

In the natural environment, organisms have to cope with a large number of stresses. Comparison of the gene expression response to different stresses across different yeast species shows that transcriptomic stress response can be linked to the lifestyle of the studied species.
Abstract
Defining how organisms respond to environmental change has always been an important step toward understanding their adaptive capacity and physiology. Variation in transcription during stress has been widely described in model species, especially in the yeast Saccharomyces cerevisiae, which helped to shape general rules regarding how cells cope with environmental constraints, as well as to decipher the functions of many genes. Comparison of the environmental stress response (ESR) across species is essential to obtaining better insight into the common and species-specific features of stress defense. In this context, we explored the transcriptional landscape of the yeast Lachancea kluyveri (formerly Saccharomyces kluyveri) in response to diverse stresses, using RNA sequencing. We investigated variation in gene expression and observed a link between genetic plasticity and environmental sensitivity. We identified the ESR genes in this species and compared them to those already found in S. cerevisiae. We observed common features between the two species, as well as divergence in the regulatory networks involved. Of interest, some changes were related to differences in species lifestyle. Thus we were able to decipher how adaptation to stress has evolved among different yeast species. Finally, by analyzing patterns of coexpression, we were able to propose potential biological functions for 42% of genes and also annotate 301 genes for which no function could be assigned by homology. This large data set allowed for the characterization of the evolution of gene regulation and provides an efficient tool for assessing gene function.
INTRODUCTION
Cells are exposed to a variety of environmental stresses and must be able to respond appropriately to them. Whole-genome transcriptomic analysis is one way in which to elucidate how cells trigger specific events or respond to environmental changes and identify the genes involved. The transcriptomic response in the yeast Saccharomyces cerevisiae to different stresses has been described by thorough case-by-case studies (Alexandre et al., 2001; Morano et al., 2012; Kasavi et al., 2014), some related to industrial conditions (Rossignol et al., 2003; Smart, 2007) and others providing a broader global view of stress response by comparing multiple conditions (Gasch et al., 2000; Causton et al., 2001). These studies have led to a description of the general rules governing yeast cell adaptation to stressful conditions. Two types of stress response have been observed, one corresponding to a specific response involving genes with functions related to the tested stress, and the other to a more general response observed regardless of the type of stress involved. The latter case, known as the environmental stress response (ESR), was first described by Gasch et al. (2000), who analyzed the expression response to 13 different types of stress (Gasch et al., 2000). For S. cerevisiae, the positive ESR genes, which are up-regulated in response to all stresses, are involved in oxidation processes, as well as in stress signaling, and include genes regulated by protein kinase A (PKA; Cullen and Sprague, 2012; Vaidyanathan et al., 2014) and the Msn2p/Msn4p network (Martínez-Pastor et al., 1996; Morano et al., 2012). The negative ESR category includes genes involved in essential processes assuring optimal cell growth, such as ribosome biogenesis and fermentation (Gasch et al., 2000; Brauer et al., 2008).
The description of the transcriptomic response to various conditions has also allowed for the identification of the functions for many genes (Ashburner et al., 2000; Nieduszynski and Liti, 2011; Andersson Rasmussen et al., 2014). With the ever-increasing stock of available fully sequenced species, there is a tremendous amount of new genes yet to be associated with a function (Génolevures Consortium et al., 2009; Scannell et al., 2011). High-throughput transcriptomic analysis is a powerful tool for resolving this problem. Automated approaches have been developed in yeast to propose gene function according to their variation in expression, based on the hypothesis that genes with similar patterns of expression across several conditions are likely involved in the same biological processes (Ashburner et al., 2000; Brauer et al., 2008; Caudy et al., 2013). Within S. cerevisiae, this approach was applied by using expression data in conjunction with computational function prediction to find mitochondrially localized proteins and revealed 47 genes previously not believed to be linked to the mitochondria (Hess et al., 2009). Another study based on the same principle improved the annotation of the CBS 7001 Saccharomyces uvarum isolate, formerly named Saccharomyces bayanus (Almeida et al., 2014). It compared S. uvarum gene expression to S. cerevisiae expression data to propose involvement in pathways for each gene (Caudy et al., 2013; Guan et al., 2013).
The exploration of expression patterns in response to diverse environmental stresses has focused on a limited number of yeast species, including S. cerevisiae (Gasch, 2007; Slavov et al., 2012). The comparison of environmental stress response among species is now feasible and essential in order to have a better insight into the features of stress defense, as well as for the functional annotation of genes. With this in mind, we characterized the gene expression landscape of Lachancea kluyveri (strain CBS 3082) across 20 environmental conditions using RNA sequencing. Of interest, L. kluyveri belongs to a clade not closely related to S. cerevisiae or the Saccharomyces genus (Génolevures Consortium et al., 2009; Jung et al., 2012; Friedrich et al., 2015). This species is preduplicated, that is, it diverged from the S. cerevisiae lineage before its ancestral whole-genome duplication >100 million years ago (Wolfe and Shields, 1997). Hence L. kluyveri is characterized by physiological differences from this main model yeast species. Of note, it represents a transitional state between the respiratory lifestyle of the Kluyveromyces clade and the respirofermentative one of S. cerevisiae. For example, this species lacks the capacity to be “petite” (loss of mitochondria; Hagman et al., 2013, 2014). Therefore completing a description of regulation perturbation due to stresses in this species would allow for the description of how stress response has evolved in parallel with genomic and physiological change. Finally, annotation of the L. kluyveri reference genome revealed that 761 genes had only low similarities to their S. cerevisiae orthologues and 466 had no orthologue (Supplemental Figure S1; Génolevures Consortium et al., 2009; Vakirlis et al., 2016). Consequently, improving the current genome annotation by high-throughput transcriptomic analysis is of great interest in this species.
By analyzing genomic expression patterns in L. kluyveri in response to diverse environmental stresses, we described the change of expression related to specific environments. We also developed a method to define a relevant list of ESR genes for any species by using a relatively small gene expression data set and taking advantage of the high-quality list of ESR genes defined by Gasch et al. (2000). We obtained an exhaustive list of ESR genes and compared this set of genes to that already determined for S. cerevisiae to provide an overview of how distantly related yeasts cope with stress. We observed that several differences can be linked to lifestyle. In addition, this large data set provided a powerful basis for deciphering gene function. Using a similar approach to previous work in S. cerevisiae and S. uvarum (Hess et al., 2009; Caudy et al., 2013), we applied a pipeline using Gene Ontology (GO) term enrichment to coexpressed genes in order to propose putative processes for 42% of the L. kluyveri genes, of which 301 did not previously have a reliably assigned function.
RESULTS
Transcriptional landscape in response to environmental changes in L. kluyveri
To assess the expression response to environmental change in L. kluyveri, we sequenced the total mRNA of the reference strain CBS 3082 during exponential growth under 20 conditions, including various carbon sources, temperatures, ionic stresses, and drugs (Supplemental Table S1). These stresses induce a reduction of growth rate ranging between 1% (growth at 37°C) and 50% (glycerol; Supplemental Table S1). We first analyzed the similarity among transcriptomic patterns by a principal component analysis and observed nonclustered dispersion, which indicated that the stresses had diverse influences on expression (Supplemental Figure S2). Of interest, three conditions (glycerol, cobalt sulfate, and calcium chloride) cluster separately from the others, possibly indicating a stronger stress effect, with a larger effect on transcript abundance.
We then completed a hierarchical clustering (HC) analysis, consisting of grouping genes that displayed the same pattern of variation across conditions. Only genes involved in significant clustering linkage were displayed, which reduced noise and allowed for the description of only relevant groups (HC height < 2.2; see Materials and Methods). Ultimately, a subset of 2289 genes was used in the final HC (Supplemental Figure S3). Functional enrichment within each cluster was identified in biological processes, including respiration, lipid and nitrogen metabolism, transcription, and ribosome biogenesis. This observation indicated that genes that display the same transcriptomic response to environmental changes are likely to be involved in the same biological processes. By a differential analysis comparing each condition to the complete reference medium (yeast extract 1%, peptone 2%, glucose 2% [YPD]), we observed which pathways were up- or down-regulated in each case (Supplemental Figure S4). Most of the affected pathways were clearly in agreement with the specific condition tested. As an example, growth on the carbon sources galactose and glycerol was responsible for overexpression of genes involved in aerobic respiration on the order of an approximately ∼1 log-fold change (log2FC) compared with the control medium (e.g., for COX13, log2FC = +1.02, and for SDH3, log2FC = +1.24), whereas the minimal limited medium (yeast nitrogen base) activated genes involved in amino acid and lipid biosynthesis (e.g., for ARG8, log2FC = +1.93, for LEU4, log2FC = +3.92, and for MET6, log2FC = +1.92) while simultaneously reducing the expression of nutrient transporters (e.g., for BAP2, log2FC = −1.78, and for GAP1, log2FC = −1.22).
We also compared the effect of environment and genetic background on the regulation of gene expression. For this purpose, we integrated into the analysis our previous description of expression variation across natural isolates within L. kluyveri (Brion et al., 2015). For each gene, we measured the genetic plasticity (GP) of its expression, which is calculated as the SD of mRNA abundance across different L. kluyveri strains (24 haploid isolates of different mating type), all measured in the same standard condition (YPD). A high value indicated that the transcription was less conserved within the species. The environmental sensitivity (ES) of each gene was also calculated using the SD in mRNA abundance among the various conditions. We observed significant positive correlation between GP and ES (R = 0.50, p < 10−15; Figure 1), indicating that genes triggered by environmental change display higher levels of GP. However, some genes harbored a greater GP than ES (log(GP/ES) > 0.7; 456 genes), with functional enrichment found related to mating (p = 6.8 × 10−5), flocculation (p = 4.4 × 10−5), and meiotic recombination (p = 3.7 × 10−5). Conversely, the genes with a higher ES than GP (log(ES/GP) > 0.7; 133 genes) were involved in rRNA processing (p = 2.0 × 10−13), fatty acid β oxidation (p = 5.9 × 10−7), and iron ion homeostasis (p = 9.9 × 10−5). These processes are known to be under environmental control and correspond to cell metabolic adaptation to stress (Gasch et al., 2000).
FIGURE 1:
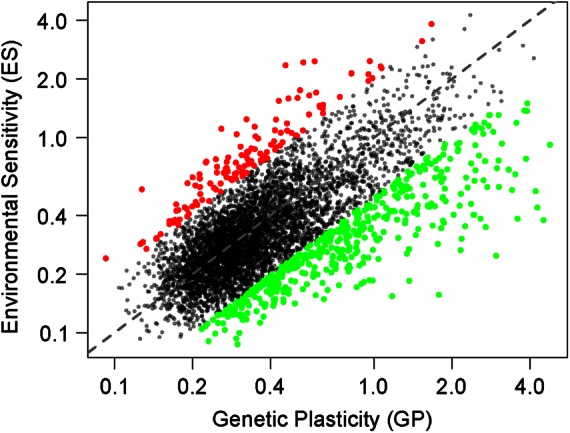
Comparison of L. kluyveri gene expression variation across different environments and genetic backgrounds. Environmental sensitivity (ES) is measured as the standard variation in expression level (log2 of the normalized read number) across conditions. Genetic plasticity (GP) is the standard variation in expression level (log2 of the normalized read number) across strains. Green dots correspond to genes with a higher GP than ES and were selected for GO term enrichment. Red dots correspond to genes with a higher ES than GP and were selected for GO term enrichment. Axes are on a logarithmic scale.
General environmental stress response in L. kluyveri
The genes involved in the ESR were defined as those differentially expressed regardless of the type of stress. In S. cerevisiae, Gasch et al. (2000) characterized these genes by analyzing the kinetics of the transcriptomic profile changes under 13 different stresses across >100 arrays. They used a clustering analysis approach to define the set of genes that responded in a “stereotypical manner” to all of the stress inductions. Because the intensity of the stress among our conditions varied (see growth rate, Supplemental Table S1) and we did not perform a kinetic test during the stress response, we chose another approach, based on the pattern of gene expression. We used the list of 868 S. cerevisiae ESR genes published by Gasch et al. (2000; www-genome.stanford.edu/yeast_stress) to define those in L. kluyveri (see Materials and Methods). From the list, we disregarded the genes absent in L. kluyveri and those that displayed an expression pattern too divergent from the average ESR expression behavior, indicating that they are not controlled by the stress level in L. kluyveri. We controlled such that these genes are all up-regulated (positive ESR) or down-regulated (negative ESR) compared with the reference condition and disregarded those that did not demonstrate a strong shift in expression. The remaining genes constituted the common ESR shared by S. cerevisiae and L. kluyveri. We obtained the L. kluyveri–specific ESR genes by identifying those with an expression pattern similar to the set of common ESRs (see Materials and Methods and Figure 2, A and B). We obtained 590 negative and 290 positive ESR genes (Figure 2, A–C).
FIGURE 2:

Definition of ESR genes in L. kluyveri using S. cerevisiae ESR. (A) Comparison of the sets of ESR genes in L. kluyveri (horizontal rectangle) and S. cerevisiae (vertical rectangle). The areas are proportional to the corresponding number of genes (indicated inside the boxes). The GO term enrichment is indicated in the boxes. CR, coregulated; DR, down-regulated; UR, up-regulated. The New box corresponds to the L. kluyveri ESR genes whose orthologues are not among S. cerevisiae ESR genes. (B) Schematic of the principle of the method used to define L. kluyveri ESR genes based on S. cerevisiae ESR genes. (C) Clustering of all L. kluyveri ESR gene expressions after normalization with the reference medium YPD (yellow, up-regulated; blue, down-regulated).
The average log2FC values for positive and negative ESR genes across all conditions were highly correlated, although they were calculated independently (R2 = 0.94, p = 9.3 × 10−13; Figure 2C and Supplemental Figure S5). The conditions that induced the highest stress levels (i.e., high average log2FC) included glycerol as a carbon source (2%), cobalt sulfate (0.25 mM), and calcium chloride (100 mM), and the conditions leading to very low stress levels were nickel sulfate (10 mM), arsenic (0.05 mM), and lower temperature (23°C).
We also studied the expression variation among negative and positive ESR genes and found that both groups displayed higher environmental sensitivity than the rest of the genome (t test, p < 10−14; Figure 3A). However, they did not display the same behavior concerning genetic plasticity, with reduced plasticity observed in negative ESR (t test, p < 10−14; Figure 3B) and higher plasticity in positive ESR genes (t test, p = 5 × 10−6; Figure 3B). This indicates that the negative ESR genes tend to have conserved levels of expression across strains, which is explained by enrichment of housekeeping genes (e.g., ribosome biogenesis and translation) being highly conserved within and across species. The positive ESR genes displayed a higher level of expression variation across strains, which suggests that strains underwent adaptation to differential stresses by modifying the expression of genes generally activated during stress.
FIGURE 3:
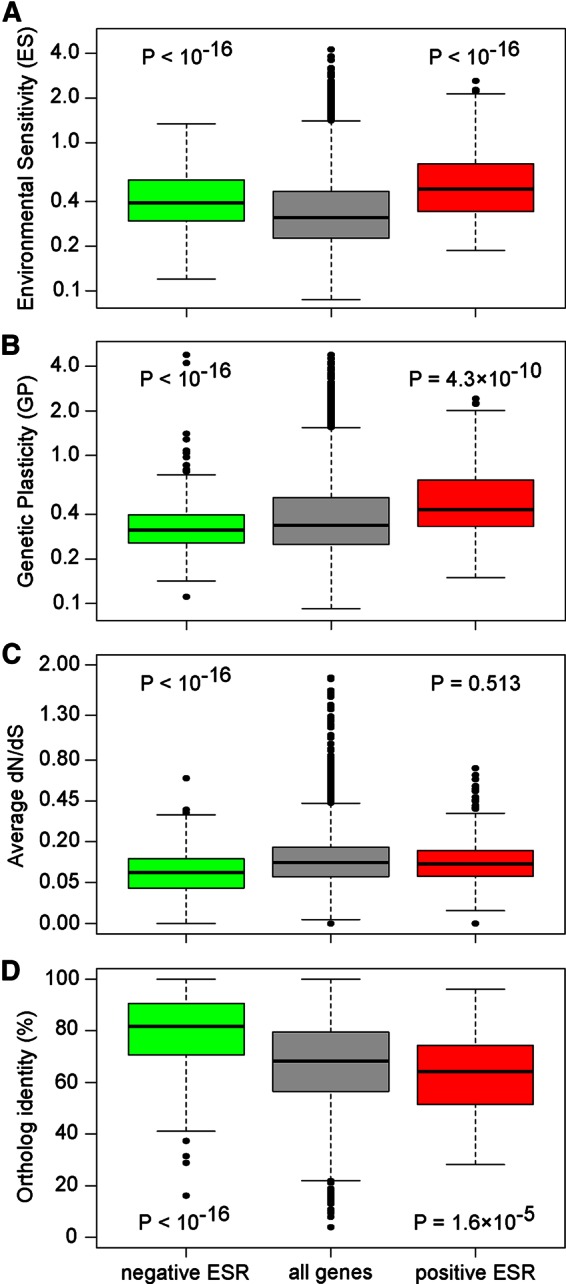
Specific behavior of the ESR genes. Green, negative ESR; red, positive ESR. The p values correspond to a t test compared with all genes. (A) Environmental sensitivity (axis on a log scale). (B) Genetic plasticity (axis on a log scale). (C) Intraspecies protein sequence variation (measured by dN/dS; axis on a square root scale). (D) L. kluyveri–S. cerevisiae protein sequence conservation (percentage of identity).
Comparison of ESR genes among yeast species
To describe the similarities and differences in ESR in the two species, we used GO term enrichment based on the S. cerevisiae gene annotations and all S. cerevisiae genes as background set, allowing for a qualitative overview of the functions involved. In total, 367 negative ESR genes are shared between the two species and are involved in ribosome biogenesis (p < 10−14) and translation (p < 10−14). The same enrichments were found for the 226 S. cerevisiae negative ESR genes not in L. kluyveri, as well as for the 223 new ESR genes identified in this species. However, more specifically, in L. kluyveri, we observed a loss of the ESR expression pattern for the pyruvate decarboxylase (PDC-like) genes, whereas those involved in mRNA transport displayed this pattern, contrary to what was found in S. cerevisiae (p = 2.0 × 10−6; Gasch et al., 2000).
There were 94 positive ESR genes shared between the two species, among which we observed a weak enrichment for genes involved in the metabolism of energy reserves (e.g., glycogen and trehalose; p = 8.6 × 10−7) and a high number of uncharacterized proteins (30 genes; p = 6.0 × 10−4). There were 212 S. cerevisiae positive ESR genes not present in L. kluyveri, displaying no orthologue (76 genes), or having a pattern different from the ESR expression pattern (136 genes). The last-named group was involved in the oxidation-reduction process (p = 2.0 × 10−10) and cellular response to oxidative stress (p = 1.3 × 10−9). In L. kluyveri, the 196 newly identified positive ESR genes did not display significant process enrichment; however, among them, we observed genes involved in autophagy (ATG9, ATG15, ATG21, ATG32, and CIS1 orthologues).
Considering that S. cerevisiae ESR genes were not defined using correlation, we needed to determine the extent to which, within S. cerevisiae, the novel ESR genes defined in L. kluyveri were coexpressed with the previously defined S. cerevisiae ESR genes. For each orthologue of these new ESR genes, we measured the average of all pairwise correlations with the S. cerevisiae ESR genes (Supplemental Figure S6). We observed that 83% of positive ESR genes and 66% of negative ESR genes newly identified in L. kluyveri had orthologues in S. cerevisiae that were not highly similar to the ESR expression pattern of this species (Pearson’s R < 0.54, threshold defined by permutation; see Materials and Methods). These results implied that the integration of these genes among L. kluyveri ESR genes is supported by a change in their regulatory mechanisms across species.
The main characteristics of the L. kluyveri ESR were the absence of an oxidative stress response, which is present in the positive S. cerevisiae ESR, and the absence of PDC11 and PDC12, orthologues to PDC1 and PDC5, which are involved in alcoholic fermentation and are present in the negative S. cerevisiae ESR. To determine whether these differences were specific to L. kluyveri or S. cerevisiae, we identified the ESR genes of a third species, S. uvarum, using the approach developed here in conjunction with previously published expression data across several stress inductions (Caudy et al., 2013). In this species, the ESR genes include the main pyruvate decarboxylase PDC1 and 13 of the 22 genes involved in the cellular response to oxidative stress that are part of the S. cerevisiae ESR (59%; Figure 4A). This confirmed that the high representation of oxidative stress response genes in positive ESRs is a characteristic shared across Saccharomyces species. However, whereas the L. kluyveri ESR does not include most of the oxidative stress response genes, such as HSP12, PRX1, GPX1, and OXR1 orthologues, we still observed a few of them having a high correlation with the average ESR gene expression pattern (e.g., for GRE3, R = 0.89; for UGA2, R = 0.88, and for TPS1, R = 0.84), but these genes are pleiotropic and have other functions than oxidative stress response.
FIGURE 4:
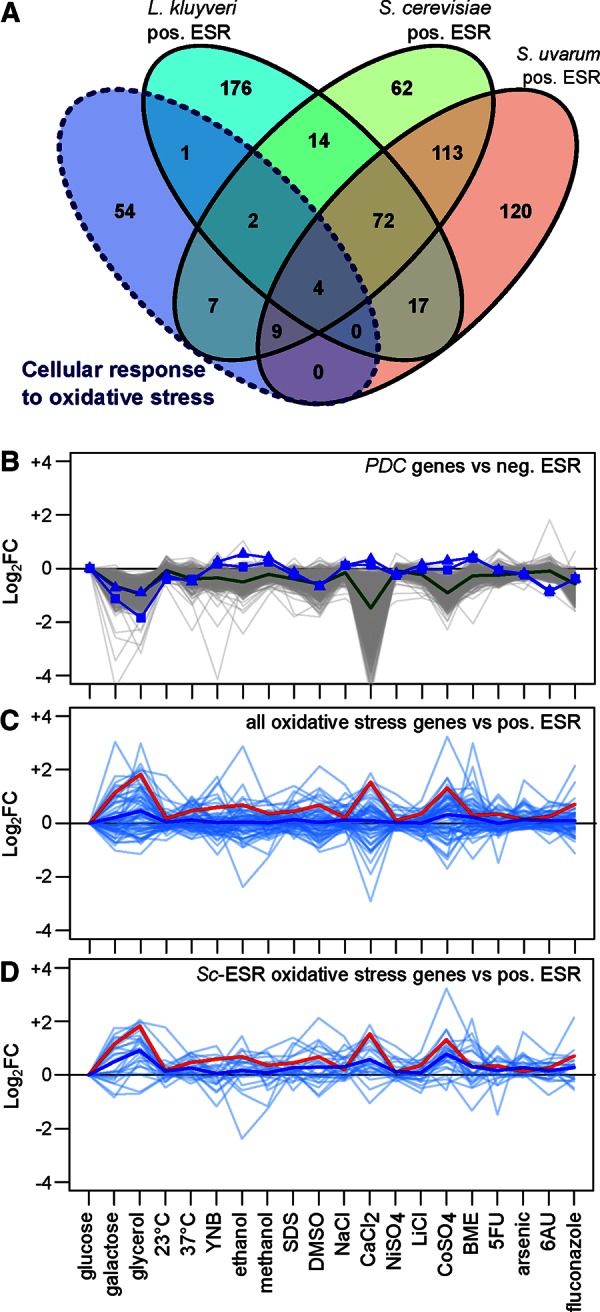
Description of the main differences between S. cerevisiae, S. uvarum, and L. kluyveri ESRs. (A) Venn diagram comparison of the ESRs of the three species and the number of genes involved in the cellular response to oxidative stress for each. The number of genes might be biased by the annotation depth of each species. (B) Comparison of expression patterns of PDC genes to negative ESR genes in L. kluyveri (PDC11: squares, R = 0.00; PDC12: triangles, R = 0.23; green, average negative ESR behavior, gray: all negative ESR behavior). (C) Comparison of expression patterns of genes involved in cellular response to oxidative stress and positive ESR genes in L. kluyveri (blue, average behavior of all cellular responses to oxidative stress orthologues, R = 0.15; red, average positive ESR behavior; light blue, all cellular response to oxidative stress individual behavior). (D) Comparison of expression patterns of the genes involved in the cellular response to oxidative stress that are also included in S. cerevisiae ESR and the positive L. kluyveri ESR genes (blue, average behavior of cellular response to oxidative stress genes included in S. cerevisiae ESR; R = 0.43; red, average positive ESR behavior; light blue, all cellular responses to oxidative stress individual behavior).
We compared the pattern of expression of the PDC and oxidative stress response genes to that of the ESR genes in order to characterize the different behaviors of these biological processes. We observed that PDC11 and PDC12 had a very low correlation with the negative ESR gene pattern (R = 0.00 and 0.23, respectively). Genes inferred to be involved in the cellular response to oxidative stress also have a low average correlation with positive ESR (R = 0.15), and this value increases (R = 0.43) when only oxidative stress response genes in the S. cerevisiae ESR are taken into account; this effect is due to the pleiotropic genes described earlier (Figure 4, B–D).
The loss of both PDC-like genes and oxidative stress response genes in the L. kluyveri ESR can be linked to the transitional aspects of this species compared with the complete respirofermentative lifestyle of S. cerevisiae and S. uvarum (Hagman et al., 2013). The final list of ESR genes is given in Supplemental Table S2.
Evolution of the ESR genes within L. kluyveri and across the Saccharomycotina subphylum
The ESR genes play a role in coping with changes in environmental conditions and thus in adaptation. Therefore we specifically focused on the evolutionary history of ESR genes. First, we compared the ratio of the densities of nonsynonymous and synonymous mutations (dN/dS) between the ESR genes and the whole genome. We observed stronger purifying selection, as measured by dN/dS, for the negative ESR genes. This might be linked to the involvement of the majority of these genes in fundamental cell processes (such as ribosome biosynthesis). Such protein conservation could also be associated with high expression levels, as reported previously (Pál et al., 2001; Wilke and Drummond, 2006; Brion et al., 2015). No specific selection pressure on the positive ESR was identified (Figure 3C).
To evaluate sequence conservation of the ESR genes at the interspecies scale, we calculated the percentage identity between the protein sequences of L. kluyveri and S. cerevisiae orthologues. We observed that conservation is high among negative ESR genes, with half displaying >80% identity, whereas positive ESR genes were less conserved, as the majority shared ∼65% identity (Figure 3D). Moreover, only five genes without an orthologue in S. cerevisiae were found among the negative ESR genes. In contrast, 34 genes without an orthologue, including three L. kluyveri–specific genes (ORFans; Vakirlis et al., 2016) were found among the positive ESRs, which are more than would have been expected by chance (Fisher’s exact test, p = 0.031). The fact that positive ESR genes are less conserved across species, with many not present in all Saccharomycotina yeast, indicates that genes activated in stressful conditions are under species-specific selection pressures. Therefore, among these genes, many have unknown functions, confirming the need to propose potential roles for them.
Use of gene expression patterns to infer biological function of genes
There are ∼6000 annotated sequences in L. kluyveri, including 456 noncoding RNAs (Supplemental Figure S1). Most of the open reading frames (ORFs) are functionally annotated based on orthology with the S. cerevisiae genome. For 75% of the ORFs (4081), the orthologue identity level is >50%, which indicates a reliable annotation. However, 466 genes do not share sufficient similarity with any S. cerevisiae gene to allow for functional assignment. For 329 of these genes, orthologues were found in other species, and, in an additional 137, no direct orthologues were observed (Génolevures Consortium et al., 2009). Furthermore, a study of genome evolution across 10 Lachancea species allowed for the detection of 74 Lachancea-specific genes and 70 ORFans in L. kluyveri (Vakirlis et al., 2016). This high number of uncharacterized genes is a unique characteristic of L. kluyveri.
Our large data set is a promising tool that can be used to enhance gene annotation. To propose a potential biological process for each gene by examining patterns of coexpression, we set up a simple automated pipeline involving three steps (Figure 5A). From a single gene, we first obtained the list of genes with which it is coexpressed among multiple strains and various conditions. Second, from that list, we compiled a set of S. cerevisiae orthologues and performed a GO term enrichment analysis. The GO terms with the highest scores were proposed to represent processes in which the gene might be involved. The third step consisted of comparing the GO terms proposed by our approach and the annotated biological process of the originally identified S. cerevisiae orthologue. The reliability of this approach was evaluated using S. cerevisiae expression data across different strains (see Materials and Methods and Figure 5B).
FIGURE 5:

Automated approach to assigning pathways to genes. (A) The three steps of the pipeline for pathway proposal. Steps 2 and 3 are completed using the topGO and GOsim packages of R Bioconductor, respectively. (B) Comparison of the success rates using this approach in S. cerevisiae and L. kluyveri. H, genes highly conserved in L. kluyveri compared with S. cerevisiae. WA, genes weakly annotated, corresponding to genes with few or no similarities to the S. cerevisiae genome. Values in parentheses correspond to the number of genes used for the analysis. (C) Pie chart summarizing the biological process superfamily associated with the assigned GO terms for 301 of the weakly annotated genes.
Applying this approach to our L. kluyveri transcriptomic data allowed us to propose putative biological processes for 2269 genes, which represent 42% of those tested. Successful GO term assignment was easier to obtain for genes that were highly conserved across species (assignment for 65% of them). These genes are generally involved in large networks that allow for numerous coexpressed genes and efficient enrichment analysis. Nevertheless, we proposed a potential GO term for 301 of the 1176 genes that previously had no reliable annotation (26%; Figure 5B), which includes various biological processes (respiration, ion resistance, mating, expression; Figure 5C). Among the 74 Lachancea-specific genes and 70 ORFans (Vakirlis et al., 2016), 11 and one were associated with a putative process, respectively. Specifically, the ORFan SAKL0F02200g might be involved in glycogen metabolism (p = 8.75 × 10−5).
Functional validation of inferred annotations
To functionally validate our approach, we selected seven genes from those with an inferred biological process (Supplemental Table S3) that were potentially easy to test by deletion. Most of these genes did not display a clear function based on homologies, with four of them having no orthologue in S. cerevisiae and three displaying low similarity level with their S. cerevisiae orthologue. Four of the genes selected were involved in respiration (electron transport chain), one in mitochondrial organization, one in mating (pheromone maturation), and one in iron-ion homeostasis. No reduction in the growth rate was observed on nonfermentable sources after the deletion of the four genes potentially involved in respiration (unpublished data). However, a simple growth test using a deletion mutant is probably not enough to reject the hypothesis that these genes are involved or not in respiration. By contrast, we observed a phenotypic change after the deletion of the three other genes in the reference strain CBS 3082.
The deletion of the gene SAKL0F11264g (potentially involved in mitochondrial organization) reduced growth capacity immediately after the transformation, which was apparent from the observation of smaller colonies on the selective medium. The transformation was performed several times to confirm this phenotype. We tested the growth of the deletion strain on glucose and nonfermentable carbon sources. Compared to the wild type, the growth of the deletion strain on glucose is effectively lower, and growth on glycerol, although still observed, was dramatically reduced (Figure 6A). We specifically tested the mitochondrial abundance and activity using a combination of MitoTracker Green (which stained all mitochondria) and MitoTracker DeepRed (which stained active mitochondria) by fluorescence microscopy. Our images suggested that the mitochondria abundance is approximately the same in the wild-type and deletion strains. However, a reduced signal of MitoTracker DeepRed was observed for the deleted strains, indicating that the mitochondria are less active (Figure 6B). Consistent with this, protein domain detection using Pfam (Finn et al., 2014) highlighted a mitochondrial domain in this gene (PF08213, p = 5.9 × 10−12).
FIGURE 6:

Functional effect of the deletion of the gene SAKL0F11264g potentially involved in mitochondrial organization. (A) Cellular growth of the deletion strain (blue) compared with the wild type (black). (B) Mitochondrial staining in the two strains, using a combination of MitoTracker Green (labels all mitochondria) and MitoTracker DeepRed (labels only active mitochondria). The images are representative of the entire slide analyzed.
The expression of the gene SAKL0H23584g is correlated with genes expressed in MATa strains. Therefore we attempted to test mating efficiency with or without this gene. We immediately noticed that the agglutination generally induced after mixing MATa and MATα strains was not observed after the deletion of SAKL0H23584g (Supplemental Figure S7). This agglutination has been thoroughly described in S. cerevisiae (de Nobel et al., 1995; Zhao et al., 2001; Dranginis et al., 2007), and the three cell wall proteins involved have been identified (a-agglutinin Aga2p/Aga1p and α-agglutinin Sag1p). In L. kluyveri, these three proteins are also present: Sakl0H21142p (51% identity with Aga2p), Sakl0A00572p (31% identity with 38% of the Aga1p sequence), and Sakl0G12320p (49.9% identity with Sag1p). The molecular function of SAKL0H23584g is difficult to assess because no domains, and in particular no glycosylphosphatidylinositol anchor signal, was detected in its protein sequence (Pfam and PredGPI; Pierleoni et al., 2008). This gene is present in only two species of the Lachancea clade: L. kluyveri and Lachancea mirantina (LAMI0G05864g). Approximately 40% identity was also found with the uncharacterized Yarrowia lipolytica protein Yali0B22836p. In contrast to flocculation, the agglutination phenomenon has thus far been strictly linked to mating, and, in this case, addition of EDTA does not restore the cultures to a homogeneous mix of cells. Although we were not able to observe a clear effect on mating efficiency (unpublished data), the effect on agglutination confirmed a link between SAKL0H23584g and mating processes.
Because the gene SAKL0B02266g was associated with metal-ion homeostasis, we tested the growth capacity of the deleted strain in various media containing a range of metallic (Fe2+, Cu2+, Ni2+, Co2+) and nonmetallic (Li+, Na+) ion concentrations. We observed a reduction in Cu2+ and Ni2+ sensitivity in the deletion strain compared with the wild type, indicating that this gene is indeed involved in metallic-ion resistance (Supplemental Figure S8). Only one transmembrane fragment was observed using TMHMM (Sonnhammer et al., 1998), which could indicate that this protein itself is not a transporter but might act as a sensor and/or interact with other membrane proteins. A version of this gene was found in all species of Lachancea clade, as well as in other preduplicated yeasts (Kluyveromyces lactis, Zygosaccharomyces rouxii) and shared 30% identity with insect papilin, a protein that interacts with a metalloprotease.
DISCUSSION
Transcriptional response to stress in L. kluyveri
In this study, we compiled a large transcriptomic data set describing cell response to various conditions in a yeast distantly related to the reference species, S. cerevisiae. This new insight into L. kluyveri gene regulation is a valuable asset for understanding the evolution of regulatory mechanisms within the Lachancea clade and across the Saccharomycotina species. The analyses presented here, together with our previous study on intraspecific expression variation in this species (Brion et al., 2015), provides a thorough description of gene regulation and evolution. We obtained direct evidence for a link between the genetic plasticity and environmental sensitivity of gene expression, which we also observed using S. cerevisiae expression data obtained from previous work (R = 0.42, p < 10−15; Gasch et al., 2000; Skelly et al., 2013). We investigated the reason for this correlation and observed than genes displaying low ES and GP correspond to housekeeping genes: transcription (p = 3.1 × 10−8) and protein transport (p < 10−14); of interest, other housekeeping genes (ribosome biogenesis) are sensitive to environmental conditions. Among the genes displaying high ES and GP, we observed genes not conserved across species, indicating a link between expression behavior and evolution history, as reported in our previous work (Brion et al., 2015). The others genes with high ES and GP are enriched for functions of transport (p = 10−12), detoxification (p = 5.7 × 10−5), and metabolism of various C compounds (p = 10−6). In S. cerevisiae, mutations have already been highlighted as responsible for variations in drug transporter expression among strains, allowing them to adapt to various stressful conditions (Brion et al., 2013). Therefore the regulation of genes activated during environmental change tends to be highly affected by intraspecies genetic variation.
Our data allow for the characterization of specific changes in gene expression in response to individual stresses and highlight the pathways triggered to cope with these conditions. Most of the responses observed are similar to those described in other species—for example, in the case in which fluconazole was found to activate the ergosterol biosynthetic pathway (Song et al., 2003; Maertens, 2004; Supplemental Figure S4). Furthermore, our approach allowed us to define a set of genes involved in the general ESR, which was composed of 590 down-regulated and 290 up-regulated genes in L. kluyveri. It is important to note that the set of ESR genes defined could also slightly vary according to the method used. Here we analyzed growth under continuous stress in batch cultures, whereas the initial definition of ESR genes in S. cerevisiae used expression kinetics during stress induction. Therefore the approach of Gasch et al. (2000) took into account instantaneous expression responses to stresses that we might have overlooked using the present methods. For these reasons, we opted for a method of ESR definition based on the highly reliable S. cerevisiae ESR list. Indeed, an independent method based only on our data provided less reliable data (Supplemental Figure S9). We characterized the pathways involved in this general response and analyzed how they have evolved. Comparison of the ESR among species is now feasible and essential in order to obtain better insight into the features of stress defense, as well as for functional gene annotation.
Differences in ESR between yeast species
Using the data set we generated, we focused on the changes in stress responses among species. The ESR we defined allowed us to elucidate a clear relationship between stress response and species “life history”: S. cerevisiae and S. uvarum are clearly established respirofermentative species, whereas L. kluyveri is characterized by a transitional lifestyle common among preduplicated yeast (Hagman et al., 2013). Indeed, fewer positive ESR genes in L. kluyveri are involved in the oxidative stress response than in both Saccharomyces species. The most striking example is the loss of ESR expression pattern for genes related to peroxidase activity (including PRX1, CTT1, HYR1, and CCP1 orthologues). The influence of lifestyle can also be observed among the negative ESR genes, with the absence of ESR expression pattern for PDC11 and PDC12, which code for key enzymes in the fermentation process. During stress, L. kluyveri and most likely the strictly respiratory yeast species do not modify their energetic metabolism and therefore do not have to cope with variation in oxidative stress. A comparison of ESR between S. pombe and S. cerevisiae did not highlight pronounced differences concerning oxidative stress responses (Gasch, 2007). However, despite the extremely high genetic divergence between these two species, they both show respirofermentative behavior. Of interest, a strong influence of metabolic phases and oxygen consumption was observed in the ESR expression of these two species (Slavov et al., 2012). Therefore it appears very likely that differences in lifestyle are responsible for our observations.
In S. cerevisiae, the regulatory mechanism of the positive ESR has been carefully described, allowing for a thorough comparison with L. kluyveri. In the model species, a portion of the ESR genes are under the control of the stress response element (STRE), to which the transcription factor Msn2/4p binds (Martínez-Pastor et al., 1996). However, in L. kluyveri, the MSN2 orthologue, SAKL0B11330g, shares only 39% of protein identity, and no orthologue has been detected for MSN4. Of the 23 STRE genes in the S. cerevisiae ESR, only four have been consistently conserved in the L. kluyveri ESR. The action of PKA, which participates in the control of the stress response in S. cerevisiae, is attributed to a set of three isoforms (Santangelo, 2006). Because L. kluyveri did not undergo whole-genome duplication, only two of these isoforms are present (TPK2 and TPK3 orthologues). Although they are highly expressed, neither of these isoforms is in the positive ESR in L. kluyveri, in contrast to S. cerevisiae. Therefore some PKA targets are also not part of the L. kluyveri ESR (e.g., HSP12 and YAK1 orthologues). However, most of the PKA controls occur at the posttranslational level, and the full implications of the expression behavior of TPK2 and TPK3 could not be fully addressed here. It is necessary to investigate more thoroughly how modifications of these regulatory genes affect networks, using molecular approaches such as those developed in Howard et al. (2014) and Sorrells et al. (2015). However, our results indicate that the regulatory mechanisms underlying stress response tend to be different among species.
The more pronounced differences in the sets of genes in the ESR across species are mostly found in the positive ESR, whereas the negative ESR displays only few changes. This can be linked directly to the involvement of these genes in fundamental functions in all species, such as gene expression, ribosome synthesis, and tRNA synthesis (Gasch et al., 2000, 2007; Brauer et al., 2008). These functions are also under the control of the growth rate, and, as already reported, it is difficult to distinguish the effect of stress from that of growth arrest on transcriptome in this type of analysis (Slavov et al., 2012). Consequently, these negative ESR genes are highly conserved within and across species, and their expression is weakly influenced by genetic background, whereas the positive ESR genes have lower levels of expression conservation. The high genetic plasticity of positive ESR gene expression observed in L. kluyveri and S. cerevisiae agrees with previous analyses of how gene–environment interaction affects expression (Eng et al., 2010) and indicates that their regulatory mechanism could be easily perturbed in different strains. Therefore we hypothesized that the exact set of positive ESR genes might change within a species, whereas the negative ESR genes might be mostly identical among strains.
Proposal of gene annotation using expression data
Our extensive set of expression data allowed us to use an automated approach to predict a biological process for 42% of genes. Of great interest, 301 previously uncharacterized L. kluyveri genes now have a potential role in some pathways. In a small set of follow-up experiments, nearly half of our predictions were validated. This method has limits: we can propose GO terms only for genes that are involved in large enough networks (small networks being subject to noise and generally displaying low enrichment p value). Moreover, based on RNA abundance data alone, this approach does not ensure that the proteins are active in the cell. Consequently, deletion of genes we predicted to be involved in respiration had no effect on fitness on nonfermentable carbon sources. We can enhance this approach by increasing the number of conditions tested or implementing tools such as Pfam (Finn et al., 2014) and TMHMM (Sonnhammer et al., 1998) to take into account detected protein domains. However, the method used here is advantageous because it is easily implemented and can be applied to large sets of expression data. The functional validation of three of the associations we predicted between genes and potential pathways indicates that this type of approach can be used, to some extent, to uncover new gene functions. The improvement of gene annotation in L. kluyveri was of great interest, as many genes did not match to orthologues in other species. Moreover, the proposal of gene function through a method other than homology can highlight new differences among species and provide a path to unraveling the evolution of gene function.
Conclusion
Providing a large data set on gene expression for a species distantly related to the well-studied S. cerevisiae satisfied two main objectives. First, by completing the description of L. kluyveri stress response, we observed similarities and differences with the reference species and revealed the effect that the species lifestyle has on the expression response to stress. Second, patterns of gene expression allowed us to propose putative involvement in processes for genes for which no orthologue had previously been functionally described. The results obtained here are useful for further genetic investigation of L. kluyveri and other Saccharomycotina species.
Materials and Methods
Strain and growth conditions
The expression analyses were performed using the L. kluyveri reference strain CBS 3082_a (MATa). Transcriptomic profiling was completed in mid exponential growth in batch cultures for 20 media listed in Supplemental Table S1. Before the growth test, the strain was plated on solid YPD and incubated for 2–3 d at 30°C and then transferred to 10 ml of liquid YPD for overnight growth at 30°C. Growth tests for RNA sampling in each condition were performed in flasks with 30 ml of medium at 30°C and monitored with OD600 of 600 nm. For each condition, biological replicates of two independent cultures were done. The definition of the stress conditions (e.g., compound concentration) was based on previous L. kluyveri dose–response assay using microcultivation (Supplemental Figure S10).
Growth tests for functional analysis of deletion mutants were performed using a microcultivation strategy as previously described (Jung et al., 2016). Briefly, precultures were transferred in a 96-well microplate containing different media (150 μl) and cultivated in a microplate reader (Infinite F200; Tecan, Männedorf, Switzerland) at 30°C. The plate was shaken for 10 min between absorbance recordings at 600 nm for 48 h.
RNA sampling
Cells were sampled by filtering 7 ml of medium from the mid exponential growth phase at an OD600 between 0.350 and 0.450 (the time points of sampling are given in Supplemental Table S1). The cells on the filter were frozen in liquid nitrogen and stored at −80°C until processing. The RNA extraction was performed as previously described (Brion et al., 2015). Briefly, the extraction was completed using hot aqua-phenol (AQUAPHEO01; MP Biomedicals, Santa Ana, CA) followed by centrifugation using a Phase Lock Gel tube (2302830; 5prime, Hilden, Germany). After precipitation in ethanol, RNA was purified using the RNeasy kit (74104; Qiagen, Venlo, Netherlands) and treated with DNase (18068-015; Invitrogen, Carlsbad, CA).
RNA sequencing and data processing
RNA sequencing was completed as previously described (Brion et al., 2015). Briefly, sequencing was performed at the Gene Core Facility, EMBL (Heidelberg, Germany), using the Illumina HiSeq2000 platform with 50–base pair nonoriented single-end reads. Reads were cleaned, filtered, trimmed, and aligned using FastQC report, CutAdapt (Martin, 2011), and TopHat, version 2.0.8b (Trapnell et al., 2009). The alignment rate ranged from 98.4 to 99%, and the number of mapped reads was 8–30 million. We used the most recent annotation file of L. kluyveri (GRYC, http://gryc.inra.fr/) to define the intragenic regions and performed the read count with HTseq-count (Anders et al., 2015). Between 88 and 93% of the mapped reads fell in the annotated regions. Raw data are available from the European Nucleotide Archive (www.ebi.ac.uk/ena) under accession number PRJEB10946.
We filtered out genes with low expression levels (number of reads was <32 for all samples) and performed intersample normalization using Bioconductor DEseq2 (Love et al., 2014) on R (R Core Team, 2014). We verified data stability by obtaining the Spearman correlation coefficient between biological replicates, which was consistently >0.96. The average values of gene expression between replicates were used for all subsequent analyses. The final data set is given in Supplemental Table S4. We also calculated log2FC for the expression of each gene in the 19 stress-inducing media compared with that in the reference medium (YPD).
Hierarchical clustering analysis of expression profiles
The identification of genes included in significantly distinct clusters was performed using standardized fold-change (log2FC), that is, SD corrected. An initial HC using all genes with the R hclust function (Euclidean distance, complete linkage, stats package) was done. For each cluster, the height (or distance) was measured as the maximum distance across all elements. We performed permutations of expression levels across conditions for all 5600 genes, followed by a new HC repeated 10 times in order to define a height threshold of 2.2, allowing for <56 clustering linkages due to chance (1%). All genes involved in clusters with height <2.2 in the first HC were used for the final HC, which was completed with Cluster 3.0 (Euclidean distance, complete linkage) and JavaTreeView.
ESR gene identification
The principle of the method is described in Figure 2B. We obtained the list of S. cerevisiae ESR genes from Gasch et al. (2000) and compiled a set of the corresponding orthologues in L. kluyveri. We considered the positive and negative ESR gene lists separately. We computed the pairwise Spearman correlation values of gene expression and disregarded genes that had an average pairwise correlation to all other genes <0.3. To conserve only genes that react stereotypically to stress, we disregarded those displaying an average log2FC across all conditions <0.2 (for positive ESR genes) and >−0.2 (for negative ESR genes). We considered the remaining genes to be the common set of ESR shared between L. kluyveri and S. cerevisiae. We calculated the pairwise Spearman correlation of expression for all of the genes in L. kluyveri compared with the list of shared ESR genes and determined that genes with an average pairwise Spearman correlation >0.54 were novel ESR genes in L. kluyveri. This last threshold corresponded to a false discovery rate of 1%, defined by performing 100 permutations of expression levels across conditions for each gene before correlation calculation.
To assess the reliability of our approach, we calculated the expression correlation between the negative and positive ESR genes and obtained an average pairwise Spearman correlation of −0.6, with all values <−0.25, indicating that our two sets of genes display opposite behaviors. As a second control, we observed a consistent overlap of 55% between the most down-regulated genes (average log2FC < −0.4) and the negative ESR and 60% between the up-regulated genes (average log2FC > 0.4) and the positive ESR (Fisher’s exact test, p < 10−12 for both; Supplemental Figure S11). These partial overlaps were expected, as major changes in expression could also be due to condition-specific stress responses.
Orthologue identity level and dN/dS
Functional annotation of the L. kluyveri genome was completed by performing a blastP similarity search against the S. cerevisiae genome to propose an orthologue for each gene. If no orthologue was detected in S. cerevisiae, the blastP search was extended to other yeast genomes (Génolevures Consortium et al., 2009; Vakirlis et al., 2016). From the list of orthologue pairs between L. kluyveri and S. cerevisiae, a local alignment and a global protein sequence alignment were performed between each gene and its orthologue using the EMBOSS Water and EMBOSS Needle algorithms, respectively (Rice et al., 2000). The orthologue identity level was calculated as a factor between the percentage identity with the aligned sequences and the ratio of the aligned sequences to the total protein size. The dN/dS ratios were determined previously (Brion et al., 2015) and were the average values obtained across the 28 sequenced strains calculated with the YN00 model (PAML package, version 4.5) based on the multiple sequence alignment of each gene (Yang, 2007).
GO term enrichment
The functional enrichments performed for the clustering analysis, environmental sensitivity/genetic plasticity balance (ES/GP), and ESR definition were run on the FunSpec website with a significance threshold of 10−4 with orthologue annotation against the entire S. cerevisiae genome as background (Robinson et al., 2002).
Automated assignment of biological processes to genes
For each gene, we generated a list of other genes that are coexpressed (Spearman correlation >0.65) across conditions and strains, using previously obtained data (Brion et al., 2015). From that list, reliable S. cerevisiae orthologues were extracted (only those with a similarity >50%). By performing 100 permutations of expression levels across conditions for each of the 5600 genes before correlation calculation, we determined that only 2.1% of the genes had >2 coexpressed genes by chance. From the list of orthologues, we performed a GO term enrichment using topGO of the R Bioconductor package, with the classic algorithm and Fisher statistical test (Alexa and Rahnenfuhrer, 2010) with org.Sc.sgd.db as the database. The three GO terms with the lowest p values were obtained and assigned to the initial gene and were generally involved in related pathways. By 10 permutations of the pairwise correlation values for each of the 5600 genes before enrichment analysis, we determined that a p threshold of 10−4 allowed for a false discovery rate of <2% and disregarded all cases of enrichment based on fewer than three genes. When the initial gene had a defined orthologue in S. cerevisiae, we extracted the annotated GO terms for the orthologue and compared them to the three we proposed, if significant. To do so, we used GOsim from the R Bioconductor package, with the relevance method and using org.Sc.sgd.db as the database. Similarity scores ranged between 0 and 1, and we arbitrarily chose a threshold of 0.60 to denote significant similarities.
We evaluated the reliability of this approach by applying this method to S. cerevisiae expression data previously obtained for 23 strains (Skelly et al., 2013). Among the 5278 genes analyzed with our pipeline, 4447 had ≥2 coexpressed genes, allowing us to perform enrichment (84%). Of these genes, 1834 were categorized as harboring at least one GO term enrichment with p < 10−4 (34.7%). There were 656 genes for which we detected correspondence between our results and the annotation (36%). We investigated why this rate was low and found that of the genes with low levels of correspondence, ∼33% had unclear functions. For the remaining two-thirds, GOsim occasionally provided a low similarity score for related processes.
Gene deletions in L. kluyveri
We deleted genes by inserting the KanMX gene from the plasmid p42Neo (Treusch et al., 2015), providing resistance to the G418 antibiotic. Because the recombination rate is low in L. kluyveri, the homologous regions of the inserts used were 500 base pairs in length. The two homologous regions and the KanMX fragments were amplified separately by PCR and joined by PCR fusion (primers listed in Supplemental Table S5). For the transformation, cells were harvested at the end of the exponential phase (OD600 of 1.5) in 100 ml of YPD, pelleted, and treated for 30 min at 37°C with 25 mM dithiothreitol in 50 mM KH2PO4 buffer, pH 7.5. Competent cells were washed twice in STM buffer (270 mM sucrose, 10 mM Tris-HCl, 1 mM MgCl2, pH 7.5) and resuspended in 0.2 ml of STM buffer. The transformation was performed by electroporation (electroporator 2510; Eppendorf, Hamburg, Germany) at 1500 V using a mixture of 50 μl of cells and 1 μg of DNA in a 2-mm electroporation cuvette. Immediately after electroporation, cells were resuspended in 1 ml of YPD and incubated for 1 h. Then, all cells were spread on a YPD agar plate containing 200 μg/l G418. The ploidy of the transformed strains was determined using flow cytometry and propidium iodine dye (protocol from Delobel and Tesnière, 2014), and the deletion of the gene was validated by PCR (primers listed in Supplemental Table S5).
MitoTracker staining and microscopy
To stain mitochondria, MitoTracker Green (which labels all mitochondria) and MitoTracker DeepRed (which only labels active mitochondria; Thermo Fisher Scientific, Waltham, MA) were added simultaneously to a culture of cells during exponential growth, each at a final concentration of 100 nM. After incubation for 1 h, cells were pelleted and resuspended in water. The cells were immediately observed using a Zeiss Axio Observer fluorescence microscope with differential interference contrast/Nomarski 100× objective in oil. MitoTracker Green was detected using filter set 13 for green fluorescent protein (excitation 470 nm, emission 505–530 nm) and MitoTracker DeepRed with filter set Cy5 (excitation 650 nm, emission 660–740 nm). Approximately six images were taken randomly for each preparation. Image processing was carried out using GIMP2.0.
Supplementary Material
Acknowledgments
We thank the two anonymous reviewers for helpful suggestions and comments, as well as Kelle Freel for invaluable advice. We also thank Gilles Fischer, Ingrid Lafontaine, and Nikolaos Vakirlis for providing the list of Lachancea-specific genes and ORFans. We are most grateful to the GeneCore sequencing team (EMBL, Heidelberg, Germany) and the Plateforme BioImage (IBMP-CNRS, Strasbourg, France). This work was supported by Agence Nationale de la Recherche Grant 2010-BLAN-1606-05 and ANR Young Investigator Grant 2011-JSV6-004-01 (J.S.). We also thank the Université de Strasbourg (IdEx 2012 Attractivité) for financial support.
Abbreviations used:
- ES
environmental sensitivity
- ESR
environmental stress response
- GO
Gene Ontology
- GP
genetic plasticity
- HC
hierarchical clustering
- PKA
protein kinase A
- STRE
stress response element.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-12-0816) on March 23, 2016.
REFERENCES
- Alexa A, Rahnenfuhrer J. 2010. topGO: Enrichment analysis for Gene Ontology. R Package version 2.16.0. Available at https://bioconductor.riken.jp/packages/3.1/bioc/html/topGO.html (accessed July 2014)
- Alexandre H, Ansanay-Galeote V, Dequin S, Blondin B. Global gene expression during short-term ethanol stress in Saccharomyces cerevisiae. FEBS Lett. 2001;498:98–103. doi: 10.1016/s0014-5793(01)02503-0. [DOI] [PubMed] [Google Scholar]
- Almeida P, Gonçalves C, Teixeira S, Libkind D, Bontrager M, Masneuf-Pomarède I, Albertin W, Durrens P, Sherman DJ, Marullo P, et al. A Gondwanan imprint on global diversity and domestication of wine and cider yeast Saccharomyces uvarum. Nat Commun. 2014;5:4044. doi: 10.1038/ncomms5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson Rasmussen A, Kandasamy D, Beck H, Crosby SD, Björnberg O, Schnackerz KD, Piškur J. Global expression analysis of the yeast Lachancea (Saccharomyces) kluyveri reveals new URC genes involved in pyrimidine catabolism. Eukaryot Cell. 2014;13:31–42. doi: 10.1128/EC.00202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, Botstein D. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell. 2008;19:352–367. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion C, Ambroset C, Sanchez I, Legras J-L, Blondin B. Differential adaptation to multi-stressed conditions of wine fermentation revealed by variations in yeast regulatory networks. BMC Genomics. 2013;14:681. doi: 10.1186/1471-2164-14-681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion C, Pflieger D, Friedrich A, Schacherer J. Evolution of intraspecific transcriptomic landscapes in yeasts. Nucleic Acids Res. 2015;43:4558–4568. doi: 10.1093/nar/gkv363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudy AA, Guan Y, Jia Y, Hansen C, DeSevo C, Hayes AP, Agee J, Alvarez-Dominguez JR, Arellano H, Barrett D, et al. A new system for comparative functional genomics of Saccharomyces yeasts. Genetics. 2013;195:275–287. doi: 10.1534/genetics.113.152918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA. Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen PJ, Sprague GF. The regulation of filamentous growth in yeast. Genetics. 2012;190:23–49. doi: 10.1534/genetics.111.127456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delobel P, Tesnière C. A simple FCM method to avoid misinterpretation in Saccharomyces cerevisiae cell cycle assessment between G0 and sub-G1. PLoS One. 2014;9:e84645. doi: 10.1371/journal.pone.0084645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nobel H, Pike J, Lipke PN, Kurjan J. Genetics of a-agglutunin function in Saccharomyces cerevisiae. Mol Gen Genet. 1995;247:409–415. doi: 10.1007/BF00293141. [DOI] [PubMed] [Google Scholar]
- Dranginis AM, Rauceo JM, Coronado JE, Lipke PN. A biochemical guide to yeast adhesins: glycoproteins for social and antisocial occasions. Microbiol Mol Biol Rev. 2007;71:282–294. doi: 10.1128/MMBR.00037-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng KH, Kvitek DJ, Keles S, Gasch AP. Transient genotype-by-environment interactions following environmental shock provide a source of expression variation for essential genes. Genetics. 2010;184:587–593. doi: 10.1534/genetics.109.107268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich A, Jung P, Reisser C, Fischer G, Schacherer J. Population genomics reveals chromosome-scale heterogeneous evolution in a protoploid yeast. Mol Biol Evol. 2015;32:184–192. doi: 10.1093/molbev/msu295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP. Comparative genomics of the environmental stress response in ascomycete fungi. Yeast (Chichester Engl) 2007;24:961–976. doi: 10.1002/yea.1512. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Génolevures Consortium. Souciet JL, Dujon B, Gaillardin C, Johnston M, Baret PV, Cliften P, Sherman DJ, Weissenbach J, Westhof E, et al. Comparative genomics of protoploid Saccharomycetaceae. Genome Res. 2009;19:1696–1709. doi: 10.1101/gr.091546.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Dunham MJ, Troyanskaya OG, Caudy AA. Comparative gene expression between two yeast species. BMC Genomics. 2013;14:33. doi: 10.1186/1471-2164-14-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagman A, Säll T, Compagno C, Piskur J. Yeast “make-accumulate-consume” life strategy evolved as a multi-step process that predates the whole genome duplication. PLoS One. 2013;8:e68734. doi: 10.1371/journal.pone.0068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagman A, Säll T, Piškur J. Analysis of the yeast short-term Crabtree effect and its origin. FEBS J. 2014;281:4805–4814. doi: 10.1111/febs.13019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DC, Myers CL, Huttenhower C, Hibbs MA, Hayes AP, Paw J, Clore JJ, Mendoza RM, Luis BS, Nislow C, et al. Computationally driven, quantitative experiments discover genes required for mitochondrial biogenesis. PLoS Genet. 2009;5:e1000407. doi: 10.1371/journal.pgen.1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard CJ, Hanson-Smith V, Kennedy KJ, Miller CJ, Lou HJ, Johnson AD, Turk BE, Holt LJ. Ancestral resurrection reveals evolutionary mechanisms of kinase plasticity. eLife. 2014;3 doi: 10.7554/eLife.04126. doi: 10.7554/eLife.04126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung PP, Friedrich A, Reisser C, Hou J, Schacherer J. Mitochondrial genome evolution in a single protoploid yeast species. G3 (Bethesda) 2012;2:1103–1111. doi: 10.1534/g3.112.003152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung PP, Sigwalt A, Ohnuki S, de Montigny J, Ohya Y, Schacherer J. Large-scale survey of intraspecific fitness and cell morphology variation in a protoploid yeast species. 2016. G3 (Bethesda), g3.115.026682. [DOI] [PMC free article] [PubMed]
- Kasavi C, Eraslan S, Arga KY, Oner ET, Kirdar B. A system based network approach to ethanol tolerance in Saccharomyces cerevisiae. BMC Syst Biol. 2014;8:90. doi: 10.1186/s12918-014-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens JA. History of the development of azole derivatives. Clin Microbiol Infect. 2004;10:1–10. doi: 10.1111/j.1470-9465.2004.00841.x. [DOI] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. [Google Scholar]
- Martínez-Pastor MT, Marchler G, Schüller C, Marchler-Bauer A, Ruis H, Estruch F. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE) EMBO J. 1996;15:2227–2235. [PMC free article] [PubMed] [Google Scholar]
- Morano KA, Grant CM, Moye-Rowley WS. The response to heat shock and oxidative stress in Saccharomyces cerevisiae. Genetics. 2012;190:1157–1195. doi: 10.1534/genetics.111.128033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieduszynski CA, Liti G. From sequence to function: Insights from natural variation in budding yeasts. Biochim Biophys Acta. 2011;1810:959–966. doi: 10.1016/j.bbagen.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pál C, Papp B, Hurst LD. Highly expressed genes in yeast evolve slowly. Genetics. 2001;158:927–931. doi: 10.1093/genetics/158.2.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierleoni A, Martelli PL, Casadio R. PredGPI: a GPI-anchor predictor. BMC Bioinformatics. 2008;9:392. doi: 10.1186/1471-2105-9-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2014. The R Project for Statistical Computing. Available at https://www.r-project.org/ (accessed July 2014) [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Robinson MD, Grigull J, Mohammad N, Hughes TR. FunSpec: a web-based cluster interpreter for yeast. BMC Bioinformatics. 2002;3:35. doi: 10.1186/1471-2105-3-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol T, Dulau L, Julien A, Blondin B. Genome-wide monitoring of wine yeast gene expression during alcoholic fermentation. Yeast. 2003;20:1369–1385. doi: 10.1002/yea.1046. [DOI] [PubMed] [Google Scholar]
- Santangelo GM. Glucose signaling in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2006;70:253–282. doi: 10.1128/MMBR.70.1.253-282.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell DR, Zill OA, Rokas A, Payen C, Dunham MJ, Eisen MB, Rine J, Johnston M, Hittinger CT. The awesome power of yeast evolutionary genetics: new genome sequences and strain resources for the Saccharomyces sensu stricto genus. G3 (Bethesda) 2011;1:11–25. doi: 10.1534/g3.111.000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly DA, Merrihew GE, Riffle M, Connelly CF, Kerr EO, Johansson M, Jaschob D, Graczyk B, Shulman NJ, Wakefield J, et al. Integrative phenomics reveals insight into the structure of phenotypic diversity in budding yeast. Genome Res. 2013;23:1496–1504. doi: 10.1101/gr.155762.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov N, Airoldi EM, van Oudenaarden A, Botstein D. A conserved cell growth cycle can account for the environmental stress responses of divergent eukaryotes. Mol Biol Cell. 2012;23:1986–1997. doi: 10.1091/mbc.E11-11-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart KA. Brewing yeast genomes and genome-wide expression and proteome profiling during fermentation. Yeast. 2007;24:993–1013. doi: 10.1002/yea.1553. [DOI] [PubMed] [Google Scholar]
- Song JL, Lyons CN, Holleman S, Oliver BG, White TC. Antifungal activity of fluconazole in combination with lovastatin and their effects on gene expression in the ergosterol and prenylation pathways in Candida albicans. Med Mycol. 2003;41:417–425. doi: 10.1080/1369378031000137233. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol. 1998;6:175–182. [PubMed] [Google Scholar]
- Sorrells TR, Booth LN, Tuch BB, Johnson AD. Intersecting transcription networks constrain gene regulatory evolution. Nature. 2015;523:361–365. doi: 10.1038/nature14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treusch S, Albert FW, Bloom JS, Kotenko IE, Kruglyak L. Genetic mapping of MAPK-mediated complex traits across S. cerevisiae. PLoS Genet. 2015;11:e1004913. doi: 10.1371/journal.pgen.1004913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan PP, Zinshteyn B, Thompson MK, Gilbert WV. Protein kinase A regulates gene-specific translational adaptation in differentiating yeast. RNA. 2014;20:912–922. doi: 10.1261/rna.044552.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakirlis N, Sarilar V, Drillon , Agier N, Meyniel JP, Blanpain L, Carbone A, Devillers H, Dubois K, Fleiss A, et al. Reconstruction of ancestral chromosome architecture and gene repertoire reveals principles of genome evolution in a model yeast genus. Genome Res. 2016 doi: 10.1101/gr.204420.116. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke CO, Drummond DA. Population genetics of translational robustness. Genetics. 2006;173:473–481. doi: 10.1534/genetics.105.051300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Shields DC. Molecular evidence for an ancient duplication of the entire yeast genome. Nature. 1997;387:708–713. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zhao H, Shen ZM, Kahn PC, Lipke PN. Interaction of alpha-agglutinin and a-agglutinin, Saccharomyces cerevisiae sexual cell adhesion molecules. J Bacteriol. 2001;183:2874–2880. doi: 10.1128/JB.183.9.2874-2880.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.