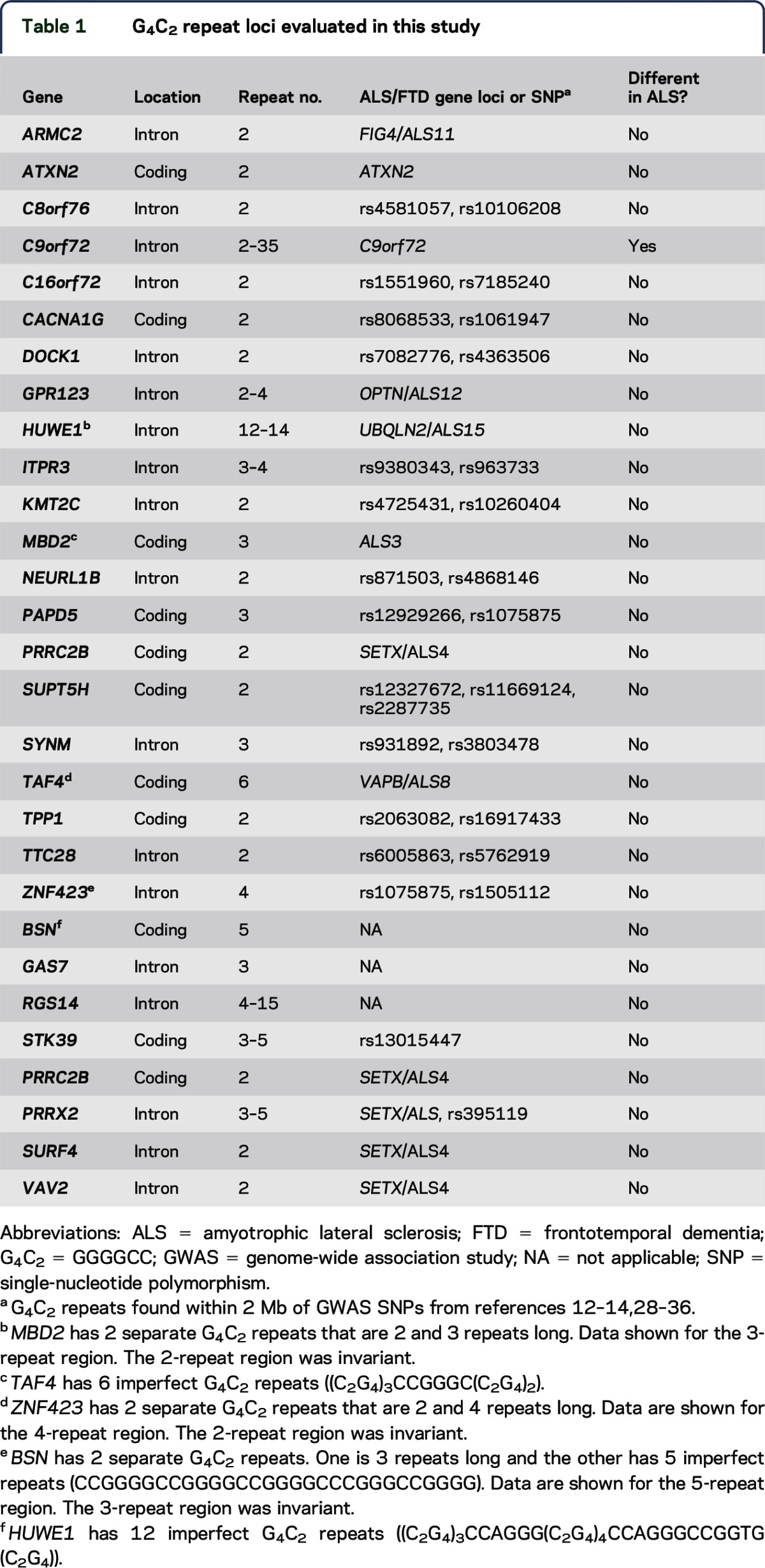
We performed a BLAST search (National Center for Biotechnology Information) against the human genome for G4C2 repeat sequences using a sequence of 5 G4C2 pure repeats (GGGGCCGGGGCCGGGGCCGGGGCCGGGGCC) as a start query. The identified repeat loci were overlaid with published genetic loci associated with ALS or FTD (see a recent review8 and table 1). We constrained our analysis to repeat loci within 2 mega base pairs (Mb) of either the mapped critical region for an ALS or FTD candidate gene/locus or with single-nucleotide polymorphisms (SNPs) that achieved statistical significance on GWAS in sporadic ALS cohorts. Additional candidate repeats located more than 2 Mb outside of disease-associated loci were identified by requiring at least 3 pure repeats in a gene with abundant neuronal expression in brain based on BioGPS and Proteomic DB database analysis.9,10 Three additional candidate genes with G4C2 repeats within the previously identified critical region of ALS4 but missed by our initial in silico analysis were added after we obtained access to case samples.
Table 1.
G4C2 repeat loci evaluated in this study
G4C2 repeat determination.
G4C2 repeat numbers in the longer allele were determined by repeat-primed PCR as previously reported,11 followed by capillary electrophoresis and fragment analysis. The primer sequences are included in table e-2. The individual reverse primers for each candidate gene were designed using Primer 3 tools (http://biotools.umassmed.edu/bioapps/primer3_www.cgi) and were labeled with either 6-FAM or 5-HEX fluorescent dye. The PCR products were diluted in highly deionized formamide (HiDi formamide; ThermoFisher Scientific, Waltham, WA) containing size standard ROX-1000 (BioVenture, TN). Fragment analysis was performed at the University of Michigan Sequencing Core facility on an ABI 3730 Sequencer. The data were analyzed using GeneMarker (SoftGenetics, PA) to determine the maximal repeat number.
Sanger sequencing.
For samples with fewer than 35 G4C2 repeats in C9orf72, DNA flanking the repeat regions was PCR-amplified with the following primers: C9orf72 forward: 5′-CCG CAG CCT GTA GCA AGC-3′ and C9orf72 reverse: 5′-AGT CGC TAG AGG CGA AAG C-3′ using the same thermal cycling program as the repeat-primed PCR. The PCR products were gel purified and subjected to Sanger sequencing at the University of Michigan Sequencing Core facility to determine the exact repeat number.
For samples with a 31-nucleotide insertion in the VAV2 gene, DNA flanking the repeat region was PCR-amplified with the following primers: VAV2 forward: 5′-GCC CAG GAC AGG AGG CCT CAG CA-3′ and VAV2 reverse: 5′- CTC AGG GCC GGG AGG AAG CAC CT-3′ using the same thermal cycling program as the repeat-primed PCR. The PCR products were gel-purified and subjected to Sanger sequencing as described above. For repeats at Huwe1 and RGS14, PCR primers flanking the repeat regions were used to determine specific repeat sizes (see supplemental data).
Southern blot confirmation of repeat expansions at C9orf72.
For Southern blot analysis, 25-μg aliquots of lymphoblast genomic DNA were obtained for 5 selected Coriell samples. Two additional lymphoblast cell lines were obtained from the Coriell Institute and were grown up to 15 million cells, and approximately 100 μg of lymphoblast genomic DNA was extracted using a DNA isolation kit. Ten fibroblast cell lines with G4C2 repeat expansion and 2 control fibroblast lines (1 ALS sample without expansion and 1 control) were obtained from the University of Michigan ALS Patient Biorepository and 100 μg of fibroblast genomic DNA was extracted from 15 million cells.
Statistical analysis.
Two-tailed Fisher exact tests or χ2 tests were performed to test for association of repeat length with ALS and to verify genotype frequencies in the VAV2 gene. For RGS14 repeat length, a 2-tailed nonparametric t test was performed to compare the median repeat size differences. For the correlation of C9orf72 repeat size and patient age at onset of ALS, the Pearson correlation test was performed.
RESULTS
We first conducted an in silico experiment to identify all human genes containing at least 2 G4C2 repeats. Our rationale was that transcribed repeats at such loci could become unstable and lead to disease-causing expansions, unless the chromosomal context of the C9orf72 repeat was critical to disease pathogenesis. Our initial analysis revealed that short G4C2 repeats are quite common in the human genome, with 344 identified as intragenic repeats. To narrow our window of potential sites of repeat expansion to a number that we could readily assay, we compared this in silico list of repeat loci with genetic loci implicated in ALS or FTD, either through critical region mapping in familial forms or by GWAS. We reasoned that utilization of GWAS conducted in patient populations similar to our cohort as well as known genetic loci in rare families (which have subsequently been shown to contribute to a percentage of sporadic cases) would identify most repeat expansions that confer a substantial contribution to the genetic burden of ALS in the United States. Consistent with this idea, the C9orf72 locus was identified on multiple GWAS surveys in ALS and FTD in sporadic populations prior to identification of the causative repeat.12–14 Using this approach, we identified 21 candidate genes, including C9orf72, containing at least 2 G4C2 repeats that are located within 2 Mb of known ALS-FTD loci or loci identified in GWAS by at least 2 associated SNPs (figure 1A).
Figure 1. G4C2 repeat numbers from controls and patients with ALS.

(A) Flow chart of candidate genes selection for this study. (B–H) Graphs show the distribution of repeat sizes from ALS cases and controls for 7 genes. *Significant (p < 0.05) difference in the distribution of repeats larger than 33 times between cases and controls by Fisher exact test. ALS = amyotrophic lateral sclerosis; G4C2 = GGGGCC.
We developed a modified fluorescent fragment analysis after repeat-primed PCR to evaluate instability at each of these loci, as previously described for C9orf72 repeat expansions.11 We also assessed repeat size at C9orf72. These assays were tested on control samples, and the actual repeat sizes were confirmed by standard PCR and Sanger sequencing. All 21 assays demonstrated accurate detection of repeat size, based on comparison with Sanger sequencing (data not shown). This assay readily detected G4C2 expansions at C9orf72 in samples from ALS cases known to harbor the repeat.
We applied these assays to a sample collection from the University of Michigan ALS Patient Repository containing 199 ALS cases, of which 17 had a family history of ALS, and 136 healthy controls (University of Michigan). The demographic characteristics of this collection are shown in table e-1. Of note, there were no differences in the age at sample collection, sex composition, or racial/ethnic makeup between ALS cases and controls, and both cases and controls had similar frequencies of a family history of a non-ALS neurodegenerative condition (table e-1). In addition, we used samples from Coriell Cell Repository panel NDPT026, containing 86 sporadic ALS cases, for additional screening for repeat expansions in C9orf72 (Coriell Institute). Consistent with published findings, we observed a wide variation of G4C2 repeat length at C9orf72 in both cases and controls (table 1, table e-3, figure 1B). Pathogenic repeat expansions were observed in 47% of the familial cases and 8.8% (17/182) of sporadic ALS cases in the University of Michigan ALS Patient Repository (figure 1B and table e-3). C9orf72 repeat expansions were observed at a similar frequency (6/86; 7.0%) in ALS samples from the Coriell Institute. To validate these findings and determine the actual repeat expansion size in cases with low repeat numbers, genomic PCR and selective Southern blotting analysis were performed. One sample contained exactly 32 repeats in the longer allele, which was confirmed by Sanger sequencing. Other samples exhibited a single band by genomic PCR reflecting the shorter, PCR-amplifiable allele. Southern blotting on genomic DNA extracted from patient-derived fibroblasts or lymphocytes in a subset of cases demonstrated a wide range of pathologic repeat expansion sizes from 200 to more than 1,500 repeats (figure e-1). Repeat expansion size from peripheral blood samples or cultured cells did not correlate with clinical age at onset (R = 0.173 in Pearson correlation test, p = 0.572, figure e-2), consistent with previous reports.15
In contrast to C9orf72, G4C2 repeats assayed at other loci exhibited little or no variance in repeat number in the University of Michigan ALS Patient Biorepository samples. Of the 20 novel repeats analyzed, 16 showed no variance at all across the cohort (80%). Four genes displayed small variations in repeat number (table 1), but none of these cases contained expansions of more than 2 repeats greater than the prevalent alleles. The degree of repeat length variation was not significantly different between control and ALS groups, demonstrating a lack of association between repeat number variation and ALS/FTD in our cohort (figure 1, B–E, p < 0.001 for C9orf72, p = 0.76 for GPR123, p = 0.69 for HUWE1, p = 0.374 for ITPR3; χ2 test for C9orf72 and ITPR3, and Fisher exact test for GPR123 and HUWE1 used for the rest for different allele frequencies).
To expand the initial screening we selected 4 additional candidate genes that are highly expressed in brain and contain at least 3 G4C2 repeats in controls. In these candidate genes, no large repeat expansions were found in ALS or control groups (table 1, figure 1, F and G). However, an intronic repeat in RGS14 displayed a much larger variation than those in other loci studied, with between 4 and 15 repeats present in both controls and ALS cases. The distribution of repeat lengths in RGS14 was independent of clinical ALS (figure 1F) or C9orf72 expansion status (control 8.49 ± 1.74 vs ALS 8.1 ± 1.93, p = 0.082, 2-tailed student t test).
As a third approach, we examined additional loci harboring G4C2 repeats located near the ALS4 locus in patients with this disorder. ALS4 is a form of juvenile ALS with very slow progression that is thought to result from dominant mutations in SETX.6 The mechanism of these dominant mutations is unclear, because recessive mutations in SETX cause a distinct neurodegenerative disorder, ataxia with oculomotor apraxia type 2, which does not include motor neuron degeneration.16 Within the critical region defined by linkage analysis,6 there are 4 genes containing short G4C2 repeats: PRRX2, PCCR2B, SURF4, and VAV2 (figure 2A). To investigate whether a cryptic repeat expansion might provide an alternative explanation for the ALS4 phenotype, we analyzed repeat numbers in 2 patients with ALS4 and an unaffected control from 2 previously described ALS4 families.6,7 No repeat expansions were observed (figure 2B). Of these 4 loci, only 1 (PRRC2B) was included in our initial repeat screen in the University of Michigan ALS Patient Repository. Assessment of the additional 3 loci in this larger cohort identified no repeat expansions, only a minor repeat length variance in gene PRRX2 (table 1, figure 1H). We did observe an amplicon length variant of 31 bp in VAV2 by fragment analysis in the ALS4 family (figure 2B). Sanger sequencing identified it as a 31-bp intronic insertion in the longer allele that contains an imperfect duplication of intronic sequence (GCCGGGGCCGTGTGGCCCTCACGCAGTGACC). This insertion was common in controls and ALS cases in the University of Michigan ALS Patient Biorepository panel (figure 2C), and appears to be nonpathogenic (p = 0.398, χ2 test).
Figure 2. Analysis of G4C2 repeats near the ALS4 locus.
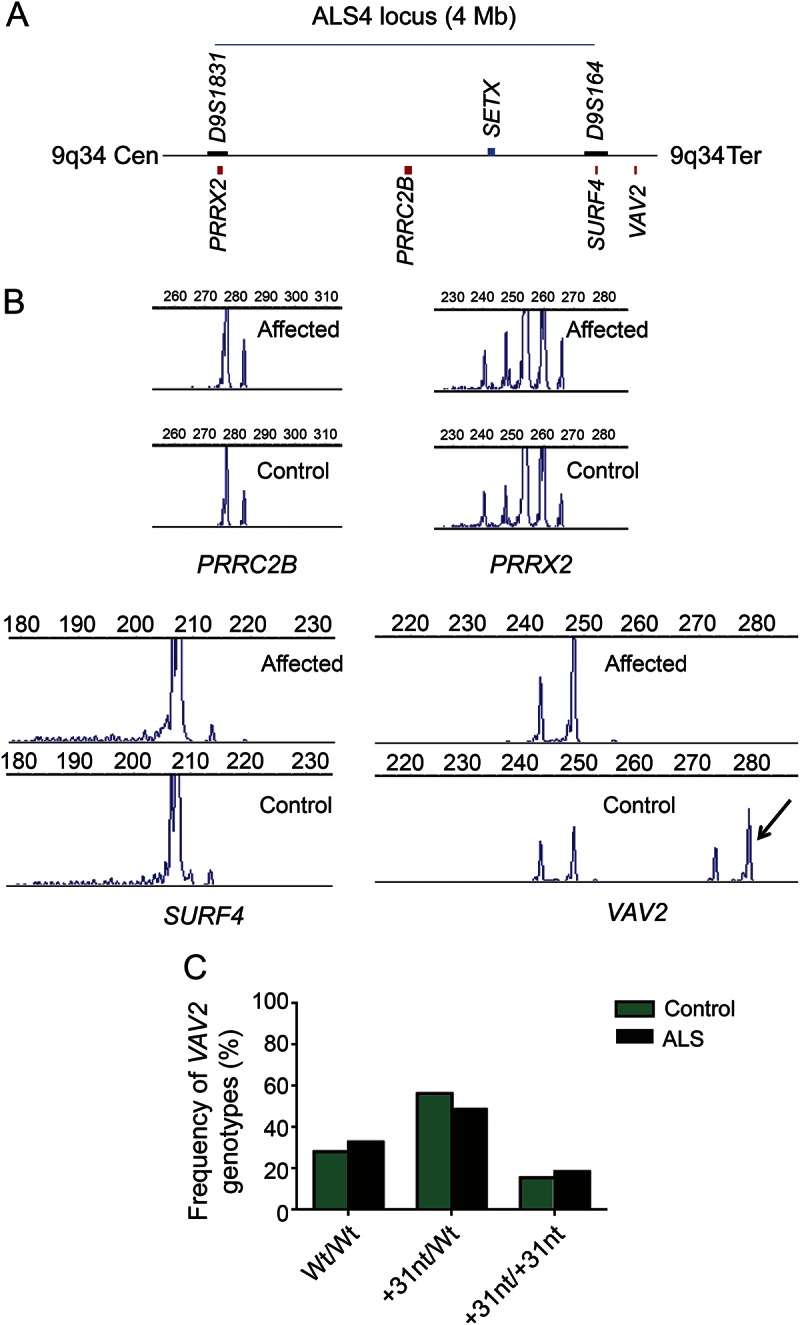
(A) Schematic of the ALS4 locus defined by chromosome markers D9S1831 and D9S164 (black boxes)6 drawn to scale showing the location of 4 G4C2 repeat-containing genes (red boxes) and the proposed disease-causing gene SETX (blue box). (B) Chromatograms showing the G4C2 repeat peaks in the 4 G4C2 repeat-containing genes from an affected ALS4 case and a control sibling. One control had a 31-bp insertion in the VAV2 gene (double peak marked with an arrow). (C) Genotypes of controls and ALS cases in the University of Michigan ALS Patient Biorepository for the 31-bp insertion in gene VAV2. Both groups had a similar distribution of genotypes carrying the 31-bp insertion allele. ALS = amyotrophic lateral sclerosis; Cen = centromere; G4C2 = GGGGCC; nt = nucleotide; Ter = telomere; Wt = wild type.
DISCUSSION
G4C2 repeat expansions in C9orf72 are the most common known cause of ALS and FTD. Here we tested whether G4C2 repeat instability at other loci could be a common cause of ALS. Our results confirmed that G4C2 repeat expansions and instability are common at the C9orf72 locus in both familial and sporadic ALS cases in 2 patient cohorts (University of Michigan ALS Patient Repository and the NDPT026 ALS collection from Coriell). However, for 27 other genomic loci that harbor G4C2 repeats, no pathogenic G4C2 expansions were identified. Thus, repeat instability at these alternative loci are unlikely to be major contributors to the genetic burden in ALS in the United States.
This study is not exhaustive in the number of repeat loci assayed or the number of patient and familial cohorts examined. Therefore, we cannot rule out G4C2 repeat expansions as rare causes of ALS. For practical reasons, we assayed fewer than 10% of the G4C2 repeats located near protein-coding genes and a much smaller fraction of the GC-rich repeats in the human genome. Specifically, we focused our analysis on repeats located adjacent to loci already implicated in ALS or FTD. Our rationale for this choice was that utilization of GWAS conducted in similar patient populations as well as known genetic loci in rare families (which have subsequently been shown to contribute to a percentage of sporadic cases) would colocalize with repeat expansions that confer a substantial contribution to the genetic burden of ALS in the United States. Consistent with this idea, the C9orf72 locus was identified on multiple GWAS surveys in ALS and FTD in sporadic populations prior to identification of the causative repeat. Moreover, we reasoned that cryptic repeat expansions might have escaped identification during the initial analyses of these loci, several of which lack an identified causative gene or a clear pathogenic mechanism for the currently implicated gene. As such, our findings that none of these alternative loci harbor repeat expansions in 199 ALS cases suggest that G4C2 repeat expansions at non-C9orf72 loci are unlikely to be a common cause of ALS in our patient population, given that most common highly penetrant mutations would be detected by this approach. However, it is important to acknowledge that for highly penetrant loci identified in familial ALS cases, it might be necessary to examine the original published cohorts, which might carry rare or private mutations. We took this step for ALS4 and found no G4C2 repeat instability at 4 neighboring loci. Application of this approach to other isolated pedigrees could more definitively rule out expansions at these genetically implicated loci.
The lack of G4C2 repeat expansions at alternative gene loci suggests that there could be something about the C9orf72 locus that promotes instability. The vast majority (75%) of the other hexanucleotide repeats studied here exhibited no variation among 335 individuals, and variance in repeat size was largely absent from the 1,000-genome database for the 321 intragenic G4C2 repeat loci not assayed (data not shown). With one exception, the repeat size variants observed were limited to 1 or 2 repeats. Studies in vitro suggest that stretches of G4C2 as short as 4 repeats are capable of forming intramolecular G quadruplexes, which might contribute to repeat instability through activation of DNA mismatch repair pathways and mistemplating.17 In general, our data are consistent with this threshold, since only genes with at least 4 repeats had variation of more than 1 repeat. However, some of the G4C2 repeats we studied were larger than this threshold but were invariant, suggesting that the length of the repeat is insufficient to explain instability. While future work will be needed to determine which factors drive selective instability, it may reflect specific aspects of the GC-rich sequence surrounding the repeat at the C9orf72 loci. This concept is supported by the finding that all pathologic repeat expansions at C9orf72 observed to date have occurred on a single haplotype background.18
Our approach here focused solely on the role of G4C2 repeat expansions in ALS. However, G4C2 repeat expansions in genes other than C9orf72 may be enriched in neurologic disorders other than ALS. Phenotypes of polyglutamine disorders associated with CAG repeat expansions, for example, are not all the same.19 In this context, it is interesting to note that the C9orf72 repeat expansion exhibits neurologic symptoms in addition to motor neuron disease and frontotemporal cognitive dysfunction, which are still being explored.11,20
We observed noteworthy repeat instability in both cases and controls in one other gene: a G4C2 intronic repeat in RGS14 with a mean repeat length of 9 and a range of 4–14 repeats. RGS14 encodes a member of the regulator of G-protein signaling family of proteins. It is highly expressed in the developing brain and has known functions in hippocampal learning and synaptic plasticity.21 Although we observed no association between the size of this repeat and clinical ALS, our study cannot rule out a role for expansion in rare ALS families or other racial/ethnic populations. As such, reevaluation of this locus may be worthy of further study in familial ALS, FTD, and other neurodegenerative disorders.
Other repeat expansions are implicated in causing or modifying motor neuron disease phenotypes. A GGCCTG repeat expansion in NOP56 causes spinocerebellar ataxia type 3622 and has motor neuron disease as a clinical feature. Moderate CAG repeat expansions (encoding polyglutamine) in ATXN223 and GCG repeat expansions (encoding polyalanine) in NIP1A24 are associated with an increased risk of ALS. However, other known pathogenic CAG and CGG repeat expansions do not influence the risk of ALS or FTD, suggesting that the observed effects may be gene specific rather than repeat sequence specific.25–27
We screened 28 loci harboring G4C2 repeats near known ALS/FTD loci in 199 patients with ALS and did not find pathogenic repeat expansions other than that previously reported in C9orf72. While future studies in larger cohorts and select families are still needed, our results suggest that G4C2 repeat expansions outside the C9orf72 gene are not a common cause of ALS, supporting the importance of identifying cis elements that explain the genetic instability at the C9orf72 locus.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all patients who provided the samples for this study and Blake Swihart and Jayna Duell, RN, for assistance in study coordination. The authors thank Suzzane Genik for technical assistance in fragment analysis and Zixing Wang for assistance on data processing. They thank the University of Michigan ALS Patient Biorepository for access to patient fibroblasts. The authors thank Michio Hirano for providing patient DNA and Phillip Chance and David Cornblath for helpful discussions. This repository is funded by the Center for Disease Control (CDC) Agency for Toxic Substances and Disease Registry (contract 200-2013-56856), the Program for Neurology Research & Discovery, and the A. Alfred Taubman Medical Research Institute. The authors also thank colleagues in P.K.T.'s laboratory for help in experiments and data interpretation and proofreading of the manuscript.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- FTD
frontotemporal dementia
- G4C2
GGGGCC
- GWAS
genome-wide association studies
- SNP
single-nucleotide polymorphism
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
F.H., M.H.M., D.Z., B.C.C., and P.K.T. conceptualized and designed the study. F.H., J.M.J., and C.F.-R. performed the experiments. F.H., J.M.J., M.H.M., B.C.C., and P.K.T. analyzed the data. M.H.M., E.L.F., S.A.G., and B.C.C. provided patient samples. F.H. and P.K.T. wrote the manuscript. All authors revised the manuscript.
STUDY FUNDING
This project was funded by NIH R01GM24872 to M.H.M.; grants from the Centers for Disease Control and Prevention (200-2013-56856) and the A. Alfred Taubman Medical Research Institute to E.L.F.; and grants from the Michigan Alzheimer's Disease Center at the University of Michigan, NIH R01NS08681001, and the Department of Veterans Affairs BLRD 1I21BX001841 to P.K.T. The funders played no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
DISCLOSURE
Drs. He and Jones report no disclosures. Dr. Figueroa-Romero has received research support from the Katherine Rayner Fund and the A. Alfred Taubman Medical Research Institute. Dr. Zhang receives intramural funding support from the NIH. Dr. Feldman receives funding support from Novo Nordisk, the NIH, the Juvenile Diabetes Research Foundation, the US-Israel Binational Science Foundation, the American Diabetes Association, and the A. Alfred Taubman Medical Research Institute. Dr. Goutman receives research funding support from Neuralstem, Inc; the Centers for Disease Control and Prevention/Agency for Toxic Substances and Disease Registry; and the Agency for Toxic Substances and Disease Registry (contract #200-2013-56856). Dr. Meisler receives funding support from the NIH. Dr. Callaghan receives funding support from Impeto Medical Inc and the NIH, has received travel/speaker honoraria from the American Academy of Neurology and the World Federation of Neurology, performs medical consultations for Advance Medical, consults for a PCORI grant, has received honoraria from the British Medical Journal, has worked with the ALS Association, and has acted as a consultant for medical legal cases. Dr. Todd receives funding support from the NIH, the Veterans Administration, the National Ataxia Foundation, the National Fragile X Foundation, and the Muscular Dystrophy Association; serves as a paid consultant for Denali Therapeutics and for an NIH grant; serves as an unpaid consultant for the National Fragile X Foundation; has received travel/speaker honoraria from the American Academy of Neurology, the National Fragile X Foundation, and the Japanese Society of Neurochemistry; has received publishing royalties from UpToDate; and has received license fee payments for antibodies developed by his group from Millipore for distribution to scientists and commercial groups, and for vectors and antibodies developed by his group from Denali Therapeutics. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gami P, Murray C, Schottlaender L, et al. A 30-unit hexanucleotide repeat expansion in C9orf72 induces pathological lesions with dipeptide-repeat proteins and RNA foci, but not TDP-43 inclusions and clinical disease. Acta Neuropathol 2015;130:599–601. [DOI] [PubMed] [Google Scholar]
- 4.Orr HT. Cell biology of spinocerebellar ataxia. J Cell Biol 2012;197:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Disease. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 6.Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 2004;74:1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirano M, Quinzii CM, Mitsumoto H, et al. Senataxin mutations and amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2011;12:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marangi G, Traynor BJ. Genetic causes of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challenges. Brain Res 2015;1607:75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu C, Macleod I, Su AI. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res 2013;41:D561–D565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilhelm M, Schlegl J, Hahne H, et al. Mass-spectrometry-based draft of the human proteome. Nature 2014;509:582–587. [DOI] [PubMed] [Google Scholar]
- 11.Meisler MH, Grant AE, Jones JM, et al. C9ORF72 expansion in a family with bipolar disorder. Bipolar Disord 2013;15:326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laaksovirta H, Peuralinna T, Schymick JC, et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol 2010;9:978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shatunov A, Mok K, Newhouse S, et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol 2010;9:986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Es MA, Veldink JH, Saris CG, et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet 2009;41:1083–1087. [DOI] [PubMed] [Google Scholar]
- 15.van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol 2013;12:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moreira MC, Klur S, Watanabe M, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet 2004;36:225–227. [DOI] [PubMed] [Google Scholar]
- 17.Reddy K, Schmidt MH, Geist JM, et al. Processing of double-R-loops in (CAG)·(CTG) and C9orf72 (GGGGCC)·(GGCCCC) repeats causes instability. Nucleic Acids Res 2014;42:10473–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci 2007;30:575–621. [DOI] [PubMed] [Google Scholar]
- 20.Cooper-Knock J, Shaw PJ, Kirby J. The widening spectrum of C9ORF72-related disease; genotype/phenotype correlations and potential modifiers of clinical phenotype. Acta Neuropathol 2014;127:333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vellano CP, Lee SE, Dudek SM, Hepler JR. RGS14 at the interface of hippocampal signaling and synaptic plasticity. Trends Pharmacol Sci 2011;32:666–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi H, Abe K, Matsuura T, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 2011;89:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010;466:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blauw HM, van Rheenen W, Koppers M, et al. NIPA1 polyalanine repeat expansions are associated with amyotrophic lateral sclerosis. Hum Mol Genet 2012;21:2497–2502. [DOI] [PubMed] [Google Scholar]
- 25.Figley MD, Thomas A, Gitler AD. Evaluating noncoding nucleotide repeat expansions in amyotrophic lateral sclerosis. Neurobiol Aging 2014;35:936.e1–936.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee T, Li YR, Chesi A, et al. Evaluating the prevalence of polyglutamine repeat expansions in amyotrophic lateral sclerosis. Neurology 2011;76:2062–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groen EJ, van Rheenen W, Koppers M, et al. CGG-repeat expansion in FMR1 is not associated with amyotrophic lateral sclerosis. Neurobiol Aging 2012;33:1852.e1–1852.e3. [DOI] [PubMed] [Google Scholar]
- 28.Dunckley T, Huentelman MJ, Craig DW, et al. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N Engl J Med 2007;357:775–788. [DOI] [PubMed] [Google Scholar]
- 29.Schymick JC, Scholz SW, Fung HC, et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol 2007;6:322–328. [DOI] [PubMed] [Google Scholar]
- 30.Cronin S, Berger S, Ding J, et al. A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum Mol Genet 2008;17:768–774. [DOI] [PubMed] [Google Scholar]
- 31.van Es MA, van Vught PW, Blauw HM, et al. Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat Genet 2008;40:29–31. [DOI] [PubMed] [Google Scholar]
- 32.Chio A, Schymick JC, Restagno G, et al. A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Hum Mol Genet 2009;18:1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landers JE, Melki J, Meininger V, et al. Reduced expression of the kinesin-associated protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 2009;106:9004–9009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iida A, Takahashi A, Kubo M, et al. A functional variant in ZNF512B is associated with susceptibility to amyotrophic lateral sclerosis in Japanese. Hum Mol Genet 2011;20:3684–3692. [DOI] [PubMed] [Google Scholar]
- 35.Kwee LC, Liu Y, Haynes C, et al. A high-density genome-wide association screen of sporadic ALS in US veterans. PLoS One 2012;7:e32768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng M, Wei L, Zuo X, et al. Genome-wide association analyses in Han Chinese identify two new susceptibility loci for amyotrophic lateral sclerosis. Nat Genet 2013;45:697–700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.