

Abstract
Human serum transferrin (sTf) is a protein that mediates the transport of iron from blood to cells. Assisted by the synergistic anion carbonate sTf transports Fe(III) by binding the metal ion in a closed conformation. Previous studies suggest sTf’s role as a potential transporter of other metals like titanium. Ti is a widely used metal in colorants, foods, and implants. A substantial amount of Ti is leached into blood from these implants. However, the fate of the leached Ti and its transport into the cells is not known. Understanding Ti interaction with sTf assumes a greater significance with our ever increasing exposure to Ti in the form of implants. Based on in vitro studies it was speculated that transferrin can bind Ti(IV) assisted by a synergistic anion. However, the role and identity of the synergistic anion(s) and the conformational state in which sTf binds Ti(IV) are not known. Here we have solved the first X-ray crystal structure of a Ti(IV)-bound sTf. We find that sTf binds Ti(IV) in an open conformation with both carbonate and citrate as synergistic anions at the metal binding sites, an unprecedented role for citrate. Studies with cell lines suggest that Ti(IV)-sTf is transported into cells and that sTf and citrate regulate the metal’s blood speciation and attenuate its cytotoxic property. Our results provide the first glimpse into the citrate-transferrin synergism in the regulation of Ti(IV) bioactivity and offers insight into the future design of Ti(IV)-based anticancer drugs.
Graphical Abstract

INTRODUCTION
Titanium is a ubiquitous metal that predominantly exists as Ti(IV) in our oxidizing environment. As Ti(IV), it is highly susceptible to hydrolysis and exists at very low concentrations (femtomolar) in water due to precipitation as titanium dioxide (TiO2), a white solid.1 TiO2 is the major form of Ti(IV), commonly used as the pigment of white paint. Ti(IV) can easily enter into the human body via foods and liquids or as TiO2 particles in toothpastes or the paint dust that we breathe. These dust particles are the reason that Ti is found at its highest levels in the lungs.2 There is no known natural function for Ti in people. Nonetheless, it displays excellent potential for multiple uses in the medical field. Following initial promise of two Ti(IV) compounds (titanocene dichloride and budotitane) as anticancer agents, several Ti(IV) compounds are in development for this application to overcome the limitations of the platinum(II) drugs,3 which are one of the major drugs in the market but suffer from a narrow spectrum of effect, many side effects, and a rapidly acquired resistance by cancer cells.
Ti has been extremely valuable in its use in prosthetics.4–6 Ti’s property of osseointegration, the ability to integrate and be structurally accepted by bone without the requirement of soft tissue connection, demonstrates that it can play structure and templating roles in biology.4 This property in addition to its general biocompatibility, high corrosion resistance, low specific gravity, and weak magnetism are the reasons why Ti has been widely applied in the development of prosthetics.5 On average, hundreds of thousands of prosthetics are implanted in people every year.4 With increasing life expectancy our dependence on Ti for prosthetic use is bound to increase. Current evidence suggests that the body’s interaction with Ti-containing implants extends beyond a simple passive, biocompatible one. The Ti reacts with biological fluids and leaches into the circulatory system leading to Ti(IV) levels in the blood elevating to high nanomolar levels,7 nearly 50 times greater than people with no implants. The leached, soluble Ti(IV) is found to be almost 100% serum transferrin (sTf) bound.7 The long term effect of this pool of Ti(IV)-bound sTf is not clear.
A textbook presentation of sTf highlights its function as a primary agent that binds circulating plasma iron in a bioavailable Fe(III) form for delivery into mammalian cells. It is typically 30% Fe(III) saturated 8. A lesser studied property of sTf is its function as a noniron metal transporter. This function has been proposed to occur in targeted efforts to deliver certain metals as therapeutics (chromium, bismuth, gallium, indium, ruthenium, titanium, vanadium) and during environmental increases in metals resulting in cellular toxicity (aluminum, lanthanides, and actinides).9,10 There is some evidence for endogenous transport of manganese(III) by sTf.9,11 The coordination of sTf to all metals is generally thought to be identical. STf is a bilobal, 80 kDa glycoprotein with its N-and C-lobes divided into two subdomains (N1 and N2, and C1 and C2) that form two Fe(III) binding sites. Fe(III) is coordinated by an aspartic acid from the N1- or C1-subdomain, a tyrosine in the hinge near the N2- or C2-subdomain, another tyrosine in the N2- or C2-subdomain, and a histidine in the hinge near the N1- or C1-subdomain (Fig. 1A). The coordination is completed by the synergistic anion carbonate, which coordinates in a bidentate form, or a carbonate-like molecule with certain size limitations.12 The carbonate is stably bound to an arginine residue (Arg124 and Arg456, at the N- and C-site, respectively) and is a requirement for the specific binding of Fe(III) to sTf.13 Fe(III) binding changes the protein’s lobes from an open to a closed conformation,14 significantly stabilizing the protein.15 Although the coordination of all metal ions is presumed to be identical to that of Fe(III), it is reasoned that the difference in metal ion affinity to sTf stems from a difference in the Lewis acidity of the ions, with stronger Lewis acidic metals having a higher affinity to the Lewis basic residues of the binding site.16 Vanadium, which is extremely hydrolysis prone, is known to differ in its coordination to sTf by providing its own synergistic anion in the form of an oxo moiety in place of carbonate.17
Figure 1.

Metal coordinating ligands in Fe (III) bound and Ti(IV) serotransferrin crystal structures, (a) Metal binding site in the C-lobe of serotransferrin (3QYT) showing the iron binding residues (yellow) and the synergistic anion carbonate (blue). (b) N-site structures showing other anions (SO42− and NTA3−) maintaining metal bound sTf in a semi-open conformation (3QYT and 4H0W). (c) The C-site of Ti-bound serotransferrin. In contrast to A, in Ti-sTf in place of the His-585 and Asp-392 a bidentate synergistic citrate anion provides the necessary contacts to fulfill the coordination of Ti(IV). The Ti(IV) is in an open conformation.
A recent crystallographic study challenges the consensus on sTf metal binding by presenting metal ions bound in a semi-open conformation.18 These conformations were achieved with the anions sulfate and nitrilotriacetate (NTA), binding the metal ions Fe(III) and Bi(III), respectively, at positions in the N-site that prevented the coordination of two or three of the protein residues and thus restricted lobe closure (Fig. 1B). In these structures, carbonate was bound to the metal ions at its typical Arg binding site. These structures were likely the product of crystallization conditions and may not reflect biological relevance.
How non-carbonate, physiological anions work in synergism with sTf to transport noniron metals is an unexplored area. This is an extremely important question especially as it relates to the biological fate of Ti(IV)-bound sTf formed from Ti-implant leaching. Small molecules must be involved in both the chelation of Ti(IV) during or following the leaching process or else the metal would completely precipitate, and in the delivery of the metal ion to sTf. The bioactive anion citrate is believed to contribute to the solubilization and binding of Ti released from implants.19,20 As of yet no association has been made between citrate chelation of Ti(IV) and Ti(IV) delivery to sTf. This is likely because citrate’s role in metal transport has not been well characterized in humans11,21,22 although it has been more extensively studied in bacteria.23,24 In the context of Fe-sTf transport, citrate is unable to substitute for carbonate as a synergistic anion.12 Rather, at high concentrations, it can compete with sTf for binding of the metal.12,25 This is quite distinct from the synergistic anion role that citrate can play for the ferric binding protein A (FbpA), which has a metal binding site similar to sTf.24
In this work, we investigate the molecular mechanism that the body uses to bind and transport Ti(IV) released in vivo by focusing on the interaction of citrate and sTf in this process. An x-ray structure of a Ti(IV)-bound sTf complex illuminates the unprecedented role of citrate as a synergistic anion for Ti(IV) coordination facilitating an open conformation. 13C NMR spectroscopy and elemental and molecular quantitative studies further reveal the molecular details of the Ti(IV) coordination. Calorimetric and cytotoxicity studies inform on the stability of the Ti(IV)-sTf complex and its impact on the blood speciation, transport, and activity of the metal. In exploring the synergistic regulation of Ti(IV) by sTf and citrate, this work provides new insight into the improved design of Ti(IV) anticancer agents. More broadly, it also elucidates on the structural requirements for the sTf anion-dependent cellular uptake of nonferric metals in the body.
RESULTS AND DISCUSSION
Citrate participates in Ti(IV) transport by sTf
To understand the role that citrate plays in the leaching of Ti from Ti-containing implants,19,20 a model for the blood speciation (pH 7.4) of Ti(IV) citrate was proposed (Supplementary Fig. 1). Blood is the first tissue that the implant comes into contact and it constitutes a major component of the peri-implant space particularly because of the blood requirement for bone healing.26 This Ti(IV) citrate blood speciation was modeled using Ti(IV) citrate formation constants27 and the blood levels of citrate (100 μM)28 and Ti(IV) (maximum) following release from implants (0.28 μM).7 The results indicate that citrate helps the Ti to leach by forming very stable complexes with the metal, of which 89% of the speciation consists of the Ti(IV) tricitrate complex ([Ti(citrate)3]8−). In this complex, the three citrates coordinate as bidentate ligands via the α-hydroxyl and α-carboxyl oxygens (Fig. S1). The Ti(IV) tricitrate complex is expected to be short-lived in blood because of the lability of the complex and the strong affinity that sTf has for the metal.29 The complex has previously been shown to rapidly deliver Ti(IV) to physiological amounts of sTf (30–60 μM) at its two metal binding sites, producing a ligand to metal charge transfer (LMCT) absorbance at 321 nm with an extinction coefficient of ~10,000 M−1cm−1 based on Ti(IV) concentration.29,30 This delivery is nearly stoichiometric at citrate concentrations as high as 10 mM.29 Saturation of the Ti(IV) coordination sites by the three citrates appears to be a prerequisite for yielding this Ti(IV)2-sTf complex because it prevents metal hydrolysis.27 This is true of low micromolar levels of the extremely aqueous unstable Ti(IV) source, titanocene dichloride, which would precipitate as Ti(IV) oxide species at pH 7.431 but becomes converted to Ti(IV) citrate species in the presence of 100 μM citrate. When sTf is also present in these solutions, the Ti(IV)2-sTf complex forms with the expected characteristic spectroscopic signals.
Citrate is a synergistic anion for sTf binding of Ti(IV) and facilitates an open conformation
The role of citrate as more than a delivery agent of Ti(IV) to sTf was examined by 13C isotopic labeling of the citrate (Supplementary Fig. 1).32 The α-carboxyl carbon was labeled ((1′-13C)Citrate) because it experiences a significant downfield shift of Δδ 8.6 when the Ti(IV) coordinates 33 STf was Ti(IV) saturated with an excess of [Ti((1′-13C)Citrate)3]8− in a pH 7.4 buffer containing 2 mM 13C-citrate and 5 mM 13C-bicarbonate. The 13C NMR spectrum (Fig. 2) of the dialyzed Ti(IV)-bound protein (~1 mM) was compared with the spectra for (1′-13C)Citrate and 13C-bicarbonate in the presence and absence of apo-sTf and [Ti((1′-13C)Citrate)3]8− (Fig. S2). The chemical shifts for (1′-13C) Citrate and 13C-bicarbonate were identical in the controls. Three 13C chemical shifts appear in the Ti(IV)-bound protein sample. These are the shift at 187.52 ppm attributed to Ti(IV)-coordinated citrate and at 165.23 and 165.50 ppm attributed to coordinated carbonate30,34,35 at the metal binding site. Complementary experiments were performed to quantify the stoichiometry of Ti(IV) and citrate bound to sTf. The binding stoichiometry was determined to be sTf:Ti:citrate 1:1.91±0.07:1.68±0.04, indicating that for every bound Ti(IV) there is one molecule of citrate. These results suggest an unprecedented synergistic anion role for citrate in the sTf metal binding. An extensive previous study on synergistic anions and binding of Fe(III) showed that citrate could not substitute for carbonate as a synergistic anion.12 Instead the ligand competes with sTf for Fe(III) coordination.12,25 In the Ti(IV) situation citrate displays an entirely different behavior and rather than substitute for carbonate, it serves as an additional synergistic anion for sTf binding of Ti(IV). In this model for Ti(IV) coordination to sTf (Ti2-sTf-(CO3)2(Citrate)2), Ti(IV) binds to both tyrosines as indicated by the presence of the LMCT absorbance, and as revealed by the 13C NMR data, to both carbonate and citrate (Fig. 1C). The citrate prevents coordination of the Asp and His moieties and thus would facilitate a metal-bound open conformation. This unusual coordination can be rationalised because Ti(IV) is a strong Lewis acid and would more favorably bind to the stronger Lewis base citrate oxygens of the α-alkoxyl and α-carboxyl groups. That the Fe(III) coordination is distinct can also be rationalized on similar grounds because Fe(III) is a weaker Lewis acid relative to Ti(IV) and would have a higher stability coordinated to the His and Asp.16 To confirm the unusual model for Ti(IV) coordination to sTf, crystallization studies were performed.
Figure 2.
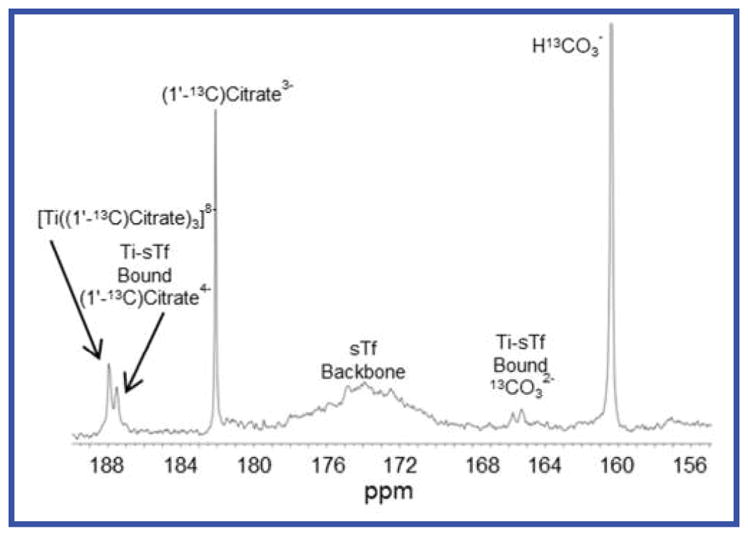
Citrate and carbonate serve as synergistic anions for sTf binding of Ti(IV). The proton-decoupled 13C NMR spectra of 1 mM transferrin in the presence of two equivalents of Ti(IV) and 13C isotopically labeled bicarbonate (5 mM) and citrate (2 mM). The 165.2 and 165.5 ppm signals are due to bound carbonate at the metal binding site and the 187.5 ppm signal is due to the bound citrate.
The crystal structure of Ti(IV)-bound sTf was solved to a resolution of 2.68 Å by the molecular replacement method using the human apotransferrin structure (Protein Data Bank (PDB) accession code 2HAU) as the search model (Table 1).25 The protein crystallized with two molecules in an asymmetric unit (PDB 5DYH) having an open conformation similar to the apo-sTf structure (Fig. 3A). Ti(IV) is present only at the C-lobe binding site (Fig. S3) coordinated in a mode identical to the proposed solution structure (Fig. 1C) showing both the carbonate and citrate serving as bidentate synergistic anions as confirmed by an omit map (Fig. S4). The 100 mM citrate concentration used for crystallization appears to be responsible for this partially Ti(IV)-bound species. A similar metal displacement effect by high citrate levels in the crystallization process yielded the apo-sTf structures (PDB 2HAU and 2HAV) from samples that were initially Fe(III) saturated.25 Nine metal-free citrate molecules bind to the protein dimer (Fig. S5), a finding similar to the apo-sTf structures.
Table 1.
Data collection and refinement statistics (Molecular replacement).
| Ti(IV) human serum transferrin* | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 88.28, 102.02, 197.90 |
| α, β, γ (°) | 90.00, 90.00, 90.00 |
| Resolution (Å) | 49.39-2.68 (2.78-2.68)† |
| Rsym | 0.09 (0.78) |
| I/σI | 33.0 (2.2) |
| Completeness (%) | 96 (84) |
| Redundancy | 2.0 (2.0) |
| Refinement | |
| Resolution (Å) | 49 - 2.68 |
| No. reflections | 93,205 |
| Rwork/Rfree | 0.20/0.24 |
| No. atoms | |
| Protein | 10375 |
| Ligand | 153 |
| Water | 0 |
| B-factors | |
| Protein | 97.95 |
| Ligand | 147.44 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.670 |
A single crystal was used for structure determination.
Highest resolution shell is shown in parenthesis.
Figure 3.

STf binds metals in both open and closed conformations mediated by anions. (a) Overlapping ribbon diagram of the apo-sTf (PDB Code: 2-HAU, red) and Ti-sTf (blue) crystal structures. The two structures closely resemble in their conformation (RSMD = 1.217). (b) C (left) and N (right) binding sites of the Ti-sTf protein crystals. (c) CO32− polar contacts as seen in the C-site of the sTf-Fe(III) bound form (3QYT).
The Ti(IV)-bound C-site reveals citrate serving two distinct functions (Fig. 3B). The citrate behaves as a synergistic anion that facilitates a metal-bound open conformation. A semi-open conformation has been previously observed with nitrilotriacetate and sulfate (Fig. 1B),18 however, these structures may not represent a physiological state but likely are a product of crystallization conditions. They were obtained via the use of the soaking-in method of concentrated solutions of Fe(sulfate)2− and Bi(NTA) applied to crystals of Fe2-sTf, in which both Fe(III) ions were in the typical closed conformations.18 No insight into the possibility of metal uptake by an open conformation for metal-bound sTf was offered in this work. However, the Ti(IV)-bound open conformation exists at endogenous citrate levels and pH, and suggests the physiological relevance of non-carbonate anions participating in sTf metal transport.
In the crystal structure of the Ti(IV)-bound sTf a second citrate (metal-free) binds near the metal binding site and operates as a non-synergistic anion36,37 by inducing structural changes that weaken the metal affinity to the protein. This second citrate binds at high mM levels and forms a hydrogen bond with the carbonate-binding Arg residue (R456). This interaction partly inhibits R456 interaction with the carbonate and is likely why the carbonate and metal dissociate from the N-site. In the Fe(III)-bound closed conformation structure, carbonate has multiple polar contacts with R456 in addition to other amino acids (Fig. 3C). At the N-site, one citrate is found directly in the metal binding site and another near the R456 but it does not engage in hydrogen bonding, an effect that may be due to reorganization following loss of the carbonate and metal ions. The Ti(IV)-bound sTf structure obtained is a transitory structure between a metal-bound and metal-free form. A physiologically relevant structure would be expected to have both Ti(IV) in an open conformation because Ti(IV) titration into both sites leads to comparable increase in the UV Vis LMCT absorbance.
The open conformation of Ti(IV)-bound sTf impacts protein stability and cellular metal uptake and release
The new model for Ti(IV) sTf coordination (Ti2-sTf-(CO3)2(Citrate)2) requires a reconsideration of several aspects of its thermodynamics. STf binding of Fe(III) results in a major stabilization of the protein due to its closed conformation. The melting temperature (Tm) of the C- and N-sites increase by 30 and 19°C, respectively, as monitored by differential scanning calorimetry (DSC).15 Binding of Ti(IV) in an open conformation should produce lesser stabilization and DSC was used to measure this effect (Fig. S6). The addition of one equivalent of Ti(IV) exhibits a preferential binding of Ti(IV) to the C-site, similar to Fe(III), and an increase of its Tm by 18.5°C At saturation of the protein, the Tm of the N-site increases by 5°C and the thermogram resembles a one transition profile. While the Ti2-sTf complex is less thermostable than Fe2-sTf, the binding of Ti(IV) does significantly stabilize sTf on account of locking the N2 and C2 subdomains into a fixed position due to coordination to the two tyrosines. In the Fe(III) version of the protein (PDB code 2HAU), metal binding to these two subdomains contributes most to sTf stabilization.25
The original determination of the Ti(IV) affinity to sTf29 rested on the assumption that Ti(IV) coordinates exactly like Fe(III) does. Our crystal structure, like the DSC data, suggests that Ti(IV) has a higher affinity for the C-site than it does for the N-site. In vivo studies with the blood of people with Ti implants have shown that Ti(IV) appears exclusively bound to the N-site.38 In the absence of conclusive Ti(IV) preferential site studies, the affinity constants were recalculated as site nonspecific constants using the previous data29 (Table S1). The affinity constants at pH 7.4, log KSite 1 = 35.8 and log KSite 2 = 34.7, are extremely high. Even in the presence of 100 mM citrate, 45 μM sTf remains approximately 60% Ti(IV) saturated.
Comparing the relative affinities of Ti(IV) and Fe(III) to sTf is difficult because of their different coordination modes, nonetheless some key differences in their interaction with sTf are important in the context of their delivery into cells. Transferrin uses an endocytotic process for intracellular delivery of metals through interaction with its receptor (TfR). The iron binding structural reorganization of sTf is believed to be important for sTf recognition by the TfR at the cell membrane to initiate endocytosis.39 However, a recent mass spectrometric experiment reveals that the apoprotein is very capable of forming a stable complex with the TfR at neutral pH and indicates that a metal induced structural reorganization is not a requirement.40 Some have argued that only a closed conformation of both lobes will facilitate metal-bound sTf to transport the metal into cells. This was the argument proposed for the physiological uptake of plutonium(IV) via the sTf route.10 The authors argue that the only way Pu(IV) can enter into cells is by being bound in a particular mixed-metal formulation where Pu(IV) is present at the C-site and Fe(III) is present at the N-site because in this formulation both metals are in a closed-conformation. Interestingly, the authors never take into consideration the role of synergistic anions in the composition of Pu(IV)-bound sTf The lack of this information makes it difficult to elucidate the physical reality of the closed-conformation requirement. However, Ti2-sTf, despite its open conformation, has been shown to have a high affinity to the two protein binding sites of TfR1 (KD1 = 6.3 nM and KD2 = 410 nM).41 A Ti(IV) uptake experiment was performed on A549 cells, known to overexpress TfR1, to determine if Ti2-sTf could deliver the metal into cells. An uptake of 3.3±0.6 femtogram Ti/cell was determined in the soluble fraction of cell lysates, which was found to be statistically significant versus apo-sTf, Fe2-sTf, and media only controls (Fig. 4a). This finding complements a positron emission tomography study performed on EMT-6 tumor-bearing BALB/c mice that showed 45 Ti(IV)-sTf uptake into the tumor cells following Ti-citrate administration.42 These results demonstrate that TfR is able to recognize an open-conformation of metal-bound sTf
Figure 4.

Transferrin delivers Ti(IV) into cells and controls its cytotoxicity with the aid of citrate. (a) A549 cells exhibit an elevated Ti(IV) content when treated with Ti2-sTf versus treatment with apo-sTf, Fe2-sTf, and media alone (control) (N=5). (b) The viability of the cell lines A549 and MRC when treated with [TiOHBED]− (0.1 mM) alone versus with [TiOHBED]− and citrate (0.01–1 mM), apo-sTf (0.03 mM), or citrate (1 mM) and apo-sTf (0.03 mM) combined. The results demonstrate that the combination of apo-sTf and citrate attenuate the cytotoxicity of [TiOHBED]−. [Fe(Citrate)25−] = 0.1 mM. Student’s t-test, **, p-value <0.01,*, p-value <0.05.
One aspect of Ti(IV) delivery into cells that is not well understood is its release from the endosome. Within the endosome, the pH decreases to 5.5 and metal affinity to sTf can dramatically change. At this pH, 100 μM citrate has been reported to deplete sTf entirely of Fe(III).43 This finding was confirmed in a side by side dialysis of Fe2-sTf and Ti2-sTf against pH 5.5 buffered solutions containing 100 μM citrate. The Fe(III) was completely removed but the Ti(IV) remained entirely bound. In the case of Fe(III), the appropriate priming by the receptor is required so that chelation of Fe(III) and/or reduction of the metal to Fe(II) results in iron release from sTf and ultimate transport out of the endosome and into the cytosol for trafficking and storage.36,44 The reduction of Ti(IV) as a release mechanism from sTf within the endosome is not feasible.45 The higher affinity to sTf that Ti(IV) possesses versus Fe(III) at pH 5.5 suggests that a powerful chelator will have to remove Ti(IV). Sadler et al. proposed that such a ligand could be ATP34 although no in vivo studies have been performed to confirm a chelation release mechanism.
Citrate and sTf work in synergism to regulate Ti(IV) cytotoxicity
A more prevalent pool of Ti(IV)-bound sTf in people with Ti implants suggests a greater uptake of Ti(IV) into cells. A study was conducted that determined the Ti(IV) released from implants must reach concentrations greater than 100 μM to exhibit any significant cytotoxicity,46 an amount that largely exceeds the actual levels released. More importantly, Ti2-sTf-(CO3)2(Citrate)2 exhibits no cytotoxicity even at 100 μM.47 STf may be responsible for maintaining nontoxic Ti(IV) speciation in blood, with citrate critical to this speciation. This finding has important implications on the design of Ti(IV) anticancer therapeutics.
The two lead Ti(IV) compounds (titanocene dichloride and budotitane) that advanced to clinical trials demonstrated poor efficacy in patients presumably because of their hydrolytic instability. Nonetheless, much effort has been devoted to the development of different families of Ti(IV) compounds as anticancer agents. For these newer compounds the identity of the ligands is crucial to their activities.48–50 An anticancer Ti(IV) complex was synthesized with the chelator N,N′-di(o-hydroxybenzyl)-ethylenediamine-N,N′-diacetic acid (HBED), to exploit its high affinity for Fe(III).50 The [TiO(HBED)]− complex is cytotoxic against A549 (cancerous) and MRC5 (noncancerous) human lung cells and is believed to operate by releasing Ti(IV) into cells due to its higher affinity for Fe(III), thus depleting the cells of Fe(III) and facilitating Ti(IV) to reach its intracellular target(s). The complex is quite stable in solution and has been shown to be unable to deliver Ti(IV) directly to sTf, however this study was conducted in the absence of citrate.47 A citrate concentration dependence study was performed in the reaction between 90 μM [TiO(HBED)]− and 45 μM sTf. No metal exchange occurred in the absence of citrate but 26±1.7% Ti(IV) saturation occurred at 100 μM citrate and 71±11% saturation occurred at 10 mM concentration. These results suggest that physiologic citrate can effectively compete with micromolar levels of HBED for binding of Ti(IV) to facilitate delivery to sTf.
The impact of this competition on the cytotoxicity of [TiO(HBED)]− against A549 and MRC5 cells was examined. MTT assays were performed in DMEM media supplemented with 10% fetal bovine serum. FBS contains 22.5 to 27.5 μM sTf and is 55 to 92% Fe(III) saturated.51 A citric acid assay measured 509±76 μM citrate in FBS. This amounts to significant levels of citrate and Fe(III)-bound sTf supplied by FBS in the MTT assay. Supplementation of citrate up to 1 mM into the media slightly improved the viability of MRC5 cells treated with 100 μM [TiO(HBED)]− but had no effect on the A549 cells (Fig. 4b). Addition of 30 μM apo-sTf substantially improved the viability of both cells. For the MRC5 cells, the improvement was even greater than the addition of 100 μM Fe(citrate)25−, which is proposed to counter the depleting of intracellular Fe(III) by HBED.50 The combination of 30 μM sTf and 1 mM citrate had the greatest impact on improving the viabilities of both cell lines from 6–12% to nearly 50% viable. The effect of supplemented citrate alone is probably low because the cell lines might employ the FBS provided sTf to take advantage of Fe(III)-bound to it. When citrate and apo-sTf are supplemented together, the two work together to dissociate [TiO(HBED)]− and capture the Ti(IV). This finding sheds new light as to why the two lead Ti(IV) compounds may have failed in clinical trials. Valentine et al. previously suggested that the formulation for administering titanocene dichloride into patients transforms the compound into a different Ti(IV) complex with decreased cytotoxicity.52 While the suggestion is plausible, it is more likely that following administration of these compounds into patients, they quickly dissociated converting into the Ti2-sTf-(CO3)2(Citrate)2 complex, which we have previously demonstrated to not show any cytotoxicity at micromolar concentrations.47 Our own work demonstrating the rapid sTf binding of Ti(IV) from titanocene dichloride in the presence of physiological levels of citrate and carbonate supports this theory. Future efforts in the design of Ti(IV) anticancer agents must take into consideration the citrate and sTf content in the cell media when assessing the cytotoxicity of these agents.
CONCLUSIONS
In summary, while Ti leaching from implants is a concern in terms of the material stability in a biological environment, surprisingly the actual released Ti(IV) pool does not concentrate to toxic levels. Our structural, binding, thermal denaturation, and cellular studies suggest that citrate controls the toxicity of Ti(IV) upon release by transporting it to sTf, remaining bound as a synergistic anion in an open protein conformation, and providing a highly stabilizing coordination for the metal. This interaction may be part of a molecular mechanism already in effect in vivo for all sources of soluble Ti(IV) in people, in which citrate and sTf work in synergism to transform Ti(IV) into a nontoxic form that is able to use the Fe(III) transport pathway to enter into cells without inhibiting intracellular iron dependent pathways (Fig. 5). This work demonstrates the importance of evaluating the influence of physiological anions on transferrin trafficking of nonferric metal ions and also highlights the need to evaluate their contribution for the effective design of nonferric metal therapeutics.
Figure 5.

The proposed mechanism of sTf mediated cellular delivery of Ti from Ti-implants and its LMW compounds as regulated by citrate. Citrate binds Ti(IV) released from implants or from LMW compounds that enter the bloodstream and then delivers the Ti(IV) to sTf. Ti(IV) bound sTf is recognized by the transferrin receptor (TfR), which transports the metal into the cell via endocytosis. Ti(IV) saturation of sTf may not be a requirement. A chelator (L, unknown) then removes Ti(IV) from the protein complex and the metal is released into the cytoplasm by a transporter (unknown).
Supplementary Material
Acknowledgments
We would like to thank Prof. Alan Saghatelian, Yahaira M. Cruz, and Aleannette López for their valuable input to this manuscript. A.D.T., S.S., Y.D., and SAL-R. are supported by the NTH SC1 (5SC1CA190504-02) provided by the NIGMS and NCI. A.D.T. is also supported by funding from the Puerto Rico Science, Technology, and Research Trust (Agreement No. 2013-000019), the University of Puerto Rico Score Stabilization Grant, and the Department of Chemistry at UPR RP. N.N. is supported by the Department of Biological Sciences at Purdue University and by the National Institute of Allergy and Infectious Diseases (1K22AI113078-01). M.S. is supported by the NSF IFN graduate fellowship (EPS-01002410).We thank the staff at the Advanced Photon Source at Argonne National Laboratory (ID22 SER-CAT beamline).
Footnotes
Notes
The authors declare no competing financial interest.
Detailed experimental section, supplementary figures, and table (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Knauss KG, Dibley MJ, Bourcier WL, Shaw HF. Appl Geochem. 2001;16:1115. [Google Scholar]
- 2.Shi HB, Magaye R, Castranova V, Zhao JS. Part Fibre Toxicol. 2013;10:15. doi: 10.1186/1743-8977-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buettner KM, Valentine AM. Chem Rev. 2012;112:1863. doi: 10.1021/cr1002886. [DOI] [PubMed] [Google Scholar]
- 4.Sansone V, Pagani D, Melato M. Clin Cases Miner Bone Metab. 2013;10:34. doi: 10.11138/ccmbm/2013.10.1.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oshida Y. Biomaterials Science: An Introduction to Materials in Medicine. 3. Elsevier Inc; 2013. [Google Scholar]
- 6.Barry NPE, Sadler PJ. Chem Commun. 2013;49:5106. doi: 10.1039/c3cc41143e. [DOI] [PubMed] [Google Scholar]
- 7.Nuevo-Ordonez Y, Montes-Bayon M, Blanco-Gonzalez E, Paz-Aparicio J, Raimundez JD, Tejerina JM, Pena MA, Sanz-Medel A. Anal Bioanal Chem. 2011;401:2747. doi: 10.1007/s00216-011-5232-8. [DOI] [PubMed] [Google Scholar]
- 8.Williams J, Moreton K. Biochem J. 1980;185:483. doi: 10.1042/bj1850483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vincent JB, Love S. Biochim Biophys Acta-Gen Subj. 2012;1820:362. doi: 10.1016/j.bbagen.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Jensen MP, Gorman-Lewis D, Aryal B, Paunesku T, Vogt S, Rickert PG, Seifert S, Lai B, Woloschak GE, Soderholm L. Nat Chem Biol. 2011;7:560. doi: 10.1038/nchembio.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crossgrove JS, Allen DD, Bukaveckas BL, Rhineheimer SS, Yokel RA. Neurotoxicology. 2003;24:3. doi: 10.1016/s0161-813x(02)00089-x. [DOI] [PubMed] [Google Scholar]
- 12.Schlabach MR, Bates GW. J Biol Chem. 1975;250:2182. [PubMed] [Google Scholar]
- 13.Bates GW, Schlabach MR. J Biol Chem. 1975;250:2177. [PubMed] [Google Scholar]
- 14.Noinaj N, Easley NC, Oke M, Mizuno N, Gumbart J, Boura E, Steere AN, Zak O, Aisen P, Tajkhorshid E, Evans RW, Gorringe AR, Mason AB, Steven AC, Buchanan SK. Nature. 2012;483:53. doi: 10.1038/nature10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin LN, Mason AB, Woodworth RC, Brandts JF. Biochemistry. 1994;33:1881. doi: 10.1021/bi00173a035. [DOI] [PubMed] [Google Scholar]
- 16.Li HY, Sadler PJ, Sun HZ. Eur J Biochem. 1996;242:387. doi: 10.1111/j.1432-1033.1996.0387r.x. [DOI] [PubMed] [Google Scholar]
- 17.Harris WR, Carrano CJ. J Inorg Biochem. 1984;22:201. doi: 10.1016/0162-0134(84)80029-x. [DOI] [PubMed] [Google Scholar]
- 18.Yang N, Zhang HM, Wang MJ, Hao Q, Sun HZ. Sci Rep. 2012;2:999. doi: 10.1038/srep00999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruneel N, Helsen JA. J Biomed Mater Res. 1988;22:203. doi: 10.1002/jbm.820220305. [DOI] [PubMed] [Google Scholar]
- 20.Silwood CJL, Grootveld M. Biochem Biophys Res Commun. 2005;330:784. doi: 10.1016/j.bbrc.2005.03.047. [DOI] [PubMed] [Google Scholar]
- 21.Sturrock A, Alexander J, Lamb J, Craven CM, Kaplan J. J Biol Chem. 1990;265:3139. [PubMed] [Google Scholar]
- 22.Kaplan J, Jordan I, Sturrock A. J Biol Chem. 1991;266:2997. [PubMed] [Google Scholar]
- 23.Lensbouer JJ, Doyle RP. Crit Rev Biochem Mol Biol. 2010;45:453. doi: 10.3109/10409238.2010.504701. [DOI] [PubMed] [Google Scholar]
- 24.Weaver KD, Gabricevic M, Anderson DS, Adhikari P, Mietzner TA, Crumbliss AL. Biochemistry. 2010;49:6021. doi: 10.1021/bi902231c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wally J, Halbrooks PJ, Vonrhein C, Rould MA, Everse SJ, Mason AB, Buchanan SK. J Biol Chem. 2006;281:24934. doi: 10.1074/jbc.M604592200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuzyk PRT, Schemitsch EH. Indian J Orthop. 2011;45:108. doi: 10.4103/0019-5413.77129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collins JM, Uppal R, Incarvito CD, Valentine AM. Inorg Chem. 2005;44:3431. doi: 10.1021/ic048158y. [DOI] [PubMed] [Google Scholar]
- 28.Tomisek AJ, Winkler EM, Natelson S. Clinical chemistry. 1975;21:730. [PubMed] [Google Scholar]
- 29.Tinoco AD, Valentine AM. J Am Chem Soc. 2005;127:11218. doi: 10.1021/ja052768v. [DOI] [PubMed] [Google Scholar]
- 30.Tinoco AD, Incarvito CD, Valentine AM. J Am Chem Soc. 2007;129:3444. doi: 10.1021/ja068149j. [DOI] [PubMed] [Google Scholar]
- 31.Toney JH, Marks TJ. J Am Chem Soc. 1985;107:947. [Google Scholar]
- 32.Winkel C, Buitenhuis EG, Lugtenburg J. Recl Trav Chim Pays-Bas-J Roy Neth Chem Soc. 1989;108:51. [Google Scholar]
- 33.Deng YF, Zhou ZH, Wan HL. Inorg Chem. 2004;43:6266. doi: 10.1021/ic0496018. [DOI] [PubMed] [Google Scholar]
- 34.Guo ML, Sun HZ, McArdle HJ, Gambling L, Sadler PJ. Biochemistry. 2000;39:10023. doi: 10.1021/bi000798z. [DOI] [PubMed] [Google Scholar]
- 35.Messori L, Orioli P, Banholzer V, Pais I, Zatta P. FEBS Lett. 1999;442:157. doi: 10.1016/s0014-5793(98)01651-2. [DOI] [PubMed] [Google Scholar]
- 36.Steere AN, Byrne SL, Chasteen ND, Mason AB. Biochim Biophys Acta-Gen Subj. 2012;1820:326. doi: 10.1016/j.bbagen.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marques HM, Watson DL, Egan TJ. Inorg Chem. 1991;30:3758. [Google Scholar]
- 38.Nuevo-Ordonez Y, Montes-Bayon M, Gonzalez EB, Sanz-Medel A. Metallomics. 2011;3:1297. doi: 10.1039/c1mt00109d. [DOI] [PubMed] [Google Scholar]
- 39.Cheng Y, Zak O, Alsen P, Harrison SC, Walz T. Cell. 2004;116:565. doi: 10.1016/s0092-8674(04)00130-8. [DOI] [PubMed] [Google Scholar]
- 40.Leverence R, Mason AB, Kaltashov IA. Proc Natl Acad Sci U S A. 2010;107:8123. doi: 10.1073/pnas.0914898107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tinoco AD, Eames EV, Valentine AM. J Am Chem Soc. 2008;130:2262. doi: 10.1021/ja076364+. [DOI] [PubMed] [Google Scholar]
- 42.Vavere AL, Welch MJ. J Nucl Med. 2005;46:683. [PubMed] [Google Scholar]
- 43.Gumerov DR, Kaltashov IA. Anal Chem. 2001;73:2565. doi: 10.1021/ac0015164. [DOI] [PubMed] [Google Scholar]
- 44.Dhungana S, Taboy CH, Zak O, Larvie M, Crumbliss AL, Aisen P. Biochemistry. 2004;43:205. doi: 10.1021/bi0353631. [DOI] [PubMed] [Google Scholar]
- 45.Siburt CJP, Lin EM, Brandt SJ, Tinoco AD, Valentine AM, Crumbliss AL. J Inorg Biochem. 2010;104:1006. doi: 10.1016/j.jinorgbio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 46.Soto-Alvaredo J, Blanco E, Bettmer J, Hevia D, Sainz RM, Chaves CL, Sanchez C, Llopis J, Sanz-Medel A, Montes-Bayon M. Metallomics. 2014;6:1702. doi: 10.1039/c4mt00133h. [DOI] [PubMed] [Google Scholar]
- 47.Tinoco AD, Thomas HR, Incarvito CD, Saghatelian A, Valentine AM. Proc Natl Acad Sci U S A. 2012;109:5016. doi: 10.1073/pnas.1119303109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schur J, Manna CM, Deally A, Koster RW, Tacke M, Tshuva EY, Ott I. Chem Commun. 2013;49:4785. doi: 10.1039/c3cc38604j. [DOI] [PubMed] [Google Scholar]
- 49.Immel TA, Grutzke M, Spate AK, Groth U, Ohlschlager P, Huhn T. Chem Commun. 2012;48:5790. doi: 10.1039/c2cc31624b. [DOI] [PubMed] [Google Scholar]
- 50.Parks TB, Cruz YM, Tinoco AD. Inorg Chem. 2014;53:1743. doi: 10.1021/ic4028749. [DOI] [PubMed] [Google Scholar]
- 51.Kakuta K, Orino K, Yamamoto S, Watanabe K. Comp Biochem Physiol A-Physiol. 1997;118:165. doi: 10.1016/s0300-9629(96)00403-3. [DOI] [PubMed] [Google Scholar]
- 52.Buettner KM, Snoeberger RC, Batista VS, Valentine AM. Dalton Trans. 2011;40:9580. doi: 10.1039/c1dt10805k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.