

Abstract
The nuclear receptors REV-ERB (consisting of REV-ERBα and REV-ERBβ) and retinoic acid receptor-related orphan receptors (RORs; consisting of RORα, RORβ and RORγ) are involved in many physiological processes, including regulation of metabolism, development and immunity as well as the circadian rhythm. The recent characterization of endogenous ligands for these former orphan nuclear receptors has stimulated the development of synthetic ligands and opened up the possibility of targeting these receptors to treat several diseases, including diabetes, atherosclerosis, autoimmunity and cancer. This Review focuses on the latest developments in ROR and REV-ERB pharmacology indicating that these nuclear receptors are druggable targets and that ligands targeting these receptors may be useful in the treatment of several disorders.
Nuclear receptors are generally classified as ligand-regulated transcription factors, as many members of the nuclear receptor superfamily serve as receptors for physiological ligands, including steroid hormones, lipids and fatty acids. The nuclear receptor superfamily is one of the primary classes of therapeutic drug targets for human disease. Among the drugs that target nuclear receptors are the anti-inflammatory glucocorticoids, steroidal contraceptives and hormone replacement therapies, as well as the fibrate class of lipid-lowering agents. Members of the nuclear receptor family have a conserved modular domain structure (FIG. 1a). The binding of ligands to a region called the ligand-binding domain (LBD) causes a conformational change in this domain, which results in a cascade of downstream events. For some nuclear receptors, such as the glucocorticoid receptor and other steroid receptors, these events include dissociation from heat shock proteins and translocation of the receptor from the cytoplasm to the nucleus. Subsequent to ligand binding, the conformational change in the receptor facilitates the recruitment of transcriptional co-regulatory proteins to receptor-specific gene promoter complexes to activate or repress transcription. However, many other nuclear receptors, such as thyroid hormone receptors and peroxisome proliferator-activated receptors (PPARs), are localized in the nucleus regardless of whether or not they are bound to a ligand and constitutively interact with DNA response elements1-3.
Figure 1. Structure of the RORs and REV-ERBs.

a | The general organizational structure of members of the nuclear receptor superfamily. b | Structure of the REV-ERBs. c | Structure of the retinoic acid receptor-related orphan receptors (RORs). Numbers above each receptor represent the amino acid position. Percentages indicate amino acid identity within a particular domain relative to either REV-ERBα or RORα. A/B, C, D, E and F refer to classically defined regions in the nuclear receptor domain structure. DBD, DNA-binding domain; LBD, ligand-binding domain.
When various hormones such as thyroid hormones and the steroid hormones (oestrogens, progestins, glucocorticoids, androgens and mineralocorticoids) were identified, it was not known that they targeted members of the nuclear receptor superfamily; indeed, they were identified before the existence of the superfamily was even known. Even today, the physiological ligands are known for only half of the nuclear receptor superfamily (of which there are 48 members in humans). The development of drugs that target ligand-regulated nuclear receptors led to the design of many therapeutic compounds, which prompted substantial interest in the identification of either natural or synthetic ligands for the orphan members of the superfamily that could be used as chemical tools to probe receptor function and to understand the potential therapeutic value of these receptors. In many cases, these efforts have led to the development of synthetic ligands with pharmacokinetic and pharmacodynamic profiles that are appropriate for their testing as therapeutic modulators in several animal models of disease. This chemical biology strategy has been successful in characterizing several orphan receptors as potential drug targets, including PPARδ, liver X receptor (LXR), retinoid X receptor and the farnesoid X receptor (FXR; also known as bile acid receptor)4-6.
The chemical biology strategy has recently been applied to additional orphan receptors, including REV-ERBs and retinoic acid receptor-related orphan receptors (RORs). These two classes of nuclear receptors share many of the same target genes and thus have substantial overlap in functions that are known to include regulation of the circadian rhythm, metabolism and immune function7-10.
In this Review, we discuss the physiological and pathological roles of these two classes of nuclear receptors and the discovery of their natural ligands, as well as the development of synthetic ligands targeting these receptors and their use in cellular and in vivo models of disease. Finally, we highlight potential future directions for therapeutics targeting REV-ERB and ROR for the treatment of autoimmune diseases, central nervous system (CNS) disorders, diabetes and obesity.
Roles of REV-ERB and ROR
REV-ERBs
The REV-ERBs acquired their unusual name owing to the unique genomic organization of NR1D1, which encodes REV-ERBα. REV-ERBα is encoded by the opposite DNA strand of the ERBA (also known as THRA) oncogene11-13, and hence its name is derived from ‘reverse strand of ERBA’. ERBA encodes the thyroid hormone receptor-α and thus REV-ERBα is encoded by sequences of DNA on the opposite strand of the gene that encodes thyroid hormone receptor-α. Both REV-ERBα and the closely related REV-ERBβ (encoded by NR1D2), which was identified a few years after REV-ERBα, have an atypical LBD that lacks the carboxy-terminal activation function 2 (AF2) region14-16 (FIG. 1b). Because the AF2 region recognizes co-activators that are necessary for transcriptional activation, REV-ERBα and REV-ERBβ are generally characterized as being unable to activate transcription. Indeed, the REV-ERBs are constitutive repressors of transcription owing to their constant binding of co-repressors such as the nuclear receptor co-repressor 1 (NOR1)17.
The recruitment of co-repressors to the target gene by a nuclear receptor (via the DNA response element) leads to repression of the target gene owing to histone deacetylation and condensation of chromatin17,18. Unlike many other nuclear receptors that function as obligate heterodimers (either as homodimers or as heterodimers with retinoid X receptor) and recognize two copies of a core sequence of nucleotides that are organized in either a palindromic or a repeated manner (termed the half site), REV-ERBs typically function as monomers and recognize a half site that consists of a single 5′ extended AGGTCA sequence7. However, REV-ERB homodimers have been reported to occur under some conditions18,19. REV-ERBs have overlapping patterns of temporal and spatial expression, which is consistent with our current understanding of their substantial overlapping functions. Both REV-ERBα and REV-ERBβ are widely expressed throughout the body and, interestingly, both receptors have a circadian pattern of expression that is essential for their role in the circadian regulation of transcription20-23.
RORs
The three members of the ROR subfamily — RORα, RORβ and RORγ — have sequence similarities to the retinoic acid receptor24-27 and each receptor can constitutively activate transcription through the ligand-independent recruitment of transcriptional co-activators (FIG. 1b). RORα is widely expressed in many tissues, including cerebellar Purkinje cells, the liver, thymus, skeletal muscle, skin, lung, adipose tissue and kidney28,29. RORγ has a similar broad pattern of expression but is observed at very high levels within the thymus. RORβ has a more restricted pattern of expression relative to the other RORs, and is found in regions of the CNS that are involved in the processing of sensory information, the retina and the pineal gland30.
There is considerable overlap in the DNA response elements that are recognized by REV-ERBs and RORs, and both receptors are often co-expressed in the same tissues31. Because RORs constitutively activate transcription, whereas REV-ERBs repress transcription, the balance of ROR and REV-ERB activity is crucial for the dynamic regulation of target genes containing the DNA response elements that are responsive to both classes of receptors (FIG. 2). Owing to the substantial overlap in expression patterns as well as the target genes that are regulated by these receptors, REV-ERBs and RORs are often involved in the regulation of similar physiological processes, as outlined below.
Figure 2. Molecular mechanism of action of the RORs and REV-ERBs.

Retinoic acid receptor-related orphan receptors (RORs) and REV-ERBs are involved in transcriptional regulation and are regulated by ligands. Haem functions as a ligand for REV-ERBs, whereas sterols (cholesterol, cholesterol sulphate and various oxysterols) function as ligands for RORs. Both classes of receptors recognize a similar DNA response element, typically denoted as a ROR response element. ROR activates transcription (via recruitment of transcriptional co-activators), whereas REV-ERB silences transcription (via recruitment of transcriptional co-repressors). REV-ERB functions as a ligand-dependent transcriptional repressor (haem binding is required for the recruitment of the co-repressor and transcriptional repression), whereas ROR typically functions as a constitutive activator of transcription, and the binding of oxysterol ligands results in decreased activity. AF1, activation function 1; DBD, DNA-binding domain; LBD, ligand-binding domain.
Regulation of the circadian rhythm
Circadian rhythms have an essential role in the sleep–wake cycle, feeding behaviour and metabolism, as well as in the control of body temperature, blood pressure and renal function32. The circadian rhythm is generated by feedback loops in the expression patterns of genes encoding proteins that make up the so-called molecular clock (FIG. 3). Heterodimers of two transcription factors, brain and muscle ARNT-like 1 (BMAL1; also known as ARNTL) and circadian locomotor output cycles protein kaput (CLOCK), induce the expression of the cryptochrome genes (CRY1 and CRY2) and the period circadian clock genes (PER1, PER2 and PER3) genes. As CRY and PER proteins reach crucial levels, they repress the stimulatory effect of the CLOCK–BMAL1 dimer on the expression of their respective genes. The dynamic interplay between the opposing circadian patterns of expression and the opposing transcriptional activity of RORs and REV-ERBs, resulting in the positive and negative regulation of gene transcription, is readily apparent in this feedback loop as both classes of receptors have been shown to regulate BMAL1 expression7.
Figure 3. Role of RORs and REV-ERBs in regulation of the mammalian clock.

a | The core mammalian clock is composed of a heterodimer of the transcription factors circadian locomotor output cycles protein kaput (CLOCK) and brain and muscle ARNT-like 1 (BMAL1) (known as the positive arm), which activate the transcription of period circadian clock (PER) and cryptochrome (CRY) genes via E box sequences within their promoters. PER and CRY proteins (known as the negative arm) form dimers and directly interact with the CLOCK–BMAL1 heterodimers, thus suppressing their activity. This feedback loop follows a 24-hour rhythm where peak expression of the CLOCK–BMAL1 complex is 12 hours out of phase with peak PER and CRY expression. A retinoic acid receptor-related orphan receptor (ROR) response element within the BMAL1 promoter is responsive to both ROR and REV-ERB (encoded by the genes NR1D1 and NR1D2); ROR activates the transcription of BMAL1, whereas REV-ERB suppresses its transcription. The expression of ROR and REV-ERB also oscillates in a circadian manner (12 hours out of phase with one another), reinforcing the core circadian oscillator. The REV-ERB promoter also contains an E box, allowing direct regulation of NR1D1 and NR1D2 transcription by BMAL1–CLOCK. PER2 has also been demonstrated to directly interact with REV-ERB at REV-ERB-responsive promoters and to regulate its activity. b | The expression of PER and CRY as well as BMAL1 and CLOCK oscillates over the course of 24 hours. REV-ERB and ROR expression also undergoes circadian oscillations.
As well as RORs and REV-ERBs, various other nuclear receptors have been implicated in the modulation and/or regulation of the circadian rhythm. Over half of the nuclear receptor superfamily members are expressed in a circadian manner33, and given their role as transcription factors this probably leads to rhythmic expression of their target genes. Other direct links between nuclear receptor activity and circadian clock function have been identified. These include the direct interaction of the glucocorticoid receptor with CRY1 and CRY2, which mediates the rhythmic repression of glucocorticoid receptor transcriptional activity; this effect is essential for normal glucocorticoid signalling, which follows a clear circadian pattern34. Additionally, PER2 has been shown to interact with PPARα and REV-ERBα at promoter sites to regulate their transcriptional activity35.
REV-ERBα represses the transcription of BMAL1 (REFS 22,36) through its actions on two DNA response elements that are located in the BMAL1 promoter. The circadian feedback loop shows additional complexity given that REV-ERBα expression is itself regulated by BMAL1–CLOCK heterodimers via E box DNA response elements found within the Nr1d1 promoter37,38. Nr1d1−/− mice have aberrant expression of Bmal1 and alterations in the period and phase of their circadian locomotor behaviour36. Nr1d2−/− mice have a much more subtle circadian phenotype, but the Nr1d1−/−Nr1d2−/− double knockout mice are arrhythmic39 and have a similar phenotype to Bmal1−/− mice40, Cry1−/−Cry2−/− mice41 and Per1−/−Per2−/− mice42. Indeed, the expression of genes encoding the REV-ERBs is driven by E-box DNA response elements in their promoter elements, which are similar to those that drive the circadian expression of the CRY and PER genes. These data suggest that the genes that encode REV-ERBs should be considered as core clock genes per se rather than components of an accessory loop that merely modulates the pattern of expression of the core clock genes.
In contrast to the REV-ERBs, RORs stimulate BMAL1 expression43. Mice with a loss-of-function mutation in RORα (Rorasg/sg mice; also known as staggerer mice) have alterations in the circadian oscillator, indicating an essential role for this receptor in normal circadian function43. REV-ERBα (the repressor) and RORα (the activator) are expressed in an oscillatory fashion 12 hours out of phase with each other, leading to alternating activation and repression of BMAL1 expression36,43. RORβ-null mice also have a circadian deficit with a longer period (τ) than wild-type mice44,45, and RORγ has also been implicated in the regulation of the circadian rhythm46. Given that RORs and REV-ERBs are regulated by ligands, synthetic ligands that act at these nuclear receptors could be used to modulate the circadian rhythm as well as to treat diseases that are associated with disrupted circadian rhythms, such as sleep disorders, metabolic disease and behavioural disorders. The initial studies that have aimed to test this hypothesis are addressed below.
Regulation of metabolism
Circadian rhythms are intricately linked to the regulation of metabolism, and genetic perturbations of core clock genes lead to a range of abnormal metabolic phenotypes in mice, including obesity, dyslipidaemia and glucose intolerance47-51. In humans, circadian disruption caused by shift work52-54 or manipulated under controlled conditions causes metabolic disturbances55,56. The role of RORs and REV-ERBs in the regulation of metabolic pathways is well characterized. Both receptors are crucial components of the clock that link the core circadian oscillator to the regulation of clock-controlled genes, which in turn regulate metabolic pathways.
Loss-of-function studies both in vitro and in vivo clearly demonstrate that REV-ERBs have a crucial role in lipid metabolism. REV-ERBα-null mice have dyslipidaemia with elevated levels of very-low-density lipoprotein (VLDL) triglyceride and increased serum levels of apolipoprotein C3 (APOC3)57,58. Rorasg/sg mice have the opposite phenotype with reduced APOC3 expression and lowered triglyceride levels58. APOA1, a component of high-density lipoprotein (HDL), is also regulated by both REV-ERBα and RORα59. The expression of several genes involved in lipid metabolism was suppressed in a myocyte cell line expressing a dominant negative form of REV-ERBβ60; these genes included fatty acid translocase (FAT; also known as CD36), fatty acid binding protein 3 (FABP3; also known as MDGL), FABP4 (also known as ALBP), mitochondrial uncoupling protein 3 (UCP3; also known as SLC25A9), sterol regulatory element-binding transcription factor 1 (SREBF1) and stearoyl-CoA desaturase (SCD). Although it is unclear whether these are direct target genes of REV-ERBβ, this study clearly demonstrates that REV-ERBβ is involved in the regulation of genes that are involved in fatty acid and lipid absorption, energy expenditure and lipogenesis in muscle.
Hepatic glucose metabolism is regulated by REV-ERBα, which directly regulates the expression of the genes encoding the gluconeogenic enzymes phosphoenolpyruvate carboxykinase (PCK) and glucose-6-phosphatase (G6PC)61. Mice that are deficient in either REV-ERBα or both REV-ERBα and REV-ERBβ have increased plasma glucose levels39,62. This phenotype has been examined in more detail in REV-ERBα-null mice, yet no alteration in insulin sensitivity was noted62. In contrast to the REV-ERBα- and REV-ERBβ-deficient mice, both the Rora−/− mice and Rora−/−Rorg−/− mice have reduced blood glucose levels63. Rorasg/sg mice also have improved insulin sensitivity with increased glucose uptake in skeletal muscle64.
REV-ERB is a key regulator of the oxidative capacity of skeletal muscle and mitochondrial biogenesis65. REV-ERBα-null mice had reduced mitochondrial content and oxidative function, which resulted in reduced exercise capacity65. RORα and RORγ are both expressed in skeletal muscle. The expression of a dominant negative form of RORα in muscle resulted in altered expression of genes involved in lipid metabolism66. Rorasg/sg mice develop muscular atrophy but the exact mechanism underlying this is unclear.
REV-ERBs are also involved in adipogenesis. REV-ERBα expression is highly induced during adipogenesis67, and overexpression of REV-ERBα in 3T3-L1 cells results in increased expression of markers of adipogenesis, such as FABP4, PPARγ and CCAAT/enhancer binding protein-α (C/EBPα), as well as an increase in lipid accumulation68. Furthermore, overexpression of REV-ERBα in these cells synergized with the effects of a PPARγ ligand to increase markers of adipogenesis68. Although REV-ERBα expression is required for adipogenesis in cell-based models, REV-ERBα deficiency in vivo is associated with increased adiposity and increased weight gain owing to a high-fat diet62. This apparent discrepancy may be due to a dual role for REV-ERBα in adipogenesis, where REV-ERBα expression is increased in the initial stages of adipogenesis but the protein is degraded in the late stages of the process to allow for efficient development of the fat cells69. Interestingly, the degradation of REV-ERBα in late-stage adipogenesis seems to be dependent on gradually increasing levels of the natural ligand for REV-ERBα70,71.
The phenotypes of the knockout mice described above are consistent with the often opposing roles of REV-ERB and ROR. Rorasg/sg mice have reduced adipose mass and are less susceptible to weight gain on a high-fat diet72. RORγ also seems to be a negative regulator of adipocyte differentiation, and mice deficient in RORγ are resistant to weight gain induced by a high-fat diet73 and have increased sensitivity to insulin. REV-ERBα-deficient mice have substantial hepatic steatosis74, whereas Rorasg/sg mice seem to be less susceptible to hepatic steatosis72. Given the diverse roles of these nuclear receptors in the regulation of metabolism, it is clear that ligands that modulate REV-ERB and ROR activity may hold utility in treatment of several metabolic disorders, including obesity, type 2 diabetes and atherosclerosis.
Regulation of immune function
RORα and RORγt (an isoform of RORγ, encoded by RORC) are crucial for the development of T helper 17 cells (TH17 cells), which have an essential role in the development of many autoimmune disorders, including multiple sclerosis, psoriasis and rheumatoid arthritis75,76. Overexpression of RORγt in naive CD4+ T cells is sufficient to drive the induction and development of TH17 cells77. Furthermore, the development of TH17 cells is impaired in Rorc−/− mice77. Mice that are deficient in RORα and RORγ lack TH17 cells altogether and are resistant to the development of autoimmune diseases78. These data suggest that the development of ROR-targeted inhibitors with the potential to suppress TH17 cell development might hold utility in the treatment of autoimmune diseases.
The knowledge of the role that REV-ERBs have in the regulation of the immune system is not as well developed as for RORγ. However, REV-ERBα has been demonstrated to regulate the production and release of the pro-inflammatory cytokine interleukin-6 (IL-6) in macrophages79. Additionally, genome-wide analysis of REV-ERBα- and REV-ERBβ-binding sites in macrophages revealed that these receptors were involved in the complex regulation of target genes, which suggests that REV-ERBα and REV-ERBβ have an important role in this cell type80. Given the opposing roles of the RORs and REV-ERBs, it is likely that REV-ERBs may directly repress TH17 cell development. In fact, TH17 cell differentiation is altered in REV-ERBα-null mice81, and further work will be required to determine the exact role of these receptors in the regulation of TH17 cell function and autoimmunity. Knockdown of Nr1d1 in haematopoietic cells followed by bone marrow transplantation into LDL receptor (LDLR)-null mice revealed that REV-ERB has a crucial role in the development of atherosclerosis82. Atherosclerotic plaque development was increased in these mice, but lipid levels were unaffected. This effect was attributed to altered macrophage function, as overexpression of REV-ERBα led to increased levels of anti-inflammatory M2 macrophages82. These data suggest that increasing the repressive activity of REV-ERB may be useful for the treatment or prevention of atherosclerosis.
Endogenous ligands for REV-ERB and ROR
The RORs and REV-ERBs were both initially identified as orphan receptors, and at the time it was not clear that these nuclear receptors were regulated by small-molecule ligands. The subsequent identification of endogenous ligands for these proteins provided clear evidence that a chemical biology approach could be taken to design synthetic ligands with the ability to regulate the activity of these receptors.
Identification of haem as a physiologically relevant REV-ERB ligand
Studies using the Drosophila melanogaster orthologue of REV-ERB suggested that the human receptor might bind to haem83. Indeed, direct binding of haem to REV-ERBs was demonstrated using several biochemical and biophysical methods, including mutation studies that identified a key amino acid residue in REV-ERBα that was necessary for haem binding61,84, transcriptional repressor function and repression of target gene transcription61,84. Moreover, reduction of intracellular haem levels decreased REV-ERB-mediated repression of REV-ERB target genes (that is, delta-aminolevulinate synthase 1 (ALAS1), BMAL1, elongation of very-long-chain fatty acid elongase 3 (ELOVL3), G6PC and PCK), decreased the interaction between REV-ERB and the NCOR–HDAC3 (histone deacetylase 3) co-repressor complex in cells, and impaired the recruitment of NCOR to REV-ERB target gene promoters61,84. These studies, together with additional biophysical studies examining the affinity of haem for REV-ERB, suggested that haem acts as a bone fide ligand for the REV-ERBs.
Adipogenesis is regulated by haem in a REV-ERB-dependent manner71. Intracellular haem levels are increased during adipogenesis, and inhibition of haem biosynthesis has been shown to inhibit adipogenesis. Increased REV-ERB levels reduce intracellular haem levels through direct modulation of PPARγ co-activator 1α (PGC1α; also known as PPARGC1A) levels85,86, suggesting a mechanism by which REV-ERBs regulate the biosynthesis of their endogenous ligand, haem. Haem may also have an important role in the regulation of the circadian function of REV-ERB. Haem levels and REV-ERB expression are regulated in a circadian manner, which suggests that oscillations in the availability of haem may regulate the transcriptional activity of REV-ERB87.
The crystal structures of REV-ERBs in the apo structure form (FIG. 4a) and bound to haem (FIGS 4b,c) have provided some insight into the molecular details of haem coordination by REV-ERBs, and indicated how REV-ERB might be targeted by synthetic ligands. The structure of the haem-bound LBD of REV-ERBβ (FIG. 4b,c) revealed that the REV-ERB ligand-binding pocket is located in the same structural region as other nuclear receptors88. The structure revealed that haem is coordinated by two key residues, a histidine residue on helix 11 and a cysteine residue on helix 3. Mutations in these residues prevent haem from binding to REV-ERB and result in loss of REV-ERB activity61,84,88,89. The ligand-binding pocket undergoes a large expansion to accommodate the haem ligand, and the bulky hydrophobic residues that make up the inaccessible pocket in the apo structure were found to form key hydrophobic interactions with the porphyrin ring of the haem molecule. However, porphyrin-like synthetic ligands have not been pursued because many proteins use haem as a cofactor, which could lead to potential specificity issues.
Figure 4. Structures of REV-ERB and ROR demonstrate their capacity to bind to natural ligands.

The apo structure of REV-ERBβ (part a) indicated that the putative ligand-binding pocket was filled with large hydrophobic residues and thus devoid of the space that would be necessary for a ligand to bind. However, the haem-bound REV-ERBβ structure (parts b and c) shows that its ligand-binding pocket can profoundly change its shape to accommodate haem, a large porphyrin natural ligand. Intriguingly, although studies indicate that the binding of haem to REV-ERB increases its interaction with the nuclear receptor co-repressor (NCOR)11,61, the structure of apo REV-ERB bound to an NCOR peptide (part d) indicates that the binding of haem may not be an absolute requirement for mediating the REV-ERB–NCOR interaction. The co-crystal structure of the ligand-binding domain of retinoic acid receptor-related orphan receptor-α (RORα) bound to cholesterol (parts e and f) sets the stage for other studies indicating that various cholesterol derivatives, such as 7-oxygenated sterols, may act as physiological ligands to influence ROR activity. Structures are shown as space-filling models (parts a, c, d and e), with and without transparency to allow visualization of ligands bound to the internal ligand-binding pocket (haem-bound REV-ERBβ and cholesterol-bound RORα). A snapshot of the residues mediating the interaction of haem with REV-ERB (part b) illustrates that the repositioned hydrophobic residues that were originally thought to block the ligand-binding pocket in fact cooperate in binding to the large hydrophobic porphyrin haem scaffold. Protein Data Bank (PDB) codes: apo REV-ERB, 2V0V; haem-bound REV-ERB, 3CQV; apo REV-ERB with an NCOR fragment (co-repressor nuclear receptor (CoRNR) box motif peptide, 3N00; cholesterol-bound ROR, 1N83.
The crystal structure of the LBD of the apo form of REV-ERBα complexed to an NCOR fragment (co-repressor nuclear receptor (CoRNR) box motif) (FIG. 4d) was reported, which was unexpected because cell-based studies indicate that NCOR recruitment is dependent on haem. This structure suggests that apo REV-ERBs can indeed bind to co-repressors. Notably, the authors were unable to obtain crystals for the REV-ERBα LBD complexed with an NCOR fragment in the presence of haem90. Although cell-based studies have revealed that the binding of haem to REV-ERB increases the recruitment of co-repressor proteins such as NCOR, leading to transcriptional repression61,84, these structural studies (as well as other biochemical studies) suggest that haem binding results in a loss of co-repressor binding. However, it should be noted that structural and biochemical studies use isolated LBDs and peptide fragments of the co-repressor proteins NCOR and SMRT (silencing mediator of retinoic acid and thyroid hormone receptor; also known as NCOR2), which are clearly distinct from the full-length co-repressor proteins that are present under physiological circumstances61,84,88. Thus, the precise mechanism of action of haem-dependent REV-ERB-mediated transcriptional repression remains unclear, and the discrepancies noted above may be due to differences in the conformation of the receptor when it is bound to relatively short NCOR peptide fragments versus the full-length NCORs.
REV-ERB as a gas sensor
The functions of many haem-binding proteins are regulated by small-molecule diatomic gases, such as carbon monoxide and nitric oxide. Indeed, REV-ERBs are responsive to diatomic gases, which repress REV-ERB-mediated transcription88, and are sensitive to the redox state of the iron-haem centre88,89,91. Cell lines treated with the chemical nitric oxide donor diethylenetriamine have increased expression of BMAL1, NR1D1 and NR1D2, and REV-ERB-dependent transcriptional repressor activity was suppressed, which suggests that nitric oxide acts as an antagonist of REV-ERB activity88. Similar to the results obtained in haem-dependent biochemical and cell-based studies, nitric oxide — which acts as an antagonist of REV-ERB activity in cells — increased the interaction between REV-ERB and co-repressor peptides in biochemical studies, which indicates that it acts as an agonist rather than an antagonist of REV-ERB activity in this setting88. Thus, the molecular mechanism by which a small-molecule diatomic gas regulates REV-ERB activity, as well as its physiological significance, remains unclear.
Before REV-ERB was identified as a haem-binding protein, its D. melanogaster orthologue E75 was shown to bind to haem in an obligate manner83. Nitric oxide regulated E75 activity by altering its ability to interact with its heterodimer partner, the D. melanogaster orphan nuclear receptor HR3 (DHR3), thus altering the transcriptional activity of the dimers83. Unlike REV-ERB, haem seems to function as an obligate cofactor in the LBD of E75. Given the role of nitric oxide in the regulation of the circadian rhythm92, it is possible that nitric oxide may be a physiological ligand for REV-ERB. This would lead to a unique model of nuclear receptor regulation where REV-ERB is regulated by haem levels as well as nitric oxide levels. Thus, REV-ERB may only be responsive to nitric oxide when haem levels are sufficiently elevated to occupy the LBD of the receptor.
Sterol ligands for RORs
Cholesterol has been demonstrated to act as a ligand for RORα by binding to the LBD93 of the receptor (FIG. 4e,f). Other cholesterol metabolites, including cholesterol-3-O-sulphate, also bind to the LBD of RORα94.
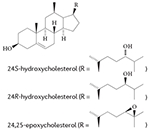
More recent studies, including mechanistic analyses, have revealed that several oxysterols are high-affinity endogenous modulators of ROR activity95-97 (TABLE 1). Oxysterol ligands bind directly to the LBD of RORα and RORγ and modulate the interaction of co-regulators. For example, 7-oxygenated sterols inhibit G6PC expression by reducing the recruitment of nuclear receptor co-activator 2 (NCOA2; also known as SRC2 or TIF2) by RORα and RORγ on the G6PC promoter96. 24S-hydroxycholestrol reduces the expression of BMAL1 by reducing SRC2 recruitment by RORα95. These actions of sterols reduce the transcriptional activity of RORs and the expression of ROR target genes in cells; through these actions, oxysterol ligands function as ROR inverse agonists. Structure-based studies have provided further evidence that cholesterol-based ligands induce a conformational change in RORα that is consistent with a reduction in the affinity of co-activators for this receptor. However, it seems that cholesterol or other sterols do not act as ligands for RORβ98.
Table 1. Natural ligands of the nuclear receptors REV-ERB and ROR.
| Ligands | Structure or scaffold | Receptor (or receptors) |
Refs |
|---|---|---|---|
|

|
REV-ERBα, REV-ERBβ |
17,84 |
|
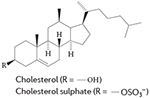
|
RORα | 93 |
|
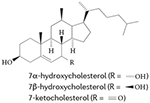
|
RORα, RORγ | 96 |
|
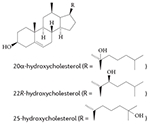
|
RORα | 97 |
|
|
RORα, RORγ | 96 |
|
|
RORβ | 99 |
|
|
RORβ | 98 |
|
|
RORα | 99 |
ROR, retinoic acid receptor-related orphan receptor.
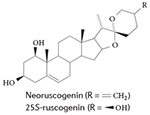
Interestingly, a recent study that screened a commercially available plant extract library for ROR ligands identified a natural product plant sterol called neoruscogenin as a RORα agonist99. In a biochemical assay measuring the recruitment of the cofactor SRC2 to RORα, neoruscogenin had an EC50 (effector concentration for half-maximum response) value of 0.11 μM. In cell-based assays, neoruscogenin induced transcription driven by a chimeric receptor, Gal4–RORα, and also increased the transcription of RORα target genes in HepG2 cells99. Although neoruscogenin had activity against the pregnane X receptor, it was selective versus other nuclear receptors and it also induced the expression of hepatic RORα target genes when it was orally administered to mice99.
Endogenous ligands for RORβ
The first ligand that was demonstrated to bind to any ROR was stearic acid, which was fortuitously discovered when the LBD of the RORβ protein that was expressed in Escherichia coli crystallized with a stearate ligand that bound to the putative ligand-binding pocket of the receptor100. However, it was unclear at the time — and is still unconfirmed at present —whether stearate is similar to a putative physiological ligand found in humans, such as a fatty acid. Because RORs are evolutionarily related to the retinoic acid receptors, which serve as receptors for retinoic acid and other metabolites, a mass spectrometry approach was used as a screening tool to determine whether retinoids may also serve as endogenous ligands for RORβ98. This approach revealed that all-trans retinoic acid (ATRA) (TABLE 1) and the synthetic retinoid ALRT 1550 (TABLE 1) bind to the RORβ LBD, which was verified by co-crystal structures of RORβ bound to these ligands. In addition to these two ligands, all-trans-4-oxoretinoic acid binds to RORβ, and all three ligands have been shown to function as inverse agonists of RORβ in a cell-based cotransfection reporter assay. Retinoids do not, however, bind to RORα or RORγ, which suggests that RORβ has a subtype-specific ligand preference compared to RORα and RORγ; this may allow for the development of subtype-specific synthetic ligands targeting these receptors.
Synthetic ligands for REV-ERB and ROR
The discovery that REV-ERBS and RORs are liganded receptors has led us and others to develop screens to find ligand scaffolds that could be used to identify potent ligands. Such potent ligands could be used as chemical tools to probe the role of REV-ERBs and RORs in physiological and disease models.
Synthetic REV-ERB ligands
REV-ERB agonist: GSK4112
The first synthetic REV-ERB-targeting ligand to be identified was 1,1-dimethylethyl N-[(4-chlorophenyl)methyl]-N-[(5-nitro-2-thienyl) methyl]glycinate101. It was first published with no other moniker and was subsequently synthesized and characterized by our group and given the name SR6452. The compound was renamed GSK4112 in a follow-up paper from the group92 that initially identified it, and we suggest that this name should now be used. GSK4112 was identified from a fluorescence resonance energy transfer (FRET) biochemical screen in which it increased the binding of an NCOR peptide to REV-ERBα in a concentration-dependent manner101. Further mechanistic studies investigated the pharmacological effects of this compound in cell-based assays71,102 (TABLE 2).
Table 2. Synthetic REV-ERB ligands.
| Compound | Structure | Comments (activity, affinity, EC50 value, IC50 value and actions) | Refs |
|---|---|---|---|
| GSK4112 (also known as SR6452) |
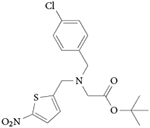
|
|
71,101, 102 |
| SR9009 |
|
|
105 |
| SR9011 |
|
|
105 |
| GSK2945 |
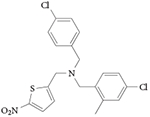
|
|
107 |
| GSK0999 |
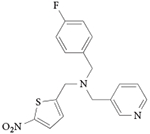
|
|
107 |
| GSK5072 |
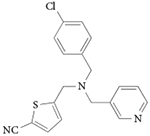
|
|
107 |
| GS2667 |

|
|
107 |
| SR8278 |
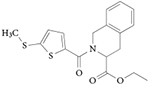
|
|
108 |
BMAL1, brain and muscle ARNT-like 1; CoRNR, co-repressor nuclear receptor; EC50, effector concentration for half-maximum response; FRET, fluorescence resonance energy transfer; IC50, half-maximal inhibitory concentration; IL-6, interleukin-6; NCOR, nuclear receptor co-repressor; PER2, period circadian clock; ROR, retinoic acid receptor-related orphan receptor; SCN, suprachiasmatic nucleus; UAS, upstream activating sequence.
GSK4112 increases the recruitment of NCOR to the BMAL1 promoter71 and increases the recruitment of HDAC3 to the G6PC promoter102, providing a mechanism for the repressive effect of the compound on REV-ERB target genes. Primary mouse hepatocytes treated with GSK4112 have decreased expression of several gluconeogenic REV-ERB target genes and a decreased hepatic glucose output, which suggests that GSK4112 has the potential to be used as an antidiabetic compound102. Additionally, the compound modulated the expression levels of several key genes involved in the circadian rhythm, providing a potential pharmacological approach for the modulation of the circadian rhythm102. As REV-ERB has been shown to be involved in the differentiation of 3T3-L1 cells to adipoctyes, the effect of the compound on adipogenesis was examined71. Both rosiglitazone (a PPARγ agonist) and GSK4112 induced the expression of several key gene markers of adipogenesis, including FABP4, PPARG, PCK, adiponectin C1Q and collagen domain containing protein (ADIPOQ), FAS and resistin (RETN), and they also increased the lipid content of the cells. Additional studies have demonstrated that rosiglitazone and GSK4112 synergistically induce adipogenesis71.
Unfortunately, GSK4112 does not have a favourable pharmacokinetic profile following intraperitoneal administration (it has a low systemic exposure) and it displayed weak efficacy in terms of REV-ERB agonism, which limits its use as a tool to probe the function of REV-ERBs in vivo. Therefore, we — and other groups — undertook a comprehensive structure–activity relationship (SAR) analysis to identify potent and efficacious REV-ERB modulators based on the GSK4112 scaffold that had pharmacokinetic and pharmacodymamic properties that would make them suitable for in vivo studies103,104. Below, we describe several key tool compounds that were identified in SAR campaigns and that led to subsequent studies providing insight into REV-ERB function in vitro and in vivo.
REV-ERB agonists: SR9009 and SR9011
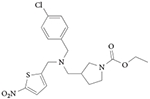
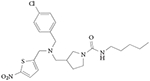
Only two REV-ERB ligands have been characterized in vivo103-105, although other compounds have been shown to be bio-available, as discussed below (TABLE 1). Both of these compounds used GSK4112 as an initial scaffold, with several modifications to improve potency, efficacy and pharmacokinetic properties (increased systemic exposure). SR9009 and SR9011 are dual synthetic agonists of REV-ERBα and REV-ERBβ, and they have been used to demonstrate that pharmacological targeting of REV-ERBs may be useful in the treatment of circadian disorders, including metabolic diseases and sleep disorders105.
SR9009 and SR9011 are three- to fourfold more potent than GSK4112, and are threefold more efficacious in driving REV-ERB repression in a reporter gene luciferase assay. In addition, they had no significant cross-activity in a specificity assay against the 46 other mem bers of the human nuclear receptor superfamily105,106. Studies to determine the effects of the compounds on circadian behaviour in mice revealed that a single intraperitoneally administered dose of either compound, given when REV-ERBα expression levels peak (that is, in the middle of the sleep period), results in loss of wheel running activity during the subsequent wakeful period. In addition, both compounds were shown to affect the circadian expression of several core clock genes in the hypothalamus of mice, including the suppression of Cry2, enhancement of Per2, a phase shift in the expression of Bmal1 and complete elimination of the circadian expression pattern of neuronal PAS domain-containing protein 2 (Npas2)105. Together, these data indicate that SR9009 and SR9011 substantially affect the circadian oscillator through the modulation of REV-ERB activity.
Consistent with the range of metabolic effects noted in REV-ERBα-null mice, pharmacological activation of REV-ERB with SR9009 and SR9011 had additional metabolic effects in mice. Most notable was the weight loss in diet-induced obese mice, which was associated with an increase in energy expenditure without alterations in locomotor behaviour or food intake105. A decrease in plasma triglycerides, total cholesterol and non-esterified fatty acids was also noted in the obese mice after treatment. These metabolic changes correlated with changes in the expression of key factors in these metabolic pathways, including decreased expression of lipogenic enzymes (fatty acid synthase (Fasn) and Scd) and cholesterologenic regulator proteins (3-hydroxy-3-methylglutaryl-CoA reductase (Hmgcr) and Srebf2), as well as increased expression of genes encoding enzymes involved in fatty acid and glucose oxidation (carnitine palmitoyltransferase 1b (Cpt1b), Ucp3, PPAR-gamma co-activator 1-beta (Ppargc1b), M2 isoform of pyruvate kinase muscle (Pkm2) and hexokinase 1 (Hk1))105.
During the development of SR9009 and SR9011, we identified almost 50 additional REV-ERB ligands, several of which had substantial in vivo exposure (including in the CNS) when administered intraperitoneally104. A more recent study has shown that the activation of REV-ERB with SR9009 in vivo leads to increased oxidative metabolism and mitochondrial biogenesis in skeletal muscle, resulting in increased exercise endurance65, which is consistent with the metabolic alterations and weight loss observed105.
Additional REV-ERB agonists
GlaxoSmithKline has also pursued the GSK4112 scaffold and recently described four additional compounds that can be used to probe REV-ERB function (TABLE 1). Four of these have sufficient pharmacokinetic properties to be used in vivo and they are orally bioavailable107. Three of these compounds have similar pharmacokinetic properties to SR9009 (compounds 10, 16 and 23), whereas one compound (compound 4) has an increased half-life, area under the curve (AUC) and maximum peak plasma concentration (Cmax). In addition, it has an approximately tenfold higher bioavailablity than the other compounds. Other than SR9009, these compounds have not yet been tested in animal models of disease.
GSK4112, SR9009 and SR9011 — and almost all of the analogues reported in REFS 104,107 — contain a nitrothiophene group, which carries a potential toxicological liability104,107. We sought to mitigate this potential toxicological liability and recently described a distinct chemical series of tetrahydroisoquinoline-based REV-ERB agonists lacking the nitrothiophene group, with potencies (that is, half-maximal inhibitory concentration (IC50) values) of ~70 nM in a REV-ERB co-transfection assay103 (TABLE 1). Although these tetrahydroisoquinoline-based REV-ERB agonists are not orally bioavailable, many of them have reasonable plasma exposure and half-lives of ~2 hours when administered intraperitoneally103. As of yet, no dedicated toxicological studies have been performed for any of the REV-ERB ligands.
Specificity of SR9009 and SR9011
Although the original report of SR9011 and SR9009 examined the specificity of these compounds in a Gal4-chimeric nuclear receptor assay panel and observed no activity other than at the REV-ERBs105, a recent report indicates that these two ligands may have some agonist activity at the LXR107. In the original paper that reported the identification of GSK4112, it was indicated that the compound did not modulate LXRα or LXRβ in reporter assays101, but it was unclear whether these data were taken from a chimeric Gal4 LBD or a full-length REV-ERB co-transfection assay. In a follow-up study, GSK4112 had an IC50 value of 5 μM in an LXRα radioligand binding assay and increased ATP-binding cassette subfamily A member 1 (ABCA1) mRNA expression in THP-1 cells, which was attributed to LXR activity107. SR9009 and SR9011 had similar activity as GSK4112 in these assays107. However, we have not observed LXR activation by either SR9011 or SR9009 in assays that use chimeric Gal4 LBD co-transfections105 or with studies that use full-length LXR co-transfections (T.B., unpublished observations). We therefore believe, as suggested by several additional studies, that these compounds do not have substantial LXR agonist activity. Treatment of HepG2 cells with SR9009 or SR9011 suppressed (rather than activated) the well-characterized LXR target gene Srebf1 (REF. 105). Additionally, when SR9009 was administered to mice, hepatic expression of Srebf1 and several other direct LXR target genes, including cytochrome P450 family 7 subfamily A polypeptide 1 (Cyp7a1), Fasn and Scd, was suppressed; this finding is inconsistent with SR9009 acting as an LXR agonist105. Although one study indicated that SR9009 and SR9011 were active in an LXR radioligand binding assay (with IC50 values of 6–13 μM), concentration–response data were not presented107.
It should also be considered that binding does not necessarily equate to modulation of receptor function and that there is often a substantial reduction in potency (ten-fold or greater) when one compares the binding potency with cell-based potency for nuclear receptor ligands. In the paper reporting that SR9009 and SR9011 may have some agonist activity at the LXR, the calculation of REV-ERB specificity was based on data from a biochemical REV-ERB NCOR recruitment assay in which SR9009 and SR9011 were inactive107, even though the compounds SR9009 and SR9011 directly interact with REV-ERB with submicromolar Kd (dissociation constant) values, based on other biochemical data (circular dichroism thermal shift assay). In our view, the specificity of SR9009, SR9011 and GSK4112 should have been calculated using a functional transcriptional response.
REV-ERB antagonist: SR8278
Only one REV-ERB antagonist has been described to date. SR8278 is a synthetic antagonist of REV-ERB activity108 (TABLE 2). SR8278 increased the activity of REV-ERBα and REV-ERBβ in a Gal4-based co-transfection reporter assay, and in an assay that used full-length REV-ERBα and luciferase reporters driven by the promoters of three REV-ERB target genes: BMAL1, PCK and G6PC. Treatment of HepG2 cells with SR8278, which express REV-ERBs endogenously, increases the expression of G6PC and PCK. A further study showed that activation of REV-ERBα by haem and GSK4112 stimulated glucose-induced insulin secretion in MIN-6 mouse insulinoma cells, which was inhibited by SR8278 (REF. 109). SR8278 was used, together with haem and GSK4112, to show that activation of the connexin 43 promoter via interaction with the transcription factor SP1 occurs in a ligand-independent manner and does not involve the LBD of REV-ERBα110.
Synthetic ROR ligands
The first report describing ligands for RORs demonstrated that melatonin and a synthetic thiazolindinedione (CGP 52608) bound to these receptors111-113 (TABLE 3). However, beyond these initial reports, ligands for these receptors have not been well validated, with the exception of a recent study suggesting that CGP 52608 activates RORα at a potency >100,000-fold weaker than originally reported (>100 μM versus 1–3 nM)114. Nevertheless, several studies have identified several synthetic ligands that target RORs and have in vitro and in vivo activity on ROR function.
Table 3. Synthetic ROR ligands.
| Compound | Structure | Comments (activity, affinity, EC50 value, IC50 value and actions) | Refs |
|---|---|---|---|
| T0901317 |
|
|
115 |
| SR1078 |
|
|
119 |
| SR3335 (also known as SR3335/ML176) |

|
|
125 |
| SR1001 |

|
|
125 |
| SR2211 |

|
|
129, 130, 143 |
| SR1555 |
|
|
130 |
| Digoxin |
|
|
131, 132 |
| Ursolic acid |
|
|
135 |
| ML209 |
|
|
144 |
| Compound 1a |

|
|
145 |
| Compound 1b: N-(4,6-dimethyl- benzo[d]thia- zol-2-yl)-3-methyl- thiophene-2- carboxamide |

|
|
145 |
| Compound 1c: N-(2-(4-ethyl- phenyl)-2H-benzo- [d][1,2,3]triazol-5-yl) propionamide |
|
|
145 |
| Inhibitor Y: N-(5-benzoyl-4-p henylthiazol-2-yl)- 2-(4-(ethylsulfonyl) phenyl)acetamide |
|
|
145 |
EC50, effector concentration for half-maximum response; FOXP3, forkhead box protein P3; FXR, farnesoid X receptor; G6PC, glucose-6-phosphatase; IL-17, interleukin-17; IL-23R, interleukin-23 receptor; IC50, half-maximal inhibitory concentration; Kd, dissociation constant; Ki, inhibition constant; LXR, liver X receptor; NCOR, nuclear receptor co-repressor; PCK, phosphoenolpyruvate carboxykinase; PXR, pregnane X receptor; RIP140, receptor-interacting protein 140; ROR, retinoic acid receptor-related orphan receptor; TH, T helper; TRAP220, thyroid hormone receptor-associated protein complex 220 kDa component; UAS, upstream activating sequence.
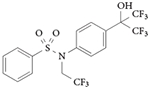
RORα and RORγ inverse agonist: T0901317
T0901317 was the first validated synthetic ligand that was shown to bind to and regulate the function of an ROR115 (TABLE 3). T0901317 was originally identified as an agonist for LXRs116, and subsequent studies demonstrated that this ligand also functions as an agonist for FXR117 and pregnane X receptor118. Only later was it determined that T0901317 was also an inverse agonist of RORα and RORγ115. As T0901317 is a nonspecific ligand for several nuclear receptors, it was used as a starting point for developing ROR-selective compounds, as described below.
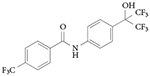
RORα and RORγ agonist: SR1078
SR1078 (TABLE 3) is a direct agonist of RORα and RORγ — it increased the expression of the ROR target genes G6PC and fibroblast growth factor 21 (FGF21) in HepG2 cells — and has no activity for FXR, LXRα and LXRβ119 in functional transcription assays. SR1078 displays acceptable pharmacokinetic properties (plasma concentrations of 3.6 μM 1 hour after an intraperitoneal injection of 10 mg per kg, and sustained levels above 800 nM 8 hours after a single injection) that allowed for a proof-of-concept analysis of the compound in animals. SR1078 was also used to characterize the FGF21 gene as a direct target gene of RORα120. For example, in vivo administration of FGF21 in rodent models of diabetes improved glucose and triglyceride levels as well as insulin sensitivity121; administration of FGF21 to diabetic non-human primates leads to a similar improvement in the metabolic profile122. Thus, small-molecule RORα agonists could be one approach for modulating the expression of this hormone, which has therapeutic potential in the treatment of obesity and diabetes.
Expression levels of RORα are decreased or down-regulated in several types of cancers and cancer cell lines, including breast, ovarian and prostate cancers123, and this has been linked to RORα-mediated regulation of the transcription factor SOX4 (which regulates tumour suppressor p53 activity). Interestingly, treating cancer cells with SR1078 leads to p53 stabilization and induces apoptosis124, which suggests that further studies are warranted to investigate the potential of this agonist in models of cancer.
RORα inverse agonist: SR3335
Additional modifications of the T0901317 and SR1078 scaffolds led to the discovery of SR3335 (TABLE 3), the first potent RORα-specific inverse agonist125. SR3335 had acceptable pharmacokinetic properties (plasma concentrations of 9 μM 30 minutes after an intraperitoneal injection of 10 mg per kg, and sustained levels above 360 nM 4 hours after a single injection), and so it was tested in a mouse model of diet-induced obesity. Mice treated with SR3335 (15 mg per kg, intraperitoneally administered twice daily) for 6 days had significantly reduced plasma glucose levels compared to vehicle control-treated animals, including a decrease in Pck, the rate-limiting enzyme in gluconeogenesis.
RORα and RORγ inverse agonist: SR1001
Previous studies with synthetic ROR ligands were limited to basic proof-of-concept in vivo studies described above. The discovery and report of SR1001 (REF. 126) (FIG. 5; TABLE 1) represented the first robust analysis demonstrating that a ROR ligand can be used to probe the role of RORs in mouse models of disease. This compound directly binds to RORα and RORγ and acts as an inverse agonist that suppresses RORα and RORγ reporter activity, decreasing the interaction between the receptor and co-activators.
Figure 5. Development of selective ROR ligands.

Following a screen of known nuclear receptor ligands against the entire nuclear receptor superfamily, the liver X receptor (LXR) agonist T0901317 was identified as a retinoic acid receptor-related orphan receptor (ROR) ligand. T0901317 has substantial promiscuity against other nuclear receptors. Various alterations in the structure led to the discovery of an agonist of RORα and RORγ (SR1078), an inverse agonist of RORα and RORγ (SR1001), and a RORγ-selective inverse agonist (SR2211). FXR, farnesoid X receptor; PXR, pregnane X receptor.
Consistent with the role of RORα and RORγt in TH17 cell development78, SR1001 inhibited the differentiation of splenocytes that were cultured under conditions to produce TH17 cells, and this inhibition was associated with inhibited expression of several cytokines, including IL17A, IL17F, IL21 and IL22 (FIG. 6). SR1001 inhibited IL-17 protein production and secretion in splenocytes as well as in human peripheral blood mononuclear cells. Notably, SR1001 did not affect the differentiation of splenocytes into inducible regulatory T cells (TReg cells) or other TH cell lineages, including TH1 cells and TH2 cells, and so it specifically targets TH17 cells. In a mouse model of TH17-mediated multiple sclerosis127,128, SR1001 treatment (25 mg per kg administered intraperitoneally) delayed the onset and clinical severity of disease and was associated with reduced expression of the cytokines Il17a, Il21 and Il22.
Figure 6. ROR inverse agonists alter TH cell development.

T helper 1 (TH1), TH2, TH17 and regulatory T (TReg) cells develop from naive CD4+ TH cells. The differentiation of naive CD4+ TH cells into these effector CD4+ T cells is initiated via an interaction of dendritic cells with naive CD4+ TH cells. Effector cell types are defined by their production of specific cytokines, function, modulation of distinct signalling pathways and the expression of distinct transcription factors. Retinoic acid receptor-related orphan receptor (ROR) inverse agonists suppress TH17 cell differentiation and function. The RORγ inverse agonist SR1555 promotes TReg cell differentiation as well. ATRA, all-trans retinoic acid; FOXO3, forkhead box protein O3; GATA3, GATA-binding protein 3; IFNγ, interferon-γ; IL, interleukin; STAT, signal transducer and activator of transcription; TBX21, T-box protein 21 (T-bet); TGFβ1, transforming growth factor-β1.
SR1001 targets both RORα and RORγ, and both of these receptors are required for the development of TH17 cell-mediated autoimmune diseases77,78. However, it is clear that RORα has additional activities, such as a more robust regulation of circadian rhythm compared to RORγ, as well as a role in cerebellar development. We therefore pursued the development of RORγ-specific inverse agonists to determine whether these compounds may hold utility in suppressing TH17 cell development. This led to the identification of two RORγ-specific ligands, SR2221 (REF. 129) and SR1555 (REF. 130), which are discussed below.
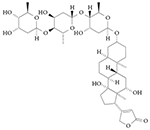
RORγ inverse agonist: digoxin
Digoxin (TABLE 3) was identified as an inhibitor of the transcriptional activity of RORγ in a screen of over 4,000 compounds using a D. melanogaster cell-based reporter system131, and was subsequently shown to occupy the ligand-binding pocket of RORγ132. In a mouse model of multiple sclerosis, digoxin delayed the onset and reduced the severity of disease progression. However, digoxin is toxic to humans at the concentrations required for influencing RORγ activity and it is known to raise intracellular calcium levels by interacting with the (Na+ + K+)ATPase133,134. Although the toxic effects limit its use as a tool and potential therapeutic compound, this proof-of-concept study provided evidence that RORγ-targeted ligands can be used to probe the activity of RORγ in vivo.
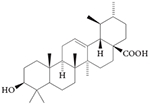
RORγ inverse agonist: ursolic acid
Ursolic acid (TABLE 3) was identified in a cell-based screen as a compound that inhibited TH17 cell development and the expression of Il17 in TH17 cells135. Structural similarity between ursolic acid and hydroxycholesterols led the authors to posit that ursolic acid functions through the RORs. RORγ was confirmed as the target of ursolic acid by carrying out biochemical assays and studies showing that ursolic acid inhibited Il17 and Il17f expression only when T cells were differentiated via overexpression of RORγt. Although ursolic acid delays the onset and decreases the severity of disease symptoms in a model of multiple sclerosis, it has many other cellular targets, including the liver kinase B1 (LKB1; also known as STK11)–AMP-activated protein kinase (AMPK) pathway136, the NFE2-related factor 2 (NRF2) pathway137, nuclear factor-κB (NF-κB)138,139, signal transducer and activator of transcription 3 (STAT3)140,141 and glucocorticoid receptor142, which suggests that it would not be well suited as a RORγ-selective probe in vivo.
RORγ inverse agonist: SR2211
Additional work on the SR1001 scaffold, directed at designing RORγ-selective inverse agonists, led to the discovery of SR2211 (TABLE 3), a compound that displays exquisite selectivity for RORγ over RORα in both biochemical and cell-based assays, with a Ki (inhibition constant) value of 105 nM at RORγ and no detectable binding to RORα129. In EL-4 cells, SR2211 treatment repressed the expression of Il17a and Il23R, as well as intracellular IL-17 protein levels, and suppressed TH17 cell differentiation130. This compound was recently shown to be active in a mouse model of collagen-induced rheumatoid arthritis; SR2211 administered twice daily for 15 days by intraperitoneal injection significantly reduced joint inflammation in these mice143.
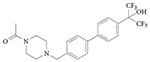
RORγ inverse agonist: SR1555
SR1555 (TABLE 3) binds to and represses the activity of RORγ. Although it is considerably less potent than SR2211, it has no RORα130 activity. Interestingly, although SR1001 (REF. 126) and SR2211 (REF. 130) specifically target and suppress TH17 cell differentiation only, SR1555 affects both TH17 cells (suppressing their differentiation) and TReg cells (increasing the frequency of inducible TReg cells)130. SR1555 is the first RORγ ligand to have an effect on TReg cells, which suggests that RORγ-specific ligands can be further optimized to have specific effects on different classes of T cells.
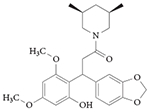
RORγ inverse agonist: ML209
A quantitative high-throughput screen of 310,000 compounds identified a series of diphenylpropanamides as selective inhibitors of RORγ activity using a cell-based RORγ reporter assay144. The initial hit displayed an IC50 value of 3.3 μM in the RORγ assay with no activity in control assays, including the RORα assay. The initial hit also inhibited TH17 cell differentiation, which indicated that this was a viable scaffold for developing SAR studies. These efforts led to the discovery of ML209 (TABLE 3), an RORγ inhibitor with an IC50 value of 0.5 μM. Notably, in a panel of 20 nuclear receptors, only weak activity was reported for oestrogen-related receptor-α (ERRα), LXRα, thyroid hormone receptor-α and thyroid hormone receptor-β. ML209 displaces 25-hydroxycholesterol from RORγ, which indicates that it binds directly to the LBD of RORγ. In RORγ-dependent TH17 cell differentiation assays, ML209 inhibited TH17 cell differentiation without affecting TH1 cells and TReg cells. The compound also inhibited the expression of the RORγ target gene Il17a and protein levels of IL-17A, but it did not affect the expression of RORα target genes, which further indicates that ML209 is specific for RORγ. The pharmacokinetic properties of ML209 were not provided in this initial report, so it is currently unknown whether this compound could be an effective tool to probe RORγ activity in vivo.
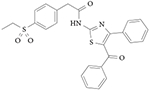
Aryl amide RORγ agonists
Several small-molecule activators of transcription driven by the IL17 promoter have been identified and shown to act as direct ligands of RORγ via a circular dichroism thermal shift assay145. Three compounds (1a, 1b and 1c) increased IL17 promoter-driven transcription in a co-transfection assay with a potency of ~100 nM (TABLE 3). Compound 1b also increases TH17 cell differentiation145. A novel inhibitor of IL17 promoter-driven transcription was identified in the same assay, but little specific information was provided with regard to its mechanism of action. Inhibitor Y (TABLE 3) was used to compete with the aryl amide agonists in the study, but it is not clear whether this compound is a direct RORγ ligand. Further studies are needed to determine the specificity of the compounds described in this study for RORγ.
Conclusions
Substantial advances have been made over the past several years in our understanding of how ligands can regulate two classes of nuclear receptors, RORs and REV-ERBs, which were once considered orphan receptors. Not only have we been able to determine the identity of endogenous, high-affinity ligands for these receptors, we have also been able to use classic methods of drug discovery to generate synthetic ligands that have been used to probe the potential of these receptors as bona fide drug targets for the treatment of several human diseases (FIG. 7). Even though synthetic ligands have only been available for use in animal models of disease for the past few years, it is clear that improved and optimized ligands may have utility for the treatment of autoimmune diseases and metabolic disorders. Given the role of these receptors in the regulation of the circadian rhythm, additional studies are under-way to investigate the potential of these compounds in targeting the central mammalian clock, with the possibility of treating behavioural and sleep disorders. Progress is being made to improve the drug-like properties of these compounds, and some of these improved compounds or their analogues have a substantially high potential to enter clinical trials in the not too distant future.
Figure 7. ROR and REV-ERB in the regulation of physiological processes.

Physiological processes are shown in pink boxes, and the potential therapeutic indications of synthetic ligands that target retinoic acid receptor-related orphan receptor (ROR) and the nuclear receptor REV-ERB are indicated in grey boxes.
Acknowledgements
The work of T.P.B. is supported by US National Institutes of Health (NIH) grants MH093429 and MH092769.
Glossary
- DNA response element
A short sequence of DNA that is specifically recognized and bound by a particular transcription factor. DNA response elements are often found in the promoter regions of genes, and confer the responsiveness of a gene to regulation by a particular transcription factor.
- E box
A particular DNA response element that is recognized by transcription factors belonging to the basic helix–loop–helix domain-containing family, such as circadian locomotor output cycles protein kaput (CLOCK) and brain and muscle ARNT-like 1 (BMAL1).
- Period (τ)
The time that elapses for one complete oscillation or cycle of a particular activity (for example, locomotor activity). Typically, the period for a circadian rhythm is almost 24 hours. In the absence of any extrinsic stimuli that act to ‘entrain’ the circadian rhythm (such as light), the period may differ; for example, mice typically have a period of slightly less than 24 hours in the absence of entrainment.
- ROR response element
A particular DNA response element that is recognized by retinoic acid receptor-related orphan receptors (RORs) and REV-ERBs.
- T helper 17 cells
(TH17 cells). A subset of TH cells that produce interleukin-17 (IL-17) and provide microbial immunity at mucosa and epithelial barriers. They have been implicated in the development of autoimmune disease.
- T cell
A type of lymphocyte that has a crucial role in cellular immunity. T cells can be distinguished from other lymphocytes based on the expression of the T cell receptor on their plasma membrane.
- Apo structure
A receptor structure that is free from a bound ligand.
- Inverse agonists
Ligands that suppress the basal activity of a receptor.
- Wheel running activity
A measure of locomotor activity as defined by rodents running on a wheel within a cage.
- Phase shift
A discrete alteration in an oscillation in locomotor activity or other measurable physiological activity along the time axis within a circadian rhythm.
- Area under the curve
(AUC). The area under the curve that is generated by plotting the concentration of a drug in plasma against time.
- Melatonin
A hormone that is produced by the pineal gland in a circadian manner and is associated with entrainment of the circadian rhythm.
- Fibroblast growth factor 21
(FGF21). A hormone that has several metabolic activities. FGF21 protects animals from diet-induced obesity and lowers blood glucose and lipid levels when administered to diabetic rodents.
- TReg cells
A subset of T cells that produce interleukin-10 (IL-10) and transforming growth factor-β (TGFβ) and have an important role in immune tolerance.
- TH1 cells
A subset of T helper (TH) cells that produces interferon-γ (IFNγ) and has an important role in cellular immunity.
- TH2 cells
A subset of T helper (TH) cells that produces interleukin-4 (IL-4), IL-5 and IL-13, and has an important role in humoral immunity.
Footnotes
Competing interests statement The authors declare no competing interests.
References
- 1.Mangelsdorf DJ, et al. The nuclear receptor superfamily — the 2nd decade. Cell. 2005;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKenna NJ, Lanz RB, O’Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr. Rev. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 3.Savkur RS, Burris TP. The coactivator LXXLL nuclear receptor recognition motif. J. Pept. Res. 2004;63:207–212. doi: 10.1111/j.1399-3011.2004.00126.x. [DOI] [PubMed] [Google Scholar]
- 4.Kliewer SA, Lehmann JM, Willson TM. Orphan nuclear receptors: shifting endocrinology into reverse. Science. 1999;284:757–760. doi: 10.1126/science.284.5415.757. [DOI] [PubMed] [Google Scholar]
- 5.Schulman IG, Heyman RA. The flip side: Identifying small molecule regulators of nuclear receptors. Chem. Biol. 2004;11:639–646. doi: 10.1016/j.chembiol.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 6.Burris TP, Busby SA, Griffin PR. Targeting orphan nuclear receptors for treatment of metabolic diseases and autoimmunity. Chem. Biol. 2012;19:51–59. doi: 10.1016/j.chembiol.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burris TP. Nuclear hormone receptors for heme: REV-ERBα and REV-ERBβ are ligand-regulated components of the mammalian clock. Mol. Endocrinol. 2008;22:1509–1520. doi: 10.1210/me.2007-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signal. 2009;7:e003. doi: 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jetten AM, Kang HS, Takeda Y. Retinoic acid-related orphan receptors α and γ: key regulators of lipid/glucose metabolism, inflammation, and insulin sensitivity. Frontiers Endocrinol. 2013;4:1. doi: 10.3389/fendo.2013.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duez H, Staels B. Rev-erb-α: an integrator of circadian rhythms and metabolism. J. Appl. Physiol. 2009;107:1972–1980. doi: 10.1152/japplphysiol.00570.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lazar MA, Hodin RA, Darling DS, Chin WW. A novel member of the thyroid steroid hormone receptor family is encoded by the opposite strand of the rat c-erbA-α transcriptional unit. Mol. Cell. Biol. 1989;9:1128–1136. doi: 10.1128/mcb.9.3.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyajima N, et al. 2 Erba homologs encoding proteins with different T3 binding-capacities are transcribed from opposite DNA strands of the same genetic-locus. Cell. 1989;57:31–39. doi: 10.1016/0092-8674(89)90169-4. [DOI] [PubMed] [Google Scholar]
- 13.Miyajima N, et al. Identification of 2 novel members of Erba superfamily by molecular-cloning - the gene-products of the 2 are highly related to each other. Nucleic Acids Res. 1988;16:11057–11074. doi: 10.1093/nar/16.23.11057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dumas B, et al. A new orphan member of the nuclear hormone receptor superfamily closely related to REV-ERB. Mol. Endocrinol. 1994;8:996–1005. doi: 10.1210/mend.8.8.7997240. [DOI] [PubMed] [Google Scholar]
- 15.Forman BM, et al. Cross talk among RORα1 and the REV-ERB family of orphan nuclear receptors. Mol. Endocrinol. 1994;8:1253–1261. doi: 10.1210/mend.8.9.7838158. [DOI] [PubMed] [Google Scholar]
- 16.Bonnelye E, et al. Rev-erb-β, a new member of the nuclear receptor superfamily, is expressed in the nervous-system during chicken development. Cell Growth Differ. 1994;5:1357–1365. [PubMed] [Google Scholar]
- 17.Yin L, Lazar MA. The orphan nuclear receptor Rev-erbα recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Mol. Endocrinol. 2005;19:1452–1459. doi: 10.1210/me.2005-0057. [DOI] [PubMed] [Google Scholar]
- 18.Harding HP, Lazar MA. The monomer-binding orphan receptor rev-erb represses transcription as a dimer on a novel direct repeat. Mol. Cell. Biol. 1995;15:6479–6479. doi: 10.1128/mcb.15.9.4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harding HP, Lazar MA. The orphan receptor rev-erba-α activates transcription via a novel response element. Mol. Cell. Biol. 1993;13:3113–3121. doi: 10.1128/mcb.13.5.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zvonic S, et al. Characterization of peripheral circadian clocks in adipose tissues. Diabetes. 2006;55:962–970. doi: 10.2337/diabetes.55.04.06.db05-0873. [DOI] [PubMed] [Google Scholar]
- 21.Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:929–937. doi: 10.1016/s0092-8674(00)81199-x. [DOI] [PubMed] [Google Scholar]
- 22.Guillaumond F, Dardente H, Giguere V, Cermakian N. Differential control of Bmal1 circadian transcription by REV-ERB and ROR nuclear receptors. J. Biol. Rhythms. 2005;20:391–403. doi: 10.1177/0748730405277232. [DOI] [PubMed] [Google Scholar]
- 23.Torra IP, et al. Circadian and glucocorticoid regulation of Rev-erbα expression in liver. Endocrinology. 2000;141:3799–3806. doi: 10.1210/endo.141.10.7708. [DOI] [PubMed] [Google Scholar]
- 24.Beckerandre M, Andre E, Delamarter JF. Identification of nuclear receptor messenger RNAs by RT-PCR amplification of conserved zinc finger motif sequences. Biochem. Biophys. Res. Commun. 1993;194:1371–1379. doi: 10.1006/bbrc.1993.1976. [DOI] [PubMed] [Google Scholar]
- 25.Giguere V, et al. Isoform specific amino-terminal domains dictate DNA binding properties of RORα, a novel family of orphan nuclear receptors. Genes Dev. 1994;8:538–553. doi: 10.1101/gad.8.5.538. [DOI] [PubMed] [Google Scholar]
- 26.Carlberg C, Vanhuijsduijnen RH, Staple JK, Delamarter JF, Beckerandre M. RZRs, a new family of retinoid-related orphan receptors that function as both monomers heterodimers. Mol. Endocrinol. 1994;8:757–770. doi: 10.1210/mend.8.6.7935491. [DOI] [PubMed] [Google Scholar]
- 27.Hirose T, Smith RJ, Jetten AM. RORγ - the 3rd member of ROR-RZR orphan receptor subfamily that is highly expressed in skeletal muscle. Biochem. Biophys. Res. Commun. 1994;205:1976–1983. doi: 10.1006/bbrc.1994.2902. [DOI] [PubMed] [Google Scholar]
- 28.Hamilton BA, et al. Disruption of the nuclear hormone receptor RORα in staggerer mice. Nature. 1996;379:736–739. doi: 10.1038/379736a0. [DOI] [PubMed] [Google Scholar]
- 29.Steinmayr M, et al. staggerer phenotype in retinoid-related orphan receptor α-deficient mice. Proc. Natl Acad. Sci. USA. 1998;95:3960–3965. doi: 10.1073/pnas.95.7.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andre E, Gawlas K, Steinmayr M, Becker-Andre M. A novel isoform of the orphan nuclear receptor ROR β is specifically expressed in pineal gland and retina. Gene. 1998;216:277–283. doi: 10.1016/s0378-1119(98)00348-5. [DOI] [PubMed] [Google Scholar]
- 31.Smith AG, Muscat GEO. Orphan nuclear receptors: therapeutic opportunities in skeletal muscle. Am. J. Physiol. Cell Physiol. 2006;291:C203–C217. doi: 10.1152/ajpcell.00476.2005. [DOI] [PubMed] [Google Scholar]
- 32.Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012;35:445–462. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang XY, et al. Cell. 2006;126:801–810. doi: 10.1016/j.cell.2006.06.050. [DOI] [PubMed] [Google Scholar]
- 34.Lamia KA, et al. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature. 2011;480:552–556. doi: 10.1038/nature10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmutz I, Ripperger JA, Baeriswyl-Aebischer S, Albrecht U. The mammalian clock component PERIOD2 coordinates circadian output by interaction with nuclear receptors. Genes Dev. 2010;24:345–357. doi: 10.1101/gad.564110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Preitner N, et al. The orphan nuclear receptor REV-ERBα controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251–260. doi: 10.1016/s0092-8674(02)00825-5. This paper describes the first characterization of a role for REV-ERB in the regulation of the circadian rhythm.
- 37.Triqueneaux G, et al. The orphan receptor Rev-erbα gene is a target of the circadian clock pacemaker. J. Mol. Endocrinol. 2004;33:585–608. doi: 10.1677/jme.1.01554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ripperger JA. Mapping of binding regions for the circadian regulators BMAL1 and CLOCK within the mouse Rev-erbα gene. Chronobiol. Int. 2006;23:135–142. doi: 10.1080/07420520500464411. [DOI] [PubMed] [Google Scholar]
- 39.Cho H, et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature. 2012;485:123–127. doi: 10.1038/nature11048. This study defines the critical overlapping role of REV-ERBα and REV-ERBβ in the control of the circadian rhythm.
- 40.Bunger MK, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vitaterna MH, et al. Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc. Natl Acad. Sci. USA. 1999;96:12114–12119. doi: 10.1073/pnas.96.21.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bae K, et al. Differential functions of mPer1, mPer2, and mPer3 in the SCN circadian clock. Neuron. 2001;30:525–536. doi: 10.1016/s0896-6273(01)00302-6. [DOI] [PubMed] [Google Scholar]
- 43.Sato TK, et al. A functional genomics strategy reveals rora as a component of the mammalian circadian clock. Neuron. 2004;43:527–537. doi: 10.1016/j.neuron.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 44.Andre E, et al. Disruption of retinoid-related orphan receptor β changes circadian behavior, causes retinal degeneration and leads to vacillans phenotype in mice. EMBO J. 1998;17:3867–3877. doi: 10.1093/emboj/17.14.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masana MI, Sumaya IC, Becker-Andre M, Dubocovich ML. Behavioral characterization and modulation of circadian rhythms by light and melatonin in C3H/HeN mice homozygous for the RORβ knockout. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;292:R2357–R2367. doi: 10.1152/ajpregu.00687.2006. [DOI] [PubMed] [Google Scholar]
- 46.Takeda Y, Jothi R, Birault V, Jetten AM. RORγ directly regulates the circadian expression of clock genes and downstream targets in vivo. Nucleic Acids Res. 2012;40:8519–8535. doi: 10.1093/nar/gks630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–1354. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell. 2008;134:728–742. doi: 10.1016/j.cell.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gamble KL, Young ME. Metabolism as an integral cog in the mammalian circadian clockwork. Crit. Rev. Biochem. Mol. Biol. 2013 doi: 10.3109/10409238.2013.786672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eckel-Mahan K, Sassone-Corsi P. Metabolism and the circadian clock converge. Physiol. Rev. 2013;93:107–135. doi: 10.1152/physrev.00016.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bass J. Circadian topology of metabolism. Nature. 2012;491:348–356. doi: 10.1038/nature11704. [DOI] [PubMed] [Google Scholar]
- 52.Li Y, Sato Y, Yamaguchi N. Shift work and the risk of metabolic syndrome: a nested case-control study. Int. J. Occupat. Environ. Health. 2011;17:154–160. doi: 10.1179/107735211799030960. [DOI] [PubMed] [Google Scholar]
- 53.Baron KG, Reid KJ, Kern AS, Zee PC. Role of sleep timing in caloric intake and BMI. Obesity. 2011;19:1374–1381. doi: 10.1038/oby.2011.100. [DOI] [PubMed] [Google Scholar]
- 54.Karlsson B, Knutsson A, Lindahl B. Is there an association between shift work and having a metabolic syndrome? Results from a population based study of 27,485 people. Occupat. Environ. Med. 2001;58:747–752. doi: 10.1136/oem.58.11.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scheer FAJL, Hilton MF, Mantzoros CS, Shea SA. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc. Natl Acad. Sci. USA. 2009;106:4453–4458. doi: 10.1073/pnas.0808180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Markwald RR, et al. Impact of insufficient sleep on total daily energy expenditure, food intake, and weight gain. Proc. Natl Acad. Sci. USA. 2013;110:5695–5700. doi: 10.1073/pnas.1216951110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raspe E, et al. Identification of Rev-erbα as a physiological repressor of apoC-III gene transcription. J. Lipid Res. 2002;43:2172–2179. doi: 10.1194/jlr.m200386-jlr200. [DOI] [PubMed] [Google Scholar]
- 58.Raspe E, Mautino G, Duez H, Fruchart JC, Staels B. Transcriptional regulation of apolipoprotein C-III gene expression by the orphan nuclear receptor Rev-erbα. Circulation. 2001;104:15–15. doi: 10.1074/jbc.M004982200. [DOI] [PubMed] [Google Scholar]
- 59.Vu-Dac N, et al. The nuclear receptors peroxisome proliferator-activated receptorα and Rev-erbα mediate the species-specific regulation of apolipoprotein A-I expression by fibrates. J. Biol. Chem. 1998;273:25713–25720. doi: 10.1074/jbc.273.40.25713. [DOI] [PubMed] [Google Scholar]
- 60.Ramakrishnan SN, Lau P, Burke LJ, Muscat GEO. Rev-erbβ regulates the expression of genes involved in lipid absorption in skeletal muscle cells — evidence for cross-talk between orphan nuclear receptors and myokines. J. Biol. Chem. 2005;280:8651–8659. doi: 10.1074/jbc.M413949200. [DOI] [PubMed] [Google Scholar]
- 61.Yin L, et al. Rev-erbα, a heme sensor that coordinates metabolic and circadian pathways. Science. 2007;318:1786–1789. doi: 10.1126/science.1150179. Along with reference 84, this study defines haem as an endogenous ligand of REV-ERB.
- 62.Delezie J, et al. The nuclear receptor REV-ERBα is required for the daily balance of carbohydrate and lipid metabolism. FASEB J. 2012;26:3321–3335. doi: 10.1096/fj.12-208751. [DOI] [PubMed] [Google Scholar]
- 63.Kang HS, et al. Gene expression profiling reveals a regulatory role for RORα and RORγ in phase I and phase II metabolism. Physiol. Genom. 2007;31:281–294. doi: 10.1152/physiolgenomics.00098.2007. [DOI] [PubMed] [Google Scholar]
- 64.Lau P, Fitzsimmons RL, Pearen MA, Watt MJ, Muscat GE. Homozygous staggerer (sg/sg) mice display improved insulin sensitivity and enhanced glucose uptake in skeletal muscle. Diabetologia. 2011;54:1169–1180. doi: 10.1007/s00125-011-2046-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Woldt E, et al. Rev-erba modulates skeletal muscle oxidative capacity by regulated mitochondrial biogenesis and autophagy. Nature Med. 2013;19:1039–1046. doi: 10.1038/nm.3213. This paper demonstrates that REV-ERB has a role in the regulation of the oxidative capacity of skeletal muscle, and also shows that SR9009 treatment could increase exercise endurance in mice.
- 66.Lau P, Nixon SJ, Parton RG, Muscat GEO. RORα regulates the expression of genes involved in lipid homeostasis in skeletal muscle cells: caveolin-3 and CPT-1 are direct targets of ROR. J. Biol. Chem. 2004;279:36828–36840. doi: 10.1074/jbc.M404927200. [DOI] [PubMed] [Google Scholar]
- 67.Chawla A, Lazar MA. Induction of REV-ERBα, an orphan receptor encoded on the opposite strand of the α thyroid hormone receptor gene, during adipocyte differentiation. J. Biol. Chem. 1993;268:16265–16269. [PubMed] [Google Scholar]
- 68.Fontaine C, et al. The orphan nuclear receptor Rev-Erbα is a peroxisome proliferator-activated receptor (PPAR) γ target gene and promotes PPARγ-induced adipocyte differentiation. J. Biol. Chem. 2003;278:37672–37680. doi: 10.1074/jbc.M304664200. [DOI] [PubMed] [Google Scholar]
- 69.Wang J, Lazar MA. Bifunctional role of Rev-erbα in adipocyte differentiation. Mol Cell. Biol. 2008;28:2213–2220. doi: 10.1128/MCB.01608-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kojetin DJ, Burris TP. A role for rev-erbα ligands in regulation of adipogenesis. Curr. Pharm. Design. 2011;17:320–324. doi: 10.2174/138161211795164211. [DOI] [PubMed] [Google Scholar]
- 71.Kumar N, et al. Regulation of adipogenesis by natural and synthetic REV-ERB ligands. Endocrinology. 2010;151:3015–3025. doi: 10.1210/en.2009-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lau P, et al. The orphan nuclear receptor, RORα, regulates gene expression that controls lipid metabolism: staggerer (sg/sg) mice are resistant to diet-induced obesity. J. Biol. Chem. 2008;283:18411–18421. doi: 10.1074/jbc.M710526200. [DOI] [PubMed] [Google Scholar]
- 73.Meissburger B, et al. Adipogenesis and insulin sensitivity in obesity are regulated by retinoid-related orphan receptor γ. EMBO Mol. Med. 2011;3:637–651. doi: 10.1002/emmm.201100172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Feng D, et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. 2011;331:1315–1319. doi: 10.1126/science.1198125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bettelli E, Oukka M, Kuchroo VK. TH-17 cells in the circle of immunity and autoimmunity. Nature Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 76.Fouser LA, Wright JF, Dunussi-Joannopoulos K, Collins M. Th17 cytokines and their emerging roles in inflammation and autoimmunity. Immunol. Rev. 2008;226:87–102. doi: 10.1111/j.1600-065X.2008.00712.x. [DOI] [PubMed] [Google Scholar]
- 77.Ivanov II, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. This study describes the crucial role of RORγ in the differentiation of TH17 cells.
- 78.Yang XXO, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gibbs JE, et al. The nuclear receptor REV-ERBα mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc. Natl Acad. Sci. USA. 2012;109:582–587. doi: 10.1073/pnas.1106750109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lam MT, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498:511–515. doi: 10.1038/nature12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yu X, et al. TH17 cell differentiation is regulated by the circadian clock. Science. 2013;342:727–730. doi: 10.1126/science.1243884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma H, et al. Increased atherosclerotic lesions in LDL receptor deficient mice with hematopoietic nuclear receptor Rev-erbα knock- down. J. Am. Heart Assoc. 2013;2:e000235. doi: 10.1161/JAHA.113.000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reinking J, et al. The Drosophila nuclear receptor E75 contains heme and is gas responsive. Cell. 2005;122:195–207. doi: 10.1016/j.cell.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 84.Raghuram S, et al. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBα and REV-ERBβ. Nature Struct. Mol. Biol. 2007;14:1207–1213. doi: 10.1038/nsmb1344. Along with reference 61, this study defines haem as an endogenous ligand of REV-ERB.
- 85.Wu N, Yin L, Hanniman EA, Joshi S, Lazar MA. Negative feedback maintenance of heme homeostasis by its receptor, Rev-erbα. Genes Dev. 2009;23:2201–2209. doi: 10.1101/gad.1825809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Estall JL, et al. PGC-1α negatively regulates hepatic FGF21 expression by modulating the heme/Rev-Erbα axis. Proc. Natl Acad. Sci. USA. 2009;106:22510–22515. doi: 10.1073/pnas.0912533106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rogers PM, Ying L, Burris TP. Relationship between circadian oscillations of Rev-erbα expression and intracellular levels of its ligand, heme. Biochem. Biophys. Res. Commun. 2008;368:955–958. doi: 10.1016/j.bbrc.2008.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pardee KI, et al. The structural basis of gas-responsive transcription by the human nuclear hormone receptor REV-ERBβ. Plos Biol. 2009;7:384–398. doi: 10.1371/journal.pbio.1000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gupta N, Ragsdale SW. Thiol-disulfide redox dependence of heme binding and heme ligand switching in nuclear hormone receptor rev-erbβ. J. Biol. Chem. 2011;286:4392–4403. doi: 10.1074/jbc.M110.193466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Phelan CA, et al. Structure of Rev-erbα bound to N-CoR reveals a unique mechanism of nuclear receptor-co-repressor interaction. Nature Struct. Mol. Biol. 2010;17:808–814. doi: 10.1038/nsmb.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marvin KA, et al. Nuclear receptors Homo sapiens rev-erb beta and Drosophila melanogaster E75 are thiolate-ligated heme proteins which undergo redox-mediated ligand switching and bind CO and NO. Biochemistry. 2009;48:7056–7071. doi: 10.1021/bi900697c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Golombek DA, Agostino PV, Plano SA, Ferreyra GA. Signaling in the mammalian circadian clock: the NO/cGMP pathway. Neurochem. Int. 2004;45:929–936. doi: 10.1016/j.neuint.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 93.Kallen JA, et al. X-ray structure of the hRORα LBD at 1.63 Å: structural and functional data that cholesterol or a cholesterol derivative is the natural ligand of RORα. Structure. 2002;10:1697–1707. doi: 10.1016/s0969-2126(02)00912-7. This study uses X-ray crystallography to show that sterols could bind to RORα.
- 94.Kallen J, Schlaeppi JM, Bitsch F, Delhon I, Fournier B. Crystal structure of the human RORα ligand binding domain in complex with cholesterol sulfate at 2.2 angstrom. J. Biol. Chem. 2004;279:14033–14038. doi: 10.1074/jbc.M400302200. [DOI] [PubMed] [Google Scholar]
- 95.Wang Y, Kumar N, Crumbley C, Griffin PR, Burris TP. A second class of nuclear receptors for oxysterols: regulation of RORα and RORγ activity by 24S-hydroxycholesterol (cerebrosterol) Biochim. Biophys. Acta. 2010;1801:917–923. doi: 10.1016/j.bbalip.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang Y, et al. Modulation of RORα and RORγ activity by 7-oxygenated sterol ligands. J. Biol. Chem. 2010;285:5013–5025. doi: 10.1074/jbc.M109.080614. This paper defines a role for oxysterols as ligands for RORα and RORγ.
- 97.Jin LH, et al. Structural basis for hydroxycholesterols as natural ligands of orphan nuclear receptor RORγ. Mol. Endocrinol. 2010;24:923–929. doi: 10.1210/me.2009-0507. This paper identifies a role for oxysterols as ligands for RORγ.
- 98.Stehlin-Gaon C, et al. All-trans retinoic acid is a ligand for the orphan nuclear receptor RORβ. Nature Struct. Biol. 2003;10:820–825. doi: 10.1038/nsb979. [DOI] [PubMed] [Google Scholar]
- 99.Helleboid S, et al. The identification of naturally occurring neoruscogenin as a bioavailable, potent, and high-affinity agonist of the nuclear receptor RORα (NR1F1) J. Biomol. Screen. 2013 doi: 10.1177/1087057113497095. http://dx.doi.org/10.1177/1087057113497095. [DOI] [PubMed] [Google Scholar]
- 100.Stehlin C, et al. X-ray structure of the orphan nuclear receptor RORβ ligand-binding domain in the active conformation. EMBO J. 2001;20:5822–5831. doi: 10.1093/emboj/20.21.5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Meng QJ, et al. Ligand modulation of REV-ERBα function resets the peripheral circadian clock in a phasic manner. J. Cell Sci. 2008;121:3629–3635. doi: 10.1242/jcs.035048. This was the first published description of a synthetic REV-ERB agonist.
- 102.Grant D, et al. GSK4112, a small molecule chemical probe for the cell biology of the nuclear heme receptor Rev-erbα. ACS Chem. Biol. 2010;5:925–932. doi: 10.1021/cb100141y. This is a description of the discovery of the first synthetic REV-ERB ligand.
- 103.Noel R, et al. Synthesis and SAR of tetrahydroisoquinolines as Rev-erbα agonists. Bioorg. Med. Chem. Lett. 2012;22:3739–3742. doi: 10.1016/j.bmcl.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shin Y, et al. Small molecule tertiary amines as agonists of the nuclear hormone receptor Rev-erbα. Bioorg. Med. Chem. Lett. 2012;22:4413–4417. doi: 10.1016/j.bmcl.2012.04.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Solt LA, et al. Regulation of circadian behavior and metabolism by synthetic REV-ERB agonists. Nature. 2012;485:62–68. doi: 10.1038/nature11030. This paper describes the discovery of SR9009 and SR9011, the first REV-ERB agonsts, which were used to define the effects of REV-ERB agonists on the circadian rhythm and metabolism in mice.
- 106.Yoo SH, et al. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl Acad. Sci. USA. 2004;101:5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Trump RP, et al. Optimized chemical probes for REV-ERBα. J. Med. Chem. 2013;56:4729–4737. doi: 10.1021/jm400458q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kojetin D, Wang Y, Kamenecka TM, Burris TP. Identification of SR8278, a synthetic antagonist of the nuclear heme receptor, REV-ERB. ACS Chem. Biol. 2011;6:131–134. doi: 10.1021/cb1002575. This study describes the first synthetic REV-ERB antagonist.
- 109.Vieira E, et al. The clock gene Rev-erbα regulates pancreatic β-cell function: modulation by leptin and high-fat diet. Endocrinology. 2012;153:592–601. doi: 10.1210/en.2011-1595. [DOI] [PubMed] [Google Scholar]
- 110.Negoro H, et al. Role of Rev-erbα domains for transactivation of the connexin43 promoter with Sp1. FEBS Lett. 2013;587:98–103. doi: 10.1016/j.febslet.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 111.Wiesenberg I, et al. Specific activation of the nuclear receptors PPARγ and RORA by the antidiabetic thiazolidinedione BRL 49653 and the antiarthritic thiazolidinedione derivative CGP 52608. Mol. Pharmacol. 1998;53:1131–1138. [PubMed] [Google Scholar]
- 112.Wiesenberg I, Missbach M, Kahlen JP, Schrader M, Carlberg C. Transcriptional activation of the nuclear receptor RZRα by the pineal-gland hormone melatonin and identification of cgp-52608 as a synthetic ligand. Nucleic Acids Res. 1995;23:327–333. doi: 10.1093/nar/23.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Missbach M, et al. Thiazolidine diones, specific ligands of the nuclear receptor retinoid Z receptor/retinoid acid receptor-related orphan receptor α with potent antiarthritic activity. J. Biol. Chem. 1996;271:13515–13522. doi: 10.1074/jbc.271.23.13515. [DOI] [PubMed] [Google Scholar]
- 114.Park Y, et al. N-methylthioureas as new agonists of retinoic acid receptor-related orphan receptor. Arch. Pharm. Res. 2012;35:1393–1401. doi: 10.1007/s12272-012-0809-0. [DOI] [PubMed] [Google Scholar]
- 115.Kumar N, et al. The benzenesulfonamide T0901317 is a novel RORα/γ inverse agonist. Mol. Pharmacol. 2010;77:228–236. doi: 10.1124/mol.109.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schultz JR, et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Houck KA, et al. T0901317 is a dual LXR/FXR agonist. Mol. Genet. Metab. 2004;83:184–187. doi: 10.1016/j.ymgme.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 118.Mitro N, Vargas L, Romeo R, Koder A, Saez E. T0901317 is a potent PXR ligand: implications for the biology ascribed to LXR. FEBS Lett. 2007;581:1721–1726. doi: 10.1016/j.febslet.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 119.Wang Y, et al. Identification of SR1078, a synthetic agonist for the orphan nuclear receptors RORA and RORG. ACS Chem. Biol. 2010;5:1029–1034. doi: 10.1021/cb100223d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang YJ, Solt LA, Burris TP. Regulation of FGF21 expression and secretion by retinoic acid receptor-related orphan receptor α. J. Biol. Chem. 2010;285:15668–15673. doi: 10.1074/jbc.M110.102160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kharitonenkov A, et al. FGF-21 as a novel metabolic regulator. J. Clin. Invest. 2005;115:1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kharitonenkov A, et al. FGF-21 regulates the metabolic state of diabetic monkeys. Diabetes. 2007;56:A668–A668. [Google Scholar]
- 123.Zhu Y, McAvoy S, Kuhn R, Smith DI. RORA, a large common fragile site gene, is involved in cellular stress response. Oncogene. 2006;25:2901–2908. doi: 10.1038/sj.onc.1209314. [DOI] [PubMed] [Google Scholar]
- 124.Wang Y, Solt LA, Kojetin DJ, Burris TP. Regulation of p53 stability and apoptosis by a ROR agonist. PloS ONE. 2012;7:e34921. doi: 10.1371/journal.pone.0034921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kumar N, et al. Identification of SR3335 (ML-176): a synthetic RORα selective inverse agonist. ACS Chem. Biol. 2011;6:218–222. doi: 10.1021/cb1002762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Solt LA, et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature. 2011;472:491–494. doi: 10.1038/nature10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xu J, Wagoner G, Douglas JC, Drew PD. Liver X receptor agonist regulation of Th17 lympocyte function in autoimmunity. J. Leukoc. Biol. 2009;86:401–409. doi: 10.1189/jlb.1008600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xu JH, Racke MK, Drew PD. Peroxisome proliferator-activated receptor-α agonist fenofibrate regulates IL-12 family cytokine expression in the CNS: relevance to multiple sclerosis. J. Neurochem. 2007;103:1801–1810. doi: 10.1111/j.1471-4159.2007.04875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kumar N, et al. Identification of SR2211: a potent synthetic RORγ-selective modulator. ACS Chem. Biol. 2012;7:672–677. doi: 10.1021/cb200496y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Solt LA, et al. Identification of a selective RORγ ligand that suppresses TH17 cells and stimulates T regulatory cells. ACS Chem. Biol. 2012;7:1515–1519. doi: 10.1021/cb3002649. This paper demonstrates that at least a subgroup of RORγ inverse agonists stimulate the differentiation of TReg cells as well as suppressing TH17 cell polarization.
- 131.Huh JR, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature. 2011;472:486–490. doi: 10.1038/nature09978. Along with reference 126, this study demonstrates that a RORγ inverse agonist suppresses TH17 cell development and has potential for the treatment of autoimmune disorders.
- 132.Fujita-Sato S, et al. Structural basis of digoxin that antagonizes RORγt receptor activity and suppresses TH17 cell differentiation and interleukin (IL)-17 production. J. Biol. Chem. 2011;286:31409–31417. doi: 10.1074/jbc.M111.254003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Eade E, Cooper R, Mitchell AR. Digoxin — time to take the gloves off? Int. J. Cardiol. 2013;164:365–367. doi: 10.1016/j.ijcard.2011.07.034. [DOI] [PubMed] [Google Scholar]
- 134.Matsui H, Schwartz A. Mechanism of cardiac glycoside inhibition of the (Na+-K+)-dependent ATPase from cardiac tissue. Biochim. Biophys. Acta. 1968;151:655–663. doi: 10.1016/0005-2744(68)90013-2. [DOI] [PubMed] [Google Scholar]
- 135.Xu T, et al. Ursolic acid suppresses IL-17 production by selectively antagonizing the function of RORγt protein. J. Biol. Chem. 2011;286:22707–22710. doi: 10.1074/jbc.C111.250407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.He Y, Li Y, Zhao T, Wang Y, Sun C. Ursolic acid inhibits adipogenesis in 3T3-L1 adipocytes through LKB1/AMPK pathway. PLoS ONE. 2013;8:e70135. doi: 10.1371/journal.pone.0070135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li L, et al. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res. 2013;1497:32–39. doi: 10.1016/j.brainres.2012.12.032. [DOI] [PubMed] [Google Scholar]
- 138.You HJ, et al. Ursolic acid enhances nitric oxide and tumor necrosis factor-α production via nuclear factor-κB activation in the resting macrophages. FEBS Lett. 2001;509:156–160. doi: 10.1016/s0014-5793(01)03161-1. [DOI] [PubMed] [Google Scholar]
- 139.Shishodia S, Majumdar S, Banerjee S, Aggarwal BB. Ursolic acid inhibits nuclear factor-κB activation induced by carcinogenic agents through suppression of IκBα kinase and p65 phosphorylation: correlation with down-regulation of cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D1. Cancer Res. 2003;63:4375–4383. [PubMed] [Google Scholar]
- 140.Lin J, et al. Ursolic acid promotes colorectal cancer cell apoptosis and inhibits cell proliferation via modulation of multiple signaling pathways. Int. J. Oncol. 2013;43:1235–1243. doi: 10.3892/ijo.2013.2040. [DOI] [PubMed] [Google Scholar]
- 141.Pathak AK, et al. Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells. Mol. Cancer Res. 2007;5:943–955. doi: 10.1158/1541-7786.MCR-06-0348. [DOI] [PubMed] [Google Scholar]
- 142.Cha HJ, et al. Ursolic acid-induced down-regulation of MMP-9 gene is mediated through the nuclear translocation of glucocorticoid receptor in HT1080 human fibrosarcoma cells. Oncogene. 1998;16:771–778. doi: 10.1038/sj.onc.1201587. [DOI] [PubMed] [Google Scholar]
- 143.Chang MR, Lyda B, Kamenecka TM, Griffin PR. Pharmacological repression of RORγ is therapeutic in the collagen induced arthritis experimental model. Arthritis Rheum. 2013 doi: 10.1002/art.38272. http://dx.doi.org/10.1002/art.38272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Huang W, et al. Probe Reports from the NIH Molecular Libraries Program [online] US National Center for Biotechnology Information; 2010. Identification of potent and selective RORγ antagonists. http://www.ncbi.nlm.nih.gov/books/NBK133441/ [PubMed] [Google Scholar]
- 145.Zhang W, et al. Increasing human Th17 differentiation through activation of orphan nuclear receptor retinoid acid-related orphan receptor γ (RORγ) by a class of aryl amide compounds. Mol. Pharmacol. 2012;82:583–590. doi: 10.1124/mol.112.078667. [DOI] [PubMed] [Google Scholar]