

Abstract

NMR-based metabolomics has shown considerable promise in disease diagnosis and biomarker discovery because it allows one to nondestructively identify and quantify large numbers of novel metabolite biomarkers in both biofluids and tissues. Precise metabolite quantification is a prerequisite to move any chemical biomarker or biomarker panel from the lab to the clinic. Among the biofluids commonly used for disease diagnosis and prognosis, urine has several advantages. It is abundant, sterile, and easily obtained, needs little sample preparation, and does not require invasive medical procedures for collection. Furthermore, urine captures and concentrates many “unwanted” or “undesirable” compounds throughout the body, providing a rich source of potentially useful disease biomarkers; however, incredible variation in urine chemical concentrations makes analysis of urine and identification of useful urinary biomarkers by NMR challenging. We discuss a number of the most significant issues regarding NMR-based urinary metabolomics with specific emphasis on metabolite quantification for disease biomarker applications and propose data collection and instrumental recommendations regarding NMR pulse sequences, acceptable acquisition parameter ranges, relaxation effects on quantitation, proper handling of instrumental differences, sample preparation, and biomarker assessment.
Keywords: NMR, urine, disease, metabolites, quantitative analysis, recommendations, standardization, quantification
1. Introduction
Metabolomics (also known as metabonomics) is the study of global metabolite profiles in biological samples such as biofluids, cell extracts, and tissues. Metabolomics can be integrated with other omics sciences such as genomics, transcriptomics, and proteomics to facilitate a more complete understanding of global biological systems. Metabolite concentrations and perturbations represent a snapshot of the metabolic dynamic that reflect the response of living systems to environmental factors, pathophysiological stimuli, or genetic modification. To characterize the vast array of metabolites found in any given biosample, metabolomics researchers must utilize a wide range analytical platforms including high-performance liquid chromatography (HPLC),1 liquid chromatography with mass spectrometry (LC–MS),2 gas chromatography with mass spectrometry (GC–MS),2a,2b,3 tandem mass spectrometry,4 and NMR spectroscopy.5 Each technique has its own advantages and disadvantages, and the choice of a given analytical platform often depends on the focus of the study and the samples.6
The use of NMR in metabolomics is particularly appealing to many researchers because of its nondestructive, quantitative nature and its ability to identify novel compounds via their unique spectral patterns.7 In NMR-based metabolomics, biological fluids such as serum, plasma, urine, saliva,8 cerebrospinal fluid,9 amniotic fluid,10 synovial fluid,11 exhaled breath condensate,12 cell extracts,13 and tissue extracts7g,14 have all proven to be particularly amenable to NMR analysis. Urine, in particular, is a very appealing biofluid for analysis because it is abundant, can be collected noninvasively, and is particularly rich in terms of its chemical diversity. Consequently, urine offers significant opportunities for data mining, data modeling, and biomarker discovery, particularly with respect to human health and disease.15 Furthermore, urine exhibits a strong phenotypic or metabotypic stability,16 which strengthens its potential for biomedical research and clinical utility.
Urine is composed primarily of small hydrophilic molecules such as sugars, organic acids, amino acids, soluble lipids, organic amines, and so on, along with inorganic salts that are small enough to have successfully passed through the body’s reticuloendothelial filtration system. While the noninvasive collection of urine is advantageous for many metabolomics applications, the major spectroscopic challenge associated with analyzing urine by NMR is the tremendous variation in its chemical concentrations. There are a number of factors, including sample collection and processing, as well as data acquisition and processing parameters, that need to be considered to enable accurate and precise quantitation of urinary metabolites by NMR-based metabolomics. Metabolic profiling of urine gives a time-averaged representation of an individual’s recent (typically within 24 h) homeostatic condition. Some metabolites may associate with individual’s physiological or pathological state, whereas others may associate with an individual’s genotype, environmental exposures, dietary habits, or drug intake17 as well as the time (season, hour of the day) of collection. Indeed, one of the most significant unresolved issues in urinary metabolomics lies in the remarkable variance in urinary excretion volumes and subsequent variations in metabolite concentrations.18 As such, it is critical that in quantitative metabolomics the inter- and intraindividual metabolite variance within the normal/control group be properly identified, defined,18 and as much as possible minimized. This may be facilitated by requiring a 12 h fast prior to urine collection or restricting the consumption of supplemental protein performance enhancing food, such as protein shakes, prior to any sample collection.19 Even with these controls in place, the variance in urine metabolite concentrations is still quite significant.
In light of these challenges, a coherent standard protocol is particularly important for the analysis of urinary biomarkers. To ensure robust and accurate quantification of potential urinary biomarkers by NMR, each step of the analytical protocol must be carefully performed and evaluated. This includes appropriate consideration for the context of the chosen application. For example, in nutritional intervention studies, it is important that a standardized diet is introduced to participants prior to the intervention and subsequent sample collection to minimize dietary effects confounding the results. Specifically, sample collection, preservation, preparation as well as instrumental optimization, NMR pulse sequence selection, and choice of acquisition parameters, data-processing parameters, peak/metabolite identification confidence, and final reporting results must be undertaken with a high level of consistency and an appropriate degree of scientific rigor to ensure the validity of the results. This process is not entirely straightforward, and much research has been devoted to refining, testing, and optimizing each step of the NMR analytical and data processing protocols.7d,20 Indeed, more than 70 papers have been published on the subject since 1990, with one of the most widely cited of these being the publication by Beckonert et al.20b While advocating the need for standardized protocols in metabolomic applications (i.e., the Metabolomics Standards Initiative),21 these authors also noted that it may be “detrimental to the exploratory nature of the subject to allow only “validated” or “approved” procedures to be used in experimental metabolism studies.” This acknowledges the importance of further research and improved methodologies for sample handling, data acquisition, and data analysis.
In a previous Review22 we proposed several recommendations regarding the standardization of the experimental conditions for using urine in NMR-based metabolomic studies. We highlighted the effects of diet, sample collection time (of day), age, gender, gut microflora, individual metabotypes, physical activity, subject selection, sample storage, salt, and pH effects as well as acquisition temperature with regard to urine metabolite composition and concentrations. We also provided recommendations regarding ethical guidelines for sample acquisition, the establishment of written SOPs, the selection of containers/consumables, patient/sample selection protocols, sample collection handling methods (centrifugation, additives, storage protocols), sample transfer methods, sample pH, chemical shift referencing, minimum sample numbers, sample randomization, magnetic field strength, optimal NMR pulse sequences, acquisition temperature, and results or reporting standards.
In this Review, we will discuss the pertinent issues regarding NMR-based urinary metabolomics with a specific emphasis on quantification for disease biomarker applications. We first review and discuss some of the key issues relating to (1) biomarker assessment, (2) urinary biomarkers and the need for metabolite quantification, (3) metabolite quantification methods for NMR, (4) examples of NMR-derived biomarkers, and (5) concentration normalization methods. Consensus recommendations will then be made regarding: concentration normalization, suitable NMR pulse sequences, acceptable parameter ranges, the effects of relaxation on quantitation, and the utility of data acquired on different instruments
2. Biomarkers and Biomarker Assessment
A biomarker is a measurable substance in a biofluid or biological tissue that can be used as an indicator of some biological perturbation caused by a disease, a change in biological state, or an environmental exposure. Biomarkers may be used for disease diagnosis, prognosis, prediction, or monitoring as well as for measuring biological responses from various drug, toxin, or environmental exposures. Biomarkers have a wide range and may include chemicals, metabolites, genes/mutations, RNA transcripts, proteins, cell counts or cell types, karyotypes, or just about any other detectable substance or measurable biological feature. As a general rule, a single biomarker often corresponds to a single medical test, with a threshold value (concentration or number) being used to distinguish between healthy and diseased states.
2.1. Biomarker Sensitivity and Specificity
The performance of a biomarker is typically evaluated by its sensitivity and specificity. Sensitivity relates to the biomarker’s ability to identify positive results and specificity relates to the biomarker’s ability to identify negative results. More specifically, sensitivity can be defined as “the proportion of patients who are known to have the disease who test positive for it”, while specificity can be defined as “the proportion of patients that are known not to have the disease who will test negative.” The mathematical definition of sensitivity and specificity is given here
 |
1 |
 |
2 |
In common medical practice, sensitivity is generally not sufficient to assess the diagnostic performance of a test where the test has no negative predictions (from a theoretical point of view, 100% sensitivity). Therefore, both sensitivity and specificity should be examined together and reported in disease diagnostic studies. Figure 1 demonstrates the assessment of biomarker performance, in the context of a biomarker’s ability to differentiate between diseased and healthy subjects.23Table 1 summarizes the relationship between positive and negative test outcomes and of true/false positives and negatives.
Figure 1.
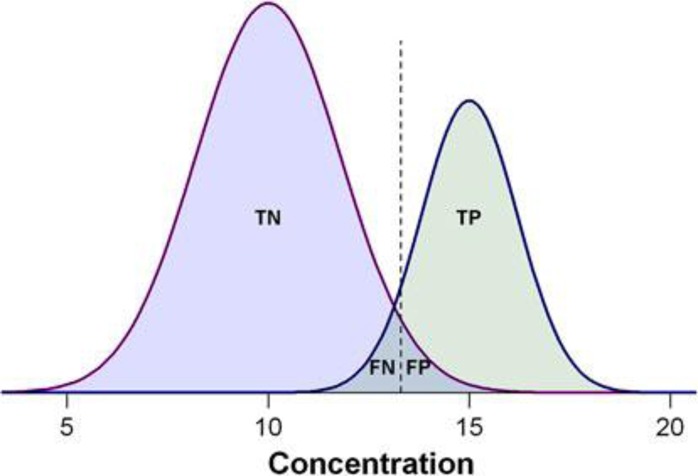
Demonstration of the biomarker prediction test with two Gaussian curves indicating the distributions of measured values, with positive cases on the right side and negative cases on the left. The dashed lines indicate the cutoff threshold of hypothetical biomarker concentration that can be used to separate positive from negative tests. The overlap between the biomarker concentrations of the two populations represents the misclassification ratio between the left-hand side of the positive cases and the right-hand side of the negative cases. TP, the number of true positives; TN, the number of true negatives; FP, the number of false positives; FN, the number of false negatives, respectively.23
Table 1. Relationship between Terms of Positive and Negative Test Outcomes.
| condition positive | condition negative | |
|---|---|---|
| positive test outcome | true positive | false positive |
| negative test outcome | false negative | true negative |
Simultaneous measurement of both sensitivity and specificity with respect to different separation threshold values is often best illustrated using a receiver operating characteristic (ROC) curve. An ROC curve shows how the sensitivity and specificity change as the classification decision boundary is varied across the range of available biomarker scores. An ROC curve is not dependent on the prevalence of a given outcome, and because it shows the performance of a biomarker test over the complete range of possible decision boundaries, it allows the optimal specificity or sensitivity to be determined posthoc. ROC curves are often summarized into a single metric known as the “area under the curve” (AUC). For a perfect biomarker test, the AUC is 1.0. An AUC of 0.5 is equivalent to randomly classifying subjects (i.e., the classifier is of no practical utility). A rough guide for assessing the utility of a biomarker based on its AUC is as follows: 0.9 to 1.0 = excellent; 0.8 to 0.9 = good to very good; 0.7 to 0.8 = fair; 0.6 to 0.7 = poor; 0.5 to 0.6 = fail. ROC curve analysis and using ROC-AUC is widely considered to be the most objective and statistically valid method for biomarker performance evaluation. A much more detailed review of ROC analysis along with general recommendations for biomarker quantification and statistical strategies for multibiomarker models is provided elsewhere.23
3. Biomarkers and Metabolite Quantification
Metabolites and metabolite concentrations can be particularly sensitive to modest physiological or subtle genetic perturbations. Observed changes to urine have historically been accredited to attempts to diagnose ailments as far back as the ancient Greek physician Hippocrates (400 BC) and the Arabian/Persian alchemist Avicenna (11th century). Indeed, as Sir Archibald Garrod (the founder of modern clinical chemistry) noted in 1908, changes in metabolite concentrations often start before the onset of clinical symptoms.24 This simple fact has served as the basis to the development of more than 180 different chemical or metabolite biomarker tests that are commonly used today (https://labtestsonline.org). Indeed, there are more approved metabolite- or chemical-based clinical tests than approved genetic, protein, or karyotype tests (https://labtestsonline.org). While most clinical tests are blood-based, an increasing number of clinical assays that use urine currently exist. The very first clinical assay (and the first genetic disease test) was designed for detecting homogentisic acid among individuals with a rare inborn error of metabolism called alkaptonuria.24 Among the most commonly used metabolite tests today are urinary glucose tests for monitoring diabetes,25 24 h urinary creatinine tests to measure kidney function,26 urinary cortisol tests to diagnose Cushing’s or Addison’s disease, urinary nitrite tests to detect bacterial infections, and urinary bilirubin tests to assess liver function (Table 2).
Table 2. Examples of Urinary Biomarkers of Disease Discovered Using NMR-Based Metabolomics in Human Studies.
| condition | comparison | biomarkers | reference |
|---|---|---|---|
| pancreatitis | pancreatitis patients vs controls | citrate | (61) |
| adenosine | |||
| bladder cancer | bladder cancer patients vs controls | hippurate | (62) |
| citrate | |||
| taurine | |||
| hepatoceullar carcinoma | hepatocellular carcinoma v cirrhosis vs noncirrhotic liver disease patients vs controls | inosine | (63) |
| indole-3-acetate | |||
| galactose | |||
| NAA | |||
| pancreatic ductal adenocarcinoma | pancreatic ductal adenocarcinoma vs controls | acetone | (76) |
| hypoxanthine | |||
| o-acetylcarnitine | |||
| dimethylamine | |||
| esophageal cancer | esophageal patients vs controls | urea | (77) |
| acetate | |||
| pantothenate | |||
| 3-hydroxyisovalerate | |||
| acetone | |||
| formate | |||
| gestational diabetes (GDM) | GDM patients vs controls | 3-hydroxyisovalerate | (78) |
| 2-hydroxyisobutyrate | |||
| neonatal health | small vs appropriate for gestational age | glycine | (65) |
| threonine | |||
| neonatal health | intrauterine growth retardation vs controls | myo-inositol | (64) |
| sarcosine | |||
| creatine | |||
| creatinine |
Almost all approved clinical chemistry tests, whether they measure chemical or protein biomarkers, are required to be quantitative or at least semiquantitative (via some ratio measurement). This requirement has allowed universal reference values for different ages and genders to be developed and applied for routine medical diagnoses. In many cases physicians and clinical chemists are required to know or memorize threshold values for a large number of medically important metabolites and proteins. Absolute quantification not only gives physicians useful reference or decision-making thresholds but also ensures consistency and reproducibility from instrument to instrument, lab to lab, city to city, and country to country. As previously stated, the issue of quantification has turned out to be an Achilles heel for many biomarker tests developed using “omics” platforms.15b,27 This is because proteomic, transcriptomic, and even most metabolomic assays were not originally designed with comparative quantitation in mind, nor were they designed to be compared accurately and precisely between different instrumental platforms.
However, there is now a growing shift in the “omics” community toward developing quantitative proteomic and quantitative metabolomic assays5g,23,28 with a long-term goal of using these quantitative assays for biomarker discovery, development, and clinical translation.23 The need for quantification, either relative or absolute, is particularly acute for metabolite-based urine analysis where individual metabolite concentration values can vary by a factor of 10 or more due to dilution, gender, diet, or diurnal effects.29 As a result there have been a number of papers, NMR-specific software tools (Chenomx, AMIX, Batman,30 Bayesil,31 etc.), as well as a number of commercial kits that have been developed (such as the BioCrates kit for mass spectrometers) to perform absolute metabolite quantification. Methods to facilitate urinary metabolite quantification, with a specific focus on NMR, are briefly reviewed in the next section.
4. Quantitation of Metabolites by NMR
NMR spectroscopy is a powerful analytical approach for both identification and quantification of analytes with superior advantages, such as being nondestructive, highly reproducible, and, most importantly, requiring minimal sample preparation.32 Indeed, NMR is inherently a quantitative technique as the intensity of an NMR signal is proportional to the concentration of detectable (usually 1H) nuclei in the receiver coil.33 The 1D 1H NMR spectra of urine samples are highly complex, with thousands of distinct signals visible in a single spectrum. Consequently, signal overlap and signal distortions from nearby (strong) peaks are often evident even in 1D 1H NMR spectra collected at 800 MHz and above. Over the past two decades, several methods had been proposed or developed to address these issues. In particular, an exhaustive technical review on this subject has recently been published by P. Giraudeau.34 This paper nicely summarizes the strengths and weaknesses of various 2D NMR methods, for example, 2D J-resolved, COSY, TOCSY, 2D INEPT, HSQC, HMBC, and so on, with regard to resolution in the second dimension and with respect to compound identification and quantification in complex mixtures. Common shortcomings identified for all (conventional) 2D NMR methods were the lack of sensitivity, the lack of speed, and the difficulties in reproducible quantitation compared to 1D NMR; however, this paper also highlighted several novel 2D NMR methods or data-processing techniques that appear to solve or at least address some of these issues. For instance, 2D 1H INADEQUATE with sparse sampling/nonlinear sampling (NLS) as proposed by Hyberts et al.35 seems to be very promising for characterizing and quantifying low abundance metabolites. More recently, another approach has been proposed,36 wherein three different collection and processing techniques were combined, including J-compensated 2D HSQC, NLS, and forward entropy (FM) reconstruction. This combination resulted in a 22-fold reduction in NMR recording time (relative to a conventional HSQC spectrum) while at the same time yielding precise metabolite quantitation in both native and lyophilized urine samples. The authors report a lower limit of detection of “tens of micromolar”; however, these types of experiments are not straightforward, and significant prior knowledge about the sample and expected spectral windows is needed to properly implement the method. Furthermore, while these approaches are promising, they have yet to be implemented in a real, nontargeted metabolomic study nor have they been comprehensively evaluated and validated. Ultrafast 2D NMR spectroscopy37 is yet another promising method; however, this approach requires sophistication in pulse field gradient performance and specific processing software, presently not available on most commercially available NMR spectrometers. Nevertheless, given its speed, resolution, and sensitivity advantages over fast heteronuclear NMR and even 1D homonuclear NMR methods, this approach may soon become the method of choice for identifying and quantifying low abundance metabolites in urine samples.
Unlike MS-based quantitative methods, which usually require expensive isotopically labeled standards and time-consuming chromatographic separations, NMR spectroscopy offers the possibility of quantifying many metabolites simultaneously without the need for any prior chromatographic separation; however, accurately and reproducibly quantifying NMR signals from different instruments at different field strengths and using different pulse sequences or pulse widths and delay times is often challenging. Indeed, despite a steady increase in the number of publications arising from NMR-based metabolomics focused on diagnostic biomarkers, only a few of these report quantified metabolite levels.23,29a,38 This paucity of quantitative data could be due to the fact that metabolite quantification is both instrumentally challenging and manually intensive work.
In terms of instrumental issues, the greatest challenge in NMR-based quantification lies in consistency and reproducibility. To obtain consistent and reliable results, identical NMR tubes must be used as well as identical instrument parameters including temperature, chemical shift referencing, tuning, shimming, magnetic field drift compensation/lock, pulse sequences, water suppression methods, acquisition times, and data-processing parameters (Table 3). These conditions must be strictly adhered to from sample to sample. This not only allows for rigorous quality assurance but also allows robust intralaboratory and interlaboratory comparison. Variations in the ionic strength of different samples can have dramatic effects on the ability to tune and match the spectrometer (assuming the probe range is sufficient), leading to profoundly different pulse widths or spectrometer responses. Specifically, what are assumed to be infinitely short “on-resonance” pulses become ineffective (much less than 90°) and contain larger and larger off-resonance pulse effects (evolution). This can have dramatic consequences regarding relaxation of the NMR signal in-between data acquisitions. In addition, some instrumental effects (e.g., field instability, temperature variations, incorrect referencing) can cause chemical shift changes. Recently, Sokolenko et al. evaluated the sources of quantification variability in NMR and determined that such seemingly mundane issues as sample insertion methods or shimming protocols as well as the choice of NMR pulse sequence could lead to significant differences in the resulting metabolite identification and quantification.39 It has also been shown that the data acquisition parameters, spectral processing parameters, and the choice of water suppression method can also substantially affect metabolite quantification results.40
Table 3. Experimental Conditions for Precise Quantitation of Urine Samples Using NMR Spectroscopy.
| sample preparation | parameters and recommended values | comments |
|---|---|---|
| sampling | overnight fasting urine collection | ensures more stable homeostatic concentrations of metabolites |
| mid stream urine collection | avoids unwanted contamination from urinary tract | |
| collecting urine sample in labeled tube containing sodium azide (NaN3) | to stop bacterial growth in samples; final concentration of 0.05% wt/vol | |
| store immediately in to −40 to –80 °C until NMR experiments are performed | helps arrest metabolic activities and sample degradation | |
| sample processing | centrifugation/filtration | centrifuge at 1000 rpm to remove the turbidity from unwanted particulates, or filter using 0.22 μ filter to remove any macromolecular content in the sample |
| phosphate buffer | phosphate buffer helps in avoid chemical shift drift that occurs due to pH variations | |
| internal reference standard; e.g., TSP or DSS | in protein/lipid free urine sample, TSP and DSS are a good choices as internal standards for quantification and normalization | |
| use of deuterated EDTA | only recommended when variation of ionic concentration urine is very large and drift in the chemical shifts is causing quantitative errors. | |
| acquisition parameters | one-dimensional gradient NOESY with water presaturation experiment. | |
| time domain points (TD): 64K | Increased resolution | |
| line broadening (lb): 0.1–0.5 Hz | ||
| relaxation delay >5.0 s | relaxation delay depends on longitudinal relaxation time (T1) of metabolite resonances; it should be five times T1 for absolute quantitation or matched to the T1 of the reference spectra used for deconvolution. | |
| acquisition time: 2.5 s | increased resolution | |
| spectral width (sw): 12 ppm | ||
| number of scan (ns): 64 | for desired S/N, more are required for diluted samples | |
| dummy scan (ds): 8 | to achieve steady state prior to acquisition | |
| excitation pulse: 90 deg | shorter pulse widths can be used for single pulse NMR analysis | |
| receiver gain (rg): optimal | either a constant RG for all or auto optimized for every sample | |
| mixing time (tm): | pulse sequence requirements for NOESY; minor loss in signal intensity due to transverse relaxation | |
| 100 ms for standard experiment | ||
| 10 ms for gradient experiment | ||
| sample temperature: 300 K | kept constant throughout the study | |
| shimming, tune, and match: for every sample | increased accuracy, precision, and reproducibility | |
| processing parameters | windowing: exponential window function with line broadening of 0.3–1.0 Hz | |
| zero filling: a factor of 2 of TD | increased resolution | |
| phase correction: manual phasing is preferred | optimal for accurate integration of peaks area | |
| baseline correction: automatic/manual | increased accuracy of peak integration | |
| chemical shift referencing | both TSP and DSS can be used for chemical shift referencing (δ 0.0), although DSS is the IUPAC standard |
As a general rule, a single excitation pulse sequence with a relatively low-power water presaturation is the default method for performing quantitative metabolite analysis of biofluids and biological extracts. Historically, quantitation in NMR required the use of long pulse delays to allow signals to fully recover, but more recently the use of a low flip angle (i.e., ≪90°) to accommodate faster signal recovery along with suitable signal correction methods has allowed much more rapid data acquisition.41 For most biological samples, there is a substantial amount of water present, so strong solvent suppression must be employed to obtain good metabolite signals. Therefore, a 1D NOESY pulse sequence with water presaturation (see ref (42) and references therein), excitation sculpting,43 or WATERGATE solvent suppression44 is preferred. Because of solute/solvent hydrogen exchange, the presaturation sequence itself can dramatically alter quantitation results. Specifically, the exchangeable proton signals from urea and other water exchangeable solutes are often suppressed by presaturation, making their quantification difficult or inconsistent. While more complex solvent suppression schemes like PURGE,45 excitation sculpting, or variations of WATERGATE46 are potentially better choices for metabolite quantification, these pulse sequences all have auxiliary regions of signal suppression, which makes them more difficult to optimize and harder to use. This added complexity can also affect interlab and intralab consistency or reproducibility. Pulsed field gradient (PFG) versions of NMR pulse sequences (e.g., 1D NOESY pulse sequence) are available, but these may lead to problems with quantification due to both lock instability and gradient ring down periods.47 These problems manifest as peak distortions (i.e., semidispersive line shape) to the internal chemical shift and quantitation reference (such as 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS)). For these reasons, gradient pulse sequences, despite their advantages,48 are not recommended.
In terms of spectral analysis, metabolite quantification requires meticulous consistency and considerable attention to detail. Prior to spectral collection, each biofluid sample must be “spiked” with an exact amount of a known quantification standard (usually a chemical shift reference standard such as tetramethylsilane (TMS) for organic solvents or trimethylsilyl propanoic acid (TSP) and 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) for water-based experiments).49 Once the NMR spectra have been collected, the data must be properly phased and baseline-corrected, and any residual interfering solvent signals must be digitally filtered. Then, individual “sentinel” peaks corresponding to the specific compounds must be identified, and the peak area(s) carefully integrated.50 This identification and quantification step (also called spectral deconvolution) can be done manually, semiautomatically, or automatically (vide infra). The concentration of a given metabolite can be directly calculated from the spiked standard using the following formula51
| 3 |
where [M] is the metabolite molar concentration, [Std] is the spiked standard’s known molar concentration, Im is the metabolite intensity, Is is the intensity of the spiked standard’s peak, Nm is the number of nuclei contributing to the metabolite peak, and Ns is the number of nuclei contributing to the spiked standard’s peak. In some cases it is not necessary to use an internal standard to quantify metabolites but rather one may use an external reference or an electronic reference signal,59,60 or even the solvent resonances.52
A key challenge with quantifying metabolites in biological samples, and especially in urine, is the considerable degree of spectral overlap seen in 1D 1H NMR spectra. For example, urine NMR spectra typically consist of >2000 detectable peaks corresponding to >200 detectable compounds.53 The positions and shapes of these peaks can vary with pH, temperature, salt concentrations, magnetic field strength, sample stability, and homogeneity. In addition, common 1H–1H dipolar or quadrupolar (e.g., 14N–1H) couplings further complicate the observations. Consequently, it is very difficult for even a trained individual to identify a single set of “sentinel” peaks that can be unambiguously identified and properly integrated for any given urine sample. To assist with this peak identification and metabolite identification process it is possible to use several approaches. One approach is spectral simplification through statistical analysis or chemometric analysis using spectral binning/alignment and statistical total correlation spectroscopy (STOCSY).54 These methods statistically align and compare the NMR spectra between two groups (diseased and healthy), and the most significantly different peaks are then identified. This leads to a reduction in the number of peaks that need to be analyzed or identified. A disadvantage of this approach is that it can lead to problems in compound identification and quantification, as the spectra have been extensively averaged or “warped” as part of the statistical processing.54
Another approach involves identifying and quantifying as many compounds as possible prior to determining any statistical differences between groups. This leaves the NMR spectra in a relatively pristine state so that compound identification and quantification is easier and more accurate; however, the process is time-consuming, as dozens of compounds and hundreds of peaks must be identified and quantified through a process known as spectral deconvolution.54a Several companies have developed spectral deconvolution tools and software (e.g., Chenomx NMRSuite from Chenomx, Inc.; AMIX from Bruker, Inc.; MnovaScreen from Mestralab Research; CRAFT or Complete Reduction to Amplitude Frequency Table from Agilent) that makes this process easier. These packages include carefully collected reference NMR spectra of hundreds of common metabolites at different field strengths and pH values. These programs and their corresponding libraries can allow 50–75 compounds to be manually identified or quantified from urine spectra in about 1–4 h; however, this process must be performed by well-trained individuals. The requirements of manual phasing, manual baseline correction, manual solvent removal, and manual spectral deconvolution often mean that quantification inconsistencies and even compound identification errors can be introduced. For example, one published study looked into these issues of consistency and reproducibility39 and found compound quantification by some of these methods is only accurate to within 15%. More recently several groups have attempted to develop fully automated methods that remove any human variability in spectral processing and compound identification.30b,55 These methods appear to be very promising, especially for simpler biofluids such as serum, saliva, or cerebrospinal fluid, but they have yet to be shown to be effective for biofluids as complex as urine.
Sample-to-Sample Normalization
The large differences in metabolite concentrations observed in different urine samples can complicate comparisons between samples and may lead to false conclusions. Thus, normalizing NMR spectra is a critical step for eliminating systematic errors. In addition to normalizing concentrations relative to creatinine (which is commonly done in clinical practice), several other normalization methods have been developed, including total integral intensity (usually the total spectral area). This is one of the most commonly used methods in NMR.56 Other methods have been proposed as an alternative to the total intensity method, including probabilistic quotient normalization (PQN)57 and histogram matching (HM),58 which effectively make the data more suitable to identifying potential metabolic biomarkers for disease.
Normalization can be problematic due to the potential influence that the method might have on compound identification or quantification. While one would hope that compound identification is unaffected (see later), the relative proportions certainly might. This assumes that normalization would occur only on the quantitation of unambiguous peaks (i.e., no overlap) and would apply to all such positively identified metabolites. This might seem obvious, but it needs to be explicitly stated as normalization can also refer to frequency-dependent compensation for NMR pulse sequence effects, for example, T1-dependent changes. Any changes to the NMR pulse sequence or parameters will perturb peak heights. Any compensation for pulse sequence differences would also affect the peak ratios for atoms in the same molecule as well as the overall metabolite quantitation. For example, a compensation for different presaturation power levels would correct amplitudes differently close to the carrier position versus the edges of the spectra. A compound containing resonances both close to and far from the water resonance would then have an intramolecular integration ratio that would not make sense and could have its identification changed or moved to an ambiguous/unassigned grouping.
This is not a problem unique to NMR. Mass spectrometry (MS) has been dealing with this issue for years. In MS, control samples are introduced into the sample queue to compensate for changes in the chromatographic steps and equipment necessary to separate metabolites. Specifically, quality control samples are submitted at regular intervals or samples are combined and run as reference samples for comparison. Interestingly, in NMR, the use of control or calibration samples is not regularly done. While chromatographic columns are not normally used in NMR, batched samples run on robotic sample handling systems must often sit for lengthy periods of time (hours) prior to loading and spectral acquisition. Control samples placed at the beginning, middle, and end of a long sample-loading run would therefore reveal any potential degradation/storage/handling problems or (less likely) changes occurring to the spectrometer/console/probe.59 Compared with the extensive efforts used in quality control for MS, the use of quality control for normalization in NMR is, in our opinion, still under-developed.
5. Examples of Potential NMR-Derived Urinary Biomarkers
There is an abundance of literature linking urinary biomarkers with human disease states.7e,15b,60 The analytical approaches used to collect these data are certainly diverse, and this diversity serves to demonstrate the complexity associated with developing standardized approaches for urinary biomarker quantification. Recently Bouatra et al.15a collated and critically evaluated the existing information on human urine to establish a comprehensive and electronically accessible human urine metabolome database (http://www.urinemetabolome.ca). This database includes quantitative concentrations of metabolites in normal and abnormal (i.e., disease-associated) urine samples and represents a significant development and resource for biomarker identification and quantification. This database also serves as a benchmarking and cross-referencing tool for future metabolomics approaches and will no doubt aid in efforts aimed at standardizing metabolomic approaches. While it is impractical to cover all biomarker examples described in the human urine metabolome database, it is perhaps useful to highlight some of the studies where NMR-based metabolomics was used to identify novel or potentially important biomarkers in urine. These are summarized in Table 2 and explained in more detail below.
As can be seen in Table 2, urinary biomarkers have been identified for a wide range of conditions, ranging from cancer to neonatal conditions to pancreatic disorders. For example, Lusczek and colleagues identified a useful set of potentially discriminating metabolites in the urine of pancreatitis patients compared with healthy controls.61 Although the metabolites identified in this study cannot be conclusively defined as biomarkers of the disease, they do have the potential to become biomarkers once additional studies are carried out to validate their findings, ideally using a larger patient cohort. In a similar study aimed at exploring other kinds of pancreatic disorders, Davis et al. used NMR-based metabolomics investigation on urine samples from age- and gender-matched patients with pancreatic ductal adenocarcinoma, compared with a healthy control group.76 These authors were able to easily differentiate between those with cancer and those in the control group (using both ROC curves and area under the curve [AUROC] calculations) using the set of strongly diagnostic metabolites listed in Table 2. As with the previous study, further validation using a larger cohort is needed to confirm the result. In another cancer-based study, urinary 1H NMR spectra of bladder cancer patients versus noncancer controls (healthy and those with urinary tract infections or bladder stones) were used to discriminate between the two groups, with taurine showing significant elevation in the urine of bladder cancer patients;62 however, the authors of this study were not able to discriminate between different disease stages possibly due to cancer-specific metabolic alterations or the low sensitivity of their particular instrument.62 Urinary biomarkers were also used to successfully and accurately diagnose a cohort West African patients with hepatocellular carcinoma.63 In this particular study the authors not only calculated ROC and AUROC values, they actually validated their findings using second larger cohort, a very good practice indeed. The metabolite biomarker panel identified in this study was also shown to perform much better than serum alpha-fetoprotein, a protein biomarker that is traditionally used for hepatocellular carcinoma diagnosis.63
Another interesting set of studies highlighted in Table 2 concerns the application of NMR-based urinary metabolomics to neonatal diagnoses. One very interesting NMR-based study used urine samples from neonates to compare those with intrauterine growth retardation versus full-term normal-weight controls to better define the metabolic patterns associated with this pathology. The authors identified myo-inositol, sarcosine, creatine, and creatinine as key metabolites that clearly differentiated between the two groups.64 While the initial findings are promising, the results will need to be further validated on a larger cohort. A more recent study on influence of early nutritional metabolic programming and long-term health in infants was carried out by Moltu et al. using NMR-based urinary metabolomics. In this intervention study, one group received significantly higher amounts of enhanced postnatal nutrition compared with the control group. We concluded that the enhanced nutrition did not appear to affect the urinary profiles to an extent exceeding the individual variation.65 This particular study is a good example of a well-conducted nutritional intervention study.
While the list of examples highlighted here (and in Table 2) is not exhaustive, it does highlight some of the common issues and challenges with respect to designing, implementing, and validating NMR-based urinary metabolomic biomarker studies. On the basis of these examples as well as an extensive review of the literature and a detailed assessment of the best practices conducted in our own laboratories and elsewhere, we have developed a set of consensus recommendations. These are summarized below.
6. Recommendations for Sample Collection and Processing
The identification of new biomarkers, along with their validation and translation into practical clinical applications, requires standardized preanalytical procedures for sample handling, sample stabilization, sample transport, and sample storage. Depending on the procedures employed, the detectable metabolites may be affected differentially by residual enzymatic activities or spontaneous chemical reactions that may alter the NMR profile.66 These alterations could seriously bias the results of studies based on samples having different collection, treatment, and storage histories. Other factors that may affect the concentration levels of urine metabolites include drug administration, health conditions, diet, physical activities, and environmental stressers. Thus, it is crucial for any quantitative urinary analysis to standardize sample collection conditions. Our recommendations are as follows:
Overnight Fasting
Overnight fasting prior to urine collection gives a more stable homeostatic picture of an individual’s urinary metabolome. Consequently, we recommend that all urine samples should be collected the morning after overnight fasting to reduce the effects of diurnal variations.67
Midstream Urine
Collecting midstream urine is recommended to avoid contamination from epithelial cells and bacteria from the urinary tract. Sampling conditions should be similar in control groups including age- and gender-matched groups for comparative analysis.
Medical Procedures
Medical procedures (including drug intake) performed before sample collection should be recorded and properly taken into account as medical treatments could induce significant changes in metabolites levels. All medical procedures prior to sample collection should be reported.
Aliquoting, Centrifugation, and Filtering Prior to Storage
Urine samples should be processed and aliquoted within 2 h from the time of collection but preferably faster. Samples must be kept refrigerated at 4 °C before processing and must not be frozen prior to processing to avoid possible cell breakage.20d Before aliquoting and long-term storage, urine samples should be centrifuged at 1000–3000 RCF (5 min at 4 °C) and (optionally) filtered using a 0.22 μm filter20d to remove cells and other particulates.
Storage
For long-term storage, urine samples should be stored at −80 °C. If possible, for very long-term storage, it is better to use liquid nitrogen vapor.20d Appropriately labeled cryovials should be used to store urine samples.
Preservatives
The addition of micromolar quantities of inorganic bacteriostatic agents such as sodium azide (to limit bacterial growth) is appropriate. The use of externally added organic preservatives (such as EDTA or glycerol) is strongly discouraged due to their possible interference with metabolite signals.
Diet Monitoring
Certain metabolites may be affected significantly by dietary intake. If possible, dietary information should be collected for 1 to 2 days before sampling. For example, taurine increases significantly after the consumption of taurine containing diet and energy drinks.68 To reduce lifestyle-related variations, and to the extent experimental design allows, a standardized diet is recommended for donors at least 1 day before sample collection.
Native Samples
Performing urine metabolomics on native (unadulterated, unprocessed) samples provides more reproducible and precise data. Extraction procedures, along with multistep sampling processes, increase the chance of metabolite loss. They also introduce analytical and operator errors.20d,69
Buffering
An example basic protocol would be to mix 630 μL of human urine with 70 μL of phosphate buffer (prepared in 2H2O) to minimize the drift in chemical shift due to pH variations. A concentrated KH2PO4 solution (1.5 M, pH 7.0) is best. If the variation in pH between the samples is still significant after the addition of the phosphate buffer, sample pH may be adjusted by adding NaOH or HCl accordingly.15b
Final Centrifugation
Centrifugation at 12 000g for at least 10 min at 4 °C to remove any suspended particles is strongly encouraged.20d
Chemical Shift Referencing
A chemical shift referencing standard such as TSP or DSS should always be added prior to NMR collection. Because the peak is easily resolved and unique, chemical shift standards can also be used for metabolite quantification. Note that DSS and TSP do bind proteins and lipids, so if these macromolecules are present to any significant degree, they may give rise to errors in quantitation. A final TSP or DSS concentration ranging between 0.1 and 0.5 mM is sufficient for most urine samples.
7. Recommendations for Spectral Acquisition and Processing
In addition to the recommendations for spectral acquisition that have been proposed in our previous reference paper,22 there are a number of instrumental, acquisition, and data-processing parameters that can significantly affect quantitative accuracy and precision. These parameters need to be optimized prior to conducting quantitative analysis of urine samples. Here we provide recommendations and justifications related to these issues.
7.1. Acquisition of Urinary NMR Data
Selection of Magnetic Field
NMR-based metabolomics studies on urine benefit from the use of the highest accessible magnetic field strengths. Presently, most NMR-based metabolomic studies are conducted using 600 MHz NMR spectrometers, as these instruments are relatively abundant and offer a good compromise between the cost, sensitivity, and resolution needed for metabolomics experiments. If the purpose of the analysis is the quantification or the identification of low abundance metabolites, we recommend using more sophisticated 2D experiments, higher fields (e.g., 800–1000 MHz), or increased sensitivity “cold-probes” or “cryoprobes”.
Pulse Sequence and General Setup
Automated pulse width calibration70 is strongly recommended for any NMR-based metabolomic study of urine. This is because urine samples exhibit considerable variability in salt concentrations, which substantially affect pulse widths. Pulse width calibration also helps to compensate for differences in sample volume or drift/decay in NMR hardware performance. For example, if components of the spectrometer or probe begin to fail, then pulse width calibrations will immediately indicate a problem and prevent wasted time and resources. Most modern NMR spectrometers have the option of sample handling robots for sample insertion into the spectrometer. Most NMR instruments also support autopulse calibration, autoshim, and auto tune/match prior to data acquisition. If available, these automated approaches should be used for all urine-based metabolomics studies to reduce human error and ensure maximum consistency and reproducibility.
Water Suppression
As previously detailed, a 1D NOESY pulse sequence with water presaturation, is the most commonly used NMR pulse sequence for metabolomics studies. While there are other excellent solvent suppression techniques such as excitation sculpting, or WATERGATE, the incorporation of pulsed field gradients can easily result in inconsistent spectrometer lock performance, the introduction of artifacts, and irreproducible solvent suppression; however, a presaturation pulse if done inconsistently, can also dramatically alter quantitation results due to hydrogen exchange with the solvent during the long saturation period(s).71 We therefore strongly recommend the use of an absolutely consistent presaturation period (e.g., 1 s) with equally consistently delivered and calibrated power (e.g., 80 Hz gamma B1 induced field). Of note, the exchangeable hydrogen signal from urea’s NH2- groups and other water exchangeable signals can also be suppressed by time-shared multifrequency saturation-based sequences,72 but these also introduce additional regions of suppression that can alter quantitation. Therefore, these types of sequences are not recommended for metabolomics.
Sample Temperature Control
Variations in the sample temperature during spectral acquisition can significantly affect the precision and reproducibility of NMR data. A calibrated sample temperature of ∼298 K (25 °C) for urine is recommended during NMR spectral acquisition. If a robotic sample handling system is being used, where large numbers of samples are prepared in advance and stored at the instrument, the samples should be kept below room temperature (∼ 5 °C) while waiting for sample insertion.29a,73 Samples should be prewarmed outside or in the probe for 5–10 min before spectral acquisition to ensure proper temperature equilibration, especially if a cooling rack is used to maintain refrigerated samples. While samples will begin to show thermal equilibrium via lock monitoring in 60 to 120 s, several minutes of equilibration are often necessary for convection currents to settle.
NMR Tubes
Higher quality NMR tubes yield higher quality spectra. Therefore, we recommend that the highest quality tubes with the lowest camber (straightest), thinnest, most consistent glass wall widths be used. Slightly curved tubes will wobble while spinning, which can lead to spinning side bands (extra peaks). Most NMR spectroscopists use 5 mm tubes, requiring volumes of 400–600 μL of material. If less material is available, the use of 3 mm or even 1.7 mm tubes is possible. Likewise, solvent-matched (Shigemi) tubes may also be used for low volume situations; however this option can increase the cost significantly. Prior to their use, NMR tubes should be cleaned to remove any film. Cleaned tubes tend to sheet refluxed material back down into the bottom of the tube, thus reaching equilibrium faster. Dirty or stock tubes with the film remaining from manufacturing will bead solvent further up the tube, effectively shortening the sample length and changing the magnetic field homogeneity.
7.2. Recommendations for Optimized Instrumental Parameters
Time Domain Points (Bruker TD or Agilent np)
Modern NMR spectrometers with high-speed/high-memory computers and digital oversampling can easily collect 64K or more data points. Considering the expected spectral windows and the desired resolution for NMR-based metabolomics work, time domain acquisitions ≥32K points are recommended for quantitative analysis of urine samples.
Repetition Time (TR)
This is also known as the relaxation delay or waiting period prior to the first hard excitation pulse. We define this period as the total time spent in acquiring a single scan spectrum, including the acquisition time and acquisition delays prior to the next excitation/acquisition. This period strongly affects the absolute quantitation of metabolites. Ideally, TR should be five times the longest T1 in the sample. This typically provides enough time for complete relaxation of all resonances between every scan, resulting in good quantitation. In metabolomics studies, using a TR that is five times the longest T1 in a sample requiring 128 scans would result in a sacrifice of enormous amounts of instrumental time that would dramatically increase the cost of the overall study. As a result, a shorter repetition time (e.g., 2–4 s) is often used for many NMR studies.69 The key is that the total relaxation time used is consistent from experiment to experiment. For absolute quantitation of metabolites, a T1 correction factor for spectra recorded over a shorter TR time can be determined and applied to compensate for the effect of incomplete relaxation.74 Application of this correction requires evaluation of the T1 for all resonances of interest in a representative sample prior to quantitation, assuming that the T1 of the resonances would be the same in all samples for the study. Alternately, using reference spectra of compounds of known concentration that have been collected using the same TR and using these reference spectra to deconvolute the mixture (as done with Chenomx, AMIX, Batman, or Bayesil) would also allow one to accurately determine concentrations.
Number of Scans/Signal to Noise Ratio
The better the signal-to-noise (S/N) is for a NMR spectrum, the more reliable the absolute quantitation. The simplest way to increase (S/N) in NMR is to increase the number of scans (also known as transients); however, adding more scans must be balanced by cost or time considerations. The S/N also depends on other factors like the magnetic field strength, stability of the spectrometer, probe type/quality, sample concentration, sample volume, excitation flip angle, and so on. If time and resources permit, maximizing each of these factors (field strength, stability, probe quality, sample concentration, sample volume, and flip angle) will yield significant gains in S/N without the need for additional acquisition time. Thirty-two to sixty-four scans are often considered enough for a normal urine sample on a 600 MHz (or greater field strength) instrument. Dilute urine will require more scans to achieve the desired S/N for quantitative NMR analysis. More scans (or higher fields) are also required to detect and quantify minor components (<10 uM) in urine or very dilute samples. Use of a cryogenically cooled probe can significantly (by a factor of 2–4 times) increase the S/N for the sample.
Receiver Gain (RG)
NMR experiments can be run either on a constant receiver gain setting (optimized to a standard sample) or with an automatically optimized receiver gain for every sample. The former is preferred for pattern recognition analysis of urine NMR data; however, it does not work on samples where concentration variation between samples is substantial. In these cases the auto receiver gain setting may be required for every sample prior to acquisition, or an efficiently low gain would be required.
Shimming
Automatic shimming options in newer NMR spectrometers have made shimming much easier and faster for metabolomic analysis. NMR shims can be optimized by watching the line shape and width at half-height of the reference signal (such as DSS or TSP) or another common low-molecular-weight component. For DSS/TSP, observing the intensity and symmetry of the silicon (29Si) satellite signal is also a good option to optimize the shimming process. The line width of chemical shift standards such as DSS or TSP should be shimmed to between 0.5 and 1.0 Hz. Adjustment of the field and lock position/phase during the shimming process can also improve the quality of the spectra.
Tuning and Matching
Probe tuning and matching (which is equivalent to tuning a radio) are essential to getting high-quality NMR spectra. Many NMR instruments are tuned for salt-free solvents (chloroform or D2O); however, buffered urine samples typically have high salt concentrations. This can lead to poor performance (low S/N, long pulses, poor solvent suppression, etc.) on high-field NMR spectrometers that have not previously been tuned to accommodate high salt samples. These difficulties can become more evident when a user is acquiring data on a cryogenically cooled NMR probe. Ideally, manual probe tuning and matching for a given sample or solvent type (using a representative sample) should be performed prior to spectral acquisition for a large number of solvent-similar samples. Autotune capabilities and match accessories along with salt tolerant NMR flat sample tubes can help mitigate this problem.
7.3. Data Processing
Proper data processing can improve the S/N, resolution, visual appearance, and the integration accuracy of 1D NMR spectra. Several simple data processing techniques may be employed, some of which are automatic while others require some manual effort and skill. Our recommendations are as follows:
Windowing or Apodization
A window function should be applied to the time domain data prior to Fourier transformation to improve the appearance of the spectra and emphasize either S/N or resolution (both are not possible). In a urine 1D spectrum, a free induction decay (FID) can be multiplied by an exponential window function. The artificially increased decay rate of an exponential windowing function (which minimizes later time points in the FID) will reduce the noise and broaden the resonance lines. On the contrary, a window function that enhances later time points in the FID will enhance resolution but at the expense of increasing spectral noise. Line broadening between 0.3 and 1.0 Hz is recommended for quantitative analysis of urine samples. The exact value will depend on the digital resolution of the experiment. Line broadening equal to the digital resolution is generally recommended, as it provides no penalty in spectral separation but does improve general spectral appearance.
Zero Filling
FID data should be zero-filled by a factor of 2 (i.e., twice the number of experimentally collected data points) before Fourier transformation to reduce noise and improve the visual appearance of the spectra. Zero filling does not increase the actual spectral resolution but functions to better interpolate between real data points in the original spectrum, thereby improving line-fitting and peak position determination. While often over emphasized, zero filling is still helpful for many NMR spectroscopists and does not require additional spectrometer time. Extending zero filling past a factor of 2 does little for the spectra, takes up more storage space, and slows software calculations.
Phasing
Phasing is one of the most critical yet one of the more error-prone steps in performing quantitative NMR. Errors in phasing can cause significant errors in absolute and relative peak area measurements.75 Manual phasing is preferred over automatic phasing because there is a greater chance of distortion among lower intensity signals during automatic phase correction. Vertical expansion should be increased as much as possible during manual phasing to make the proper adjustments for smaller signals. Water peaks can be ignored during phasing, and the water signal should be removed (through postprocessing digital filtration) from the spectra to enable further qualitative and quantitative analysis. Experimental errors in the pulse sequence can often manifest as apparent “phase” distortions but cannot be corrected with either frequency-dependent or frequency-independent corrections. Dispersive peak characteristics, especially on very intense peaks, should be carefully examined and noted in case the NMR parameters/setup need to be altered.
Base Line Correction
Proper baseline corrections are necessary for quantitative accuracy of peak integration and curve-fitting methods applied to calculate peak areas. Most baseline corrections are performed semiautomatically through manual identification of baseline regions, followed by a computer-generated spline fit to the baseline regions. Several third-party software packages can also perform automated baseline correction.50
Chemical Shift Referencing
Every spectrum needs to be properly referenced to an internal chemical shift standard before qualitative/quantitative analysis. In many cases, because the precise concentration of the standard is known, the chemical shift standard can also be used to determine the concentration of other compounds by comparing the relative peak areas. Both trimethylsilyl propanoic acid (TSP) and 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) can be used for chemical shift referencing in water (δ 0.0), although DSS is the IUPAC standard. While other peaks may be used in place of an internal standard if they are very insensitive to pH/ionic strength variations, this practice is definitely not recommended for most applications (water has been successfully used as a concentration reference).52 Chemical shift referencing to solvent peaks (i.e., water) should be avoided because its peak position is sensitive to pH, salt, and temperature effects.
Concluding Remarks
Despite the growing number of publications focused on diagnostic metabolite biomarkers appearing in the scientific literature, a surprisingly small number of these reports provide information on quantified metabolite levels, especially in urine. This situation indicates that metabolite identification and quantification in urine is still considered a challenging task. For metabolite biomarkers, accurate identification and quantification is essential for advancing biomarker discoveries to clinical practice. As noted throughout this review, NMR spectroscopy offers a robust route to the identification and quantification of metabolites. In this regard, we believe quantitative NMR-based metabolomics represents a superb (albeit under-used) platform for the discovery, development, and translation of metabolite biomarkers to clinical practice. In this Review, we focused on the use of urine as a biofluid for biomarker discovery and biomarker applications. In particular, we explained how biomarkers should be evaluated or assessed. We also elaborated on several methods for metabolite identification, quantification and normalization by NMR and provided examples of a number of newly discovered urinary metabolite biomarkers. We further discussed a number of the most significant issues or challenges regarding the experimental aspects of quantitative, NMR-based urinary metabolomics. To address these experimental challenges, several consensus recommendations were provided. These included best-practice recommendations regarding: (1) sample collection, (2) sample processing and storage, (3) NMR data acquisition, and (4) NMR instrument setup and NMR data processing. Detailed justifications were provided for each of these recommendations. We believe that if these recommendations are followed, they will help members of the NMR metabolomics community better validate and translate their biomarker discoveries from the lab into clinical practice.
Acknowledgments
We thank Dr. Virginia Unkefer from KAUST for her assistance and helpful remarks. This work was supported by King Abdullah University of Science and Technology (KAUST). Funding for this work from the NIH (R01GM085291) is gratefully acknowledged and European Commission (FP 7) COSMOS Grant EC312941.
The authors declare no competing financial interest.
References
- a Al-Talla Z. A.; Akrawi S. H.; Emwas A. H. M. Solid state NMR and bioequivalence comparison of the pharmacokinetic parameters of two formulations of clindamycin. Int. J. Clin. Pharmacol. Ther. 2011, 49 (7), 469–476. 10.5414/CP201478. [DOI] [PubMed] [Google Scholar]; b Al-Talla Z. A.; Akrawi S. H.; Tolley L. T.; Sioud S. H.; Zaater M. F.; Emwas A. H. Bioequivalence assessment of two formulations of ibuprofen. Drug Des., Dev. Ther. 2011, 5, 427–33. 10.2147/DDDT.S24504. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Li J.; Wang S.; Wang M.; Shi W.; Du X.; Sun C. The toxicity of 3-chloropropane-1,2-dipalmitate in Wistar rats and a metabonomics analysis of rat urine by ultra-performance liquid chromatography-mass spectrometry. Chem.-Biol. Interact. 2013, 206 (2), 337–345. 10.1016/j.cbi.2013.10.004. [DOI] [PubMed] [Google Scholar]; d Nie C. Y.; Han T.; Zhang L.; Li Y.; Liu H.; Xiao S. X.; Li Y.; Kang H.; Liu S. Y. Cross-sectional and dynamic change of serum metabolite profiling for Hepatitis B-related acute-on-chronic liver failure by UPLC/MS. Journal of Viral Hepatitis 2014, 21 (1), 53–63. 10.1111/jvh.12122. [DOI] [PubMed] [Google Scholar]; e Szultka M.; Krzeminski R.; Walczak J.; Jackowski M.; Buszewski B. Pharmacokinetic study of amoxicillin in human plasma by solid-phase microextraction followed by high-performance liquid chromatography-triple quadrupole mass spectrometry. Biomed. Chromatogr. 2014, 28 (2), 255–264. 10.1002/bmc.3014. [DOI] [PubMed] [Google Scholar]; f Lan K.; Zhang Y.; Yang J.; Xu L. Simple quality assessment approach for herbal extracts using high performance liquid chromatography-UV based metabolomics platform. Journal of Chromatography A 2010, 1217 (8), 1414–1418. 10.1016/j.chroma.2009.12.031. [DOI] [PubMed] [Google Scholar]; g Liang X.; Zhang L.; Zhang X.; Dai W.; Li H.; Hu L.; Liu H.; Su J.; Zhang W. Qualitative and quantitative analysis of traditional Chinese medicine Niu Huang Jie Du Pill using ultra performance liquid chromatography coupled with tunable UV detector and rapid resolution liquid chromatography coupled with time-of-flight tandem mass spectrometry. J. Pharm. Biomed. Anal. 2010, 51 (3), 565–571. 10.1016/j.jpba.2009.09.015. [DOI] [PubMed] [Google Scholar]; h Zheng S.; Yu M.; Lu X.; Huo T.; Ge L.; Yang J.; Wu C.; Li F. Urinary metabonomic study on biochemical changes in chronic unpredictable mild stress model of depression. Clin. Chim. Acta 2010, 411 (3–4), 204–209. 10.1016/j.cca.2009.11.003. [DOI] [PubMed] [Google Scholar]
- a Ciborowski M.; Lipska A.; Godzien J.; Ferrarini A.; Korsak J.; Radziwon P.; Tomasiak M.; Barbas C. Combination of LC-MS- and GC-MS-based Metabolomics to Study the Effect of Ozonated Autohemotherapy on Human Blood. J. Proteome Res. 2012, 11 (12), 6231–6241. 10.1021/pr3008946. [DOI] [PubMed] [Google Scholar]; b Liu M.-L.; Zheng P.; Liu Z.; Xu Y.; Mu J.; Guo J.; Huang T.; Meng H.-Q.; Xie P. GC-MS based metabolomics identification of possible novel biomarkers for schizophrenia in peripheral blood mononuclear cells. Mol. BioSyst. 2014, 10 (9), 2398–2406. 10.1039/C4MB00157E. [DOI] [PubMed] [Google Scholar]; c Raji M.; Amad M.; Emwas A. H. Dehydrodimerization of pterostilbene during electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2013, 27 (11), 1260–6. 10.1002/rcm.6571. [DOI] [PubMed] [Google Scholar]
- a Emwas A.-H. M.; Al-Talla Z. A.; Kharbatia N. M. Sample collection and preparation of biofluids and extracts for gas chromatography-mass spectrometry. Methods Mol. Biol. (N. Y., NY, U. S.) 2015, 1277, 75–90. 10.1007/978-1-4939-2377-9_7. [DOI] [PubMed] [Google Scholar]; b Emwas A.-H. M.; Al-Talla Z. A.; Yang Y.; Kharbatia N. M. Gas chromatography-mass spectrometry of biofluids and extracts. Methods Mol. Biol. (N. Y., NY, U. S.) 2015, 1277, 91–112. 10.1007/978-1-4939-2377-9_8. [DOI] [PubMed] [Google Scholar]
- a Guo J.; Zhang M.; Elmore C. S.; Vishwanathan K. An integrated strategy for in vivo metabolite profiling using high-resolution mass spectrometry based data processing techniques. Anal. Chim. Acta 2013, 780, 55–64. 10.1016/j.aca.2013.04.012. [DOI] [PubMed] [Google Scholar]; b Huang Y.; Tian Y.; Li G.; Li Y.; Yin X.; Peng C.; Xu F.; Zhang Z. Discovery of safety biomarkers for realgar in rat urine using UFLC-IT-TOF/MS and H-1 NMR based metabolomics. Anal. Bioanal. Chem. 2013, 405 (14), 4811–4822. 10.1007/s00216-013-6842-0. [DOI] [PubMed] [Google Scholar]; c Vadla N. C.; Davalagar V. D.; Sripadi P. Detection and characterization of N-alkyl diethanolamines and N-2-alkoxyethyl diethanolamines in milk by electrospray ionization mass spectrometry. Metabolomics 2013, 9 (3), 623–630. 10.1007/s11306-012-0492-7. [DOI] [Google Scholar]; d Allard E.; Backstrom D.; Danielsson R.; Sjoberg J. R.; Bergquist J. Comparing Capillary Electrophoresis - Mass Spectrometry Fingerprints of Urine Samples Obtained after Intake of Coffee, Tea, or Water. Anal. Chem. 2008, 80 (23), 8946–8955. 10.1021/ac801012y. [DOI] [PubMed] [Google Scholar]; e Chen Y.-H.; Chen C.-H.; Lin C.-J.; Chen C.-C. Metabonomic Study with a High Performance Liquid Chromatography Coupling to a Triple Quadruple Mass Spectrometer to Identify Biomarkers from Urine of High-fat Fed and Streptozotocin Treated Rats. J. Food Drug Anal. 2009, 17 (1), 28–35. [Google Scholar]; f Cho S.-H.; Choi M. H.; Kwon O. S.; Lee W.-Y.; Chung B. C. Metabolic significance of bisphenol A-induced oxidative stress in rat urine measured by liquid chromatography-mass spectrometry. J. Appl. Toxicol. 2009, 29 (2), 110–117. 10.1002/jat.1387. [DOI] [PubMed] [Google Scholar]
- a Kim J. W.; Ryu S. H.; Kim S.; Lee H. W.; Lim M.-s.; Seong S. J.; Kim S.; Yoon Y.-R.; Kim K.-B. Pattern Recognition Analysis for Hepatotoxicity Induced by Acetaminophen Using Plasma and Urinary H-1 NMR-Based Metabolomics in Humans. Anal. Chem. 2013, 85 (23), 11326–11334. 10.1021/ac402390q. [DOI] [PubMed] [Google Scholar]; b Wang B.; Goodpaster A. M.; Kennedy M. A. Coefficient of variation, signal-to-noise ratio, and effects of normalization in validation of biomarkers from NMR-based metabonomics studies. Chemom. Intell. Lab. Syst. 2013, 128, 9–16. 10.1016/j.chemolab.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; c He C.-C.; Dai Y.-Q.; Hui R.-R.; Hua J.; Chen H.-J.; Luo Q.-Y.; Li J.-X. NMR-based metabonomic approach on the toxicological effects of a Cimicifuga triterpenoid. J. Appl. Toxicol. 2012, 32 (2), 88–97. 10.1002/jat.1633. [DOI] [PubMed] [Google Scholar]; d Calvani R.; Miccheli A.; Capuani G.; Miccheli A. T.; Puccetti C.; Delfini M.; Iaconelli A.; Nanni G.; Mingrone G. Gut microbiome-derived metabolites characterize a peculiar obese urinary metabotype. Int. J. Obes. 2010, 34 (6), 1095–1098. 10.1038/ijo.2010.44. [DOI] [PubMed] [Google Scholar]; e Liu Y.; Huang R.; Liu L.; Peng J.; Xiao B.; Yang J.; Miao Z.; Huang H. Metabonomics study of urine from Sprague-Dawley rats exposed to Huang-yao-zi using H-1 NMR spectroscopy. J. Pharm. Biomed. Anal. 2010, 52 (1), 136–141. 10.1016/j.jpba.2009.12.026. [DOI] [PubMed] [Google Scholar]; f Zhang S.; Nagana Gowda G. A.; Ye T.; Raftery D. Advances in NMR-based biofluid analysis and metabolite profiling. Analyst 2010, 135 (7), 1490–1498. 10.1039/c000091d. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Wishart D. S. Quantitative metabolomics using NMR. TrAC, Trends Anal. Chem. 2008, 27 (3), 228–237. 10.1016/j.trac.2007.12.001. [DOI] [Google Scholar]; h Zhang S.; Nagana Gowda G. A.; Asiago V.; Shanaiah N.; Barbas C.; Raftery D. Correlative and quantitative H-1 NMR-based metabolomics reveals specific metabolic pathway disturbances in diabetic rats. Anal. Biochem. 2008, 383 (1), 76–84. 10.1016/j.ab.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emwas A.-H. M. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol. Biol. (N. Y., NY, U. S.) 2015, 1277, 161–93. 10.1007/978-1-4939-2377-9_13. [DOI] [PubMed] [Google Scholar]
- c Smolinska A.; Blanchet L.; Buydens L. M. C.; Wijmenga S. S. NMR and pattern recognition methods in metabolomics: From data acquisition to biomarker discovery: A review. Anal. Chim. Acta 2012, 750, 82–97. 10.1016/j.aca.2012.05.049. [DOI] [PubMed] [Google Scholar]; d Heather L. C.; Wang X.; West J. A.; Griffin J. L. A practical guide to metabolomic profiling as a discovery tool for human heart disease. J. Mol. Cell. Cardiol. 2013, 55, 2–11. 10.1016/j.yjmcc.2012.12.001. [DOI] [PubMed] [Google Scholar]; e Wei R. Metabolomics and Its Practical Value in Pharmaceutical Industry. Curr. Drug Metab. 2011, 12 (4), 345–358. 10.2174/138920011795202947. [DOI] [PubMed] [Google Scholar]; f Zulyniak M. A.; Mutch D. M. Harnessing Metabolomics for Nutrition Research. Curr. Pharm. Biotechnol. 2011, 12 (7), 1005–1015. 10.2174/138920111795909113. [DOI] [PubMed] [Google Scholar]; g Nageeb A.; Al-Tawashi A.; Emwas A.-H.; Al-Talla Z.; Al-Rifai N. Comparison of Artemisia annua Bioactivities between Traditional Medicine and Chemical Extracts. Curr. Bioact. Compd. 2014, 9 (4), 324–332. 10.2174/157340720904140404151439. [DOI] [PMC free article] [PubMed] [Google Scholar]; a Emwas A.-H. M.; Antakly T.; Saoudi A.-H.; Al-Ghamdi S.; Serai H. Magnetic Resonance Spectroscopy and Imaging in Breast Cancer Prognosis and Diagnosis. Applications of NMR Spectroscopy 2015, 3, 4–35. 10.2174/97816810806281150301. [DOI] [Google Scholar]; b Emwas A.-H. M.; Merzaban J. S.; Serrai H.; Atta-ur-Rahman M. I. C. Theory and Applications of NMR-Based Metabolomics in Human Disease Diagnosis. Applications of NMR Spectroscopy 2015, 1, 93–130. 10.2174/97816080596211150101. [DOI] [Google Scholar]
- Aimetti M.; Cacciatore S.; Graziano A.; Tenori L. Metabonomic analysis of saliva reveals generalized chronic periodontitis signature. Metabolomics 2012, 8 (3), 465–474. 10.1007/s11306-011-0331-2. [DOI] [Google Scholar]
- Wishart D. S.; Lewis M. J.; Morrissey J. A.; Flegel M. D.; Jeroncic K.; Xiong Y.; Cheng D.; Eisner R.; Gautam B.; Tzur D.; Sawhney S.; Bamforth F.; Greiner R.; Li L. The human cerebrospinal fluid metabolome. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2008, 871 (2), 164–173. 10.1016/j.jchromb.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Graca G.; Duarte I. F.; Goodfellow B. J.; Carreira I. M.; Couceiro A. B.; Domingues M. d. R.; Spraul M.; Tseng L.-H.; Gil A. M. Metabolite profiling of human amniotic fluid by hyphenated nuclear magnetic resonance spectroscopy. Anal. Chem. 2008, 80 (15), 6085–6092. 10.1021/ac800907f. [DOI] [PubMed] [Google Scholar]
- Lacitignola L.; Fanizzi F. P.; Francioso E.; Crovace A. H-1 NMR investigation of normal and osteoarthritic synovial fluid in the horse. Veterinary and Comparative Orthopaedics and Traumatology 2008, 21 (1), 85–88. 10.3415/VCOT-06-12-0101. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.; Miniati M.; Monti S.; Tenori L. Phenotyping COPD by H-1 NMR metabolomics of exhaled breath condensate. Metabolomics 2014, 10 (2), 302–311. 10.1007/s11306-013-0572-3. [DOI] [Google Scholar]
- Bertini I.; Hu X.; Luchinat C. Global metabolomics characterization of bacteria: pre-analytical treatments and profiling. Metabolomics 2014, 10 (2), 241–249. 10.1007/s11306-013-0571-4. [DOI] [Google Scholar]
- Cacciatore S.; Hu X.; Viertler C.; Kap M.; Bernhardt G. A.; Mischinger H.-J.; Riegman P.; Zatloukal K.; Luchinat C.; Turano P. Effects of Intra- and Post-Operative Ischemia on the Metabolic Profile of Clinical Liver Tissue Specimens Monitored by NMR. J. Proteome Res. 2013, 12 (12), 5723–5729. 10.1021/pr400702d. [DOI] [PubMed] [Google Scholar]
- a Bouatra S.; Aziat F.; Mandal R.; Guo A. C.; Wilson M. R.; Knox C.; Bjorndahl T. C.; Krishnamurthy R.; Saleem F.; Liu P.; Dame Z. T.; Poelzer J.; Huynh J.; Yallou F. S.; Psychogios N.; Dong E.; Bogumil R.; Roehring C.; Wishart D. S. The Human Urine Metabolome. PLoS One 2013, 8 (9), e73076. 10.1371/journal.pone.0073076. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Emwas A.-H. M.; Salek R. M.; Griffin J. L.; Merzaban J. NMR-based metabolomics in human disease diagnosis: applications, limitations, and recommendations. Metabolomics 2013, 9 (5), 1048–1072. 10.1007/s11306-013-0524-y. [DOI] [Google Scholar]
- a Assfalg M.; Bertini I.; Colangiuli D.; Luchinat C.; Schaefer H.; Schuetz B.; Spraul M. Evidence of different metabolic phenotypes in humans. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (5), 1420–1424. 10.1073/pnas.0705685105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bernini P.; Bertini I.; Luchinat C.; Nepi S.; Saccenti E.; Schaefer H.; Schuetz B.; Spraul M.; Tenori L. Individual Human Phenotypes in Metabolic Space and Time. J. Proteome Res. 2009, 8 (9), 4264–4271. 10.1021/pr900344m. [DOI] [PubMed] [Google Scholar]
- Travagli V.; Zanardi I.; Bernini P.; Nepi S.; Tenori L.; Bocci V. Effects of Ozone Blood Treatment on the Metabolite Profile of Human Blood. Int. J. Toxicol. 2010, 29 (2), 165–174. 10.1177/1091581809360069. [DOI] [PubMed] [Google Scholar]
- Ryan D.; Robards K.; Prenzler P. D.; Kendall M. Recent and potential developments in the analysis of urine: A review. Anal. Chim. Acta 2011, 684 (1–2), 17–29. 10.1016/j.aca.2010.10.035. [DOI] [PubMed] [Google Scholar]
- Pinto J.; Domingues M. R. M.; Galhano E.; Pita C.; Almeida M. d. C.; Carreira I. M.; Gil A. M. Human plasma stability during handling and storage: impact on NMR metabolomics. Analyst 2014, 139 (5), 1168–1177. 10.1039/c3an02188b. [DOI] [PubMed] [Google Scholar]
- a Abriata L. A. Utilization of NMR spectroscopy to study biological fluids and metabolic processes: Two introductory activities. Concepts Magn. Reson., Part A 2012, 40A (4), 171–178. 10.1002/cmr.a.21235. [DOI] [Google Scholar]; b Beckonert O.; Keun H. C.; Ebbels T. M. D.; Bundy J. G.; Holmes E.; Lindon J. C.; Nicholson J. K. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2 (11), 2692–2703. 10.1038/nprot.2007.376. [DOI] [PubMed] [Google Scholar]; c Beltran A.; Suarez M.; Rodriguez M. A.; Vinaixa M.; Samino S.; Arola L.; Correig X.; Yanes O. Assessment of Compatibility between Extraction Methods for NMR- and LC/MS-Based Metabolomics. Anal. Chem. 2012, 84 (14), 5838–5844. 10.1021/ac3005567. [DOI] [PubMed] [Google Scholar]; d Bernini P.; Bertini I.; Luchinat C.; Nincheri P.; Staderini S.; Turano P. Standard operating procedures for pre-analytical handling of blood and urine for metabolomic studies and biobanks. J. Biomol. NMR 2011, 49 (3–4), 231–243. 10.1007/s10858-011-9489-1. [DOI] [PubMed] [Google Scholar]; e Bingol K.; Brueschweiler R. Multidimensional Approaches to NMR-Based Metabolomics. Anal. Chem. 2014, 86 (1), 47–57. 10.1021/ac403520j. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Foxall P. J. D.; Spraul M.; Farrant R. D.; Lindon L. C.; Neild G. H.; Nicholson J. K. 750 MHz 1H-NMR spectroscopy of human blood plasma. J. Pharm. Biomed. Anal. 1993, 11 (4–5), 267–276. 10.1016/0731-7085(93)80017-U. [DOI] [PubMed] [Google Scholar]; g Garde A. H.; Hansen A. M.; Kristiansen J.; Knudsen L. E. Comparison of uncertainties related to standardization of urine samples with volume and creatinine concentration. Ann. Occup. Hyg. 2004, 48 (2), 171–179. 10.1093/annhyg/meh019. [DOI] [PubMed] [Google Scholar]
- Fiehn O.; Robertson D.; Griffin J.; van der Werf M.; Nikolau B.; Morrison N.; Sumner L. W.; Goodacre R.; Hardy N. W.; Taylor C.; Fostel J.; Kristal B.; Kaddurah-Daouk R.; Mendes P.; van Ommen B.; Lindon J. C.; Sansone S.-A. The metabolomics standards initiative (MSI). Metabolomics 2007, 3 (3), 175–178. 10.1007/s11306-007-0070-6. [DOI] [Google Scholar]
- Emwas A.-H.; Luchinat C.; Turano P.; Tenori L.; Roy R.; Salek R. M.; Ryan D.; Merzaban J. S.; Kaddurah-Daouk R.; Zeri A. C.; Gowda G. A. N.; Raftery D.; Wang Y.; Brennan L.; Wishart D. S. Standardizing the experimental conditions for using urine in NMR-based metabolomic studies with a particular focus on diagnostic studies: a review. Metabolomics 2015, 11 (4), 872–894. 10.1007/s11306-014-0746-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J.; Broadhurst D. I.; Wilson M.; Wishart D. S. Translational biomarker discovery in clinical metabolomics: an introductory tutorial. Metabolomics 2013, 9 (2), 280–299. 10.1007/s11306-012-0482-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrod A. E. The croonian lectures on inborn errors of metabolism. Lancet 1908, 172 (4427), 1–7. 10.1016/S0140-6736(01)78482-6. [DOI] [Google Scholar]
- Marshall S. L.; Edidin D. V.; Arena V. C.; Becker D. J.; Bunker C. H.; Gishoma C.; Gishoma F.; LaPorte R. E.; Kaberuka V.; Ogle G.; Sibomana L.; Orchard T. J. Glucose control in Rwandan youth with type 1 diabetes following establishment of systematic, HbA1c based, care and education. Diabetes Res. Clin. Pract. 2015, 107 (1), 113–122. 10.1016/j.diabres.2014.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decrease in Urinary Creatinine Excretion in Early Stage Chronic Kidney Disease. PLoS One 2014, 9 (11), e111949. 10.1371/journal.pone.0111949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberring J.; Ciborowski P. Biomarker discovery and clinical proteomics. TrAC, Trends Anal. Chem. 2010, 29 (2), 128–140. 10.1016/j.trac.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagana Gowda G. A.; Gowda Y. N.; Raftery D. Expanding the limits of human blood metabolite quantitation using NMR spectroscopy. Anal. Chem. 2015, 87 (1), 706–15. 10.1021/ac503651e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Saude E. J.; Adamko D.; Rowe B. H.; Marrie T.; Sykes B. D. Variation of metabolites in normal human urine. Metabolomics 2007, 3 (4), 439–451. 10.1007/s11306-007-0091-1. [DOI] [Google Scholar]; b Schnackenberg L. K.; Sun J.; Espandiari P.; Holland R. D.; Hanig J.; Beger R. D. Metabonomics evaluations of age-related changes in the urinary compositions of male Sprague Dawley rats and effects of data normalization methods on statistical and quantitative analysis. BMC Bioinf. 2007, 8, S3. 10.1186/1471-2105-8-S7-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hao J.; Liebeke M.; Astle W.; De Iorio M.; Bundy J. G.; Ebbels T. M. D. Bayesian deconvolution and quantification of metabolites in complex 1D NMR spectra using BATMAN. Nat. Protoc. 2014, 9 (6), 1416–1427. 10.1038/nprot.2014.090. [DOI] [PubMed] [Google Scholar]; b Hao J.; Astle W.; De Iorio M.; Ebbels T. M. D. BATMAN-an R package for the automated quantification of metabolites from nuclear magnetic resonance spectra using a Bayesian model. Bioinformatics 2012, 28 (15), 2088–2090. 10.1093/bioinformatics/bts308. [DOI] [PubMed] [Google Scholar]
- Ravanbakhsh S.; Liu P.; Bjordahl T. C.; Mandal R.; Grant J. R.; Wilson M.; Eisner R.; Sinelnikov I.; Hu X.; Luchinat C.; Greiner R.; Wishart D. S. Accurate, Fully-Automated NMR Spectral Profiling for Metabolomics. PLoS One 2015, 10 (5), e0124219. 10.1371/journal.pone.0124219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Abuhijleh A. L.; Abu Ali H.; Emwas A.-H. Synthesis, spectral and structural characterization of dinuclear rhodium (II) complexes of the anticonvulsant drug valproate with theophylline and caffeine. J. Organomet. Chem. 2009, 694 (22), 3590–3596. 10.1016/j.jorganchem.2009.07.031. [DOI] [Google Scholar]; b Blindauer C. A.; Emwas A. H.; Holy A.; Dvorakova H.; Sletten E.; Sigel H. Complex formation of the antiviral 9- 2-(phosphonomethoxy)ethyl adenine (PMEA) and of its N1, N3, and N7 deaza derivatives with copper(II) in aqueous solution. Chem. - Eur. J. 1997, 3 (9), 1526–1536. 10.1002/chem.19970030922. [DOI] [Google Scholar]; c Di Gangi I. M.; Vrhovsek U.; Pazienza V.; Mattivi F. Analytical metabolomics-based approaches to pancreatic cancer. TrAC, Trends Anal. Chem. 2014, 55, 94–116. 10.1016/j.trac.2013.12.006. [DOI] [Google Scholar]
- a Mattar S. M.; Emwas A. H.; Calhoun L. A. Spectroscopic studies of the intermediates in the conversion of 1,4,11,12-tetrahydro-9,10-anthraquinone to 9,10-anthraquinone by reaction with oxygen under basic conditions. J. Phys. Chem. A 2004, 108 (52), 11545–11553. 10.1021/jp040280v. [DOI] [Google Scholar]; b Lam C.-W.; Law C.-Y.; Sze K.-H.; To K. K.-W. Quantitative metabolomics of urine for rapid etiological diagnosis of urinary tract infection: Evaluation of a microbial-mammalian co-metabolite as a diagnostic biomarker. Clin. Chim. Acta 2015, 438, 24–28. 10.1016/j.cca.2014.07.038. [DOI] [PubMed] [Google Scholar]; c Simmler C.; Napolitano J. G.; McAlpine J. B.; Chen S.-N.; Pauli G. F. Universal quantitative NMR analysis of complex natural samples. Curr. Opin. Biotechnol. 2014, 25, 51–59. 10.1016/j.copbio.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraudeau P. Quantitative 2D liquid- state NMR. Magn. Reson. Chem. 2014, 52 (6), 259–272. 10.1002/mrc.4068. [DOI] [PubMed] [Google Scholar]
- Hyberts S. G.; Takeuchi K.; Wagner G. Poisson-Gap Sampling and Forward Maximum Entropy Reconstruction for Enhancing the Resolution and Sensitivity of Protein NMR Data. J. Am. Chem. Soc. 2010, 132 (7), 2145–2147. 10.1021/ja908004w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R. K.; Sinha N. Fast and Accurate Quantitative Metabolic Profiling of Body Fluids by Nonlinear Sampling of H-1-C-13 Two-Dimensional Nuclear Magnetic Resonance Spectroscopy. Anal. Chem. 2012, 84 (22), 10005–10011. 10.1021/ac302457s. [DOI] [PubMed] [Google Scholar]
- a Pathan M.; Akoka S.; Tea I.; Charrier B.; Giraudeau P. ″Multi-scan single shot″ quantitative 2D NMR: a valuable alternative to fast conventional quantitative 2D NMR. Analyst 2011, 136 (15), 3157–3163. 10.1039/c1an15278e. [DOI] [PubMed] [Google Scholar]; b Ludwig C.; Marin-Montesinos I.; Saunders M. G.; Emwas A. H.; Pikramenou Z.; Hammond S. P.; Gunther U. L. Application of ex situ dynamic nuclear polarization in studying small molecules. Phys. Chem. Chem. Phys. 2010, 12 (22), 5868–71. 10.1039/c002700f. [DOI] [PubMed] [Google Scholar]
- Da Silva L.; Godejohann M.; Martin F.-P. J.; Collino S.; Burkle A.; Moreno-Villanueva M.; Bernhardt J.; Toussaint O.; Grubeck-Loebenstein B.; Gonos E. S.; Sikora E.; Grune T.; Breusing N.; Franceschi C.; Hervonen A.; Spraul M.; Moco S. High-resolution quantitative metabolome analysis of urine by automated flow injection NMR. Anal. Chem. 2013, 85 (12), 5801–9. 10.1021/ac4004776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolenko S.; McKay R.; Blondeel M. J. L.; Lewis M. J.; Chang D.; George B.; Aucoin M. G. Understanding the variability of compound quantification from targeted profiling metabolomics of 1D-1H-NMR spectra in synthetic mixtures and urine with additional insights on choice of pulse sequences and robotic sampling. Metabolomics 2013, 9, 887–903. 10.1007/s11306-013-0503-3. [DOI] [Google Scholar]
- Bharti S. K.; Roy R. Quantitative H-1 NMR spectroscopy. TrAC, Trends Anal. Chem. 2012, 35, 5–26. 10.1016/j.trac.2012.02.007. [DOI] [Google Scholar]
- a Hoffmann M. M.; Sobstyl H. S.; Seedhouse S. J. T-1 relaxation measurement with solvent suppression. Magn. Reson. Chem. 2008, 46 (7), 660–666. 10.1002/mrc.2228. [DOI] [PubMed] [Google Scholar]; b Lauridsen M.; Maher A. D.; Keun H.; Lindon J. C.; Nicholson J. K.; Nyberg N. T.; Hansen S. H.; Cornett C.; Jaroszewski J. W. Application of the FLIPSY pulse sequence for increased sensitivity in H-1 NMR-based metabolic profiling studies. Anal. Chem. 2008, 80 (9), 3365–3371. 10.1021/ac702563u. [DOI] [PubMed] [Google Scholar]
- McKay R. T. How the 1D-NOESY Suppresses Solvent Signal in Metabonomics NMR Spectroscopy: An Examination of the Pulse Sequence Components and Evolution. Concepts Magn. Reson., Part A 2011, 38A (5), 197–220. 10.1002/cmr.a.20223. [DOI] [Google Scholar]
- a Nguyen B. D.; Meng X.; Donovan K. J.; Shaka A. J. SOGGY: Solvent-optimized double gradient spectroscopy for water suppression. A comparison with some existing techniques. J. Magn. Reson. 2007, 184 (2), 263–274. 10.1016/j.jmr.2006.10.014. [DOI] [PubMed] [Google Scholar]; b Roumestand C.; Canet D. Extending the excitation sculpting concept for selective excitation. J. Magn. Reson. 2000, 147 (2), 331–339. 10.1006/jmre.2000.2206. [DOI] [PubMed] [Google Scholar]; c Jerschow A. Unwanted signal leakage in excitation sculpting with single axis gradients. J. Magn. Reson. 1999, 137 (1), 206–214. 10.1006/jmre.1998.1652. [DOI] [PubMed] [Google Scholar]
- Piotto M.; Saudek V.; Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 1992, 2 (6), 661–665. 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- Simpson A. J.; Brown S. A. Purge NMR: Effective and easy solvent suppression. J. Magn. Reson. 2005, 175 (2), 340–346. 10.1016/j.jmr.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Adams R. W.; Holroyd C. M.; Aguilar J. A.; Nilsson M.; Morris G. A. ″Perfecting″ WATERGATE: clean proton NMR spectra from aqueous solution. Chem. Commun. 2013, 49 (4), 358–360. 10.1039/C2CC37579F. [DOI] [PubMed] [Google Scholar]
- Price W. S. Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational diffusion: Part II. Experimental aspects. Concepts Magn. Reson. 1998, 10 (4), 197–237. . [DOI] [Google Scholar]
- Mulder F. A. A.; Spronk C.; Slijper M.; Kaptein R.; Boelens R. Improved HSQC experiments for the observation of exchange broadened signals. J. Biomol. NMR 1996, 8 (2), 223–228. 10.1007/BF00211169. [DOI] [PubMed] [Google Scholar]
- Markley J. L.; Bax A.; Arata Y.; Hilbers C. W.; Kaptein R.; Sykes B. D.; Wright P. E.; Wuthrich K. Recommendations for the presentation of NMR structures of proteins and nucleic acids - IUPAC-IUBMB-IUPAB Inter-Union Task Group on the Standardization of Data Bases of Protein and Nucleic Acid Structures Determined by NMR Spectroscopy. J. Biomol. NMR 1998, 12 (1), 1–23. 10.1023/A:1008290618449. [DOI] [PubMed] [Google Scholar]
- Mercier P.; Lewis M. J.; Chang D.; Baker D.; Wishart D. S. Towards automatic metabolomic profiling of high-resolution one-dimensional proton NMR spectra. J. Biomol. NMR 2011, 49 (3–4), 307–323. 10.1007/s10858-011-9480-x. [DOI] [PubMed] [Google Scholar]
- Holzgrabe U.; Deubner R.; Schollmayer C.; Waibel B. Quantitative NMR spectroscopy - Applications in drug analysis. J. Pharm. Biomed. Anal. 2005, 38 (5), 806–812. 10.1016/j.jpba.2005.01.050. [DOI] [PubMed] [Google Scholar]
- Mo H.; Raftery D. Solvent Signal as an NMR Concentration Reference. Anal. Chem. 2008, 80 (24), 9835–9839. 10.1021/ac801938j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouatra S.; Aziat F.; Mandal R.; Guo A. C.; Wilson M. R.; Knox C.; Bjorndahl T. C.; Krishnamurthy R.; Saleem F.; Liu P.; Dame Z. T.; Poelzer J.; Huynh J.; Yallou F. S.; Psychogios N.; Dong E.; Bogumil R.; Roehring C.; Wishart D. S. PLoS One 2013, 8, e73076. 10.1371/journal.pone.0073076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fonville J. M.; Maher A. D.; Coen M.; Holmes E.; Lindon J. C.; Nicholson J. K. Evaluation of Full-Resolution J-Resolved H-1 NMR Projections of Biofluids for Metabonomics Information Retrieval and Biomarker Identification. Anal. Chem. 2010, 82 (5), 1811–1821. 10.1021/ac902443k. [DOI] [PubMed] [Google Scholar]; b Cloarec O.; Dumas M. E.; Craig A.; Barton R. H.; Trygg J.; Hudson J.; Blancher C.; Gauguier D.; Lindon J. C.; Holmes E.; Nicholson J. Statistical total correlation spectroscopy: An exploratory approach for latent biomarker identification from metabolic H-1 NMR data sets. Anal. Chem. 2005, 77 (5), 1282–1289. 10.1021/ac048630x. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Zhang S.; Ragg S.; Raftery D.; Vitek O. Identification and quantification of metabolites in H-1 NMR spectra by Bayesian model selection. Bioinformatics 2011, 27 (12), 1637–1644. 10.1093/bioinformatics/btr118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bollard M. E.; Stanley E. G.; Lindon J. C.; Nicholson J. K.; Holmes E. NMR-based metabonomic approaches for evaluating physiological influences on biofluid composition. NMR Biomed. 2005, 18 (3), 143–162. 10.1002/nbm.935. [DOI] [PubMed] [Google Scholar]; b Brindle J. T.; Antti H.; Holmes E.; Tranter G.; Nicholson J. K.; Bethell H. W. L.; Clarke S.; Schofield P. M.; McKilligin E.; Mosedale D. E.; Grainger D. J. Rapid and noninvasive diagnosis of the presence and severity of coronary heart disease using 1 H-NMR-based metabonomics. Nat. Med. 2002, 8 (12), 1439–1445. 10.1038/nm1202-802. [DOI] [PubMed] [Google Scholar]; c Waters N. J.; Waterfield C. J.; Farrant R. D.; Holmes E.; Nicholson J. K. Metabonomic deconvolution of embedded toxicity: application to thioacetamide hepato-and nephrotoxicity. Chem. Res. Toxicol. 2005, 18 (4), 639–654. 10.1021/tx049869b. [DOI] [PubMed] [Google Scholar]; d Ringeissen S.; Connor S. C.; Brown H. R.; Sweatman B. C.; Hodson M. P.; Kenny S. P.; Haworth R. I.; Mcgill P.; Price M. A.; Aylott M. C.; et al. Potential urinary and plasma biomarkers of peroxisome proliferation in the rat: identification of N-methylnicotinamide and N-methyl-4-pyridone-3-carboxamide by 1H nuclear magnetic resonance and high performance liquid chromatography. Biomarkers 2003, 8 (3-4), 240–271. 10.1080/1354750031000149124. [DOI] [PubMed] [Google Scholar]
- Dieterle F.; Ross A.; Schlotterbeck G.; Senn H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 2006, 78 (13), 4281–4290. 10.1021/ac051632c. [DOI] [PubMed] [Google Scholar]
- Torgrip R. J. O.; Åberg K.; Alm E.; Schuppe-Koistinen I.; Lindberg J. A note on normalization of biofluid 1D 1 H-NMR data. Metabolomics 2008, 4 (2), 114–121. 10.1007/s11306-007-0102-2. [DOI] [Google Scholar]
- Dunn W. B.; Broadhurst D. I.; Atherton H. J.; Goodacre R.; Griffin J. L. Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 2011, 40 (1), 387–426. 10.1039/B906712B. [DOI] [PubMed] [Google Scholar]
- a Kamal M. A.; Priyamvada S.; Anbazhagan A. N.; Jabir N. R.; Tabrez S.; Greig N. H. Linking Alzheimer’s disease and type 2 diabetes mellitus via aberrant insulin signaling and inflammation. CNS Neurol. Disord.: Drug Targets 2014, 13 (2), 338–46. 10.2174/18715273113126660137. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dawiskiba T.; Deja S.; Mulak A.; Zabek A.; Jawien E.; Pawelka D.; Banasik M.; Mastalerz-Migas A.; Balcerzak W.; Kaliszewski K.; Skora J.; Barc P.; Korta K.; Pormanczuk K.; Szyber P.; Litarski A.; Mlynarz P. Serum and urine metabolomic fingerprinting in diagnostics of inflammatory bowel diseases. World Journal of Gastroenterology 2014, 20 (1), 163–174. 10.3748/wjg.v20.i1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Deja S.; Barg E.; Mlynarz P.; Basiak A.; Willak-Janc E. H-1 NMR-based metabolomics studies of urine reveal differences between type 1 diabetic patients with high and low HbAc1 values. J. Pharm. Biomed. Anal. 2013, 83, 43–48. 10.1016/j.jpba.2013.04.017. [DOI] [PubMed] [Google Scholar]; d Vazquez-Fresno R.; Llorach R.; Marinic J.; Tulipani S.; Garcia-Aloy M.; Espinosa-Martos I.; Jimenez E.; Rodriguez J. M.; Andres-Lacueva C. Urinary metabolomic fingerprinting after consumption of a probiotic strain in women with mastitis. Pharmacol. Res. 2014, 87, 160–165. 10.1016/j.phrs.2014.05.010. [DOI] [PubMed] [Google Scholar]; e Nagrath D.; Caneba C.; Karedath T.; Bellance N. Metabolomics for mitochondrial and cancer studies. Biochim. Biophys. Acta, Bioenerg. 2011, 1807 (6), 650–663. 10.1016/j.bbabio.2011.03.006. [DOI] [PubMed] [Google Scholar]; f Wang X.; Zhang A.; Han Y.; Wang P.; Sun H.; Song G.; Dong T.; Yuan Y.; Yuan X.; Zhang M.; Xie N.; Zhang H.; Dong H.; Dong W. Urine Metabolomics Analysis for Biomarker Discovery and Detection of Jaundice Syndrome in Patients With Liver Disease. Mol. Cell. Proteomics 2012, 11 (8), 370–380. 10.1074/mcp.M111.016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusczek E. R.; Paulo J. A.; Saltzman J. R.; Kadiyala V.; Banks P. A.; Beilman G.; Conwell D. L. Urinary 1H-NMR metabolomics can distinguish pancreatitis patients from healthy controls. Pancreatology 2013, 14 (2), 161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S.; Roy R.; Singh S.; Kumar P.; Dalela D.; Sankhwar S. N.; Goel A.; Sonkar A. A. Taurine - a possible fingerprint biomarker in non-muscle invasive bladder cancer: A pilot study by 1H NMR spectroscopy. Cancer biomarkers: section A of Disease markers 2010, 6 (1), 11–20. 10.3233/CBM-2009-0115. [DOI] [PubMed] [Google Scholar]
- Ladep N. G.; Dona A. C.; Lewis M. R.; Crossey M. M.; Lemoine M.; Okeke E.; Shimakawa Y.; Duguru M.; Njai H. F.; Fye H. K.; Taal M.; Chetwood J.; Kasstan B.; Khan S. A.; Garside D. A.; Wijeyesekera A.; Thillainayagam A. V.; Banwat E.; Thursz M. R.; Nicholson J. K.; Njie R.; Holmes E.; Taylor-Robinson S. D. Discovery and validation of urinary metabotypes for the diagnosis of hepatocellular carcinoma (HCC) in West Africans. Hepatology 2014, 60, 1291. 10.1002/hep.27264. [DOI] [PubMed] [Google Scholar]
- Dessi A.; Atzori L.; Noto A.; Visser G. H.; Gazzolo D.; Zanardo V.; Barberini L.; Puddu M.; Ottonello G.; Atzei A.; De Magistris A.; Lussu M.; Murgia F.; Fanos V. Metabolomics in newborns with intrauterine growth retardation (IUGR): urine reveals markers of metabolic syndrome. J. Matern.-Fetal Neonat. Med. 2011, 24 (Suppl 2), 35–9. 10.3109/14767058.2011.605868. [DOI] [PubMed] [Google Scholar]
- Moltu S. J.; Sachse D.; Blakstad E. W.; Strommen K.; Nakstad B.; Almaas A. N.; Westerberg A. C.; Ronnestad A.; Braekke K.; Veierod M. B.; Iversen P. O.; Rise F.; Berg J. P.; Drevon C. A. Urinary metabolite profiles in premature infants show early postnatal metabolic adaptation and maturation. Nutrients 2014, 6 (5), 1913–30. 10.3390/nu6051913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Saude E. J.; Slupsky C. M.; Sykes B. D. Optimization of NMR analysis of biological fluids for quantitative accuracy. Metabolomics 2006, 2 (3), 113–123. 10.1007/s11306-006-0023-5. [DOI] [Google Scholar]; b Rasmussen L. G.; Savorani F.; Larsen T. M.; Dragsted L. O.; Astrup A.; Engelsen S. B. Standardization of factors that influence human urine metabolomics. Metabolomics 2011, 7 (1), 71–83. 10.1007/s11306-010-0234-7. [DOI] [Google Scholar]; c Pelantova H.; Buganova M.; Anyz J.; Zelezna B.; Maletinska L.; Novak D.; Haluzik M.; Kuzma M. Strategy for NMR metabolomic analysis of urine in mouse models of obesity- from sample collection to interpretation of acquired data. J. Pharm. Biomed. Anal. 2015, 115, 225–235. 10.1016/j.jpba.2015.06.036. [DOI] [PubMed] [Google Scholar]
- Walsh M. C.; Brennan L.; Malthouse J. P. G.; Roche H. M.; Gibney M. J. Effect of acute dietary standardization on the urinary, plasma, and salivary metabolomic profiles of healthy humans. Am. J. Clin. Nutr. 2006, 84 (3), 531–539. [DOI] [PubMed] [Google Scholar]
- Lombardini J.; Schaffer S.; Azuma J.; Odle J.; Glass E.; Czarnecki-Maulden G.; Baker D., Urinary Excretion of Taurine as a Function of Taurine Intake: Potential for Estimating Taurine Bioavailability in the Adult Cat. In Taurine; Springer: 1992; Vol. 315, pp 55–62. [DOI] [PubMed] [Google Scholar]
- Beckonert O.; Keun H. C.; Ebbels T. M. D.; Bundy J.; Holmes E.; Lindon J. C.; Nicholson J. K. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2 (11), 2692–2703. 10.1038/nprot.2007.376. [DOI] [PubMed] [Google Scholar]
- a Yao S.; Keizer D. W. Nutation frequency modulation on NMR signal of nuclear spins in chemical exchange with solvent water under the BEST conditions. Magn. Reson. Chem. 2014, 52 (4), 190–194. 10.1002/mrc.4045. [DOI] [PubMed] [Google Scholar]; b Wu P. S. C.; Otting G. Rapid pulse length determination in high-resolution NMR. J. Magn. Reson. 2005, 176 (1), 115–119. 10.1016/j.jmr.2005.05.018. [DOI] [PubMed] [Google Scholar]
- Grzesiek S.; Bax A. The importance of not saturating water in protein NMR. Application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc. 1993, 115 (26), 12593–12594. 10.1021/ja00079a052. [DOI] [Google Scholar]
- Prost E.; Sizun P.; Piotto M.; Nuzillard J. M. A simple scheme for the design of solvent-suppression pulses. J. Magn. Reson. 2002, 159 (1), 76–81. 10.1016/S1090-7807(02)00003-4. [DOI] [PubMed] [Google Scholar]
- a Rist M.; Muhle-Goll C.; Görling B.; Bub A.; Heissler S.; Watzl B.; Luy B. Influence of Freezing and Storage Procedure on Human Urine Samples in NMR-Based Metabolomics. Metabolites 2013, 3 (2), 243–258. 10.3390/metabo3020243. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Beckonert O.; Keun H. C.; Ebbels T. M. D.; Bundy J.; Holmes E.; Lindon J. C.; Nicholson J. K. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2 (11), 2692–2703. 10.1038/nprot.2007.376. [DOI] [PubMed] [Google Scholar]
- Bharti S.; Sinha N.; Joshi B.; Mandal S.; Roy R.; Khetrapal C. Improved quantification from 1H-NMR spectra using reduced repetition times. Metabolomics 2008, 4 (4), 367–376. 10.1007/s11306-008-0130-6. [DOI] [Google Scholar]
- Wishart D. S. Quantitative metabolomics using NMR. TrAC, Trends Anal. Chem. 2008, 27 (3), 228–237. 10.1016/j.trac.2007.12.001. [DOI] [Google Scholar]
- Davis V. W.; Schiller D. E.; Eurich D.; Bathe O. F.; Sawyer M. B. Pancreatic ductal adenocarcinoma is associated with a distinct urinary metabolomic signature. Ann. Surg Oncol 2013, 20 (Suppl 3), S415–23. 10.1245/s10434-012-2686-7. [DOI] [PubMed] [Google Scholar]
- Davis V. W.; Schiller D. E.; Eurich D.; Sawyer M. B. Urinary metabolomic signature of esophageal cancer and Barrett’s esophagus. World journal of surgical oncology 2012, 10, 271. 10.1186/1477-7819-10-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz S. O.; Pinto J.; Graca G.; Duarte I. F.; Barros A. S.; Galhano E.; Pita C.; Almeida M. d. C.; Goodfellow B. J.; Carreira I. M.; Gil A. M. Metabolic biomarkers of prenatal disorders: an exploratory NMR metabonomics study of second trimester maternal urine and blood plasma. J. Proteome Res. 2011, 10 (8), 3732–42. 10.1021/pr200352m. [DOI] [PubMed] [Google Scholar]