

Abstract
Key points
Acid‐sensing ion channels (ASICs) act as neurotransmitter receptors by responding to synaptic cleft acidification.
We investigated how ASIC1a homomers and ASIC1a/2a heteromers respond to brief stimuli, jumping from pH 8.0 to 5.0, approximating the time course of neurotransmitter in the cleft.
We find that ASICs deactivate surprisingly fast in response to such brief stimuli from pH 8.0 to 5.0, whereas they desensitize comparatively slowly to prolonged activation.
The combination of unusually fast deactivation with slow desensitzation enables recombinant ASIC1a homomers and ASIC1a/2a heteromers, as well as native ASICs of sensory neurons, to follow trains of such brief pH 8.0 to 5.0 stimuli at high frequencies.
This capacity for high‐frequency signalling persists under a physiological pH of 7.4 with ASIC1a/2a heteromers, suggesting that they may sustain postsynaptic responses when other receptors desensitize.
Abstract
The neurotransmitter‐gated ion channels that underlie rapid synaptic transmission are often subjected to bursts of very brief neurotransmitter release at high frequencies. When challenged with such short duration high‐frequency stimuli, neurotransmitter‐gated ion channels generally exhibit the common response of desensitization. Recently, acid‐sensing ion channels (ASICs) were shown to act as neurotransmitter‐gated ion channels because postsynaptic ASICs can be activated by the transient acidification of the synaptic cleft accompanying neurotransmission. In the present study, we examined the responses of recombinant ASIC1a homomers, ASIC1a/2a heteromers and native ASICs from sensory neurons to 1 ms acidification stimuli, switching from pH 8.0 to 5.0, as either single pulses or trains of pulses at physiologically relevant frequencies. We found that ASIC deactivation is extremely fast and, in contrast to most other neurotransmitter‐gated ion channels, ASICs show no desensitization during high‐frequency stimulus trains under these conditions. We also found that accelerating ASIC desensitization by anion substitution can induce depression during high‐frequency trains. When using a baseline physiological pH of 7.4, the ASIC1a responses were too small to reliably measure, presumably as a result of steady‐state desensitization. However, ASIC1a/2 heteromers gave robust responses when using a baseline pH of 7.4 and were also able to sustain these responses during high‐frequency stimulus trains. In conclusion, we report that the slow desensitization and fast deactivation of ASIC1a/2a heteromers enables them to sustain postsynaptic responses to bursts at high frequencies at a physiological pH that may desensitize other receptors.
Key points
Acid‐sensing ion channels (ASICs) act as neurotransmitter receptors by responding to synaptic cleft acidification.
We investigated how ASIC1a homomers and ASIC1a/2a heteromers respond to brief stimuli, jumping from pH 8.0 to 5.0, approximating the time course of neurotransmitter in the cleft.
We find that ASICs deactivate surprisingly fast in response to such brief stimuli from pH 8.0 to 5.0, whereas they desensitize comparatively slowly to prolonged activation.
The combination of unusually fast deactivation with slow desensitzation enables recombinant ASIC1a homomers and ASIC1a/2a heteromers, as well as native ASICs of sensory neurons, to follow trains of such brief pH 8.0 to 5.0 stimuli at high frequencies.
This capacity for high‐frequency signalling persists under a physiological pH of 7.4 with ASIC1a/2a heteromers, suggesting that they may sustain postsynaptic responses when other receptors desensitize.
Abbreviations
- ASIC
acid‐sensing ion channel
- CHO
Chinese hamster ovary
- DRG
dorsal root ganglia
- Egfp
enhanced green fluorescent protein
- LTP
long‐term potentiation
- NGIC
neurotransmitter‐gated ion channel
Introduction
Acid‐sensing ion channels (ASICs) are cation‐selective pH‐gated ion channels belonging to the degenerin/epithelial Na+ channel family (Waldmann & Lazdunski, 1998). Of the six ASIC subunits identified, ASIC1a, ASIC2a and ASIC2b show widespread expression throughout the central and peripheral nervous systems, whereas ASIC1b and ASIC3 are largely restricted to the peripheral nervous system (Wemmie et al. 2006; Kellenberger & Schild, 2015). Consistent with their wide distribution, ASICs are implicated in a variety of pathological conditions, such as neurotoxicity following ischaemia, anxiety and pain (Xiong et al. 2004; Wemmie et al. 2006; Baron & Lingueglia, 2015; Deval & Lingueglia, 2015; Kellenberger & Schild, 2015; Lin et al. 2015).
ASICs are also heavily clustered at postsynaptic densities (Wemmie et al. 2004) where they interact with the postsynaptic scaffolding proteins PSD‐95 (Zha et al. 2009) and PICK1 (Baron et al. 2002 a) and affect synaptic spine morphology (Zha et al. 2006). In addition, ASICs are important for long‐term potentiation (LTP) in both the amygdala (Chiang et al. 2015) and the hippocampus (Wemmie et al. 2002; but see also Wu et al., 2013). These findings suggest that ASICs represent more than an extracellular ‘acid alarm’ for noxious stimuli and may also impact neuronal signalling under physiological conditions. In particular, ASICs have been proposed to respond to the transient acidification of the synaptic cleft that accompanies neurotransmission (Krishtal et al. 1987; Waldmann et al. 1997; Wemmie et al., 2002, 2008). This acidification, arising from the release of protons stored in synaptic vesicles, can reduce the cleft sufficiently to activate postsynaptic ASICs (Miesenbock et al. 1998; Palmer et al. 2003; Wemmie et al. 2008; Grunder & Pusch, 2015). Recently, two studies have confirmed that ASICs do respond to the brief synaptic cleft acidification during neurotransmission and contribute to excitatory postsynaptic currents in both the lateral amygdala (Du et al. 2014) and nucleus accumbens core (Kreple et al. 2014). Thus, protons can act as neurotransmitters and their cognate receptor is the ASIC. These seminal studies shed light on the synaptic roles of ASICs and open new avenues of inquiry in fear memory and drug addiction research.
The evidence that ASICs contribute to synaptic responses also prompts new biophysical investigations. Almost all previous studies of ASICs have used comparatively long acidic applications, generally ≥1–5 s. However, if the clearance of protons in the cleft is comparable to that of other neurotransmitters, synaptic ASICs would only experience acidification for ∼1 ms (Clements et al. 1992; Jones & Westbrook, 1995, 1996; Beato, 2008; Wemmie et al. 2008). To date, no study has investigated the responses of ASICs to such rapid synaptic‐like pH changes. In the present study, we explored the kinetic properties of recombinant and native ASICs to such brief 1 ms stimuli in outside‐out patches. This approach avoids complicating factors such as presynaptic adaptions (Zucker & Regehr, 2002; Cho & Askwith, 2008), the geometry of and clearance from the synaptic cleft (Clements et al. 1992; Franks et al. 2003; Graydon et al. 2014) or metabotropic/cell signalling influences (Gao et al. 2005; Smith et al. 2007; Kellenberger & Schild, 2015) and allowed us to measure the fundamental biophysical capabilities of ASIC1a and ASIC1a/2a in responses to well‐defined stimuli. In addition, because synaptic transmission often occurs in rapid bursts of high‐frequency firing (Gray & McCormick, 1996; Cooper, 2002; Saviane & Silver, 2006), we examined the response of both native and recombinant ASICs to trains of 1 ms pH jumps from pH 8.0 to 5.0 at high frequencies (up to 50 Hz). We report that, under these conditions, ASICs exhibit extremely fast deactivation kinetics, approaching the limit of rapid solution exchange. Moreover, we find that this rapid deactivation, when paired with the slow desensitization of ASICs, allows these channels to follow high‐frequency trains of stimuli with great fidelity and no desensitization. Importantly, this capacity to follow stimulus trains persisted at physiological pH 7.4 for ASIC1a/2a, whereas ASIC1a responses were too small to reliably measure under these same conditions. The capacity of the heteromeric ASIC for sustained synaptic transmission may contribute to its roles in learning, fear memory and drug addiction.
Methods
Ethical approval
The present study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Mice were housed, handled and killed in accordance with approved institutional animal care and use committee (IACUC) protocols at the University of Texas Health Science centre in Houston. Mice were killed using an overdose of isoflurane followed by decapitation.
Cell culture, mutagenesis and transfection
Chinese hamster ovary (CHO) cells were maintained in Ham's F‐12 Nutrient Mix (Invitrogen/Life Technologies, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (Sigma‐Aldrich, St Louis, MO, USA) and penicillin/streptomycin (Invitrogen/Life Technologies). Cells were passaged upon reaching ∼90% confluence, every 2–3 days. Each construct was confirmed by sequencing (Genewiz, Inc., South Plainfield, NJ, USA). For transfections, CHO cells were plated in poly‐d‐lysine‐coated 35 mm dishes and transfected 24–48 h later. Cells were transfected with rat ASIC1a and enhanced GFP (GFPS65T) at a ratio of 8:1 μg of cDNA/10 ml of media for the ASIC1a experiments. For heteromeric experiments, rat ASIC1a, rat ASIC2a and enhanced green fluorescent protein (eGFP) were transfected at a ratio of 2:6:1 μg of cDNA/10 ml of media. In GluA2 experiments, a plasmid bearing the Q version of rat GluA2‐flip followed by an internal ribosome entry site and eGFP (provided by Dr. Mark L Mayer, NIH, Bethesda, MD, USA) was transfected at a concentration of 7.5 μg of cDNA/10 ml of media. Transfections were performed using Lipofectamine 2000 (Invitrogen/Life Technologies) in accordance with the manufacturer's instructions with the media changed after 6–12 h. Electrophysiology experiments began 48–72 h after the start of transfection.
Neuronal cultures
Dorsal root ganglion (DRG) was dissected from the strain of Cbl/Blk 6 mice (>3 months of age) as described by Malin et al. (2007). Briefly, following an overdose of isoflurane and decapitation, the spinal column was removed and the thoracic DRG was exposed and extracted. The DRG was dissociated with papain followed by collagenase/dispase, plated on poly‐d‐lysine plated coverslips and stored at 37°C in F‐12 media supplemented with nerve growth factor. Experiments were performed 18–36 h after plating.
Electrophysiology
Outside‐out patches were excised from eGFP‐expressing CHO cells, or DRG neurons, using thick‐walled borosilicate glass pipettes of 3–5 MΩ resistance. Pipettes were coated in bees wax, fire polished and filled with internal solution containing (in mm): 135 CsF, 33 CsOH, 11 EGTA, 10 Hepes, 2 MgCl2 and 1 CaCl2 (pH 7.4). External solutions were composed of (in mm): 150 NaCl, 20 Hepes, 1 CaCl2, 1 MgCl2 and pH 8.0 (NaOH) unless otherwise stated. For pH values more acidic than 7.0, Hepes was replaced with Mes. In anion substitution experiments, NaCl was replaced with either NaI or NaMeSO3 as indicated. All recordings were performed at room temperature with a hold potential of −80 mV using an Axopatch 200B (Molecular Devices, Sunnyvale, CA, USA). Data were acquired at 50 or 100 kHz and filtered at 10 kHz under the control of Clampex, version 10.2 (Molecular Devices). Series resistance was routinely compensated by > 90% where the amplitude exceeded 100 pA. Rapid application was performed using home built theta‐barrel application pipettes, manufactured as described by MacLean (2015). Translation of application pipettes was achieved using a LZ150M piezo translator (Burleigh Instruments, Victor, NY, USA) with voltage commands low‐pass filtered (eight‐pole Bessel; Frequency Devices, Ottawa, IL, USA) at 180–200 Hz. At higher frequencies of piezo movement (ie. > 50 Hz), mechanical oscillations of the application pipettes gave rise to solution exchange artefacts, including systematic variations in jump duration and undesired ‘double jumps’. We therefore limited our stimulus frequency to 50 Hz, which also allowed for an exact comparison with previous work (Papke et al. 2011). Solution exchange was routinely measured at the end of each patch recording using open tip potentials, which ranged from 80 to 250 μs (10–90% rise time). In some deactivation experiments, in particular with ASIC1a/2a heteromers and ASICs from DRG neurons, the measured kinetics either approach or are at the limit of our solution exchange speed and therefore probably underestimate the true deactivation kinetics. The times between the end of one stimulation or stimulus train and the start of a subsequent stimulation or stimulus train for each receptor type were: GluA2: 4–5 s, ASIC1a, ASIC1a/2a and native ASICs: 10 s (1 or 300 ms pulse), 15 s (trains) or 30 s (5 s pulse), except for the ASIC1a/2a 5 s pulses, which had a waiting time of ≥20 s.
Statistical analysis
Data were analysed using Origin, version 8.6 (OriginLab Cop., Northampton, MA, USA) and Clampfit, version 10.2 (Molecular Devices). Desensitization and deactivation kinetics were fit with either single‐ or double‐exponential functions. Both desensitization and deactivation of AMPA receptors were fit with double‐exponential functions. In general ASIC desensitization decays were well fit with double exponentials. ASIC deactivation kinetics were well fit by single exponentials in some patches and double exponentials in others. For simplicity, we report the weighted time constants from these fits. For the measurement of pH 6/5 peak response ratios, 300 ms jumps were used because these limit desensitization and tachyphlaxis (Chen & Grunder, 2007; Li et al. 2012). During the recovery from desensitization experiments, we used a paired pulse design with a 5 s pH 5.0 application to induce desensitization, followed by a pH 5.0 test pulse for 100 ms at a variable interpulse interval. Where possible, a recovery experiment was conducted three times in each patch. Peak responses to each test pulse were normalized to the average of the preceding and subsequent desensitizing pulse peaks. Normalized test pulse responses from each patch were then fit to single‐ and double‐exponential functions and the parameters from each patch averaged to produce the mean ± SE reported in the present study. The single‐exponential function gave visibly poor fits. For all statistical tests, n was considered as a single patch. For example traces, one trace was used for desensitization experiments and between three and 10 individual traces from a single patch were averaged for the other experiments. To assess statistical significance, independent sample two‐tailed t test (or paired t tests in the case of 50 Hz trains and anion substitution experiments) were used with P < 0.05 being considered statistically significant. Multiple comparisons within a single condition, as in the anion substitution experiments, were Bonferroni corrected. All summary plots show the mean ± SEM.
Results
Desensitization and deactivation kinetics
Because ASIC1a homomers and ASIC1a/2a heteromers are the only populations reported to contribute to synaptic responses to date (Du et al. 2014; Kreple et al. 2014), we limited our recombinant experiments to these subtypes. To measure the response kinetics of ASIC1a homomers and ASIC 1a/2 heteromers to both long applications of acidic solution (ie. desensitization) and brief 1 ms applications (i.e. deactivation), outside‐out patches were excised from transfected CHO cells expressing each construct. As a control, this experiment was also carried out with the rapidly deactivating and desensitizing GluA2 AMPA subunit. As seen in Fig. 1, GluA2 responses to prolonged application of 10 mm glutamate were rapidly activating (10–90% rise time: 290 ± 70 μs, n = 8) (Fig. 1 A, left and C, left) and desensitized with a weighted time constant of 5.1 ± 0.2 ms (n = 8, Fig. 1 A, left, and B), which is consistent with previous studies (Robert et al. 2005; Carbone & Plested, 2012; MacLean et al. 2014). GluA2 deactivation kinetics were also quite rapid with a time constant of 0.70 ± 0.07 ms (n = 8) (Fig. 1 C, left, and D), similar to previous studies (Carbone & Plested, 2012). This concordance with earlier work on AMPA receptors with extremely rapid activation and deactivation highlights the reliability and precision of our experimental set‐up. Furthermore, we routinely estimated the solution exchange in each patch after the experiment using open‐tip currents with dilute extracellular solution. The upper traces in Fig. 1 C (left) illustrate our solution exchange as estimated from such an open‐tip current.
Figure 1. ASIC1a homomers and 1a/2a heteromers have slow desensitization and fast deactivation .
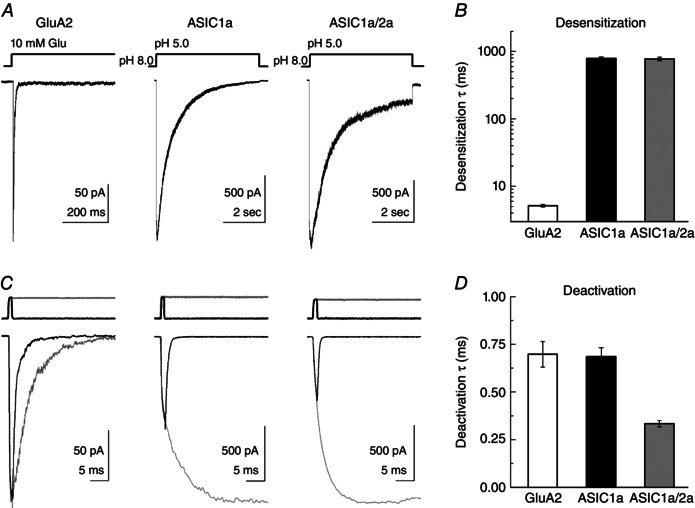
A, example responses from outside‐out patches expressing GluA2 (left), rat ASIC1a (middle) or ASIC1a/2a (right) to a 5 s agonist exposure. B, summary of weighted time constants of desensitization from each construct. C, example responses from GluA2 (left), rat ASIC1a (middle) or ASIC1a/2a (right) to a 1 ms (black traces) or 5 s agonist application (grey traces). Same patches and agonist concentrations as (A). Solution exchange current from each patch for 1 ms (black traces) or 5 s (grey traces) jumps are shown above. D, summary of deactivation time constants for each construct tested.
To measure the activation and desensitization kinetics of homomeric rat ASIC1a, outside‐out patches were jumped from pH 8.0 (to eliminate steady‐state inactivation; Babini et al. 2002) to pH 5.0 for 5 s (Fig. 1 A, middle). ASIC1a responses showed slower activation compared to AMPA receptors, with rise times of 22 ± 4 ms (n = 13) (Fig. 1 A and C, middle). As expected from previous experiments (Kusama et al. 2010), we found that ASIC1a desensitized rather slowly and almost completely to a pH 5.0 stimulus with a weighted time constant of 700 ± 60 ms (n = 13) (Fig. 1 A, middle, and B). By contrast to this relatively slow desensitization decay, the deactivation decay in response to pH 5.0 exposure for 1 ms was very fast, with a time constant of 0.69 ± 0.06 ms (n = 13) (Fig. 1 C, middle, and D). Interestingly, we also observed the peak response to a brief 1 ms application of pH 5.0 was considerably less than the peak observed from a long pulse of pH 5.0 with ASIC1a 1 ms jump peak responses being only 30 ± 5% (n = 13) of long pulse peak responses (Fig. 1 C, middle). By contrast, for GluA2 AMPA receptors, the peak response of a 1 ms jump into 10 mm glutamate was 100 ± 1% (n = 8) of the peak response from a long pulse (Fig. 1 C, left). This incomplete activation arises as the protons are removed within 1 ms, whereas full activation requires several milliseconds.
We next measured the activation, desensitization and deactivation kinetics of ASIC1a/2a heteromers. To determine whether co‐transfection of ASIC1a and 2a (in a 1:3 mass ratio) resulted in faithful expression of heteromeric ASIC1a/2a receptors, we measured the ratio of peak responses to pH 5.0 and to pH 6.0 in each patch. Our purely monomeric ASIC1a patches yielded a pH 6/5 ratio of 0.82 ± 0.02 (n = 13) (Fig. 2), whereas a purely monomeric ASIC2a population would produce negligible responses to either pH 5.0 or 6.0 (Bartoi et al. 2014). By contrast, heteromeric populations respond to pH 6.0 with ∼25% of their response to pH 5.0 (Bartoi et al. 2014); therefore, a pH 6/5 ratio of 0.25 or less would be expected of heteromeric ASIC1a/2a receptors. In keeping with faithful expression of ASIC1a/2a heteromers, all our patches in ASIC1a/2a co‐transfected dishes (18 patches) gave ratios between 0.06 and 0.27 with a mean of 0.13 ± 0.01 (n = 18) (Fig. 2). This ratio is expected of heteromers and not homomers of either type. Therefore, we conclude that our experiments accurately reflect the behaviour of ASIC1a/2a heteromers. As with homomeric ASIC1a, heteromeric ASIC1a/2a exhibited relatively slow activation (rise time: 19 ± 4 ms, n = 18) and desensitization (weighted time constant: 780 + 50 ms) (Fig. 1 A, right, and B). In 1 ms application experiments, heteromeric responses deactivated with strikingly fast kinetics (time constant: 0.32 + 0.02 ms, n = 10) (Fig. 1 C, right, and D). Interestingly, these kinetics were even faster than homomeric ASIC1a (P < 0.0001). We note that such kinetics approach the limit of resolution given our solution exchange. Therefore, ASIC1a/2a heteromers clearly deactivate faster than ASIC1a homomers, although we cannot precisely say how much faster. However, this difference in deactivation and the above use of the pH 6/5 ratio provide two means of distinguishing homomeric from heteromeric ASIC populations. These are the first set of experiments to directly asses ASIC responses to single brief well‐defined acidic stimuli, approximating the synaptic time course, and they reveal that ASICs deactivate very rapidly. However, synaptic transmission is often a barrage of brief stimuli at high frequencies (Gray & McCormick, 1996; Cooper, 2002; Saviane & Silver, 2006), and not simply a single pulse. Therefore, we next examined how ASICs respond to such a train of 1 ms stimuli at high frequencies.
Figure 2. pH 6/5 ratio reports ASIC1a/2a heteromerization .
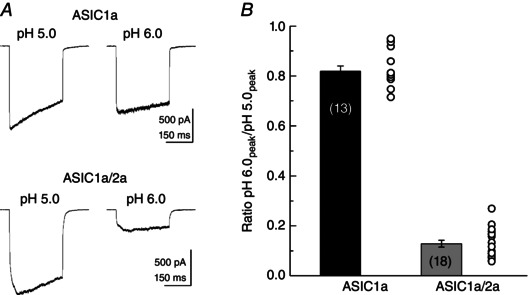
A, example responses to a 300 ms jump to the indicated pH from patches excised from ASIC1a (upper traces) or ASIC1a/2a (lower traces) transfected cells. B, summary of the peak response ratio to pH 6.0 vs. pH 5.0 in ASIC1a (black bar) or ASIC1a/2a (grey bar) transfected cells. Circles indicate the individual patch values for each condition.
Responses to high‐frequency trains of 1 ms stimuli
In an elegant study, Papke et al. (2011) compared the responses of each neurotransmitter‐gated ion channel (NGIC) family to high‐frequency stimulation. They found that representative examples of each neurotransmitter family (nictonic, GABA, glycine, glutamate and P2X receptors) desensitized during repetitive 1 ms stimulation at high frequencies (Papke et al. 2011). The ubiquity of desensitization at high frequencies suggests that it is an inbuilt feature of NGICs, possibly highlighting an important role for desensitization in shaping neuronal communication (Jones & Westbrook, 1996; Papke et al. 2011). Given that ASICs have just attained the status of NGICs (Du et al. 2014; Kreple et al. 2014), we investigated whether they also display desensitization/depression during high‐frequency stimulation. As a control for these high‐frequency experiments, we also appraised this behaviour in GluA2. In response to 1 ms applications of 10 mm glutamate at 50 Hz, GluA2 receptors desensitized to 62 ± 0.03% (n = 8) of their initial response within five pulses (Fig. 3 A, upper). Over the course of a 2 s train, this desensitization continued, with the final pulse of the train being only 51 ± 0.03% (n = 8) of the initial peak response (Fig. 3 A, lower, and D). As expected, desensitization at high frequencies is stimulus frequency‐dependent (Fig. 3 D). At 1 Hz, essentially no desensitization was observed (final pulse 0.99 ± 0.03 of first pulse, n = 7) (Fig. 3 D, blue circles) but, beginning at 10 Hz (cyan circles, final pulse 0.89 ± 0.03, n = 8) (Fig. 3 D), desensitization was statistically distinguishable (P = 0.035 comparing 1 Hz and 10 Hz) because channels are unable to unbind ligand and recover completely from desensitization before the next pulse arrives. Consequently, desensitization accumulates until an equilibrium is reached between the rate of entering desensitization and pulse frequency on the one hand and ligand unbinding and recovery on the other. This frequency dependence of the extent of desensitization is also observed in all other NGICs (Papke et al. 2011).
Figure 3. Recombinant ASICs follow high‐frequency stimuli .
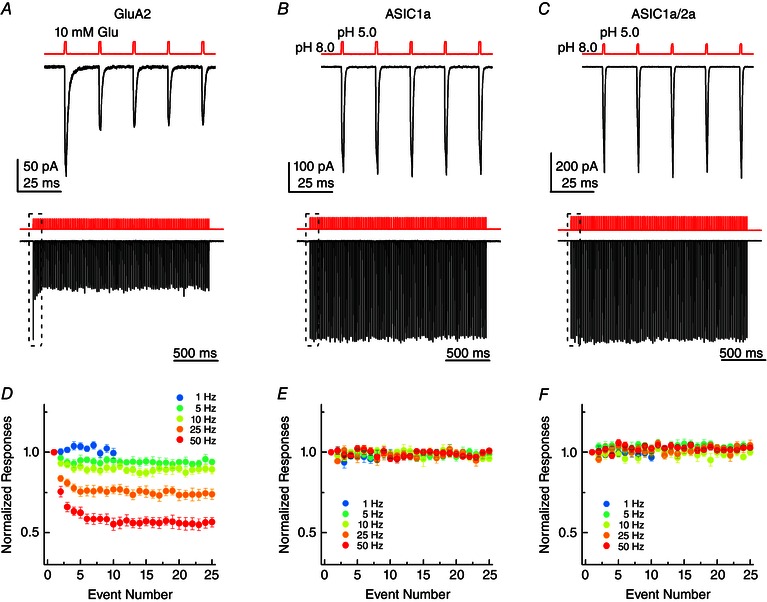
A, example responses from GluA2 to 1 ms jumps to 10 mm glutamate at 50 Hz. Upper panels: the first five responses to a train. Lower panel: the full 100 responses to the 2 s train. The upper panels are expanded views of the boxed region in the lower panels. B, example responses from ASIC1a to 1 ms jumps to pH 5.0 solution at 50 Hz, either the first five responses (upper) or all 100 responses (lower). C, example responses from ASIC1a/2a heteromers to 1 ms jumps to pH 5.0 solution at 50 Hz, either the first five responses (upper) or all 100 responses (lower). D, summary of GluA2 peak responses across patches to 1 ms 10 mm Glu jumps at the indicated stimulus frequencies, normalized to the first response amplitude. E, summary of ASIC1a peak responses across patches to 1 ms pH 5.0 jumps at the indicated stimulus frequencies, normalized to the first response amplitude. F, same plot as (D) and (E) but for ASIC1a/2a.
We next addressed how recombinant ASICs respond to these trains of 1 ms pH 8.0 to 5.0 stimuli at a range of frequencies up to 50 Hz. As seen in Fig. 3 B, ASIC1a showed no evidence of desensitization during trains of 1 ms stimuli at 50 Hz, with the peak response to the final pulse being 97 ± 3% of initial peak (n = 9) (Fig. 3 B, middle). This result was quite striking because other NGICs exhibit desensitization ranging from 50% for GluA2 (Fig. 3 A and D) to 92% for P2X2 receptors (Papke et al. 2011). Given that no desensitization was evident at 50 Hz, is it unsurprising that lower frequencies also did not provoke desensitization (Fig. 3 E). We next repeated these experiments with ASIC1a/2a heteromers and found that these too exhibited no detectable desensitization to trains of deactivation stimuli at 50 Hz (final pulse 100 ± 2% of initial peak, n = 14) or any lower frequency (Fig. 3 C and F).
We also attempted to evaluate the response of ASIC to high‐frequency trains using pH 7.4 as a baseline solution. As a result of very strong steady‐state desensitization, we were unable to obtain resolvable responses of ASIC1a at pH 7.4. However, ASIC1a/2a showed essentially no steady‐state desensitization at pH 7.4 and gave robust responses in outside‐out patches. We therefore repeated the 50 Hz stimulus using either pH 8.0 or 7.4 as the baseline solution in the same patch (Fig. 4 A). As seen in Fig. 4, altering the baseline pH from 8.0 to 7.4 produced no statistically detectable effect (final pulse: 102 ± 1% of initial peak at pH 8.0, 98 ± 3% at pH 7.4, n = 6, paired t test P = 0.286), indicating that ASIC1a/2a is capable of following trains at up to 50 Hz at physiological pH. Interestingly, we also observed that the deactivation kinetics of ASIC1a/2a were slightly slower in pH 7.4 (time constant: 0.42 ± 0.04 ms, n = 7) than pH 8.0 (time constant: 0.30 ± 0.02 ms, n = 7, P = 0.026, paired t test).
Figure 4. Heteromeric ASIC1a/2a follows high‐frequency stimuli at physiological pH .
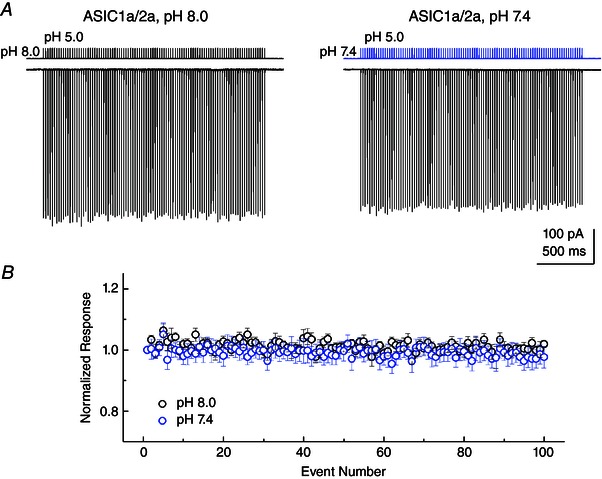
A, example responses from ASIC1a/2a heteromers to 1 ms jumps to pH 5.0 solution at 50 Hz, either from pH 8.0 (left) or pH 7.4 (right) background solution. Responses are from the same patch and the solution exchange current from this patch is shown above the traces. B, summary of ASIC1a/2a peak responses to 1 ms pH 5.0 jumps from pH 8.0 (black circles) or pH 7.4 (blue circles) baseline solution as in (A).
Heteromeric ASIC1a/2a recovery from desensitization is strongly pH‐dependent
The response of a ligand‐gated ion channel to high‐frequency stimulus is determined by the deactivation kinetics, as well as the rates of entry into and exit from desensitization (Papke et al. 2011). We report the fast deactivation kinetics of ASIC1a homomers and ASIC1a/2a heteromers, as well as their desensitization decays, which have also been described previously (Kusama et al. 2010). Similarly, the recovery from desensitization for ASIC1a has been reported previously and ranges from less than 3 s at pH 8.0 to between 5 and 13 s at pH 7.4 (Sutherland et al. 2001; Babini et al. 2002; Kusama et al. 2010). However, to our knowledge, no study has investigated the recovery from desensitization of ASIC1a/2a. To measure this property of the heteromeric receptors, we used a paired pulse protocol with a 5 s pH 5.0 conditioning pulse to induce desensitization and 100 ms pH 5.0 test pulse to measure recovery, separated by a variable interval of baseline pH lasting for between 10 ms and 20 s. Figure 5 A illustrates the results of such experiments using either pH 8.0 (Fig. 5 A, upper trace) or pH 7.4 (Fig. 5 A, lower trace) as the baseline pH. We found that the recovery of ASIC1a/2a from desensitization process was strikingly bi‐exponential, as reported for ASIC1a (Li et al. 2012), and poorly fit by a single‐exponential function (Fig. 5 C, dotted black line). Specifically, at pH 8.0, heteromeric ASIC1a/2a receptors recover to ∼85% of their initial responses (86 ± 3%, n = 6) with a fast time constant of 67 ± 5 ms and with the remaining 15% recovering with a slower time constant of 2700 ± 380 ms (n = 6) (Fig. 5 B and C). Interestingly, at pH 7.4, the recovery process is considerably slowed and also retains its bi‐exponential nature. At pH 7.4, ∼40% (42 ± 8%; n = 6) of the initial response recovers with a time constant of 320 ± 40 ms, with the remaining 60% recovering with a time constant of 2200 ± 390 ms (n = 6) (Fig. 5 B and C). Therefore, heteromeric recovery from desensitization slows at pH 7.4 as a result of a slowing of the fast component and an increased contribution from the slow component. Although we cannot exclude the possibility these two components arise from distinct heteromeric populations (ie. 1:2 ASIC1a/2a vs. 2:1 ASIC1a/2a), this is not probable because their relative contributions change between pH 8.0 and 7.4. Rather, it is more plausible that these components stem from discrete steps in the recovery process. Clearly, additional studies are required to investigate the determinants and kinetic mechanism of ASIC recovery from desensitization. However, for the purposes of the present study, it is clear that, as in the case of ASIC1a, the mechanism by which ASICs follow high‐frequency trains cannot involve rapid recovery between pulses in a train because recovery is too slow.
Figure 5. Heteromeric ASIC1a/2a recovery from desensitization is pH‐dependent .
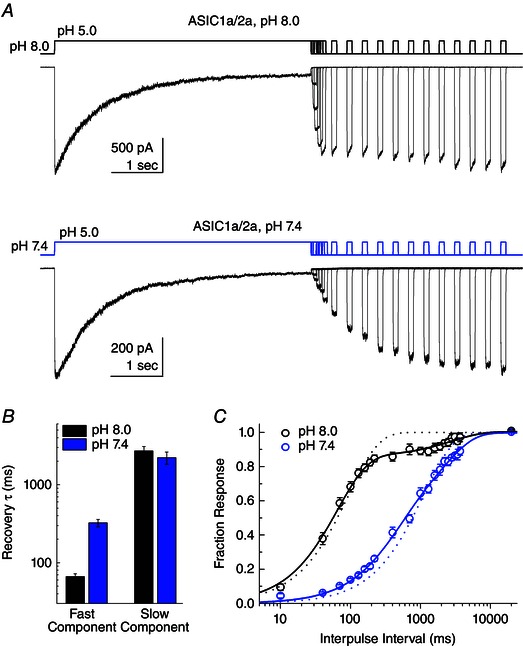
A, family of responses from patches containing ASIC1a/2a heteromers to a paired pulse protocol using either pH 8.0 (upper traces) or pH 7.4 (lower traces) as the baseline solution and pH 5.0 as the activating solution. The first pulses are 5 s in duration to induce desensitization, followed by a variable incubation in baseline pH solution before a second 100 ms test pulse using pH 5.0. B, time constants for double‐exponential fits of the recovery time course for either pH 8.0 (black bars) or pH 7.4 (blue bars) baseline solution. C, summary of recovery time course across patches for either pH 8.0 (black circles) or pH 7.4 (blue circles) baseline solution. The continuous lines show double‐exponential fits of the recovery time course for pH 8.0 (black lines) or pH 7.4 (blue lines). The dotted lines indicate fits using only a single‐exponential function for each condition.
Fast deactivation and slow desensitization set ASICs apart from other NGICs
Previous work from Papke et al. (2011) surveyed representative subtypes from each major family of NGICs and found that, although the extent was variable, desensitization accumulates during trains at 50 Hz in every case and often at much lower frequencies. It appears to be a generally shared property of NGICs. What makes ASICs an exception? We hypothesized that the unusual combination of slow desensitization and fast deactivation may enable ASICs to follow such stimuli patterns at the same time as avoiding appreciable desensitization. In essence, at the stimulus frequencies we probed, the very brief deactivation may provide minimal opportunity for the slow desensitization process to occur. In support of this interpretation, heteromeric ASIC1a/2a is able to follow high‐frequency trains at both pH 8.0 and 7.4 (Fig. 4) despite the latter pH dramatically slowing recovery from desensitization (Fig. 5). This finding suggests that appreciable desensitization does not occur during the 50 Hz train. An examination of biophysical properties for each channel type further supports the hypothesis that ASICs avoid depression during trains by combining fast deactivation with slow desensitization. In Fig. 6 A, the weighted desensitization and deactivation time constants for each NGIC in the study by Papke et al. (2011) and our own experiments are plotted on the abscissa and ordinate, respectively (ASIC1a/2a deactivation time constants are from pH 7.4 experiments, Fig. 4). Although there is considerable variation, in general, deactivation is ∼3‐ to 100‐fold faster than desensitization across all NGICs except ASICs (Fig. 6), although we note that the kinetics of ASIC1a deactivation were obtained using a background pH of 8.0, whereas all of the others reflect more physiological circumstances. Indeed, deactivation and desensitization of all other NGICs (ie. not including ASICs) shows an approximate linear correlation with a correlation co‐efficient of 0.9 (Fig. 6 A, dotted line, slope of 1.4). Including ASICs in the fit reduces the correlation co‐efficient to 0.35. Indeed, ASICs appear to be an outlier in another regard because they are insensitive to desensitization at high frequencies (Figs 3 and 4). This is also illustrated in Fig. 6 A, where each NGIC data point is colour coded by its extent of desensitization to 50 Hz stimuli. ASICs are not susceptible (Fig. 6 A, blue circles) and are furthest from the linear correlation. Figure 6 B is another representation of this relationship where the ratio of desensitization to deactivation time constants is plotted on the abscissa and the final response to a 50 Hz train plotted on the ordinate. Again, ASICs (Fig. 6 B, circles) stand apart from other NGICs with a desensitization to deactivation ratio of 1000 or greater. By contrast, other NGICs have ratios of between 3 and 170 and show strong train‐induced desensitization (Fig. 6 B). If ASICs can follow high‐frequency trains as a result of the mix of fast deactivation and slow desensitization, then accelerating desensitization should disrupt this capacity. To test this idea, we set out to experimentally manipulate desensitization using anion substitutions.
Figure 6. The slow desensitization and fast deactivation of ASICs is unique amongst NGICs .
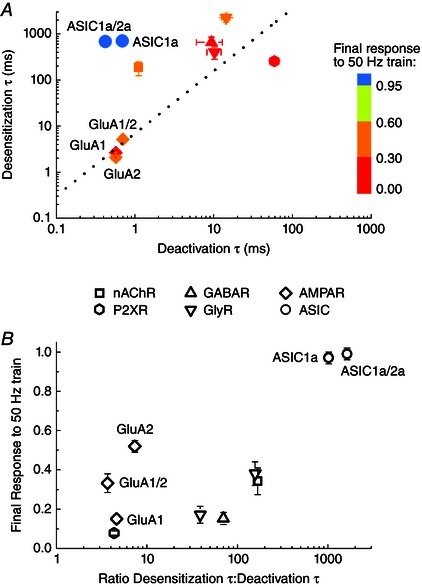
A, plot of weighted time constants for desensitization and deactivation for various NGICs. Nicotinic ACh receptors (square), GABAA receptors (upward triangle), glycine receptors (downward triangles), P2X receptors (hexagon), and the GluA1 and GluA1/2 AMPA receptors (diamonds) are all taken from Papke et al. (2011). GluA2 (diamond) and ASICs (circles) are from the present study. ASIC1a/2a data are for pH 7.4. Each receptor is coloured based on the amplitude of the final response to a 2 s 50 Hz train from either Papke et al. (2011) or the present study. The dotted line is a linear fit through the data, excluding the ASIC data points. B, plot of the ratio of desensitization time constants over deactivation time constant on the abscissa and the normalized response at the end of a 50 Hz train on the ordinate for each NGIC in (A). Note that all the NGICs, except ASICs (circles), cluster in the bottom right quadrant of strong desensitization. ASIC1a and 1a/2a are the sole occupants of the upper right quadrant.
Anion substitutions accelerate desensitization and can induce depression during high‐frequency stimulation
Structural studies of chicken ASIC1a reveal a well‐defined anion binding site formed by the two α helices of the thumb domain of one subunit and a Lys residue of an adjacent subunit (Gonzales et al. 2009). Interestingly, changing the external anion from Cl− to MeSO3 − or I− accelerates desensitization of ASIC1a (Kusama et al. 2010). Importantly, anion substitution does not alter the rates of recovery from desensitization (Kusama et al. 2010) because this process may impact sustained signalling as well (Papke et al. 2011). Although there are numerous mutations that accelerate desensitization (Cushman et al. 2007; Li et al. 2010; Springauf et al. 2011; Frey et al. 2013), anion exchanges have the added advantage that they can be performed in the same patch to improve reliability and statistical power. We therefore used MeSO3 − and I− substitutions (in both control and low pH solutions) and measured the desensitization and deactivation kinetics, as well as responses to high‐frequency stimulus trains as above. In keeping with previous work on mouse ASIC1a (Kusama et al. 2010), replacing Cl− with MeSO3 − or I− accelerated the desensitization kinetics of rat ASIC1a from 700 ± 60 ms (n = 13) in Cl− to 340 ± 20 (n = 8) and 210 ± 30 (n = 8) in MeSO3 − and I−, respectively (Fig. 7 A and B). Importantly, the difference between desensitization kinetics in MeSO3 − and I− is statistically significant (P < 0.003). Anion substitution also slowed deactivation kinetics compared to Cl− in a statistically detectable fashion (P < 0.0003 for both MeSO3 − and I−), although the effect was small. Specifically, deactivation time constants in MeSO3 − were 1.4 ± 0.1 (n = 8) and 1.2 ± 0.1 (n = 8) in I− (Fig. 7 B). When challenged with a train of 1 ms stimuli at 50 Hz, we found that MeSO3 − substitution showed no evidence of desensitization during trains (99th pulse 98 ± 2% of first peak, n = 8, Fig. 7 C and E). Interestingly, I− substitution did produce desensitization during 50 Hz trains (Fig. 7 D and E) with the 99th pulse yielding 84 ± 3% of the first pulse (n = 8, P < 0.0003). This experiment indicates that the unusually slow desensitization of ASIC1a is one component of its ability to follow trains because accelerating desensitization, at the same time as keeping other features of gating relatively constant, can induce depression during high‐frequency trains.
Figure 7. Accelerating desensitization by anionic substitution can induce depression during high‐frequency stimulation .

A, example responses to a 5 s pH 5.0 jump from patches expressing ASIC1a in NaCl solution (black trace), NaMeSO3 (purple trace) or NaI (orange trace). B, summary of desensitization and deactivation time constants from ASIC1a in either NaCl (black bars), NaMeSO3 (purple bars) or NaI (orange bars). C, example responses from ASIC1a‐containing patch to a 2 s train at 50 Hz of 1 ms pH 5.0 applications in NaMeSO3. D, responses from the same patch as in (C) but in NaI. The solution exchange current from this patch is depicted in purple for NaMeSO3 (C) and orange for Na (D). Note the onset of depression during the train in NaI but not NaMeSO3. E, summary of responses to 1 ms pH 5.0 jumps at 50 Hz in either NaCl (black circles), NaMeSO3 (purple circles) or NaI (orange circles).
Responses of native ASICs to high‐frequency stimuli
Given the surprising capacity of recombinant ASICs to follow such high‐frequency trains, we investigated whether this facility was retained in native ASIC populations. Accordingly, we measured desensitization and deactivation kinetics in excised patches from DRG cultures. We used DRGs because they are readily accessible and give rise to sizeable whole‐cell ASIC currents (∼4 nA) (Benson et al. 2002) that enable resolvable outside‐out patch responses. Previous studies have indicated pH‐activated currents from DRGs come from a mix of ASIC1a, 2a or ASIC3‐containing heteromeric receptors, with ASIC2 showing stronger expression than ASIC1a or ASIC3 (Benson et al. 2002; Xie et al. 2002; Kusama et al. 2013). Consistent with a mixed heteromeric population, pH 5.0 evoked currents from DRG excised patches showed a wide range of desensitization kinetics, with some patches showing ASIC3‐like rapid desensitization (Fig. 8 A, red trace, and B) and others showing ASIC1a‐like slow desensitization (Fig. 8 A, blue trace, and B). Despite desensitization kinetics ranging from 84 ms to 1080 ms (Fig. 8 A and B), deactivation kinetics were uniformly rapid, spanning 160 to 660 μs with a mean of 330 ± 40 μs (n = 10) (Fig. 8 B). There was a poor positive correlation between desensitization and deactivation (correlation coefficient 0.65) (Fig. 8 B, dotted line). For each patch, we also obtained a pH 6/5 ratio and found that faster desensitizing patches tended to have pH 6/5 ratios nearer to 1, whereas slower desensitizing patches showed ratios closer to 0.5 (correlation coefficient 0.73) (Fig. 9). Taken together, these measurements are consistent with a mix of ASIC heteromers, primarily ASIC2‐containing (as judged by the rapid deactivation) and ASIC3‐containing heteromers in the faster patches (Fig. 8 A, red trace) with more ASIC2‐ and ASIC1a‐containing heteromers in the slower patches (Fig. 8 a, blue trace). For each patch, we also examined the responses to 50 Hz trains of 1 ms pH 8.0 to 5.0 jumps and, in every case, found that the responses showed no evidence of desensitization (Fig. 8 C–E). Fast desensitizing patches (Fig. 8 A, red trace, and C) and slow desensitizing patches (Fig. 8 A, blue trace, and D) were all able to follow these trains with high fidelity. Indeed, we observed a surprising facilitation during such high‐frequency trains, with the final response being 110 ± 2% of the initial peak response (n = 10, P = 0.0004 compared to ASIC1a) (Fig. 8 E). Given this, we conclude that the capacity to follow high‐frequency brief pH 8.0 to 5.0 stimuli, which approximate the time course of synaptic neurotransmission, is conserved in at least some neuronal ASIC populations.
Figure 8. Native ASICs follow high‐frequency stimuli .
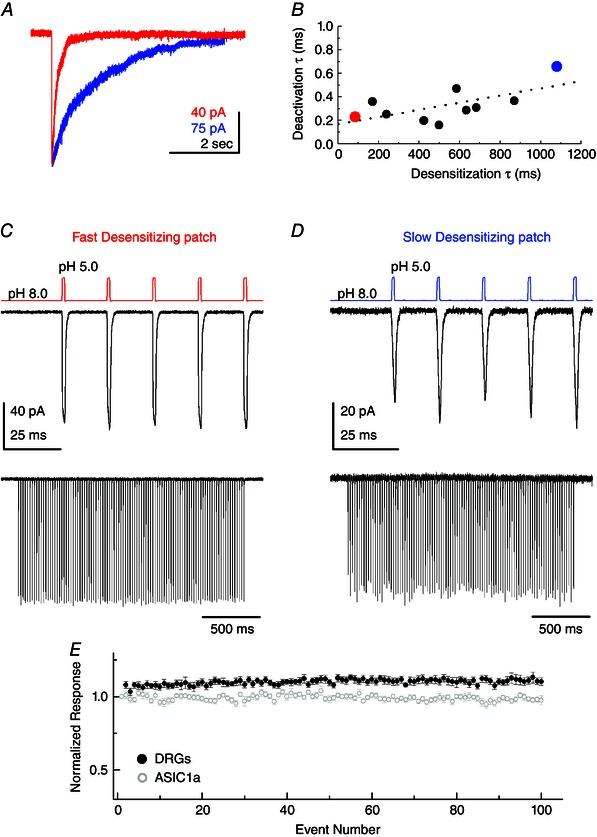
A, example responses to a 5 s pH 5.0 jump from patches excised from cultured DRG neurons. Red trace is from the fastest desensitizing patch in the dataset; blue is from the slowest. B, scatter plot of time constants of desensitization and deactivation for each patch from DRG neurons. Red data point is the fastest desensitizing patch from (A); blue data point is the slowest desensitizing patch. Note a wide spread in desensitization kinetics but minimal variance in deactivation. Dotted line is a linear fit to the data. C, responses from the fastest desensitizing patch to 1 ms pH 5.0 jumps at 50 Hz, with the first five responses in the upper panel and the whole 2 s train in the lower panel. D, same plot as in (C) but for the slow desensitizing patch from (A). E, summary of peak responses to 1 ms pH 5.0 jumps at 50 Hz, normalized to first responses, for DRG neurons (black circles) and recombinant ASIC1a (grey circles).
Figure 9. pH 6/5 ratio is correlated with desensitization kinetics in DRG patches .
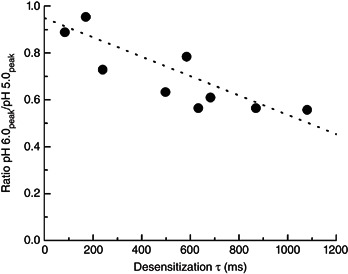
Summary of pH 6/5 ratio and desensitization time constants for each patch excised from DRG neurons. The dotted line is a linear fit of the data.
Discussion
In the present study, we observed that the deactivation kinetics of ASIC1a homomers and 1a/2a heteromers after a 1 ms pH jump are strikingly fast, amongst the fastest reported for any NGIC (Fig. 1). We also find that, unlike other reported NGICs, ASICs are able to follow trains of such 1 ms stimuli at frequencies up to 50 Hz (Fig. 3). Furthermore, heteromeric ASICs are able to follow such trains at physiological pH (Fig. 4), although ASIC1a was unable to be evaluated in this regard. Our results place ASICs in the unique position of showing very fast deactivation yet slow desensitization (Figs 1 and 6). We hypothesized that this combination enables ASICs to sustain channel activity at high frequencies. To test this, we experimentally accelerated desensitization using anion substitutions (Fig. 7) and were able to induce some desensitization during high‐frequency stimuli, supporting our hypothesis. Importantly, we also find that native ASICs from DRG neurons share the capacity to follow pH 8.0 to 5.0 stimuli at high frequencies (Fig. 8). These results not only set ASICs apart from other NGICs described previously (Papke et al. 2011), but also open up new avenues of investigation for ASIC signalling in the nervous system.
Synaptic activation of ASICs by pH
In the present study, we rapidly jumped excised patches containing ASICs from pH 8.0 to 5.0 for 1 ms (deactivation) or 5 s (desensitization) (for pH 7.4 experiments, see Figs 4 and 5). These experimental conditions represent a balance between physiological conditions and experimental feasibility. The deactivation jump of 1 ms was chosen because it is commonly taken as the time course of neurotransmitter in the synaptic cleft (Clements et al. 1992; Jones & Westbrook, 1995, 1996; Beato, 2008). However, comparatively little is known about the mechanisms of buffering, clearance and the time course of protons in the cleft (Wemmie et al. 2008). In the absence of any high temporal resolution data, we adopted a 1 ms protocol because it at least permits the direct comparison with other NGICs (Papke et al. 2011). In preliminary experiments with homomers, we employed pH 7.4 as a ‘resting pH’ and pH 6.0 or pH 6.5 as an ‘activating pH’ for such 1 ms jumps. Although pH 6.0 is near saturating for ASIC1a (Fig. 2), it also represents a 10‐fold reduction in agonist concentration and hence a 10‐fold reduction in the first order‐rate constant for agonist binding. Consequently, minimal activation developed during the 1 ms window of pH 6.0 or pH 6.5 applications, which made peak responses to these single jumps or stimulus trains difficult to resolve convincingly. The situation was further exacerbated by steady‐state desensitization at pH 7.4, which reduced the observed response amplitude. We therefore adopted the pH 8.0 to 5.0 protocol used in the present study. How well does this approximate the synaptic waveform of protons? On the one hand, theoretical and experimental data indicate the cleft drops to around pH 6.5 (Miesenbock et al. 1998; Palmer et al. 2003; Du et al. 2014; Grunder & Pusch, 2015). On the other hand, such ‘mild’ acidification would produce essentially no activation of the synaptic ASIC1a/2a heteromers (Bartoi et al. 2014; Kreple et al. 2014). Yet, clearly, ASIC1a/2a heteromers are activated by acidification accompanying transmission (Du et al. 2014; Kreple et al. 2014). One possibile reconciliation of these observations is that ASICs may be clustered directly opposite active zone release sites. This privileged subsynaptic location would experience a much higher local proton concentration during a release event than the rest of the cleft, enabling strong acidification and transient activation. However, further work is clearly needed to determine the precise pH waveform impinging on synaptic ASICs.
Mechanism of fast deactivation
This is the first study to evaluate the deactivation kinetics of recombinant ASIC1a homomers, 1a/2a heteromers and native ASICs from DRGs after 1 ms pH jumps. The extremely fast deactivation observed, at or near the limit of rapid solution exchange, was surprising given that deactivation time courses reflect both the single channel mean open time, as well as the dissociation of the agonist. Rat ASIC1a subunits are known to have two distinct open states or modes, with mean open times of ∼37 ms for Mode 1 and 7 ms for Mode 2 (Zhang & Canessa, 2002), both of which are considerably longer lived than the 700 μs deactivation observed in the present study (Fig. 1). It may be that ASICs progress through such open states or modes of differing duration during the activation process and the brief pulse curtails this progression, revealing a short‐lived open state. It may also be that protons do not need to wait for shut states but can directly dissociate from the open states. Consistent with this latter possibility, there are no obvious structures sterically impeding proton dissociation as there are for other neurotransmitters. Ionotropic glutamate receptors, for example, hold their ligands in place by closure of a large clam‐shell shaped ligand binding domain (Karakas & Furukawa, 2014; Yelshanskaya et al. 2014), whereas GABA and nicotinic receptors secure agonists by the ‘C‐loop’, which caps the binding site (Jadey & Auerbach, 2012; Miller & Aricescu, 2014). Moreover, because protons are the smallest possible ligand, their dissociation is probably extremely rapid. Future experiments at the single channel level, as well as structure–function analysis, will help unravel how ASICs combine high affinity activation and relatively long lived open states with extremely rapid deactivation.
Why can fast desensitizing patches from DRGs operate at high frequencies?
For most ligand‐gated ion channels, the response amplitude during high‐frequency trains reflects both how quickly channels enter desensitization (or how long they reside in states leading to desensitization), as well as how readily they exit desensitization (Papke et al. 2011). However, we suggest that, for ASIC1a and ASIC1a/2a, the very brief deactivation provides too small a ‘window’ for the relatively slow or unfavourable process of ASIC desensitization to occur. Consequently, minimal desensitization develops during a train and, hence, even when recovery is slowed (Fig. 5), there is no depression. However, when desensitization is accelerated or made more favourable, depression during trains does occur, as in the case of I− substitutions (Fig. 7 D and E). However, in patches excised from DRG neurons, we found a number of fast desensitizing ASIC responses (Fig. 8), some of which had desensitization decays faster than ASIC1a in the presence of I− (Fig. 7 D and E). Yet patches from DRG neurons uniformly showed no depression during high‐frequency stimuli (Fig. 8 E). As stated above, we suggest that ASIC3‐containing receptors comprise most or all of the responses from these faster desensitizing patches based on the strong expression of ASIC3 in murine DRGs (Kusama et al. 2013), as well as the desensitization kinetics and the ratio of pH 6.0 to 5.0 matching those reported from recombinant ASIC3 (Sutherland et al. 2001; Kusama et al. 2013). So why might these putative ASIC3‐containing patches be resistant to depression during a train, whereas ASIC1a in I−, desensitizing at a comparable rate, is not? We speculate that this is a result of the faster recovery of ASIC3 from desensitization. Although recovery does not appear to be an important factor in the response of ASIC1a or ASIC1a/2a to trains, the much faster entry of ASIC3 into desensitization requires a commensurate faster recovery if depression during a train is to be avoided. Supporting this supposition, ASIC3 is known to recover from desensitization much faster than ASIC1a or 1a/2a. Specifically, time constants for recovery from desensitization (pH 7.4) for ASIC3 are in the 500 ms range (Sutherland et al. 2001), whereas ASIC1a recovers with a time constant in the range 5–13 s (Sutherland et al. 2001; Babini et al. 2002) and ASIC1a/2a with weighted time constants of approximately 1.5 s (Fig. 5). Heteromers formed from of ASIC2a and 3 may well recover exhibit even faster recovery from desensitization. Therefore, we suggest that our faster desensitizing DRG patches are primarily ASIC3‐containing heteromers and hence their accelerated recovery prevents desensitization during high‐frequency stimulation. It is interesting to speculate that the faster recovery of ASIC3 may be an adaption that allows it to retain fast desensitization yet still follow high‐frequency trains.
How might ASICs employ their unique ability?
In the present study, a baseline of pH 8.0 was used for most experiments to eliminate steady‐state inactivation. Importantly heteromeric ASIC1a/2a did not show any steady‐state inactivation at pH 7.4, enabling us to reveal that, at physiological pH, ASIC1a/2a also does not desensitize during high‐frequency stimulation (Fig. 4). Because ASIC1a/2a is one of the main ASIC populations in the brain (Baron et al. 2002 b; Askwith et al. 2004; Kreple et al. 2014), its capacity to follow such stimulus patterns at physiological pH is particularly relevant. High‐frequency stimulation is generally associated with synaptic plasticity and LTP. The strongest evidence linking ASICs, high‐frequency stimulation and plasticity derives from studies in the amygdala. ASICs are crucial for establishing fear memories (Wemmie et al. 2003; Wemmie et al. 2004; Coryell et al. 2008) and are necessary for the high‐frequency stimulus‐induced LTP in lateral amygdala synapses (Du et al. 2014; Chiang et al. 2015). Moreover, the extent of LTP correlates with the level of ASIC expression in postsynaptic neurons (Chiang et al. 2015). NMDA receptor activation is also necessary for these processes. Given all this, it is interesting to reflect that ASICs are attuned to respond to the exact stimuli that strongly inhibit NMDA receptors, namely acidification (Traynelis et al. 1995). The small size of postsynaptic ASIC currents (Du et al. 2014; Kreple et al. 2014) and their limited calcium permeability, particular for heteromers (Kellenberger & Schild, 2015), make it improbable that ASICs ‘take over’ from NMDA receptors under high‐frequency conditions. Rather, they may function to boost excitability or prolong the window of depolarization when other postsynaptic receptors, such as AMPA receptors, are compromised by desensitization. Although the pairing of the unique high‐frequency capacity of ASIC and associations with LTP are enticing, further experiments are needed to clarify how ASICs contribute to these processes.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
DM acquired and analysed data. DM and VJ designed the experiments, interpreted the results and wrote the manuscript. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by MCB1110501 NSF grant and R01GM113212 NIH grant to VJ and K99NS094761 to DMM.
Acknowledgements
We thank Dr Jin Bin Tian for providing DRG cultures; Drs Papke and Grosman for sharing data from their study; and Dr Stephan Grunder for the gift of rat ASIC1a and 2a cDNAs.
References
- Askwith CC, Wemmie JA, Price MP, Rokhlina T & Welsh MJ (2004). Acid‐sensing ion channel 2 (ASIC2) modulates ASIC1 H+‐activated currents in hippocampal neurons. J Biol Chem 279, 18296–18305. [DOI] [PubMed] [Google Scholar]
- Babini E, Paukert M, Geisler HS & Grunder S (2002). Alternative splicing and interaction with di‐ and polyvalent cations control the dynamic range of acid‐sensing ion channel 1 (ASIC1). J Biol Chem 277, 41597–41603. [DOI] [PubMed] [Google Scholar]
- Baron A, Deval E, Salinas M, Lingueglia E, Voilley N & Lazdunski M (2002. a). Protein kinase C stimulates the acid‐sensing ion channel ASIC2a via the PDZ domain‐containing protein PICK1. J Biol Chem 277, 50463–50468. [DOI] [PubMed] [Google Scholar]
- Baron A & Lingueglia E (2015). Pharmacology of acid‐sensing ion channels – physiological and therapeutical perspectives. Neuropharmacology 94, 19–35. [DOI] [PubMed] [Google Scholar]
- Baron A, Waldmann R & Lazdunski M (2002. b). ASIC‐like, proton‐activated currents in rat hippocampal neurons. J Physiol 539, 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoi T, Augustinowski K, Polleichtner G, Grunder S & Ulbrich MH (2014). Acid‐sensing ion channel (ASIC) 1a/2a heteromers have a flexible 2:1/1:2 stoichiometry. Proc Natl Acad Sci USA 111, 8281–8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beato M (2008). The time course of transmitter at glycinergic synapses onto motoneurons. J Neurosci 28, 7412–7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson CJ, Xie J, Wemmie JA, Price MP, Henss JM, Welsh MJ & Snyder PM (2002). Heteromultimers of DEG/ENaC subunits form H+‐gated channels in mouse sensory neurons. Proc Natl Acad Sci USA 99, 2338–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone AL & Plested AJ (2012). Coupled control of desensitization and gating by the ligand binding domain of glutamate receptors. Neuron 74, 845–857. [DOI] [PubMed] [Google Scholar]
- Chen X & Grunder S (2007). Permeating protons contribute to tachyphylaxis of the acid‐sensing ion channel (ASIC) 1a. J Physiol 579, 657–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PH, Chien TC, Chen CC, Yanagawa Y & Lien CC (2015). ASIC‐dependent LTP at multiple glutamatergic synapses in amygdala network is required for fear memory. Sci Rep 5, 10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH & Askwith CC (2008). Presynaptic release probability is increased in hippocampal neurons from ASIC1 knockout mice. J Neurophysiol 99, 426–441. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE & Westbrook GL (1992). The time course of glutamate in the synaptic cleft. Science 258, 1498–1501. [DOI] [PubMed] [Google Scholar]
- Cooper DC (2002). The significance of action potential bursting in the brain reward circuit. Neurochem Int 41, 333–340. [DOI] [PubMed] [Google Scholar]
- Coryell MW, Wunsch AM, Haenfler JM, Allen JE, McBride JL, Davidson BL & Wemmie JA (2008). Restoring acid‐sensing ion channel‐1a in the amygdala of knock‐out mice rescues fear memory but not unconditioned fear responses. J Neurosci 28, 13738–13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushman KA, Marsh‐Haffner J, Adelman JP & McCleskey EW (2007). A conformation change in the extracellular domain that accompanies desensitization of acid‐sensing ion channel (ASIC) 3. J Gen Physiol 129, 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deval E & Lingueglia E (2015). Acid‐sensing ion channels and nociception in the peripheral and central nervous systems. Neuropharmacology 94, 49–57. [DOI] [PubMed] [Google Scholar]
- Du J, Reznikov LR, Price MP, Zha XM, Lu Y, Moninger TO, Wemmie JA & Welsh MJ (2014). Protons are a neurotransmitter that regulates synaptic plasticity in the lateral amygdala. Proc Natl Acad Sci USA 111, 8961–8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks KM, Stevens CF & Sejnowski TJ (2003). Independent sources of quantal variability at single glutamatergic synapses. J Neurosci 23, 3186–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey EN, Pavlovicz RE, Wegman CJ, Li C & Askwith CC (2013). Conformational changes in the lower palm domain of ASIC1a contribute to desensitization and RFamide modulation. PloS ONE 8, e71733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Duan B, Wang DG, Deng XH, Zhang GY, Xu L & Xu TL (2005). Coupling between NMDA receptor and acid‐sensing ion channel contributes to ischemic neuronal death. Neuron 48, 635–646. [DOI] [PubMed] [Google Scholar]
- Gonzales EB, Kawate T & Gouaux E (2009). Pore architecture and ion sites in acid‐sensing ion channels and P2X receptors. Nature 460, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray CM & McCormick DA (1996). Chattering cells: superficial pyramidal neurons contributing to the generation of synchronous oscillations in the visual cortex. Science 274, 109–113. [DOI] [PubMed] [Google Scholar]
- Graydon CW, Cho S, Diamond JS, Kachar B, von Gersdorff H & Grimes WN (2014). Specialized postsynaptic morphology enhances neurotransmitter dilution and high‐frequency signaling at an auditory synapse. J Neurosci 34, 8358–8372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunder S & Pusch M (2015). Biophysical properties of acid‐sensing ion channels (ASICs). Neuropharmacology 94, 9–18. [DOI] [PubMed] [Google Scholar]
- Jadey S & Auerbach A (2012). An integrated catch‐and‐hold mechanism activates nicotinic acetylcholine receptors. J Gen Physiol 140, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV & Westbrook GL (1995). Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron 15, 181–191. [DOI] [PubMed] [Google Scholar]
- Jones MV & Westbrook GL (1996). The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci 19, 96–101. [DOI] [PubMed] [Google Scholar]
- Karakas E & Furukawa H (2014). Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 344, 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger S & Schild L (2015). International union of basic and clinical pharmacology. XCI. structure, function, and pharmacology of acid‐sensing ion channels and the epithelial Na+ channel. Pharmacol Rev 67, 1–35. [DOI] [PubMed] [Google Scholar]
- Kreple CJ, Lu Y, Taugher RJ, Schwager‐Gutman AL, Du J, Stump M, Wang Y, Ghobbeh A, Fan R, Cosme CV, Sowers LP, Welsh MJ, Radley JJ, LaLumiere RT & Wemmie JA (2014). Acid‐sensing ion channels contribute to synaptic transmission and inhibit cocaine‐evoked plasticity. Nat Neurosci 17, 1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishtal OA, Osipchuk YV, Shelest TN & Smirnoff SV (1987). Rapid extracellular pH transients related to synaptic transmission in rat hippocampal slices. Brain Res 436, 352–356. [DOI] [PubMed] [Google Scholar]
- Kusama N, Gautam M, Harding AM, Snyder PM & Benson CJ (2013). Acid‐sensing ion channels (ASICs) are differentially modulated by anions dependent on their subunit composition. Am J Physiol Cell Physiol 304, C89–C101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusama N, Harding AM & Benson CJ (2010). Extracellular chloride modulates the desensitization kinetics of acid‐sensing ion channel 1a (ASIC1a). J Biol Chem 285, 17425–17431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Yang Y & Canessa CM (2010). Leu85 in the beta1‐beta2 linker of ASIC1 slows activation and decreases the apparent proton affinity by stabilizing a closed conformation. J Biol Chem 285, 22706–22712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Yang Y & Canessa CM (2012). Impact of recovery from desensitization on acid‐sensing ion channel‐1a (ASIC1a) current and response to high frequency stimulation. J Biol Chem 287, 40680–40689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SH, Sun WH & Chen CC (2015). Genetic exploration of the role of acid‐sensing ion channels. Neuropharmacology 94, 99–118. [DOI] [PubMed] [Google Scholar]
- MacLean DM (2015). Constructing a rapid solution exchange system In Ionotropic Glutamate Receptor Technologies, ed. Popescu GK, pp. 165–183. Springer, New York, NY, USA. [Google Scholar]
- MacLean DM, Ramaswamy SS, Du M, Howe JR & Jayaraman V (2014). Stargazin promotes closure of the AMPA receptor ligand‐binding domain. J Gen Physiol 144, 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin SA, Davis BM & Molliver DC (2007). Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat Protoc 2, 152–160. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA & Rothman JE (1998). Visualizing secretion and synaptic transmission with pH‐sensitive green fluorescent proteins. Nature 394, 192–195. [DOI] [PubMed] [Google Scholar]
- Miller PS & Aricescu AR (2014). Crystal structure of a human GABAA receptor. Nature 512, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Hull C, Vigh J & von Gersdorff H (2003). Synaptic cleft acidification and modulation of short‐term depression by exocytosed protons in retinal bipolar cells. J Neurosci 23, 11332–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke D, Gonzalez‐Gutierrez G & Grosman C (2011). Desensitization of neurotransmitter‐gated ion channels during high‐frequency stimulation: a comparative study of Cys‐loop, AMPA and purinergic receptors. J Physiol 589, 1571–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A, Armstrong N, Gouaux JE & Howe JR (2005). AMPA receptor binding cleft mutations that alter affinity, efficacy, and recovery from desensitization. J Neurosci 25, 3752–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saviane C & Silver RA (2006). Fast vesicle reloading and a large pool sustain high bandwidth transmission at a central synapse. Nature 439, 983–987. [DOI] [PubMed] [Google Scholar]
- Smith ES, Cadiou H & McNaughton PA (2007). Arachidonic acid potentiates acid‐sensing ion channels in rat sensory neurons by a direct action. Neuroscience 145, 686–698. [DOI] [PubMed] [Google Scholar]
- Springauf A, Bresenitz P & Grunder S (2011). The interaction between two extracellular linker regions controls sustained opening of acid‐sensing ion channel 1. J Biol Chem 286, 24374–24384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland SP, Benson CJ, Adelman JP & McCleskey EW (2001). Acid‐sensing ion channel 3 matches the acid‐gated current in cardiac ischemia‐sensing neurons. Proc Natl Acad Sci USA 98, 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Hartley M & Heinemann SF (1995). Control of proton sensitivity of the NMDA receptor by RNA splicing and polyamines. Science 268, 873–876. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C & Lazdunski M (1997). A proton‐gated cation channel involved in acid‐sensing. Nature 386, 173–177. [DOI] [PubMed] [Google Scholar]
- Waldmann R & Lazdunski M (1998). H(+)‐gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Curr Opin Neurobiol 8, 418–424. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Askwith CC, Lamani E, Cassell MD, Freeman JH, Jr. & Welsh MJ (2003). Acid‐sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci 23, 5496–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemmie JA, Chen J, Askwith CC, Hruska‐Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, Freeman JH, Jr. & Welsh MJ (2002). The acid‐activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 34, 463–477. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Coryell MW, Askwith CC, Lamani E, Leonard AS, Sigmund CD & Welsh MJ (2004). Overexpression of acid‐sensing ion channel 1a in transgenic mice increases acquired fear‐related behavior. Proc Natl Acad Sci USA 101, 3621–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemmie JA, Price MP & Welsh MJ (2006). Acid‐sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci 29, 578–586. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Zha XM & Welsh MJ (2008). Acid‐sensing ion channels (ASICs) and pH in synapse physiology In Structural and Functional Organization of the Synapse, ed. Hell JW & Ehlers MD, pp. 661–681. Springer, New York, NY, USA. [Google Scholar]
- Xie J, Price MP, Berger AL & Welsh MJ (2002). DRASIC contributes to pH‐gated currents in large dorsal root ganglion sensory neurons by forming heteromultimeric channels. J Neurophysiol 87, 2835–2843. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ & Simon RP (2004). Neuroprotection in ischemia: blocking calcium‐permeable acid‐sensing ion channels. Cell 118, 687–698. [DOI] [PubMed] [Google Scholar]
- Yelshanskaya MV, Li M & Sobolevsky AI (2014). Structure of an agonist‐bound ionotropic glutamate receptor. Science 345, 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha XM, Costa V, Harding AM, Reznikov L, Benson CJ & Welsh MJ (2009). ASIC2 subunits target acid‐sensing ion channels to the synapse via an association with PSD‐95. J Neurosci 29, 8438–8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha XM, Wemmie JA, Green SH & Welsh MJ (2006). Acid‐sensing ion channel 1a is a postsynaptic proton receptor that affects the density of dendritic spines. Proc Natl Acad Sci USA 103, 16556–16561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P & Canessa CM (2002). Single channel properties of rat acid‐sensitive ion channel‐1alpha, ‐2a, and ‐3 expressed in Xenopus oocytes. J Gen Physiol 120, 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS & Regehr WG (2002). Short‐term synaptic plasticity. Annu Rev Physiol 64, 355–405. [DOI] [PubMed] [Google Scholar]