

Abstract
Schizophrenia is a neuropsychological disorder with a strong heritable component; genetic risk for schizophrenia is conferred by both common variants of relatively small effect and rare variants with high penetrance. Genetically engineered mouse models can recapitulate rare variants, displaying some behavioral defects associated with schizophrenia; however, these mouse models cannot recapitulate the full genetic architecture underlying the disorder. Patient-derived human induced pluripotent stem cells (hiPSCs) present an alternative approach for studying rare variants, in the context of all other risk alleles. Genome editing technologies, such as CRISPR-Cas9, enable the generation of isogenic hiPSC lines with which to examine the functional contribution of single variants within any genetic background. Studies of these rare variants using hiPSCs have the potential to identify commonly disrupted pathways in schizophrenia and allow for the identification of new therapeutic targets.
Keywords: Schizophrenia, CNV, mouse models, hiPSC models
Introduction
Schizophrenia is a debilitating yet relatively common psychiatric disorder, affecting approximately 1% of the world population (Health, 2013). It is uniquely characterized by positive symptoms including delusions, hallucinations and disorganized thinking. Negative symptoms, including depression and anhedonia, and cognitive symptoms such as working memory impairments are also important features in the disorder. The type and severity of symptoms presents heterogeneously within the patient population (Weinberger, 1987). Currently available antipsychotics are fairly effective at treating the positive symptoms, but have little effect on the negative or cognitive symptoms. One-third of patients do not experience any symptom amelioration after antipsychotic treatment and less than 20% of patients will return to adequate social functioning after their first psychotic episode (Robinson et al., 2004). The inability to treat a large percent of patients results in an increased risk of suicide and homelessness among schizophrenia patients. Typically, the onset of schizophrenia occurs in early adulthood, creating a high societal burden relative to many other neurological disorders with onsets much later in life. Although schizophrenia is diagnosed in early adulthood, abnormal neurodevelopmental processes can begin much earlier and it is now thought of as a neurodevelopmental disorder.
Epidemiological studies of affected families as well as twin studies have established schizophrenia to be a highly heritable disorder with heritability estimates between 50-80% (Sullivan et al., 2003). Although highly heritable, there is no one gene responsible for schizophrenia. Instead, it has been suggested that there are both many common variants with small effect sizes as well as more penetrant and difficult to detect rare variants that can contribute to the disorder. The debate underlying the genetics of schizophrenia has surrounded these two hypotheses; the common variant common disease and the rare variant common disease models (Malhotra and Sebat, 2012). It is now becoming clear that both types of genetic variation contribute to the development of schizophrenia (reviewed Chen et al., 2015), and perhaps contribute additively to symptom severity and prognosis. For example, childhood-onset schizophrenia (COS), which is a rare and severe form of the disorder, coincides with both significantly more rare CNVs (Ahn et al., 2014b) and polygenic risk burden (Ahn et al., 2014a).
This review will focus on the role of rare variants in schizophrenia, beginning with what we have learned from human genetic studies and animal models. We suggest that patient-derived human induced pluripotent stem cells (hiPSCs) provide an advantageous model with which to investigate the casual role of rare variants on the underlying cellular and molecular phenotypes of schizophrenia. Identification of common biological pathways may provide insight into the mechanisms underlying schizophrenia, allowing for development of new therapeutics. Along with studies of basic disease mechanisms, hiPSC models can support drug discovery and development through high throughput drug screening, stratification of patient populations, and improved assessment of drug toxicity.
Contribution of Copy Number Variants to Schizophrenia Genetics
Rare copy number variations (CNVs) were the first type of genetic defect associated with psychiatric illness. CNVs are often very large (>100kb) and typically occur at hotspots in the genome (Conrad et al., 2011). CNVs can arise from non-allelic homologous recombination, non-homologous end joining, fork stalling/template switching, and L1-mediated retro-transposition, which all result in large duplications or deletions within a chromosomal region (Malhotra and Sebat, 2012). Due to their size, CNVs were easy to detect using original microarray technologies; now, the application of more current genotyping platforms and new algorithms for CNV discovery has enabled identification of new de novo CNVs as well as very rare variants (Neale and Sklar, 2015). CNVs greater than 10kb occur at rate of 0.01-0.02 per generation, which is very rare compared to the estimated 30-100 single nucleotide polymorphisms per generation (Malhotra and Sebat, 2012; Mills et al., 2011). Despite their rarity, the relative contributions of CNVs to genomic variation is substantial, at least in part because CNVs can affect multiple genes and potentially disrupt many functions at one site (reviewed Hiroi et al., 2013; Rodriguez-Revenga et al., 2007).
To investigate the role of rare CNVs in schizophrenia, two main genetic approaches have been undertaken. The first approach compares the CNV burden in cases of schizophrenia to controls while the second examines the impact of de novo CNVs. The focus on rare CNVs in schizophrenia begin with cytogenetic studies, which demonstrated an enrichment of detectable chromosome abnormalities in autism and schizophrenia patients (Malhotra and Sebat, 2012). Over the last ten years there have been many, increasingly larger, studies performed which all reveal an increase in the number of CNVs in schizophrenia cases compared to a healthy control population (International and Consortium, 2008; Elliott Rees et al., 2014; Stefansson et al., 2008; Walsh et al., 2008). With tens of thousands of patients and controls now genotyped, 2.5% of patients with schizophrenia and 0.9% of controls carry one of the 11 significantly associated schizophrenia CNVs (Elliott Rees et al., 2014).
There have also been numerous studies looking for de novo CNVs by sequencing both parents and the affected child, or proband. These trio studies demonstrate that rare de novo CNV mutations also are more frequent in cases compared to controls (Kirov et al., 2011). Most recently, a large study from Szatkiewicz et al combined inherited and de novo CNVs, confirming the findings of the ISC report and further demonstrating deletions to be more enriched in schizophrenia cases than duplications (Szatkiewicz et al., 2014).
Although enrichment in CNV burden indicates an association between CNVs and schizophrenia, it is important to determine which specific CNVs are significantly associated with the disease state in order to gain insights on disease mechanism. The identification of rare CNVs is difficult because very large populations are required to analyze these rare events with sufficient statistical power. The 22q11.2 deletion was the first CNV to be convincingly implicated in schizophrenia (reviewed Karayiorgou et al., 2010; Schneider et al., 2014). Deletion at this locus is the strongest risk factor for developing schizophrenia with 25% of carriers presenting with psychosis (Warnica et al., 2014). In addition to schizophrenia, it is known that disruption of the 22q11.2 locus is the cause of most cases of DiGeorge and Velocardiofacial syndromes and it contributes to increased risk of psychiatric problems in childhood including autism, anxiety and depression (Elliott Rees et al., 2014). While deletions at this locus are variable, the most common 22q11.2 deletion is approximately 3Mb while the remaining deletions in the population are about 1.5Mb within the same region. Interestingly, duplications of 22q11.2 may prove to be protective against schizophrenia (E Rees et al., 2014).
With the recruitment of large patient cohorts and better technologies, many additional CNVs began to be associated with schizophrenia. One of the largest single CNV data-sets (6882 cases; 6216 controls), analyzed by Rees et al, demonstrated significant association between 13 of the 15 CNVs previously implicated in schizophrenia. The 1q21.1 deletion, 1q21.1 duplication, NRXN deletion, 3q29 deletion, WBS duplication, 15q11.2 deletion, AS/PWS duplication, 15q13.3 deletion, 16p13.11 duplication, 16p11.2 duplication, 17p12 deletion, 17q12 deletion and the 22q11.2 deletion all exhibited significant association with schizophrenia (Elliott Rees et al., 2014). Identification of these structural variations provides a starting point to begin examining the underlying biology of schizophrenia clinically, in animal models and in in vitro systems [Table 1].
Table 1.
Characteristics of selected schizophrenia associated CNVs in humans and findings from mouse models engineered to recapitulate each CNV. Odds ratios derived from Rees et al., 2014(Elliott Rees et al., 2014).
| CNV locus | Size (Mb) |
Odds Ratio |
Mouse Models | Major Findings of Mouse Model |
|---|---|---|---|---|
| 22q11.2 deletion |
1.24 | N/A (28.27-?) |
(Paylor and Lindsay, 2006) (Long et al., 2006) (Stark et al., 2008) (Merscher et al., 2001) |
Sensorimotor gating abnormalities Impaired paired pulse inhibition Memory and learning impairments |
| NRXN deletion |
1.11 | 9.01 (4.44- 18.29) |
(Grayton et al., 2013) (Etherton et al., 2009) (Missler et al., 2003) |
Altered social approach Reduced social investigation Reduced locomotor activity in novel environment Increased aggression Decreased EPSCs in hippocampus Decreased paired pulse inhibition Impaired neurotransmitter release Reduced synaptic calcium channel function |
| 15q13.3 deletion |
1.35 | 7.52 (3.98- 14.19) |
(Fejgin et al., 2014) | Impaired long-term spatial reference memory Decreased theta frequency in hippocampus and prefrontal cortex Increased body weight Increased aggression after mild stress |
| 16p11.2 deletion |
0.23 | 3.39 (1.21- 9.52) |
(Horev et al., 2011) | Macrocephaly Diurnal deficits |
| 16p11.2 duplication |
0.56 | 11.52 (1.73- 7.57) |
(Horev et al., 2011) | Microcephaly Diurnal deficits |
Clinical Phenotypes of Neuropsychiatric CNVs
Clinically, many schizophrenia patients present with cognitive deficits; however, it is not known whether these deficits are a primary or secondary symptom of the disorder. The cognitive effects of harboring a CNV was studied by Stefansson et al who examined patients with neuropsychiatric CNVs, unaffected carriers of neuropsychiatric CNVs and a control population; both affected and unaffected carriers of a neuropsychiatric CNV scored substantially lower than control populations without neuropsychiatric associated CNVs. As expected, schizophrenia patients carrying a neuropsychiatric CNV performed significantly worse on cognitive tests for attention and spatial working memory than the non-carrier control population. Interestingly, the neuropsychiatric CNV carrier controls scored between those two groups (Stefansson et al., 2014). These results suggest that CNVs associated with neuropsychiatric disorders may not always manifest in a diagnosis of neuropsychiatric illness, but do still confer cognitive deficits in unaffected carriers.
To link CNVs to symptoms or specific endophenotypes (defined as “quantitative, heritable, trait-related deficits typically assessed by laboratory-based methods rather than clinical observation”(Braff et al., 2006)) of schizophrenia, the genes within each CNV have been closely examined. Hypotheses on how a particular CNV contributes to a phenotype have been developed based upon the function of genes in the affected region. For example, the common deletions at the 22q11.2 locus disrupt two genes associated with microRNAs. The DGCR8 gene disrupted by this deletion is a component of the microRNA processor complex and involved in microRNA biogenesis (Han, 2004; Wang et al., 2007). Another disrupted gene is miR-185, whose target genes have been previously associated with schizophrenia (Warnica et al., 2014). The function of these affected genes has contributed to the hypothesis that the heterogeneous symptoms underlying schizophrenia could be due to many genes becoming dysregulated because of microRNA dysfunction (reviewed Forstner et al., 2013; Geaghan and Cairns, 2014).
Another widely supported hypothesis is that synaptic defects are driving schizophrenia symptoms. The identification of the single gene rare variant, deletion of NRXN1 at the 2p16.3 locus, which is a highly penetrant CNV associated with schizophrenia, supports this hypothesis (Kirov et al., 2014). NRXN1 is a very large gene (~1MB) encoding a presynaptic neuronal cell adhesion molecule (Chen et al., 2013). Deletion of this gene is present in significantly more cases than in the general population (International and Consortium, 2008; Kirov et al., 2009; Elliott Rees et al., 2014), thereby supporting the contribution of synapse disruption.
Mouse Models of Neuropsychiatric CNVs
Human behavioral and genetic studies of CNVs can only correlate important loci with symptoms or endophenotypes. Human genetic studies can then hypothesize how this region may contribution to the broader mechanism of schizophrenia. However, for large CNVs, it is extremely difficult to determine which gene disruptions are the key drivers of the endophenotype. To examine the causal contribution of a given CNV, mouse models for many of the thirteen schizophrenia associated CNVs have been developed (Horev et al., 2011; Li et al., 2009; Missler and Südhof, 1998; Missler et al., 2003; Sigurdsson et al., 2010; Tamada et al., 2010; Walz et al., 2003). These models do not encapsulate all of the symptoms of schizophrenia, but are useful in examining the common endophenotypes such as paired pulse inhibition (PPI), and anxiety (Kellendonk et al., 2009). Mouse models are also useful for examining specific neuronal defects caused by a CNV, which can only be done in humans using post mortem tissue (and which are subject to the confounds of patient studies). Both behavioral and physiological defects have been established in animal models recapitulating CNVs at 22q11.2, 16p11.2, 17p11.2, and NRXN1(Etherton et al., 2009; Horev et al., 2011; Li et al., 2009; Missler et al., 2003; Sigurdsson et al., 2010; Tamada et al., 2010; Walz et al., 2003).
To create the first mouse model of 22q11.2, the homologous chromosome 16 region in mice was first identified (Galili et al., 1997) and this region was then deleted to generate a mouse model (Paylor, 2001). Since the generation of the first mouse model of 22q11.2, this and other mice containing more comprehensive deletions of the 22q11.2 region have been extensively studied (Long et al., 2006; Mukai et al., 2008; Stark et al., 2008). These animals display defects in behavior and neuronal morphology. Behaviorally, the animals exhibit poor performance in cognitive tasks such as spatial working memory (Stark et al., 2008), and conditioned fear memory (Stark et al., 2008) as well as reduced PPI (Paylor and Lindsay, 2006; Paylor, 2001; Stark et al., 2008), an endophenotype also seen in patients with 22q11.2 deletions (Drew et al., 2011) and more generally in many SZ patients (Swerdlow et al., 2014). Defects occurs in neurogenesis and cortical development (Meechan et al., 2009) as well as interneuron migration (Meechan et al., 2012); deficits in neuronal morphology include altered excitatory/inhibitory balance, synaptic plasticity, and functional connectivity (Chun et al., 2014; Mukai et al., 2015, 2008), which are hypothesized endophenotypes of schizophrenia (Allen et al., 2009). Systematic deletions of smaller segments within the 22q11.2 locus have further identified some critical genes for specific endophenotypes (Paylor, 2001). Deletion of a 200kb segment corresponding to a portion of the mouse critical region was sufficient to decrease PPI, while deletion of other segments were sufficient to impair sensorimotor gating and context conditioned fear (Drew et al., 2011; Paylor and Lindsay, 2006). Although a few single gene deletions confer subtle defects on specific tasks, it is still not well understood what their individual contributions are to the behavioral phenotype (reviewed (Drew et al., 2011).
In addition to modeling complex CNVs, mouse models of single gene rare variants associated with schizophrenia, such as NRXN1 deletions, have been closely examined. Behaviorally, homozygous deletions of neurexin-1α result in decreased PPI, increased grooming behaviors (Etherton et al., 2009), and altered social behaviors (Grayton et al., 2013); while heterozygous deficiencies lead to perturbed novelty response (Laarakker et al., 2012). Similarly, non-social cognitive deficits have been reports in neurexin1α-knockout rats (Esclassan et al., 2015). Electrophysiologically, neurexin-1α-knockout mice show defects in synaptic calcium channel function resulting in impaired neurotransmitter release, linking behavior to a specific synaptic event (Etherton et al., 2009). Neurexin dysfunction in adult neurons is sufficient to cause aberrant behavior in mice, indicating that even functional impairment of mature circuits can trigger neuropsychiatric phenotypes (Rabaneda et al., 2014). Establishing a causal relationship between behavior and physiology is very important for determining whether a genetic defect in a mouse is relevant to a complex genetic disease with many, primarily human, attributes.
As described above, mouse models can recapitulate some behavioral and cellular phenotypes of schizophrenia. The major benefit of using mouse models is their ability to examine causal links between genetics, cellular mechanisms, and behavioral phenotypes in a complex in vivo environment. However, for investigation of complex disorders such as schizophrenia, mouse models have some disadvantages. First, it is known that many genetic variants that contribute to disease are located in the non-coding regions (Ripke et al., 2014). While mice are highly homologous to humans in the coding regions, there is much less conservation of the non-coding regions and therefore these variants are difficult to study (Batzoglou, 2000; Johnson et al., 2009). Second, mouse models can lack the complex interacting genetic factors that are known to contribute to the disorder. This is due to the difficultly of perturbed network interactions via a simple knockout or transgenic mouse. Instead, mouse models are best suited for investigating the functional significance of single variants. Finally, mouse models are currently considered the gold standard for the determination of preclinical toxicity in the drug discovery pipeline. However, it has been demonstrated that drugs which are non-toxic and effective in animals are not always non-toxic and effective in human clinical trials (Diener et al., 2008; Tall et al., 2007). Therefore development of more reliable models to test toxicity and efficacy are necessary (reviewed Grskovic et al., 2011).
Patient derived hiPSCs as a Model System
The development of patient derived induced pluripotent stem cells (hiPSCs) provides an additional model, which overcomes many disadvantages of the mouse model system (reviewed K. J. Brennand et al., 2014). Patient derived hiPSCs maintain the patients complete set of risk alleles, including variants in the coding and non-coding regions. Having the patients entire set of risk alleles enables the study of complex genetic interactions, such as the effect of genetic load from disease associated SNPs on CNV expression. New technologies such as the CRISPR/Cas9 system for gene editing can be used to identify the role of single genes. Patient derived hiPSCs can be incorporated into drug toxicity studies to examine outcomes in human cells or into drug development pipelines to help stratify patients likely to be drug responders versus non-responders (reviewed Ko and Gelb, 2014).
Traditionally, patient derived hiPSCs are generated by collecting a skin biopsy or blood sample followed by expression of the classical reprogramming factors (SOX2, OCT4, KLF4 and c-MYC) (Takahashi et al., 2007). Initially, the reprograming factors were expressed from lentiviral vectors or other DNA-integrating viruses; however, this method of expression can induce unwanted genetic alterations; therefore non-integrating methods, such as Sendai virus and synthetic mRNAs, are now widely used (Schlaeger et al., 2014) . Although non-integrating methods have reduced probability of genetic mutations, the formation of CNVs has been associated with the reprogramming process with more CNVs present in early passage hiPSCs than in late passage hiPSCs; however, these CNVs are typically lost after reprogramming and are not a major concern if multiple hiPSC clones from the same skin sample are generated and validated (Ho et al., 2015). Care must therefore be taken, in both the initial characterization and subsequent culturing of new hiPSC lines, to minimize the risk of studying potentially aberrant cells (Ronen and Benvenisty, 2012).
Once reprogrammed, hiPSCs can be differentiated into neuronal progenitor cells and neurons (Paulsen et al., 2012) [Figure 1]. The ability to produce neural progenitor cells and neurons has created a new source of brain tissue, which is often very difficult to obtain. The population of neurons formed by the standard directed differentiation approach is heterogeneous consisting varying percentages of glutamatergic, GABAergic and dopaminergic neurons along with glial populations (Kristen J. Brennand et al., 2011). Directed differentiation and induction protocols that yield more homogenous populations of differentiated neurons are beginning to emerge (Cunningham et al., 2014; Espuny-Camacho et al., 2013; Ho et al., 2015; Kriks et al., 2011; Mariani et al., 2012; Maroof et al., 2013; Nicholas et al., 2013; Shi et al., 2012). These protocols are being optimized to produce pure populations of each of the major neuronal cell types as well as glia.
Figure 1.

Representative control and schizophrenia patient derived hiPSCs and neurons. Both control and patient hiPSCs express pluripotency markers NANOG and TRA-1-60 while hiPSC-derived neurons express neuronal makers MAP2AB and βIII-TUBULIN. (Adapted with permission from (Kristen J. Brennand et al., 2011).
The hiPSC derived neuronal populations exhibit heterogeneous spatial and temporal identities. Comparison of the gene expression studies of hiPSC derived NPCs and neurons indicate that these cells most closely resemble early fetal development (K. Brennand et al., 2014; Mariani et al., 2012). Therefore, it is important to acknowledge that these neurons are not modeling a disease, which presents during early adulthood, as is the case for schizophrenia. Instead, hiPSC derived neurons are best suited for modeling disease predisposition. Studying neurons that most closely resemble the fetal developmental stage can elucidate neurodevelopmental mechanisms that confer risk for developing schizophrenia as an adult.
Using hiPSCs to Link Genetics to Cellular Phenotypes
Schizophrenia is an ideal disorder to study using hiPSC because it is a complex genetic disorder with no single causal mutation. Once neural progenitor cells or neurons are generated from patient hiPSCs, cellular phenotypes can be investigated to gain insight into the cellular mechanisms of schizophrenia. Chiang et al was the first group to derive and characterize hiPSCs from schizophrenia patients alongside controls(Chiang et al., 2011). Follow up studies by our group and others examined the neuronal phenotypes from patient hiPSC derived neurons. Patient hiPSC derived neurons display decreased neurite outgrowth and synapse formation (Kristen J Brennand et al., 2011; Robicsek et al., 2013; Wen et al., 2014) and impaired neurotransmitter release (Hook et al., 2014), as well as defects in neuronal migration (K. Brennand et al., 2014), deficits with adherens junctions (Yoon et al., 2014) and impaired mitochondrial function (K. Brennand et al., 2014; Paulsen et al., 2012; Robicsek et al., 2013).
The initial studies of hiPSC-derived neurons were limited by the availability of patient fibroblasts and therefore these findings represent a heterogeneous patient population where little is known about the patients’ genetic or clinical background. Follow up studies are beginning to follow two distinct approaches. The first is to characterize the cellular phenotype of clinically homogeneous patient cohorts. This provides the ability to learn about disease mechanisms, which are shared across patients who exhibit similar symptoms or have similar drug response profiles. This type of study may lead to commonly disrupted pathways that could be relevant across a large subset of schizophrenia patients.
The second approach is to examine a cohort of patients all carrying the same genetic mutation (Wen et al., 2014), such as a CNV (Yoon et al., 2014). This approach is similar to mouse models as it links cellular phenotypes to pathogenic risk variants. Using this approach with patient derived hiPSCs can demonstrate the role of a CNV in the context of all other variants and endogenous regulators in the patient’s genome. This approach was used to uncover a cellular phenotype of the 15q11.2 CNV. The 15q11.2 microdeletion has been associated with schizophrenia while larger duplications of this region at 15q11.2-13 or the entire chromosome 15 have both been associated with autism spectrum disorder. Moreover, microdeletions or uniparental disomy or imprinting defects at 15q11-q13 result in Angelman syndrome (AS) when maternally inherited and Prader-Willi syndrome (PWS) when paternally inherited (Kalsner and Chamberlain, 2015). hiPSCs developed from a patient with a 15q11.2 microdeletion displayed deficits in adherens junctions and apical polarity. CYFIP1, one of the 4 genes deleted in this region is involved in regulating the WAVE complex, which participates in cytoskeleton remodeling [Figure 2]. hiPSCs containing the 15q11.2 deletion displayed an 80% decrease levels of WAVE2, which was completely rescued by lentiviral expression of CYFIP1. Knockdown of CYFIP1 in a mouse model confirmed these findings to be physiologically relevant in vivo and uncovered a novel signaling cascade which regulates radial glial cells in the developing embryonic mouse cortex (Yoon et al., 2014). This approach was also taken by Adamo et al to study a CNV at 7q11.23 where deletion results in Williams-Beuren syndrome, while duplication causes 7q-microduplication syndrome. Examination of patient derived hiPSCs and the differentiated, disease relevant cell types showed dosage-dependent alterations in disease relevant transcriptional circuits (Adamo et al., 2014). This group also investigated the role of a transcription factor, GTF2l which is disrupted by the CNV and found that manipulation GTF2l caused a significant proportion of the transcription dysregulation created by the entire CNV (Adamo et al., 2014). Applying this type of study to schizophrenia may lead to discovery of important transcriptional circuits or key regulators of the disorder. A summary of studies of neuropsychiatric CNVs using hiPSCs is shown in Table 2.
Figure 2.
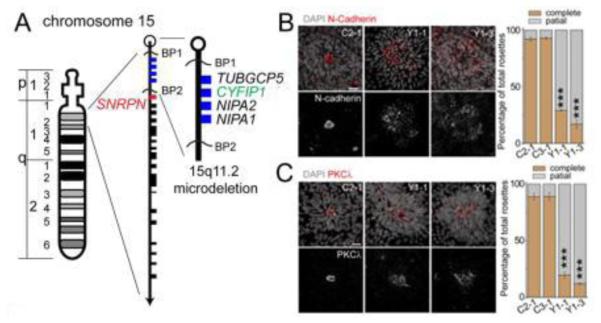
hiPSC model of the schizophrenia associated CNV 15q11.2 (Del) was engineered by Yoon et al. hiPSC derived neurons carrying the 15q11.2 microdeletion were shown to have deficits in adherens junctions and apical polarity. This work also determined that haplosufficiency of the CYFIP1 locus was casual to these defects. (Adapted with permission from (Yoon et. al, 2014)).
Table 2.
Major findings from studies of neuropsychiatric CNVs using hiPSC models.
| CNV locus |
Neuropsychi atric Condition |
hiPSC- derived cell type |
Major Findings | Referenc es |
|---|---|---|---|---|
| 22q11.2 Del |
Schizophreni a |
Neurons | Recapitulation of miRNA expression pattern expected of 22q11.2 haploinsufficiency |
(Zhao et. al, 2015) |
| 15q11.2 and DISC1 |
Schizophreni a |
Neural Progenitor Cells (NPCs) |
Defects in adherens junctions and apical polarity; Destabilization of WAVE complex due to CYFIP1 deficiency |
(Yoon et. al, 2014) |
| DISC1 | Schizophreni a |
Neurons | Impaired synaptic vesicle release; Dysregulates expression of synaptic genes implicated in psychiatric disorders |
(Wen et. al, 2014) |
| 15q11- q13.1 Dup |
Autism | Neurons | 15q11-q13.1 transcript levels did not consistently correlate with copy number |
(Germain et. al, 2014) |
| 22q13.3 | Phelan- McDermid Syndrome |
Neurons | Reduced SHANK3 expression; Defects in excitatory synaptic transmission; Rescued by SHANK3 expression and IGF1 treatment |
(Shcheglovi tov et. al, 2013) |
| MeCP2 | Rett Syndrome |
Neurons | Reduced levels of pallidin transcript | (Larimore et. al, 2013) |
| FMR1 | Fragile X | Neurons | Reduced PSD95 expression; Reduced synaptic puncta density and neurite length; Increased Ca2+ transient amplitude and frequency |
(Liu et. al, 2012) |
| 7q11.23 Del & Dup |
Williams- Beuren syndrome |
hiPSC, NPC | 7q11.23 dosage affects disease relevant transcriptional programs Significant proportion of dysregulation is due to GTF21 gene |
(Adamo et al., 2014) |
Genome Editing as a Tool for Investigating Casual Variants
A useful tool for assessing CNVs using hiPSC derived neurons is genome editing technology, such as CRISPR/Cas9 which provides a simple, efficient approach for introducing genetic mutations, correcting genetic mutations (Jiang et al., 2013; Miller et al., 2007), and even enhancing (Maeder et al., 2013) or repressing (Larson et al., 2013) gene expression (reviewed Hsu et al., 2014). In the CRISPR/Cas9 system the Cas9 endonuclease uses a sgRNA bind to its complementary target on the DNA, which then causes Cas9 to create a double stranded break in the DNA. The double stranded break can be repaired through non-homologous end joining (NHEJ) or through homology directed repair. A repair template can also be supplied allowing for the creation or repair of mutations at specific sites (Cong et al., 2013; Ran et al., 2013). Genome editing technologies have most commonly been applied to study monogenic disorders such as cystic fibrosis or Huntington’s disease. However, the CRISPR/Cas9 system has been used to edit hiPSCs, which are then differentiated into neurons to study the functional implications of a mutation or a rescued mutation. Recently, CRISPR-Cas9 was successfully used to for targeted deletion of the CGG repeats found in fragile X patients. Park et al used CRISP/Cas9 9 to delete the CGG repeats in patient derived hiPSCs and demonstrated demethylation, open chromatin conformation and transcription initiation at the FMR1 promoter in the patient cells after editing (Park et al., 2015). CRISPR/Cas9 has also been used to study Rett syndrome, which is caused by mutations in the MECP2 gene. Additionally. Ananiev et al used CRISPR/Cas9 to create isogenic controls for common and rare single nucleotide polymorphism (SNP) in the MECP2 gene implicated in Rett syndrome (Ananiev et al., 2011). This approach for genome editing allows investigation into the casual relationship between then CNV and cellular phenotypes of disease relevant cell types from human patients, as isogenic control lines can be made using CRISPR/Cas9, while the rest of the patients genome remains unchanged. Additionally, patients with the same CNV but different genetic backgrounds can help researchers understand the contribution of additional risk alleles to a cellular phenotype [Figure 3].
Figure 3.

Schematic demonstrating the utility of genome editing in modeling CNVs in hiPSC derived neurons. In the CRISPR-Cas9 system, the guide RNA is complexed with the Cas9 protein. When the guide RNA binds to complementary DNA, Cas9 will great a double stranded break into the DNA. Using this technology a hiPSC derived neuron from a control cell line can be engineered to possess the SZ-associated CNV or a CNV from a patient derived hiPSC derived neuron can be repaired by providing a wild type copy which can be inserted through homologous recombination. (Adapted with permission from Nestor et al, 2015).
It may even by possible to assess the role of single genes within a CNV assessed in patient derived hiPSCs by using CRISPR-Cas9 to replace or delete copies of single genes, while maintaining the rest of the disrupted locus. This can be done by activating the endogenous expression of the wild type allele in the case of a CNV deletion or repressing the endogenous expression of the wild type allele in the case of CNV duplication. To convert the CRISPR-Cas9 system into a system for genetic activation, the nuclease activity of Cas9 must be disrupted. This can be done by mutating the RuvC and HNH nuclease domains of Cas9. Once the nuclease activity of Cas9 has been disrupted it can be fused to the transcription-activating domain of transcription factors. The sgRNAs can then be targeted to the promoter of specific genes allowing for modulation of their expression. With optimized protocols creating pure neuronal populations, questions regarding cell specificity and temporal regulation can be incorporated into the experimental design with CRISPR technology (Konermann et al., 2014). A single type of neuron can be edited to contain repair a disease associated CNV. These neurons can then be co-cultured with another neuronal or glial population, to determine the cell autonomous effects of a CNV. These approaches could help determine the genetic elements and the cell types that are most important for patients with schizophrenia who carry a known neuropsychiatric CNV, leading to more targeted treatment of these patients.
Using hiPSCs to Query Common Biological Pathways Associated with Neuropsychiatric CNVs
Studying the role of CNVs may provide insight into genes or cellular mechanisms relevant to a broader range of schizophrenia patients. For example, it has been hypothesized that a genome wide set of common single nucleotide polymorphisms (SNPs) could act as a determinant for disorders, such a factor might differentially modify phenotypic expression of a CNV in individuals (Hiroi et al., 2013). The most recent GWAS study by the psychiatric genomics consortium has identified 108 loci significantly associated with schizophrenia (Ripke et al., 2014). Patient derived hiPSCs presents one model system to test how these variants relate to the CNVs, which are known to be strongly associated with schizophrenia. For example, the neurodevelopmental gene KCTD13, encoding the polymerase delta-interacting protein 1 (Neale and Sklar, 2015), lies in a region with significant common variant association to schizophrenia. This gene is also in a schizophrenia and autism associated duplication on chromosome 16p11.2 (McCarthy et al., 2009; Weiss et al., 2008). KCTD13 may play in important role in the disease pathophysiology because defects in the gene can confer risk to schizophrenia through common variation and it lies within a more penetrant CNV (Neale and Sklar, 2015). The role of these two genetic elements, the common variant near KCTD13 and the 16p11.2 duplication can be dissociated using hiPSC derived neurons, providing a better understanding of which of the pathways that KCTD13 is involved in is actually conferring risk for schizophrenia. Approaches such as these with incorporation of human genetic studies, mouse models, and hiPSC studies of schizophrenia associated CNVs will allow for important biological pathways to be uncovered. These pathways will provide new targets for drug design and development.
Gene set and pathway analysis has demonstrated that patients harboring CNVs are enriched in disrupted genes associated with neurodevelopment and synaptic transmission (Walsh et al., 2008). More specifically, calcium signaling and synaptic transmission dependent on the genes encoding the NMDAR and ARC complexes are implicated in the disorder (Etherton et al., 2009; Kirov et al., 2011; Purcell et al., 2014; Szatkiewicz et al., 2014). Recently, Pocklington et al further demonstrated that patient CNVs are enriched for genes involved in GABAergic neurotransmission as well as genes involved in glutamatergic signaling (Pocklington et al., 2015). These findings based on gene enrichment studies in CNV are supported by pathway analysis of the common variants. Although GABAergic and glutamatergic signaling are likely disrupted in patients, the most effective antipsychotic treatments of schizophrenia are D2 receptor antagonists (Siu et al., 2013). The mechanism of how these antipsychotics act to reduce positive symptoms of schizophrenia is unknown. Although the antipsychotics modulating dopamine transmission can reduce the positive symptoms for some patients, dopamine transmission is thought to be a downstream effect of disrupted GABAergic and glutamatergic signaling (Demjaha et al., 2014; Weinberger, 1987; Wen et al., 2010). Therefore, there is potential for the development of more targeted and effective medications that improve the negative and cognitive symptoms as well.
Using hiPSCs to Complement Drug Discovery
Patient derived hiPSCs can be used in both of the standard approaches of drug discovery. One approach is to design a drug against a known target and test the drug’s efficacy and toxicity. This approach leverages knowledge of the basic mechanism of the disease to design the therapeutic. Using patient derived hiPSCs the new therapeutic can be tested directly on the cell type of interest to quickly assess its function. Cellular assays can be used to determine efficacy and toxicity in human cells before moving into complex in vivo systems. The second approach involves high throughput screening of millions of compounds to find candidate therapeutics. Patient derived hiPSCs can serve as platform to perform these screens in a disease relevant cell type and genetic context. During these screens it may be possible to determine drug responder vs. non responder populations, which would help researchers outline the appropriate patient population before moving into a clinical trial.
The existing protocol for drug development begins pre-clinically with animal models and some in vitro screening, followed by multiphase clinical trials in humans. Animal models are very well suited for assessing the pharmacokinetics and pharmacodynamics before reaching humans. In pre-clinical studies patient derived hiPSCs may be able to provide a human in vitro component to better assess the drug’s toxicity in humans. Patient derived hiPSCs can also be incorporated into the clinical trial processes as a human in vitro component. In phase 2 trials, where the drug’s efficacy in patients is being evaluated by fixed endpoints, comparison of patient and control hiPSCs, before and after drug treatment, can be done using a cellular assay to provide a simple, specific endpoint. In this phase patient derived hiPSCs, which are drug responders may help stratify the patient population. If only certain genetic backgrounds respond, future trials can be designed to include only the subset of patients for whom the drug is efficacious. During a phase 3 trial, patient derived hiPSCs can be generated from many individuals with diverse genetic backgrounds to help establish drug toxicity in a wide range of genetic backgrounds to help reduce negative outcomes (reviewed Ko and Gelb, 2014). However, there are limitations for using hiPSCs during clinical trials, including the heterogeneity resulting from both the reprogramming process and the differentiation protocol, this type of intra-individual variation may impact the reproducibility of a clinical trial if the cellular phenotype is not robust. Additionally, some cell types are more difficult to grow in large quantities and it remains very expensive to grow large numbers of patient lines. As the technology surrounding hiPSCs generation, maintenance and differentiation expands, their utility as a disease model will grow.
Future Considerations and Conclusions
The future advancement in developing new therapeutics for schizophrenia, or any other neurological disorder, relies upon an understanding of commonly disrupted pathways. These pathways can be uncovered through careful integration of human genetic studies to identify disease related variants; mouse models to examine the function of a variant in complex in vivo environment; and patient derived hiPSC models to determine the casual role of a variant on a cellular phenotype and the contribution of complex genetic interactions. Incorporation of this type of information from studies investigating CNVs in schizophrenia has begun to implicate specific pathways, such as a genes regulating NMDAR and ARC complexes (Purcell et al., 2014; Szatkiewicz et al., 2014). This knowledge can inform the development of new therapeutics. However, the potential of hiPSCs does not end in mechanistic studies but rather continues into clinical trials where hiPSCs can be used as a complementary human in vitro component. Before the full potential of hiPSCs becomes a reality there is still a large amount of work to be done. Optimized protocols for more homogeneous populations of hiPSCs and pure populations of differentiated cells need to be fully developed and validated. This will decrease the time and cost needed to develop patient derived hiPSC models allowing for more patient lines to be examined. Better culturing techniques should be applied to form more complex neural circuitries that better resemble the human brain. This is currently an area of great interest with the development of human cortical spheroids (Paşca et al., 2015). Finally, experiments investigating complex diseases should be designed to collect multilayer data including the patient’s clinical information, genome, transcriptome, proteome, and epigenome (Schadt et al., 2014). Gathering this type of multilayered information is now possible with the advancement being made in sequencing technology and bioinformatics. From this information we can begin to build networks and identify the key drivers of complex genetic disorders such as schizophrenia.
Highlights.
The contribution of copy number variants(CNVs) to schizophrenia genetics
Summary of findings from current mouse models of CNVs
The advantages of using hiPSCs to model CNVs
The potential of hiPSCs in drug discovery, screening and toxicity testing
Acknowledgements
Kristen Brennand is a New York Stem Cell Foundation - Robertson Investigator. The Brennand Laboratory is supported by a Brain and Behavior Young Investigator Grant, National Institute of Health (NIH) grant R01 MH101454 and the New York Stem Cell Foundation
Abbreviations
- hiPSC
human induced pluripotent stem cell
- CNV
Copy number variation
- SNP
Single nucleotide polymorphism
- PPI
Paired pulse inhibition
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Information
The authors have declared that no competing interests exist.
References
- Adamo A, Atashpaz S, Germain P-L, Zanella M, D’Agostino G, Albertin V, Chenoweth J, Micale L, Fusco C, Unger C, Augello B, Palumbo O, Hamilton B, Carella M, Donti E, Pruneri G, Selicorni A, Biamino E, Prontera P, McKay R, Merla G, Testa G. 7Q11.23 Dosage-Dependent Dysregulation in Human Pluripotent Stem Cells Affects Transcriptional Programs in Disease-Relevant Lineages. Nat. Genet. 2014;47:132–141. doi: 10.1038/ng.3169. doi:10.1038/ng.3169. [DOI] [PubMed] [Google Scholar]
- Ahn K, An SS, Shugart YY, Rapoport JL. Common polygenic variation and risk for childhood-onset schizophrenia. Mol. Psychiatry. 2014a:1–3. doi: 10.1038/mp.2014.158. doi:10.1038/mp.2014.158. [DOI] [PubMed] [Google Scholar]
- Ahn K, Gotay N, Andersen TM, Anvari a a, Gochman P, Lee Y, Sanders S, Guha S, Darvasi A, Glessner JT, Hakonarson H, Lencz T, State MW, Shugart YY, Rapoport JL. High rate of disease-related copy number variations in childhood onset schizophrenia. Mol. Psychiatry. 2014b;19:568–572. doi: 10.1038/mp.2013.59. doi:10.1038/mp.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen AJ, Griss ME, Folley BS, Hawkins K. a, Pearlson GD. Endophenotypes in schizophrenia: A selective review. Schizophr. Res. 2009;109:24–37. doi: 10.1016/j.schres.2009.01.016. doi:10.1016/j.schres.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananiev G, Williams EC, Li H, Chang Q. Isogenic Pairs of Wild Type and Mutant Induced Pluripotent Stem Cell (iPSC) Lines from Rett Syndrome Patients as In Vitro Disease Model. PLoS One. 2011;6:e25255. doi: 10.1371/journal.pone.0025255. doi:10.1371/journal.pone.0025255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzoglou S. Human and Mouse Gene Structure: Comparative Analysis and Application to Exon Prediction. Genome Res. 2000;10:950–958. doi: 10.1101/gr.10.7.950. doi:10.1101/gr.10.7.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff DL, Freedman R, Schork NJ, Gottesman II. Deconstructing Schizophrenia: An Overview of the Use of Endophenotypes in Order to Understand a Complex Disorder. Schizophr. Bull. 2006;33:21–32. doi: 10.1093/schbul/sbl049. doi:10.1093/schbul/sbl049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand K, Savas JN, Kim Y, Tran N, Simone a, Hashimoto-Torii K, Beaumont KG, Kim HJ, Topol a, Ladran I, Abdelrahim M, Matikainen-Ankney B, Chao S-H, Mrksich M, Rakic P, Fang G, Zhang B, Yates JR, Gage FH. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry. 2014;20:1–8. doi: 10.1038/mp.2014.22. doi:10.1038/mp.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Landek-Salgado MA, Sawa A. Modeling Heterogeneous Patients With a Clinical Diagnosis of Schizophrenia With Induced Pluripotent Stem Cells. Biol. Psychiatry. 2014;75:936–944. doi: 10.1016/j.biopsych.2013.10.025. doi:10.1016/j.biopsych.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, McCarthy S, Sebat J, Gage FH. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. doi:10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, McCarthy S, Sebat J, Gage FH. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. doi:10.1038/nature10603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Cao F, Liu L, Wang L, Chen X. Genetic studies of schizophrenia: an update. Neurosci. Bull. 2015;31:87–98. doi: 10.1007/s12264-014-1494-4. doi:10.1007/s12264-014-1494-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Shen Y, Zhang F, Chiang C, Pillalamarri V, Blumenthal I, Talkowski M, Wu BL, Gusella JF. Molecular analysis of a deletion hotspot in the NRXN1 region reveals the involvement of short inverted repeats in deletion CNVs. Am. J. Hum. Genet. 2013;92:375–386. doi: 10.1016/j.ajhg.2013.02.006. doi:10.1016/j.ajhg.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C-H, Su Y, Wen Z, Yoritomo N, Ross CA, Margolis RL, Song H, Ming G. Integration-free induced pluripotent stem cells derived from schizophrenia patients with a DISC1 mutation. Mol. Psychiatry. 2011;16:358–360. doi: 10.1038/mp.2011.13. doi:10.1038/mp.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Westmoreland JJ, Bayazitov IT, Eddins D, Pani AK, Smeyne RJ, Yu J, Blundon JA, Zakharenko SS. Specific disruption of thalamic inputs to the auditory cortex in schizophrenia models. Science (80−. ) 2014;344:1178–1182. doi: 10.1126/science.1253895. doi:10.1126/science.1253895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science (80−. ) 2013;339:819–823. doi: 10.1126/science.1231143. doi:10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JEM, DePristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, Zilversmit M, Cartwright R, Rouleau GA, Daly M, Stone EA, Hurles ME, Awadalla P. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011;43:712–714. doi: 10.1038/ng.862. doi:10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham M, Cho J-H, Leung A, Savvidis G, Ahn S, Moon M, Lee PKJ, Han JJ, Azimi N, Kim K-S, Bolshakov VY, Chung S. hPSC-Derived Maturing GABAergic Interneurons Ameliorate Seizures and Abnormal Behavior in Epileptic Mice. Cell Stem Cell. 2014;15:559–573. doi: 10.1016/j.stem.2014.10.006. doi:10.1016/j.stem.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demjaha A, Egerton A, Murray RM, Kapur S, Howes OD, Stone JM, McGuire PK. Antipsychotic Treatment Resistance in Schizophrenia Associated with Elevated Glutamate Levels but Normal Dopamine Function. Biol. Psychiatry. 2014;75:e11–e13. doi: 10.1016/j.biopsych.2013.06.011. doi:10.1016/j.biopsych.2013.06.011. [DOI] [PubMed] [Google Scholar]
- Diener HC, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, Shuaib A, Ashwood T, Wasiewski W, Alderfer V, Hårdemark HG, Rodichok L. NXY-059 for the treatment of acute stroke: Pooled analysis of the SAINT I and II trials. Stroke. 2008;39:1751–1758. doi: 10.1161/STROKEAHA.107.503334. doi:10.1161/STROKEAHA.107.503334. [DOI] [PubMed] [Google Scholar]
- Drew LJ, Crabtree GW, Markx S, Stark KL, Chaverneff F, Xu B, Mukai J, Fenelon K, Hsu P, Gogos JA, Karayiorgou M. The 22q11.2 microdeletion: Fifteen years of insights into the genetic and neural complexity of psychiatric disorders. Int. J. Dev. Neurosci. 2011;29:259–281. doi: 10.1016/j.ijdevneu.2010.09.007. doi:10.1016/j.ijdevneu.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esclassan F, Francois J, Phillips KG, Loomis S, Gilmour G. Phenotypic characterization of nonsocial behavioral impairment in neurexin 1α knockout rats. Behav. Neurosci. 2015;129:74–85. doi: 10.1037/bne0000024. doi:10.1037/bne0000024. [DOI] [PubMed] [Google Scholar]
- Espuny-Camacho I, Michelsen K. a., Gall D, Linaro D, Hasche A, Bonnefont J, Bali C, Orduz D, Bilheu A, Herpoel A, Lambert N, Gaspard N, Péron S, Schiffmann SN, Giugliano M, Gaillard A, Vanderhaeghen P. Pyramidal Neurons Derived from Human Pluripotent Stem Cells Integrate Efficiently into Mouse Brain Circuits In Vivo. Neuron. 2013;77:440–456. doi: 10.1016/j.neuron.2012.12.011. doi:10.1016/j.neuron.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Etherton MR, Blaiss C. a, Powell CM, Südhof TC. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc. Natl. Acad. Sci. U. S. A. 2009;106:17998–18003. doi: 10.1073/pnas.0910297106. doi:10.1073/pnas.0910297106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejgin K, Nielsen J, Birknow MR, Bastlund JF, Nielsen V, Lauridsen JB, Stefansson H, Steinberg S, Sorensen HBD, Mortensen TE, Larsen PH, Klewe IV, Rasmussen SV, Stefansson K, Werge TM, Kallunki P, Christensen KV, Didriksen M. A Mouse Model that Recapitulates Cardinal Features of the 15q13.3 Microdeletion Syndrome Including Schizophrenia- and Epilepsy-Related Alterations. Biol. Psychiatry. 2014;76:128–137. doi: 10.1016/j.biopsych.2013.08.014. doi:10.1016/j.biopsych.2013.08.014. [DOI] [PubMed] [Google Scholar]
- Forstner AJ, Degenhardt F, Schratt G, Nöthen MM. MicroRNAs as the cause of schizophrenia in 22q11.2 deletion carriers, and possible implications for idiopathic disease: a mini-review. Front. Mol. Neurosci. 2013;6:47. doi: 10.3389/fnmol.2013.00047. doi:10.3389/fnmol.2013.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili N, Baldwin HS, Lund J, Reeves R, Gong W, Wang Z, Roe B. a, Emanuel BS, Nayak S, Mickanin C, Budarf ML, Buck C. a. A region of mouse chromosome 16 is syntenic to the DiGeorge, velocardiofacial syndrome minimal critical region. Genome Res. 1997;7:17–26. doi: 10.1101/gr.7.1.17. [DOI] [PubMed] [Google Scholar]
- Geaghan M, Cairns MJ. MicroRNA and Posttranscriptional Dysregulation in Psychiatry. Biol. Psychiatry. 2014:1–9. doi: 10.1016/j.biopsych.2014.12.009. doi:10.1016/j.biopsych.2014.12.009. [DOI] [PubMed] [Google Scholar]
- Grayton HM, Missler M, Collier DA, Fernandes C. Altered Social Behaviours in Neurexin 1α Knockout Mice Resemble Core Symptoms in Neurodevelopmental Disorders. PLoS One. 2013;8:e67114. doi: 10.1371/journal.pone.0067114. doi:10.1371/journal.pone.0067114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grskovic M, Javaherian A, Strulovici B, Daley GQ. Induced pluripotent stem cells — opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 2011:10. doi: 10.1038/nrd3577. doi:10.1038/nrd3577. [DOI] [PubMed] [Google Scholar]
- Han J. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. doi:10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Health, N.I. of M. The Numbers Count?: Mental Disorders in America [WWW Document]. nimh.nih.gov. 2013 URL www.nimh.nih.gov/health/publications/the-numbers-count-mental-disorders-in-america/index.shtml.
- Hiroi N, Takahashi T, Hishimoto A, Izumi T, Boku S, Hiramoto T. Copy number variation at 22q11 : from rare variants to common mechanisms of developmental neuropsychiatric disorders. Mol. Psychiatry. 2013;18:1153–1165. doi: 10.1038/mp.2013.92. doi:10.1038/mp.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S, Topol A, Brennand KJ. From “Directed Differentiatio” to “Neuronal Induction”: Modeling Neuropsychiatric Disease. Biomark. Insights. 2015;10:31. doi: 10.4137/BMI.S20066. doi:10.4137/BMI.S20066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook V, Brennand KJ, Kim Y, Toneff T, Funkelstein L, Lee KC, Ziegler M, Gage FH. Human iPSC Neurons Display Activity-Dependent Neurotransmitter Secretion: Aberrant Catecholamine Levels in Schizophrenia Neurons. Stem Cell Reports. 2014;3:531–538. doi: 10.1016/j.stemcr.2014.08.001. doi:10.1016/j.stemcr.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horev G, Ellegood J, Lerch JP, Son Y-EE, Muthuswamy L, Vogel H, Krieger AM, Buja A, Henkelman RM, Wigler M, Mills AA. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. 2011;108:17076–17081. doi: 10.1073/pnas.1114042108. doi:10.1073/pnas.1114042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. doi:10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International T, Consortium S. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. doi:10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Bikard D, Cox D, Zhang F, Marraffini L. a. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013;31:233–9. doi: 10.1038/nbt.2508. doi:10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MB, Kawasawa YI, Mason CE, Krsnik Ž, Coppola G, Bogdanović D, Geschwind DH, Mane SM, State MW, Šestan N. Functional and Evolutionary Insights into Human Brain Development through Global Transcriptome Analysis. Neuron. 2009;62:494–509. doi: 10.1016/j.neuron.2009.03.027. doi:10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsner L, Chamberlain SJ. Prader-Willi, Angelman, and 15q11-q13 Duplication Syndromes. Pediatr. Clin. North Am. 2015;62:587–606. doi: 10.1016/j.pcl.2015.03.004. doi:http://dx.doi.org/10.1016/j.pcl.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayiorgou M, Simon TJ, Gogos J. a. 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nat. Rev. Neurosci. 2010;11:402–416. doi: 10.1038/nrn2841. doi:10.1038/nrn2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellendonk C, Simpson EH, Kandel ER. Modeling cognitive endophenotypes of schizophrenia in mice. Trends Neurosci. 2009;32:347–358. doi: 10.1016/j.tins.2009.02.003. doi:10.1016/j.tins.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov G, Grozeva D, Norton N, Ivanov D, Mantripragada KK, Holmans P, Craddock N, Owen MJ, O’Donovan MC. Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum. Mol. Genet. 2009;18:1497–1503. doi: 10.1093/hmg/ddp043. doi:10.1093/hmg/ddp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov G, Pocklington A, Holmans PA, Ivanov DK, Ikeda M, Ruderfer D, Moran J, Chambert K, Toncheva D, Georgieva L, Grozeva DV, Fjodorova M, Wollerton RL, Rees E, Nikolova I, van de Lagemaat LN, Bayés À, Fernandez E, Olason PI, Böttcher Y, Komiyama NH, Collins MO, Choudhary J, Stefansson K, Stefansson H, Grant SGN, Purcell S, Sklar P, O’Donovan MC, Owen MJ. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry. 2011;142:153. doi: 10.1038/mp.2011.154. doi:10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov G, Rees E, Walters JTR, Escott-Price V, Georgieva L, Richards AL, Chambert KD, Davies G, Legge SE, Moran JL, McCarroll SA, O’Donovan MC, Owen MJ. The Penetrance of Copy Number Variations for Schizophrenia and Developmental Delay. Biol. Psychiatry. 2014;75:378–385. doi: 10.1016/j.biopsych.2013.07.022. doi:10.1016/j.biopsych.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HC, Gelb BD. Concise Review: Drug Discovery in the Age of the Induced Pluripotent Stem Cell. Stem Cells Transl. Med. 2014;3:500–509. doi: 10.5966/sctm.2013-0162. doi:10.5966/sctm.2013-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2014;517:583–8. doi: 10.1038/nature14136. doi:10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S, Shim J-W, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. doi:10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laarakker MC, Reinders NR, Bruining H, Ophoff R. a., Kas MJH. Sex-dependent novelty response in neurexin-1?? mutant mice. PLoS One. 2012;7:3–8. doi: 10.1371/journal.pone.0031503. doi:10.1371/journal.pone.0031503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson MH, Gilbert L. a, Wang X, Lim W. a, Weissman JS, Qi LS. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013;8:2180–2196. doi: 10.1038/nprot.2013.132. doi:10.1038/nprot.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HH, Roy M, Kuscuoglu U, Spencer CM, Halm B, Harrison KC, Bayle JH, Splendore A, Ding F, Meltzer L. a., Wright E, Paylor R, Deisseroth K, Francke U. Induced chromosome deletions cause hypersociability and other features of Williams-Beuren syndrome in mice. EMBO Mol. Med. 2009;1:50–65. doi: 10.1002/emmm.200900003. doi:10.1002/emmm.200900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JM, LaPorte P, Merscher S, Funke B, Saint-Jore B, Puech A, Kucherlapati R, Morrow BE, Skoultchi AI, Wynshaw-Boris A. Behavior of mice with mutations in the conserved region deleted in velocardiofacial/DiGeorge syndrome. Neurogenetics. 2006;7:247–257. doi: 10.1007/s10048-006-0054-0. doi:10.1007/s10048-006-0054-0. [DOI] [PubMed] [Google Scholar]
- Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA–guided activation of endogenous human genes. Nat. Methods. 2013;10:977–979. doi: 10.1038/nmeth.2598. doi:10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Sebat J. CNVs: Harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. doi:10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani J, Simonini MV, Palejev D, Tomasini L, Coppola G, Szekely AM, Horvath TL, Vaccarino FM. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc. Natl. Acad. Sci. 2012;109:12770–12775. doi: 10.1073/pnas.1202944109. doi:10.1073/pnas.1202944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof AM, Keros S, Tyson JA, Ying S-W, Ganat YM, Merkle FT, Liu B, Goulburn A, Stanley EG, Elefanty AG, Widmer HR, Eggan K, Goldstein PA, Anderson SA, Studer L. Directed Differentiation and Functional Maturation of Cortical Interneurons from Human Embryonic Stem Cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. doi:10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, Krause V, Kumar R. a, Grozeva D, Malhotra D, Walsh T, Zackai EH, Kaplan P, Ganesh J, Krantz ID, Spinner NB, Roccanova P, Bhandari A, Pavon K, Lakshmi B, Leotta A, Kendall J, Lee Y-H, Vacic V, Gary S, Iakoucheva LM, Crow TJ, Christian SL, Lieberman J. a, Stroup TS, Lehtimäki T, Puura K, Haldeman-Englert C, Pearl J, Goodell M, Willour VL, Derosse P, Steele J, Kassem L, Wolff J, Chitkara N, McMahon FJ, Malhotra AK, Potash JB, Schulze TG, Nöthen MM, Cichon S, Rietschel M, Leibenluft E, Kustanovich V, Lajonchere CM, Sutcliffe JS, Skuse D, Gill M, Gallagher L, Mendell NR, Craddock N, Owen MJ, O’Donovan MC, Shaikh TH, Susser E, Delisi LE, Sullivan PF, Deutsch CK, Rapoport J, Levy DL, King M-C, Sebat J. Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. doi:10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Tucker ES, Maynard TM, LaMantia A-S. Cxcr4 regulation of interneuron migration is disrupted in 22q11.2 deletion syndrome. Proc. Natl. Acad. Sci. 2012;109:18601–18606. doi: 10.1073/pnas.1211507109. doi:10.1073/pnas.1211507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Tucker ES, Maynard TM, LaMantia A-S. Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proc. Natl. Acad. Sci. U. S. A. 2009;106:16434–16445. doi: 10.1073/pnas.0905696106. doi:10.1073/pnas.0905696106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein J. a., Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. doi:10.1016/S0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Miller JC, Holmes MC, Wang J, Guschin DY, Lee Y-L, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, Gregory PD, Pabo CO, Rebar EJ. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007;25:778–785. doi: 10.1038/nbt1319. doi:10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. Others. doi:10.1038/nature09708.Mapping. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missler M, Südhof TC. Neurexins: Three genes and 1001 products. Trends Genet. 1998;14:20–26. doi: 10.1016/S0168-9525(97)01324-3. doi:10.1016/S0168-9525(97)01324-3. [DOI] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Südhof TC. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. 2003;423:939–948. doi: 10.1038/nature01755. doi:10.1038/nature01755. [DOI] [PubMed] [Google Scholar]
- Mukai J, Dhilla A, Drew LJ, Stark KL, Cao L, MacDermott AB, Karayiorgou M, Gogos J. a. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nat. Neurosci. 2008;11:1302–1310. doi: 10.1038/nn.2204. doi:10.1038/nn.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai J, Tamura M, Fénelon K, Rosen AM, Spellman TJ, Kang R, MacDermott AB, Karayiorgou M, Gordon JA, Gogos JA. Molecular Substrates of Altered Axonal Growth and Brain Connectivity in a Mouse Model of Schizophrenia. Neuron. 2015:680–695. doi: 10.1016/j.neuron.2015.04.003. doi:10.1016/j.neuron.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Sklar P. Genetic analysis of schizophrenia and bipolar disorder reveals polygenicity but also suggests new directions for molecular interrogation. Curr. Opin. Neurobiol. 2015;30:131–138. doi: 10.1016/j.conb.2014.12.001. doi:10.1016/j.conb.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Nestor MW, Phillips AW, Artimovich E, Nestor JE, Hussman JP, Blatt GJ. Human Inducible Pluripotent Stem Cells and Autism Spectrum Disorder: Emerging Technologies. Autism Res. 2015 doi: 10.1002/aur.1570. n/a–n/a. doi:10.1002/aur.1570. [DOI] [PubMed] [Google Scholar]
- Nicholas CR, Chen J, Tang Y, Southwell DG, Chalmers N, Vogt D, Arnold CM, Chen Y-JJ, Stanley EG, Elefanty AG, Sasai Y, Alvarez-Buylla A, Rubenstein JLR, Kriegstein AR. Functional Maturation of hPSC-Derived Forebrain Interneurons Requires an Extended Timeline and Mimics Human Neural Development. Cell Stem Cell. 2013;12:573–586. doi: 10.1016/j.stem.2013.04.005. doi:10.1016/j.stem.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Halevy T, Lee DR, Sung JJ, Lee JS, Yanuka O, Benvenisty N, Kim D. Reversion of FMR1 Methylation and Silencing by Editing the Triplet Repeats in Fragile X iPSC-Derived Neurons. Cell Rep. 2015:1–8. doi: 10.1016/j.celrep.2015.08.084. doi:10.1016/j.celrep.2015.08.084. [DOI] [PubMed] [Google Scholar]
- Paşca AM, Sloan S. a, Clarke LE, Tian Y, Makinson CD, Huber N, Kim CH, Park J-Y, O’Rourke N. a, Nguyen KD, Smith SJ, Huguenard JR, Geschwind DH, Barres B. a, Paşca SP. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods. 2015:12. doi: 10.1038/nmeth.3415. doi:10.1038/nmeth.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen B. da S., Maciel R. de M., Galina A, da Silveira MS, Souza C. dos S., Drummond H, Pozzatto EN, Junior HS, Chicaybam L, Massuda R, Setti-Perdigão P, Bonamino M, Belmonte-de-Abreu PS, Castro NG, Brentani H, Rehen SK. Altered Oxygen Metabolism Associated to Neurogenesis of Induced Pluripotent Stem Cells Derived From a Schizophrenic Patient. Cell Transplant. 2012;21:1547–1559. doi: 10.3727/096368911X600957. doi:10.3727/096368911X600957. [DOI] [PubMed] [Google Scholar]
- Paylor R. Mice deleted for the DiGeorge/velocardiofacial syndrome region show abnormal sensorimotor gating and learning and memory impairments. Hum. Mol. Genet. 2001;10:2645–2650. doi: 10.1093/hmg/10.23.2645. doi:10.1093/hmg/10.23.2645. [DOI] [PubMed] [Google Scholar]
- Paylor R, Lindsay E. Mouse Models of 22q11 Deletion Syndrome. Biol. Psychiatry. 2006;59:1172–1179. doi: 10.1016/j.biopsych.2006.01.018. doi:10.1016/j.biopsych.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Pocklington AJ, Rees E, Walters JTR, Han J, Kavanagh DH, Chambert KD, Holmans P, Moran JL, McCarroll SA, Kirov G, O’Donovan MC, Owen MJ. Novel Findings from CNVs Implicate Inhibitory and Excitatory Signaling Complexes in Schizophrenia. Neuron. 2015;86:1203–1214. doi: 10.1016/j.neuron.2015.04.022. doi:10.1016/j.neuron.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kähler A, Duncan L, Stahl E, Genovese G, Fernández E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PKE, Banks E, Shakir K, Garimella K, Fennell T, DePristo M, Grant SGN, Haggarty SJ, Gabriel S, Scolnick EM, Lander ES, Hultman CM, Sullivan PF, McCarroll S. a, Sklar P. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–90. doi: 10.1038/nature12975. doi:10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabaneda LG, Robles-Lanuza E, Nieto-González J, Scholl FG. Neurexin Dysfunction in Adult Neurons Results in Autistic-like Behavior in Mice. Cell Rep. 2014;8:338–346. doi: 10.1016/j.celrep.2014.06.022. doi:10.1016/j.celrep.2014.06.022. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PPD, Wright J, Agarwala V, Scott D. a, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–308. doi: 10.1038/nprot.2013.143. doi:10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees E, Kirov G, Sanders a, Walters JTR, Chambert KD, Shi J, Szatkiewicz J, O’Dushlaine C, Richards a L., Green EK, Jones I, Davies G, Legge SE, Moran JL, Pato C, Pato M, Genovese G, Levinson D, Duan J, Moy W, Göring HHH, Morris D, Cormican P, Kendler KS, O’Neill F. a, Riley B, Gill M, Corvin a, Craddock N, Sklar P, Hultman C, Sullivan PF, Gejman PV, McCarroll S. a, O’Donovan MC, Owen MJ. Evidence that duplications of 22q11.2 protect against schizophrenia. Mol. Psychiatry. 2014;19:37–40. doi: 10.1038/mp.2013.156. doi:10.1038/mp.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees E, Walters JTR, Georgieva L, Isles AR, Chambert KD, Richards AL, Mahoney-Davies G, Legge SE, Moran JL, McCarroll S. a., O’Donovan MC, Owen MJ, Kirov G. Analysis of copy number variations at 15 schizophrenia-associated loci. Br. J. Psychiatry. 2014;204:108–114. doi: 10.1192/bjp.bp.113.131052. doi:10.1192/bjp.bp.113.131052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans P. a., Lee P, Bulik-Sullivan B, Collier D. a., Huang H, Pers TH, Agartz I, Agerbo E, Albus M, Alexander M, Amin F, Bacanu S. a., Begemann M, Belliveau Jr R. a., Bene J, Bergen SE, Bevilacqua E, Bigdeli TB, Black DW, Bruggeman R, Buccola NG, Buckner RL, Byerley W, Cahn W, Cai G, Campion D, Cantor RM, Carr VJ, Carrera N, Catts SV, Chambert KD, Chan RCK, Chen RYL, Chen EYH, Cheng W, Cheung EFC, Ann Chong S, Robert Cloninger C, Cohen D, Cohen N, Cormican P, Craddock N, Crowley JJ, Curtis D, Davidson M, Davis KL, Degenhardt F, Del Favero J, Demontis D, Dikeos D, Dinan T, Djurovic S, Donohoe G, Drapeau E, Duan J, Dudbridge F, Durmishi N, Eichhammer P, Eriksson J, Escott-Price V, Essioux L, Fanous AH, Farrell MS, Frank J, Franke L, Freedman R, Freimer NB, Friedl M, Friedman JI, Fromer M, Genovese G, Georgieva L, Giegling I, Giusti-Rodríguez P, Godard S, Goldstein JI, Golimbet V, Gopal S, Gratten J, de Haan L, Hammer C, Hamshere ML, Hansen M, Hansen T, Haroutunian V, Hartmann AM, Henskens F. a., Herms S, Hirschhorn JN, Hoffmann P, Hofman A, Hollegaard MV, Hougaard DM, Ikeda M, Joa I, Julià A, Kahn RS, Kalaydjieva L, Karachanak-Yankova S, Karjalainen J, Kavanagh D, Keller MC, Kennedy JL, Khrunin A, Kim Y, Klovins J, Knowles J. a., Konte B, Kucinskas V, Ausrele Kucinskiene Z, Kuzelova-Ptackova H, Kähler AK, Laurent C, Lee Chee Keong J, Hong Lee S, Legge SE, Lerer B, Li M, Li T, Liang K-Y, Lieberman J, Limborska S, Loughland CM, Lubinski J, Lönnqvist J, Macek Jr M, Magnusson PKE, Maher BS, Maier W, Mallet J, Marsal S, Mattheisen M, Mattingsdal M, McCarley RW, McDonald C, McIntosh AM, Meier S, Meijer CJ, Melegh B, Melle I, Mesholam-Gately RI, Metspalu A, Michie PT, Milani L, Milanova V, Mokrab Y, Morris DW, Mors O, Murphy KC, Murray RM, Myin-Germeys I, Müller-Myhsok B, Nelis M, Nenadic I, Nertney D. a., Nestadt G, Nicodemus KK, Nikitina-Zake L, Nisenbaum L, Nordin A, O’Callaghan E, O’Dushlaine C, O’Neill FA, Oh S-Y, Olincy A, Olsen L, Van Os J, Endophenotypes International Consortium, P. Pantelis C, Papadimitriou GN, Papiol S, Parkhomenko E, Pato MT, Paunio T, Pejovic-Milovancevic M, Perkins DO, Pietiläinen O, Pimm J, Pocklington AJ, Powell J, Price A, Pulver AE, Purcell SM, Quested D, Rasmussen HB, Reichenberg A, Reimers M. a., Richards AL, Roffman JL, Roussos P, Ruderfer DM, Salomaa V, Sanders AR, Schall U, Schubert CR, Schulze TG, Schwab SG, Scolnick EM, Scott RJ, Seidman LJ, Shi J, Sigurdsson E, Silagadze T, Silverman JM, Sim K, Slominsky P, Smoller JW, So H-C, Spencer C. a., Stahl E. a., Stefansson H, Steinberg S, Stogmann E, Straub RE, Strengman E, Strohmaier J, Scott Stroup T, Subramaniam M, Suvisaari J, Svrakic DM, Szatkiewicz JP, Söderman E, Thirumalai S, Toncheva D, Tosato S, Veijola J, Waddington J, Walsh D, Wang D, Wang Q, Webb BT, Weiser M, Wildenauer DB, Williams NM, Williams S, Witt SH, Wolen AR, Wong EHM, Wormley BK, Simon Xi H, Zai CC, Zheng X, Zimprich F, Wray NR, Stefansson K, Visscher PM, Trust Case-Control Consortium, W. Adolfsson R, Andreassen O. a., Blackwood DHR, Bramon E, Buxbaum JD, Børglum AD, Cichon S, Darvasi A, Domenici E, Ehrenreich H, Esko T, Gejman PV, Gill M, Gurling H, Hultman CM, Iwata N, Jablensky AV, Jönsson EG, Kendler KS, Kirov G, Knight J, Lencz T, Levinson DF, Li QS, Liu J, Malhotra AK, McCarroll S. a., McQuillin A, Moran JL, Mortensen PB, Mowry BJ, Nöthen MM, Ophoff R. a., Owen MJ, Palotie A, Pato CN, Petryshen TL, Posthuma D, Rietschel M, Riley BP, Rujescu D, Sham PC, Sklar P, St Clair D, Weinberger DR, Wendland JR, Werge T, Daly MJ, Sullivan PF, O’Donovan MC. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. doi:10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robicsek O, Karry R, Petit I, Salman-Kesner N, Müller F-J, Klein E, Aberdam D, Ben-Shachar D. Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol. Psychiatry. 2013;18:1067–76. doi: 10.1038/mp.2013.67. doi:10.1038/mp.2013.67. [DOI] [PubMed] [Google Scholar]
- Robinson DG, Woerner MG, McMeniman M, Mendelowitz A, Bilder RM. Symptomatic and functional recovery from a first episode of schizophrenia or schizoaffective disorder. Am. J. Psychiatry. 2004;161:473–479. doi: 10.1176/appi.ajp.161.3.473. doi:http://dx.doi.org/10.1176/appi.ajp.161.3.473. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Revenga L, Mila M, Rosenberg C, Lamb A, Lee C. Structural variation in the human genome: the impact of copy number variants on clinical diagnosis. Genet. Med. 2007;9:600–606. doi: 10.1097/gim.0b013e318149e1e3. doi:10.1097/GIM.0b013e318149e1e3. [DOI] [PubMed] [Google Scholar]
- Ronen D, Benvenisty N. Genomic stability in reprogramming. Curr. Opin. Genet. Dev. 2012;22:444–449. doi: 10.1016/j.gde.2012.09.003. doi:10.1016/j.gde.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Schadt EE, Buchanan S, Brennand KJ, Merchant KM. Evolving toward a human-cell based and multiscale approach to drug discovery for CNS disorders. Front. Pharmacol. 2014;5:1–15. doi: 10.3389/fphar.2014.00252. doi:10.3389/fphar.2014.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaeger TM, Daheron L, Brickler TR, Entwisle S, Chan K, Cianci A, DeVine A, Ettenger A, Fitzgerald K, Godfrey M, Gupta D, McPherson J, Malwadkar P, Gupta M, Bell B, Doi A, Jung N, Li X, Lynes MS, Brookes E, Cherry ABC, Demirbas D, Tsankov AM, Zon LI, Rubin LL, Feinberg AP, Meissner A, Cowan C. a, Daley GQ. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2014;33:55–60. doi: 10.1038/nbt.3070. doi:10.1038/nbt.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Debbané M, Bassett AS, Chow EWC, Fung WLA, van den Bree MBM, Owen M, Murphy KC, Niarchou M, Kates WR, Antshel KM, Fremont W, McDonald-McGinn DM, Gur RE, Zackai EH, Vorstman J, Duijff SN, Klaassen PWJ, Swillen A, Gothelf D, Green T, Weizman A, Van Amelsvoort T, Evers L, Boot E, Shashi V, Hooper SR, Bearden CE, Jalbrzikowski M, Armando M, Vicari S, Murphy DG, Ousley O, Campbell LE, Simon TJ, Eliez S. Psychiatric Disorders From Childhood to Adulthood in 22q11.2 Deletion Syndrome: Results From the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am. J. Psychiatry. 2014;171:627–639. doi: 10.1176/appi.ajp.2013.13070864. doi:10.1176/appi.ajp.2013.13070864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Smith J, Robinson HPC, Livesey FJ. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 2012;15:477–486. doi: 10.1038/nn.3041. doi:10.1038/nn.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson T, Stark KL, Karayiorgou M, Gogos J. a, Gordon J. a. Impaired hippocampal–prefrontal synchrony in a genetic mouse model of schizophrenia. Nature. 2010;464:763–767. doi: 10.1038/nature08855. doi:10.1038/nature08855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu CO, Agid O, Remington G. Efficacy of antipsychotic drugs for schizophrenia. Lancet. 2013;382:1874. doi: 10.1016/S0140-6736(13)62616-1. doi:10.1016/S0140-6736(13)62616-1. [DOI] [PubMed] [Google Scholar]
- Stark KL, Xu B, Bagchi A, Lai W-S, Liu H, Hsu R, Wan X, Pavlidis P, Mills A. a, Karayiorgou M, Gogos J. a. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008;40:751–760. doi: 10.1038/ng.138. doi:10.1038/ng.138. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, Bjornsdottir G, Walters GB, Jonsdottir G. a, Doyle OM, Tost H, Grimm O, Kristjansdottir S, Snorrason H, Davidsdottir SR, Gudmundsson LJ, Jonsson GF, Stefansdottir B, Helgadottir I, Haraldsson M, Jonsdottir B, Thygesen JH, Schwarz AJ, Didriksen M, Stensbøl TB, Brammer M, Kapur S, Halldorsson JG, Hreidarsson S, Saemundsen E, Sigurdsson E, Stefansson K. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature. 2014;505:361–6. doi: 10.1038/nature12818. doi:10.1038/nature12818. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietiläinen OPH, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Möller H-J, Hartmann A, Shianna KV, Ge D, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Mühleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nöthen MM, Peltonen L, Collier DA, St Clair D, Stefansson K, Kahn RS, Linszen DH, van Os J, Wiersma D, Bruggeman R, Cahn W, de Haan L, Krabbendam L, Myin-Germeys I. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. doi:10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a Complex Trait. Arch. Gen. Psychiatry. 2003;60:1187. doi: 10.1001/archpsyc.60.12.1187. doi:10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Light GA, Sprock J, Calkins ME, Green MF, Greenwood TA, Gur RE, Gur RC, Lazzeroni LC, Nuechterlein KH, Radant AD, Ray A, Seidman LJ, Siever LJ, Silverman JM, Stone WS, Sugar CA, Tsuang DW, Tsuang MT, Turetsky BI, Braff DL. Deficient prepulse inhibition in schizophrenia detected by the multi-site COGS. Schizophr. Res. 2014;152:503–512. doi: 10.1016/j.schres.2013.12.004. doi:10.1016/j.schres.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatkiewicz JP, O’Dushlaine C, Chen G, Chambert K, Moran JL, Neale BM, Fromer M, Ruderfer D, Akterin S, Bergen SE, Kähler a, Magnusson PKE, Kim Y, Crowley JJ, Rees E, Kirov G, O’Donovan MC, Owen MJ, Walters J, Scolnick E, Sklar P, Purcell S, Hultman CM, McCarroll S. a, Sullivan PF. Copy number variation in schizophrenia in Sweden. Mol. Psychiatry. 2014;19:762–73. doi: 10.1038/mp.2014.40. doi:10.1038/mp.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. doi:10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tall AR, Yvan-Charvet L, Wang N. The failure of torcetrapib: Was it the molecule or the mechanism? Arterioscler. Thromb. Vasc. Biol. 2007;27:257–260. doi: 10.1161/01.ATV.0000256728.60226.77. doi:10.1161/01.ATV.0000256728.60226.77. [DOI] [PubMed] [Google Scholar]
- Tamada K, Tomonaga S, Hatanaka F, Nakai N, Takao K, Miyakawa T, Nakatani J, Takumi T. Decreased Exploratory Activity in a Mouse Model of 15q Duplication Syndrome; Implications for Disturbance of Serotonin Signaling. PLoS One. 2010;5:e15126. doi: 10.1371/journal.pone.0015126. doi:10.1371/journal.pone.0015126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King M-C, Sebat J. Rare Structural Variants Disrupt Multiple Genes in Neurodevelopmental Pathways in Schizophrenia. Science (80−. ) 2008;320:539–543. doi: 10.1126/science.1155174. doi:10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- Walz K, Caratini-Rivera S, Bi W, Fonseca P, Mansouri DL, Lynch J, Vogel H, Noebels JL, Bradley A, Lupski JR. Modeling del(17)(p11.2p11.2) and dup(17)(p11.2p11.2) contiguous gene syndromes by chromosome engineering in mice: phenotypic consequences of gene dosage imbalance. Mol. Cell. Biol. 2003;23:3646–3655. doi: 10.1128/MCB.23.10.3646-3655.2003. doi:10.1128/MCB.23.10.3646-3655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat. Genet. 2007;39:380–385. doi: 10.1038/ng1969. doi:10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnica W, Merico D, Costain G, Alfred SE, Wei J, Marshall CR, Scherer SW, Bassett AS. Copy Number Variable MicroRNAs in Schizophrenia and Their Neurodevelopmental Gene Targets. Biol. Psychiatry. 2014;77:158–166. doi: 10.1016/j.biopsych.2014.05.011. doi:10.1016/j.biopsych.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. doi:10.1001/archpsyc.1988.01800350089019. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MAR, Green T, Platt OS, Ruderfer DM, Walsh CA, Altshuler D, Chakravarti A, Tanzi RE, Stefansson K, Santangelo SL, Gusella JF, Sklar P, Wu B, Daly MJ. Association between Microdeletion and Microduplication at 16p11.2 and Autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. doi:10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Wen L, Lu Y-S, Zhu X-H, Li X-M, Woo R-S, Chen Y-J, Yin D-M, Lai C, Terry AV, Vazdarjanova A, Xiong W-C, Mei L. Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc. Natl. Acad. Sci. U. S. A. 2010;107:1211–1216. doi: 10.1073/pnas.0910302107. doi:10.1073/pnas.0910302107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Nguyen HN, Guo Z, Lalli M. a., Wang X, Su Y, Kim N-S, Yoon K-J, Shin J, Zhang C, Makri G, Nauen D, Yu H, Guzman E, Chiang C-H, Yoritomo N, Kaibuchi K, Zou J, Christian KM, Cheng L, Ross C. a., Margolis RL, Chen G, Kosik KS, Song H, Ming G. Synaptic dysregulation in a human iPS cell model of mental disorders. Nat. Lett. 2014;515:1–5. doi: 10.1038/nature13716. doi:10.1038/nature13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K-J, Nguyen HN, Ursini G, Zhang F, Kim N-S, Wen Z, Makri G, Nauen D, Shin JH, Park Y, Chung R, Pekle E, Zhang C, Towe M, Hussaini SMQ, Lee Y, Rujescu D, St. Clair D, Kleinman JE, Hyde TM, Krauss G, Christian KM, Rapoport JL, Weinberger DR, Song H, Ming G. Modeling a Genetic Risk for Schizophrenia in iPSCs and Mice Reveals Neural Stem Cell Deficits Associated with Adherens Junctions and Polarity. Cell Stem Cell. 2014;15:79–91. doi: 10.1016/j.stem.2014.05.003. doi:10.1016/j.stem.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]