

Abstract
Key points
The thalamus is a structure critical for information processing and transfer to the cortex.
Thalamic reticular neurons are inhibitory cells interconnected by electrical synapses, most of which require the gap junction protein connexin36 (Cx36).
We investigated whether electrical synapses play a role in the maturation of thalamic networks by studying neurons in mice with and without Cx36.
When Cx36 was deleted, inhibitory synapses were more numerous, although both divergent inhibitory connectivity and dendritic complexity were reduced. Surprisingly, we observed non‐Cx36‐dependent electrical synapses with unusual biophysical properties interconnecting some reticular neurons in mice lacking Cx36.
The results of the present study suggest an important role for Cx36‐dependent electrical synapses in the development of thalamic circuits.
Abstract
Neurons within the mature thalamic reticular nucleus (TRN) powerfully inhibit ventrobasal (VB) thalamic relay neurons via GABAergic synapses. TRN neurons are also coupled to one another by electrical synapses that depend strongly on the gap junction protein connexin36 (Cx36). Electrical synapses in the TRN precede the postnatal development of TRN‐to‐VB inhibition. We investigated how the deletion of Cx36 affects the maturation of TRN and VB neurons, electrical coupling and GABAergic synapses by studying wild‐type (WT) and Cx36 knockout (KO) mice. The incidence and strength of electrical coupling in TRN was sharply reduced, but not abolished, in KO mice. Surprisingly, electrical synapses between Cx36‐KO neurons had faster voltage‐dependent decay kinetics and conductance asymmetry (rectification) than did electrical synapses between WT neurons. The properties of TRN‐mediated inhibition in VB also depended on the Cx36 genotype. Deletion of Cx36 increased the frequency and shifted the amplitude distributions of miniature IPSCs, whereas the paired‐pulse ratio of evoked IPSCs was unaffected, suggesting that the absence of Cx36 led to an increase in GABAergic synaptic contacts. VB neurons from Cx36‐KO mice also tended to have simpler dendritic trees and fewer divergent inputs from the TRN compared to WT cells. The findings obtained in the present study suggest that proper development of thalamic inhibitory circuitry, neuronal morphology, TRN cell function and electrical coupling requires Cx36. In the absence of Cx36, some TRN neurons express asymmetric electrical coupling mediated by other unidentified connexin subtypes.
Key points
The thalamus is a structure critical for information processing and transfer to the cortex.
Thalamic reticular neurons are inhibitory cells interconnected by electrical synapses, most of which require the gap junction protein connexin36 (Cx36).
We investigated whether electrical synapses play a role in the maturation of thalamic networks by studying neurons in mice with and without Cx36.
When Cx36 was deleted, inhibitory synapses were more numerous, although both divergent inhibitory connectivity and dendritic complexity were reduced. Surprisingly, we observed non‐Cx36‐dependent electrical synapses with unusual biophysical properties interconnecting some reticular neurons in mice lacking Cx36.
The results of the present study suggest an important role for Cx36‐dependent electrical synapses in the development of thalamic circuits.
Abbreviations
- aCSF
artificial cerebrospinal fluid
- CCf
coupling coefficient
- Cin
input capacitance
- cIPSC
coincident IPSC
- Cx36
connexin36
- Gj
junctional conductance
- KO
knockout
- mIPSC
miniature IPSC
- Rin
input resistance
- TRN
thalamic reticular nucleus
- VB
ventrobasal nucleus
- Vj
junctional voltage
- WT
wild‐type
Introduction
Neuronal gap junctions (electrical synapses) mediate rapid, bidirectional communication. Electrical synapses may have diverse functions, including synchronizing activity (Bennett and Zukin, 2004), mediating intercellular molecular signalling (Harris, 2001) and contributing to cell adhesion (Elias et al. 2007). A long‐standing yet poorly understood issue is the role of electrical synapses during neural circuit development (Fischbach, 1972; Connors et al. 1983; Kandler and Katz, 1995, 1998 a, 1998 b; Roerig and Feller, 2000; Montoro and Yuste, 2004; Sutor and Hagerty, 2005; Li et al. 2012; Yu et al. 2012; Niculescu and Lohmann, 2013). Electrical synapses have been implicated in the proper development of chemical synaptic connections in the mammalian olfactory bulb (Maher et al. 2009), retinogeniculate system (Blankenship et al. 2011) and neuromuscular junction (Personius et al. 2007), as well as other neural systems (Mentis et al. 2002; Szabo et al. 2004; Arumugam et al. 2005; Neunuebel and Zoran, 2005; Szabo and Zoran, 2007; Todd et al. 2010). These studies suggest that electrical synapses importantly regulate network development. Most mammalian investigations have focused on glutamatergic synapses (Yu et al. 2012; Pereda, 2014), yet the majority of electrically coupled neurons in the forebrain are GABAergic. To test whether electrical synapses are important for inhibitory circuit development, we chose the thalamic reticular nucleus (TRN). The GABAergic TRN neurons are interconnected by electrical synapses (Landisman et al. 2002; Long et al. 2004) and project their axons onto thalamocortical relay neurons (Pinault, 2004). TRN‐mediated inhibition helps to shape receptive field properties of relay neurons (Lee et al. 1994) and generate certain types of rhythms associated with sleep and seizure states (Huguenard and McCormick, 2007).
Most mammalian electrical synapses depend on the neuronal gap junction protein connexin36 (Cx36) (Deans et al. 2001; Hormuzdi et al. 2001; Connors and Long, 2004). TRN neurons are densely interconnected by Cx36‐containing gap junction channels (Belluardo et al. 2000; Landisman et al. 2002; Liu and Jones, 2003), which are present at birth and strengthen over the next 2 weeks (Parker et al. 2009). Other connexins may also contribute to neuronal network maturation. Over 20 connexin genes have been identified in the mouse; around half of them are expressed in the brain, mainly in glial cells (Willecke et al. 2002; Sohl et al. 2005). Only a few connexin subtypes are expressed in central neurons, and only Cx36 has been consistently implicated in electrical coupling in the mammalian brain (Connors & Long, 2004; Pereda, 2014). Each connexin subtype, and in some cases heteromeric or heterotypic combinations of subtypes, can form gap junction channels with distinctive channel properties (Harris, 2001; Rash et al. 2013). Cx36, however, does not appear to form heterotypic or heteromeric channels (Koval et al. 2014).
Inhibitory synapses undergo extensive remodelling during development (Kim and Kandler, 2003; Hensch and Fagiolini, 2005; Kätzel and Miesenböck, 2014; Froemke, 2015). The thalamic inhibitory circuit changes rapidly and dramatically during the first two postnatal weeks (Warren and Jones, 1997; Lee et al. 2010). In the present study, we investigated whether the development of intrathalamic GABAergic synapses and circuits is affected by the deletion of Cx36. The results obtained imply that electrical synapses have a broad influence on the development of thalamic networks.
Methods
Slice preparation
All experiments were approved by the Institutional Animal Care and Use Committee of Brown University. Animals were deeply anaesthetized with propofol prior to preparation of 300 μm‐thick thalamocortical slices (Agmon and Connors, 1991) from wild‐type (WT) (postnatal ages P0 – P13) and littermate WT and Cx36 knockout (KO) FvB/C57 mice (P2 – P13) of either sex. The experimenter was blind to the genotype during the recording and analyses. Immediately following slice preparation, the slices were incubated at 32°C for 30 min and at room temperature for at least an additional 30 min before recording in a submersion chamber at 32°C. Slices were visualized on an BX50WI microscope (Olympus, Tokyo, Japan) using a CCD camera (Hamamatsu City, Japan) and infrared‐differential interference contrast optics.
The artificial cerebrospinal fluid (aCSF) bathing the slices during recording and slicing contained (in mm): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 2 MgCl2 and 10 dextrose, and was saturated with 95% O2–5% CO2. In some experiments, 6,7‐dinitroqinoxaline‐2,3‐dione (20 μm) and D‐2‐amino‐5‐phosphopentanoic acid (50 μm) were added to the aCSF to block AMPA and NMDA receptors, respectively; to isolate miniature IPSCs (mIPSCs), TTX (1 μm) was added to the aCSF to block sodium channel‐dependent spikes.
Recording and imaging
Dual whole‐cell current clamp and voltage clamp recordings were made with low resistance (2.5–4 MΩ) microelectrodes. For current clamp recordings, the intracellular solution contained (in mm): 130 K‐gluconate, 4 KCl, 2 NaCl, 0.2 EGTA, 10 Hepes, 4 ATP‐Mg, 0.3 GTP‐Tris and 14 phosphocreatine‐Tris. For voltage clamp recordings, the intracellular solution contained (in mm): 54 Cs‐gluconate, 56 CsCl, 1 CaCl2, 1 MgCl2, 10 EGTA, 10 Hepes, 5 QX‐314, 4 ATP‐Mg, 0.3 GTP‐Tris and 14 phosphocreatine‐Tris. Both solutions were adjusted to pH 7.25–7.30 (285–295 mosmol). A MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA) was used to make simultaneous recordings from pairs of closely apposed (<2 μm) cell somata in the TRN; both cells were held at −60 mV in voltage clamp or at around −60 mV in current clamp by continuous current injection. Single or pairs of relay neurons were also recorded in the ventrobasal (VB) complex. Series resistance was continually monitored and compensated (70–80% in voltage clamp) and was typically between 6 and 20 MΩ before compensation. Recordings were not corrected for liquid junction potential.
Cell capacitance was determined using the built‐in capacitance‐compensation function of the amplifier. Cell input resistance was determined in current clamp or voltage clamp by measuring the steady‐state voltage (in current clamp) or current (in voltage clamp) response from small, 600 ms current (−5 pA to −50 pA for current clamp) or voltage (−10 mV for voltage clamp) steps.
Electrical coupling was measured in voltage clamp by simultaneously stepping both cells to 0 mV and then stepping one or the other cell to −80 mV (Fig. 2 A) at the same time as recording the holding current in both cells. In current clamp, electrical coupling was measured by injecting hyperpolarizing current into one cell (the driven cell; V d) to bring it to around −100 mV, at the same time as recording the voltage deflections in the adjacent coupled cell (the follower cell; V f) and V d. The coupling coefficient (CCf) is defined as: CCf = (∆V f/∆V d) × 100.
Figure 2. Effect of Cx36 genotype on voltage‐dependent kinetics of electrical coupling in the TRN .
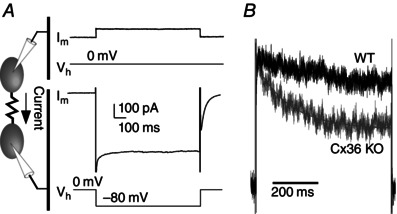
A, protocol for measuring electrical coupling in voltage clamp (see Methods). B, overlay of the junctional current from WT (black) and Cx36 KO (grey) neuron pairs. Current amplitudes are normalized.
For extracellular stimulation, a tungsten monopolar or concentric biopolar electrode (FHC, Bowdoinham, ME, USA) was placed in the TRN or VB, <200 μm from the recording site. To determine the paired‐pulse ratio, two stimuli with a 100 ms interstimulus interval were given at 0.04–0.1 Hz. IPSCs were recorded exclusively in Cs‐based internal solution at V hold = −60 mV (E Cl = −20 mV), filtered at 2 kHz and digitized at 20 kHz.
In some experiments, Alexa Fluor 633 hydrazide dye (75 μm; Invitrogen, Carlsbad, CA, USA) was added to the pipette solution for cell loading and subsequent confocal imaging. Slices with dye‐filled neurons were fixed with 4% paraformaldehyde in 0.1 m phosphate buffer for 30 min at room temperature. The slices were then moved to phosphate‐buffered saline (< 1 day) until they were mounted on slides with VectaShield (Vector Laboratories, Burlingame, CA, USA) mounting medium. An LSM510 confocal microscope (Carl Zeiss, Oberkochen, Germany) with a 20× Plan‐Neofluar (0.8 NA) objective was used for imaging. Images were scanned at 2048 × 2048 pixels. The typical step size per section through the z‐axis was 1 μm, or occasionally 0.2 μm.
Basic morphological properties of neurons were determined by counting the number of primary dendrites and by measuring the 3‐D soma surface area using convex hull analysis in NeuroExplorer (MicroBrightField Inc., Williston, VT, USA) after reconstruction in AutoNeuron (MicroBrightfield Inc.). Dendritic complexity was quantified using 2‐D Sholl analysis (Sholl, 1953) with increments of 10 μm between circles. Total dendritic length was estimated by multiplying the Sholl intersections per circle by the increment distance (10 μm) and taking the sum of all circles. All analysis was performed in ImageJ (NIH, Bethesda, MD, USA), Neuroexplorer and Zeiss LSM Image Browser.
Detection of spontaneous coincident IPSCs
IPSCs were considered coincident between two cells if the onset time of an IPSC in one cell occurred within ±1 ms of the onset time of an IPSC in the other cell. The onset time of an IPSC is the point of downward deflection at the start of the IPSC waveform, and this was measured using optimal fit parameters with Minianalysis (Synaptosoft Inc., Fort Lee, NJ, USA). Spontaneous IPSC onset times were compiled for each neuron pair from 3–5 min of continuously recorded data. Using these data, a custom Matlab (MathWorks Inc., Natick, MA, USA) algorithm was written to test for statistically significant occurrences of cIPSCs. First, the number of coincident IPSCs (cIPSCs) was calculated (see above) by comparing the compiled IPSC onset times from each neuron in a pair and finding paired values within ±1 ms. Second, the inter‐IPSC intervals were calculated from the IPSC onset times for each neuron in the pair. Third, the inter‐IPSC intervals for each neuron of a pair were measured independently, and randomly shuffled, using a MatLab randomization function, into 10,000 surrogate data sets. Fourth, each surrogate data set was scanned for cIPSCs as was carried out in step one with the real data set. IPSC coincidence was considered significant if 95% of the surrogate data sets had fewer cIPSCs than the real data set.
The expected chance occurrence of cIPSCs was then calculated. The number of IPSCs in each neuron of a pair was determined and divided by the number of milliseconds over which those events occurred to find the probability of an IPSC occurring within a 1 ms bin. This assumes that the IPSCs were independent and randomly distributed across time. The 1 ms bin probability from one neuron was then multiplied by the 1 ms bin probability in the other neuron to obtain the probability of two IPSCs occurring, one in each neuron, within 1 ms. This probability was then multiplied by the duration of the recording to estimate the expected chance occurrences of coincident IPSCs:
Expected chance cIPSCs = (α) x (β) x recording time (ms)
where α = cell A 1 ms bin probability = total IPSCs of cell A/time (ms) and β = cell B 1 ms bin probability = total IPSCs of cell B/time (ms).
Statistical analysis
Gap junctional conductance (Gj) was determined in voltage clamp by:
where V j is the transjunctional voltage and I j is the resulting junctional current. Cell pairs generally had high input resistance (>150 MΩ) and low coupling conductance (<1 nS) and the series resistance in each electrode was generally less than 6 MΩ after compensation. Therefore, minimal error in G j is expected from these factors (Van Rijen et al. 1998). In current clamp, G j was calculated as (Bennett, 1966):
where R in(x) is the input resistance of each cell in a recorded pair and R tr (transfer resistance) is the value of the voltage response in the follower cell divided by the current amplitude injected into the driven cell.
The decay time constant of the junctional conductance was determined by fitting a single exponential (Fig. 2).
The frequency of mIPSCs was determined by counting events from raw data traces using pClamp, version 10.2 (Molecular Devices). mIPSCs were also identified by template matching in pClamp. More than 50 mIPSCs were averaged to determine the mean mIPSC amplitude and decay time for each cell. mIPSC amplitude was defined as the difference between the mean holding current in a 10 ms window before the onset of the mIPSC and the mean current during a 1–2 ms window centered on the peak of the mIPSC. Falling phases of the mIPSCs were fit with a double exponential (I = A 1exp−t /τD1 + A 2 e−t /τD2), which was used to determine the weighted decay time constant from τIPSC = (τD1 A 1 + τD2 A 2)/(A 1 + A 2) (Huntsman and Huguenard, 2000).
P < 0.05 was considered statistically significant. Data are reported as the mean ± SE. Multiple linear regression analysis and Student's t test were used to compare genotype and age differences. The Kolomogorov–Smirnov two sample test was used to compare genotype differences in amplitude distribution histograms.
Results
Electrical synapses in the TRN
In many brain areas, the prevalence of electrical coupling is greatest during early development and declines with age. In the TRN, however, electrical synapses are ubiquitous as early as the day of birth (Parker et al. 2009), and coupling conductance steadily strengthens by P14–18. In the mature mouse, electrical coupling is largely dependent on Cx36 (Landisman et al. 2002). Consistent with these results, we found that the probability of electrical coupling between neuron pairs was stable across the first 2 weeks of development in the WT TRN (mean of 68.6% coupling probability, n = 105 pairs) and, in neurons from Cx36 KO mice, the coupling probability was more than five‐fold less (mean of 12.4% coupling probability, n = 129 pairs) (Fig. 1 A and B). The prevalence of electrical coupling in the KO TRN was not influenced by age. This suggests that the large majority of electrical coupling in TRN requires Cx36 at all ages tested but that non‐Cx36‐dependent electrical synapses can also form between TRN cells. These results are consistent with a study from our laboratory reporting significant dye‐coupling among TRN neurons from Cx36 KO mice (Lee et al. 2014).
Figure 1. Electrical coupling in the TRN .

A, current clamp recording of electrical coupling between two TRN neurons aged P8. The incidence (B) and mean ± SEM conductance (C) of electrical coupling between adjacent neuron pairs during development. Data in (B) and (C) are taken from 105 WT and 129 KO pairs.
Among the measurable electrical synapses that we observed, the mean coupling conductance (G j) between KO cell pairs was less than one‐third that of WT pairs (WT, 190.3 pS, n = 64; KO, 60.9 pS, n = 16) (Fig. 1 C). Thus, electrical synapses were both dramatically sparser and weaker in the absence of Cx36. Coupling between WT cells showed a significant age‐dependent increase in G j, whereas G j in the KO did not have an age‐dependent change (WT, P = 0.05; KO, P = 0.38, linear regression) (Fig. 1 C). The conductance decrease that we measured between P10–11 to P12–13 was not significant (P = 0.27, post‐hoc Tukey's honestly significant difference test) and thus may not indicate a deviation from previous results (Parker et al. 2009). Despite a much lower G j in the KO cells overall, the CCf (see Methods) were, on average, only modestly lower than in WT cells (WT, CCf = 6.8%, n = 72; KO, CCf = 4.8%, n = 16).
The product (CCf × P coupling) provides an overall measure of electrical coupling within the TRN network (Amitai et al. 2002). Collapsed across development, this measure is 4.7 for the WT and 0.6 for the KO samples, suggesting that deletion of Cx36 reduces the influence of electrical synapses by more than 85% during the first two postnatal weeks. Overall, our data suggest that electrical coupling in the TRN of WT cells remains stable and largely but not entirely dependent upon Cx36 during postnatal development.
Properties of non‐Cx36‐dependent electrical synapses
As described above, some electrical coupling remains between TRN neurons of the Cx36‐KO. The properties of non‐Cx36 gap junctions have not been described previously in central mammalian neurons, and so these were examined further. All vertebrate gap junctions have some voltage‐dependence; in every case, G j is maximal when transjunctional voltage (V j) is zero, and G j declines as V j deviates from zero. Both the steepness of the voltage‐dependence of G j and the time constant (τ) of its decline vary widely but characteristically with connexin subtype (Moreno et al. 1995; Teubner et al. 2000; Bukauskas et al. 2006). The G j for Cx36 gap junctions is almost insensitive to V j over the range of ±50 mV (Teubner et al. 2000). Other gap junction channels, including those comprised of Cx45, have a considerably steeper voltage‐dependence (Moreno et al. 1995). Because Cx45 is one of the few connexins besides Cx36 that is expressed in thalamic neurons (Willecke et al. 2002; Sohl et al. 2005), it is a prime candidate for mediating electrical coupling in the Cx36 KO.
We performed dual voltage clamp measurements in pairs of TRN neurons to measure the gating properties of their electrical synapses (Fig. 2 A). Consistent with previous work on Cx36 junctions (Teubner et al. 2000), we found that its gating was slow. In pairs of coupled neurons from the Cx36 KO, however, the τ of G j decline was much faster compared to WT junctions. During a 600 ms, 80 mV transjunctional step, the τ of G j decline was 490 ms for WT and 151 ms for KO connections (Fig. 2 B). Some invertebrate electrical synapses have distinctly asymmetric conductances when measured in each direction across the junction (Furshpan and Potter, 1959; Jaslove and Brink, 1986). The large majority of electrical synapses among vertebrate central neurons are symmetric (Connors and Long, 2004), although a few exceptions have been described (Auerbach and Bennett, 1969; Haas et al. 2011; Rash et al. 2013; Sevetson and Haas, 2015). Gap junction asymmetry may arise from heterotypic hemichannels (Phelan et al. 2008). We found that electrical synapses from Cx36‐KO neurons, but not from WT cells, often had asymmetric G j. Examples of symmetric WT and asymmetric KO junctions are shown in Fig. 3 A. In each coupled cell pair, G j was measured in each direction and a G j ratio was defined as G j(weaker)/G j(stronger). The G j ratio was significantly lower in junctions from KO than from WT cells (WT, mean G j ratio 0.85, n = 35; KO, mean G j ratio 0.53, n = 12; P = 0.006) (Fig. 3 B). One possible source of error in estimating G j asymmetry could be differences in the R in of coupled cell pairs in the KO. However, the R in ratio was not significantly different between WT and KO cell pairs (P = 0.81) and the G j ratios were not correlated with the R in ratios (Fig. 3 B). The enhanced G j asymmetry of KO junctions suggests that asymmetry might be related to the relative weakness of KO electrical synapses. We plotted the average junctional current for each cell in a pair against the G j ratio for that pair; even weak WT synapses with low junctional currents passed current quite symmetrically, whereas KO synapses tended to be much more asymmetric (Fig. 3 C). We conclude that non‐Cx36 electrical synapses in TRN are often asymmetric (i.e. rectifying).
Figure 3. Effect of Cx36 genotype on the asymmetry of electrical coupling in TRN .
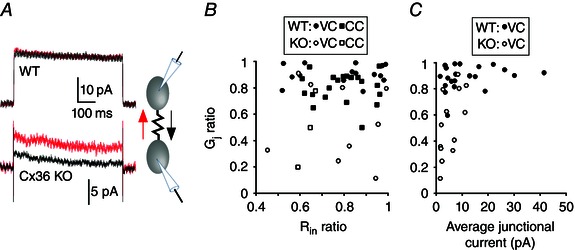
A, junctional currents induced by +80 mV and –80 mV transjunctional voltage steps (black and red traces, respectively). Junctional currents in the WT pair are similar in both directions. Junctional currents in the KO pair are direction‐dependent (i.e. asymmetric). B, relationship between junctional coupling asymmetry (G j ratio) and input resistance (R in ratio) among WT and KO neuron pairs. C, coupling asymmetry vs. mean coupling strength (pA) between WT and KO neuron pairs. Each symbol in (B) and (C) represents data from a neuron pair of the type as specified in the insets (VC, voltage clamp; CC, current clamp measurements).
Morphology and intrinsic physiology of TRN neurons
We next tested whether the absence of Cx36 in the TRN network affected the morphology and intrinsic physiology of TRN neurons. We first quantified dendritic arbors and soma surface area during early (P2–5) and later (P10–13) periods in postnatal development (Fig. 4 A) (see Methods). The complexity of dendritic arbors in TRN neurons increased during development, as indicated by an age‐dependent rise in the number of Sholl intersections (Fig. 4 B) and a larger total dendritic length (Fig. 4 C). Cx36 genotype did not significantly affect the number of primary dendrites or dendritic length (Fig. 4 C). The dendritic complexity assessed by Sholl intersection frequency was also unaffected (P2–5, P = 0.97; P10–13, P = 0.79; Kolmogorov–Smirnov test) (Fig. 4 B). At P2–5, the mean surface area of TRN somata was larger in KO cells (1961 ± 267 μm2) than in WT cells (1076 ± 153 μm2, P = 0.014), although somata did not differ significantly at older ages (P10–13, KO vs. WT, P = 0.97) (Fig. 4 C).
Figure 4. Morphology of TRN neurons by Cx36 genotype and age .
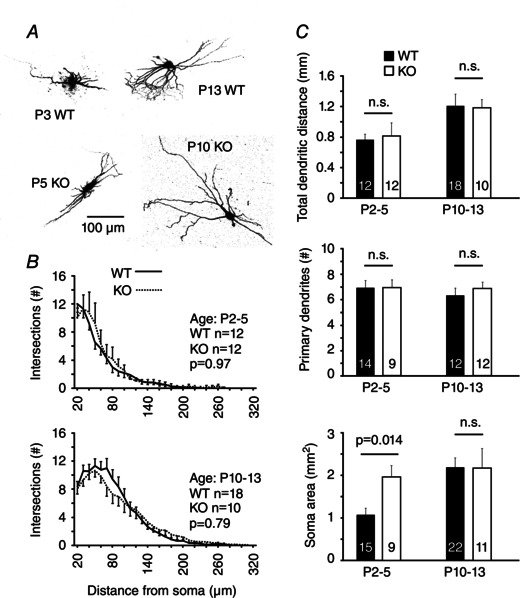
A, photomicrographs of example neurons used for morphological analysis. B, counts of Sholl intersections by distance from the soma (mean ± SEM) by genotype and age group. P values are from a Kolmogorov–Smirnov test. C, morphological characteristics determined from Sholl intersections and 3‐D reconstruction of somata. Numbers of sampled neurons for each group are indicated on the bars. n.s., not significant.
Electrical coupling among neurons can also affect their intrinsic physiological properties; in neocortical interneurons, for example, coupling accounts for around half of each cell's input conductance (the inverse of input conductance is input resistance, R in; Amitai et al. 2002). Consistent with this, we found that R in was significantly higher in KO cells than in WT cells (P < 0.001, r = 0.74, multiple linear regression) (Fig. 5 A). Between P2 and P13, the R in of TRN neurons decreased by more than five‐fold in both genotypes. Input capacitance, C in, a measure of cell surface area, increased considerably with age, which is consistent with the results of previous studies (Parker et al. 2009; Lee et al. 2010), although there was no significant difference between Cx36 genotypes (P = 0.31, r = 0.57) (Fig. 5 B). Overall, these results indicate that the input resistance is affected by the Cx36 genotype in TRN neurons and dendritic size and complexity increases dramatically with age.
Figure 5. Passive membrane properties of TRN neurons as a function of age and Cx36 genotype .

A, input resistance declined with age and was significantly higher in KO neurons (P < 0.001, r = 0.74). B, cell capacitance increased with age and was unaffected by genotype (P = 0.31, r = 0.57). P values were determined based on linear regression. A total of 305 neurons was sampled (159 WT and 146 KO neurons); n for each genotype per 2 day age group ranged from 12 to 39 neurons.
Inhibitory synaptic currents in VB relay neurons
Synaptic properties change dramatically during network development, and gap junctional coupling has often been implicated in this process (Roerig and Feller, 2000). We tested whether the synaptic inputs to VB neurons from TRN were affected by the deletion of Cx36. We first recorded GABAA‐dependent (Fig. 6 A) mIPSCs in VB cells in the presence of glutamate receptor antagonists and TTX. The mIPSC frequency increased dramatically across age until P8–9, after which it stabilized (Fig. 6 B). Cx36 genotype also affected mIPSC frequency; mIPSC frequencies were significantly higher in KO cells than in WT cells (P = 0.004, Kolmogorov–Smirnov test) (Fig. 6 B). The difference between genotypes appeared at around P4–5, although the time‐course of the age‐dependent increase in mIPSC frequency was unaffected by genotype.
Figure 6. Effects of Cx36 genotype on the properties of IPSCs in VB relay neurons .

A, examples of mIPSCs and sensitivity to picrotoxin, a GABAA receptor antagonist. B, mIPSC properties as a function of age and Cx36 genotype. mIPSC frequency is significantly higher in the KO compared to WT (P = 0.006, r = 0.53) (n = 175 neurons; 84 KO, 91 WT). The mIPSC decay rate (τIPSC, P = 0.14, r = 0.72) and amplitude (P = 0.96, r = 0.46) were not affected by genotype. Bars plot the mean ± SEM for each age and genotype group; P values are based on linear regression. C, examples of WT and KO mIPSCs compared at P2 and P13. Each trace is the average of 40 mIPSCs, with amplitudes normalized. D, mIPSC amplitude frequency histogram for P2–5 (n = 12 neurons for each genotype) and P12–13 (n = 4 neurons for each genotype). P values from the Kolmogorov–Smirnov test. E, paired‐pulse ratio of evoked IPSCs. Bar graph shows the mean ± SEM, with sample numbers specified on each bar. Inset: example trace of IPSCs from a WT cell.
The decay time constants of mIPSCs (τIPSC) became progressively faster between P0–1 and P12–13 in VB (Fig. 6 B and C), decreasing by ∼62% during this period. Cx36 genotype did not have a significant effect on τIPSC. The mIPSC amplitudes also changed with age, falling slightly after P6–9 (Fig. 6 B). Cx36 genotype influenced the distributions of mIPSC amplitudes (Fig. 6 D). At both P2–5 and P12–13, the difference in amplitude distribution was significantly shifted to larger amplitudes in the Cx36 KO (P2–5: P = 0.00001, n = 1743, n = 12 cells for each genotype; P12–13: P < 0.00001, n = 1869, n = 4 cells for each genotype; Kolmogorov–Smirnov test). These data suggest that the absence of Cx36 leads to a distribution of larger mIPSC amplitudes in VB neurons.
Evoked IPSCs were triggered with paired (100 ms interval) extracellular stimuli to test the effects of Cx36 deletion on presynaptic mechanisms. In WT‐VB neurons, the second IPSC was depressed relative to the first (paired‐pulse ratios = 0.55) (Fig. 6 E). Cx36 genotype did not have a significant effect on the paired‐pulse ratio (P = 0.96).
When recording spontaneous IPSCs simultaneously from pairs of neighbouring VB neurons (without TTX), we found that some IPSCs occurred coincidently, with onsets within ±1 ms of each other, suggesting that they may have been driven by a common presynaptic TRN neuron (Fig. 7 A). By comparing the frequency of cIPSCs in recordings to sets of randomly shuffled IPSC timings (see Methods), we found that some neuron pairs appeared to have cIPSCs above chance levels, consistent with common presynaptic inputs. At younger ages, P5–P9, almost half of WT and KO cell pairs had cIPSCs, and there was no significant difference by genotype (P = 0.34, Fisher's exact test) (Fig. 7 B). Interestingly, whereas older WT cell pairs (P10–P13) also generated cIPSCs, none of the tested KO pairs had cIPSCs (P = 0.02, Fisher's exact test) (Fig. 7 B). This result suggests that the absence of Cx36 leads to less divergence of TRN inputs onto VB neurons.
Figure 7. Coincident IPSCs from VB neuron pairs .
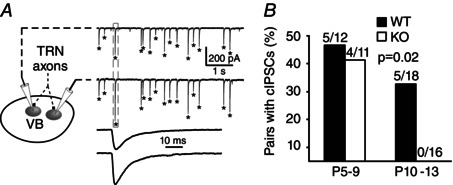
A, example of cIPSCs; asterisks indicate IPSCs that are coincident (i.e. peaks within ±1 ms of each other). The box indicates the cIPSCs shown at a fast time scale below. B, prevalence of neuron pairs with statistically significant occurrences of cIPSCs by age range (see Methods).
Taken together, greater mIPSC frequency and a lack of paired‐pulse effects implies more GABAergic release sites, and fewer cIPSCs suggests a decrease in divergent inputs on KO‐VB neurons.
Morphology and intrinsic physiology of VB neurons
We next tested whether the morphology and intrinsic physiological properties of VB neurons were affected by the deletion of Cx36. VB neurons were filled intracellularly with Alexa dye at ages P2–5 or P10–13 (Fig. 8 A). Interestingly, the frequency of Sholl intersections, and thus dendritic complexity, was significantly lower in KO neurons than WT neurons in the P10–13 age groups between 20 and 200 μm from the soma (P = 0.03, Kolmogorov–Smirnov) (Fig. 8 B), the region of greatest dendritic branching and complexity in these neurons. There was, however, no difference in intersection frequency in the younger P2–P5 age group by genotype between 20 and 140 μm from the soma (P = 0.81, Kolmogorov–Smirnov) (Fig. 8 B). The total dendritic distance measured from Sholl intersection frequency data tended to be larger in the WT than the KO but did not reach significance (P = 0.07) (Fig. 8 B). The number of primary dendrites was unaffected by genotype or age (Fig. 8 C). The 3‐D surface areas of VB somata were not significantly different by genotype (Fig. 8 C).
Figure 8. Morphology of VB neurons by Cx36 genotpye and age .
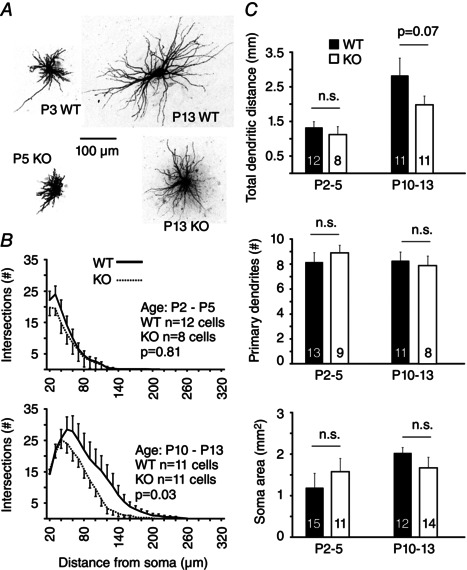
A, photomicrographs of example neurons used for morphological analysis. B, counts of Sholl intersections by distance from the soma (mean ± SEM) by genotype and age group. P values are from a Kolmogorov–Smirnov test. C, morphological characteristics determined from Sholl intersections and 3‐D reconstruction of somata. Numbers of sampled neurons for each group are indicated on the bars. n.s., not significant.
As with TRN neurons, the R in of neurons in VB sharply decreased with age (Fig. 9 A), falling by more than three‐fold; in contrast to TRN neurons, the R in of VB neurons did not differ significantly by genotype (P = 0.66, r = 0.73, multiple linear regression). C in increased considerably with age in VB cells but did not differ between Cx36 genotypes (P = 0.17, r = 0.61) (Fig. 9 B).
Figure 9. Passive membrane properties of VB neurons as a function of age and Cx36 genotype .
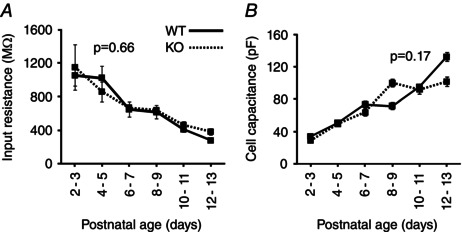
A, input resistance declined with age but was unaffected by genotype (P = 0.66, r = 0.73). B, cell capacitance increased with age but was unaffected by genotype (P = 0.17, r = 0.61). P values were determined based on linear regression. A total of 138 neurons were sampled (71 WT and 67 KO neurons); n for each genotype per 2‐day age group ranged from four to 15 neurons.
Our results indicate that the Cx36 KO genotype leads to less complex dendritic arbors of VB neurons while maintaining the same number of primary dendrites and soma size. R in decreases with age and C in increases with age, although they do not depend on the Cx36 genotype.
Discussion
The results of the present study demonstrate that the deletion of Cx36 leads to alterations in electrical coupling, neuron morphology, intrinsic physiology and inhibitory synapse functions during the early postnatal maturation of thalamic networks. This implies that electrical synapses play a significant role in the formation of inhibitory thalamic circuits.
Cx36 and electrical synapse asymmetry
The incidence of electrical coupling in the WT‐TRN remained high and steady during the first two postnatal weeks, and junctional conductance increased during this period (Fig. 1), consistent with a previous study (Parker et al. 2009). Some electrical connections remained between TRN cells in the Cx36 KO. Previous reports from our laboratory have also suggested that there is weak electrical coupling in Cx36 KO mice (Landisman et al. 2002), as well as some dye‐coupling between Cx36 KO‐TRN neurons (Lee et al. 2014). We conclude that TRN neurons can express functional gap junctions that lack Cx36, although we cannot say whether non‐Cx36 junctions are present in WT cells or occur only as a compensatory response to Cx36 deletion. In principle, the relatively low‐conductance gap junctions between KO neurons could arise from electrotonically distant connections that appear weak as a result of space clamp errors, or they may actually have reduced junctional conductance. The latter is more probable because Cx36 KO mice also have relatively small clusters of dye‐coupled TRN neurons compared to WT neurons (Lee et al. 2014). It is not clear whether electrical coupling in the KO‐TRN is relevant to network activity, as it is in the WT‐TRN (Long et al. 2004). Non‐Cx36 gap junctions might play a non‐electrical role by allowing transfer of signalling molecules among neurons (Mese et al. 2007); consistent with this, dye‐coupling is more evident than electrical coupling in the TRN of the Cx36 KO (Lee et al. 2014).
Surprisingly, we found that many electrical synapses in the KO‐TRN had asymmetric G j. Asymmetric G j (i.e. rectification) is common among coupled neurons of invertebrates and some fish (Furshpan and Potter, 1959; Auerbach and Bennett, 1969; Rash et al. 2013), although reports of rectifying junctions in the mammalian brain are few and inconsistent (Devor and Yarom, 2002; Hoge et al. 2011). Interestingly, electrical coupling asymmetry was reported between rat TRN neurons under specific physiological conditions (Haas et al. 2011) and was shown to have a substantial influence on the spike timing relationship between coupled neurons (Sevetson and Haas, 2015). Because a few cases of WT electrical coupling were modestly asymmetrical (Fig. 3 B and C), it is possible that some of the gap junctions present in the WT‐TRN are non‐Cx36‐dependent.
Asymmetric G j can occur in heterotypic gap junction channels, with each hemichannel comprised of different connexins (Barrio et al. 1992; He et al. 1999; Bukauskas et al. 2002). Heterotypic gap junctions underlie asymmetric Gj between coupled neurons of Drosophila (Phelan et al. 2008) and teleost fish (Rash et al. 2013). These results suggest that two or more connexins, neither of them Cx36, are expressed in some KO‐TRN neurons. Two possibilities are Cx45 (Sohl et al. 2005) and Cx30.2 (Kreuzberg et al. 2008; Perez et al. 2010), which are both neuronal connexins expressed in the mouse thalamus. In expression systems, Cx45 and Cx30.2 can form heterotypic and heteromeric junctions with voltage‐dependent decay kinetics intermediate between homotypic channels of either connexin alone (Kreuzberg et al. 2005; Gemel et al. 2008). These properties roughly fit the decay kinetics that we measured from electrical connections in TRN neurons of the Cx36 KO (Fig. 2 B).
Cx36 and development of passive membrane properties
During postnatal development, neuronal R in declined and C in increased in both TRN and VB, consistent with previous studies (Warren and Jones, 1997; Parker et al. 2009; Lee et al. 2010). The mean R in of TRN cells dropped six‐fold between P2 and P13, possibly as a result of the addition of membrane leak channels and increased gap junctional conductance. The mean R in of TRN neurons from KO mice was higher than that of WT cells. The presence of gap junctions tends to reduce R in in other types of neurons (Deans et al. 2001; Amitai et al. 2002; Long et al. 2002). Similar differences of R in between WT and Cx36 KO‐TRN neurons were reported by Landisman et al. (2002) in P14–P18 mice. The electrical synapses among KO TRN neurons probably do not significantly contribute to input conductance. R in of VB neurons was not influenced by Cx36 genotype, which is consistent with their weak electrical coupling postnatally (Lee et al. 2010).
The absence of most gap junctions among KO TRN neurons could have a direct effect on thalamic network function during development. A higher R in amplifies voltage responses to synaptic inputs and lengthens the membrane time constant, which affects temporal and spatial integration and attenuates high‐frequency events. This could be particularly important during development because network maturation depends strongly on the strength, and perhaps synchrony, of electrical activity (Catalano and Shatz, 1998; Dantzker and Callaway, 1998; Zhang and Poo, 2001).
Cx36 and development of inhibition and neuronal morphology
Synaptogenesis commences before birth in the mouse thalamus, and network formation continues for several weeks thereafter (Jones, 2007). We found significant increases in mIPSC frequency during the first postnatal week (Fig. 6 B), which probably reflects rapid addition of GABA‐releasing synapses. The mIPSC frequency levels out by P8, presumably as synaptogenesis ends; this is around the time when thalamic spindle oscillations begin to mature (Warren and Jones, 1997). Age also correlated strongly with mIPSC decay rate (Fig. 6 B), probably as a result of the changing subunit composition of GABAA receptors (Laurie et al. 1992). The decay rate of IPSCs in VB is an important determinant of the frequency of spindle oscillations (Bal et al. 1995; Sohal et al. 2006). Deletion of Cx36 significantly increased mIPSC frequency in VB. This could be caused by more GABAergic synapses or increased spontaneous GABA release. It is also possible that increased dendritic length constants would allow distal IPSCs to propagate to somata more readily (Carnevale and Johnston, 1982). Because the evoked inhibitory paired‐pulse ratio was not affected by genotype (Fig. 6 E), and because the mIPSC frequency did not correlate with R in (which may relate to dendro‐somatic electrotonic coupling; data not shown), release probability or R in probably do not account for this result (Prange and Murphy, 1999). We suggest that the deletion of Cx36 leads to an increased number of GABAergic release sites.
Both Cx36 and age affected the distribution of mIPSC amplitudes in VB neurons (Fig. 6 D), which suggests alterations of synaptic vesicle size or neurotransmitter concentration, or the density of postsynaptic GABAA receptors. In general, there were fewer large mIPSCs in older cells. Overall, there is more inhibition in the thalamus of the Cx36 KO than in WT thalamus, probably resulting from homeostatic changes as a result of altered synaptic and network activity in the Cx36 KO.
We also found that Cx36 affected the development of neuron morphology. Most notably, VB dendritic fields were more complex in the WT compared to the KO, and TRN somata were larger in the P2–5 KO‐TRN compared to the P2–5 WT‐TRN. Interestingly, previous work on inferior olivary neurons showed that Cx36 KO alters dendritic thickness (De Zeeuw et al. 2003), perhaps implying a role for Cx36 in the maturation of neuron morphology.
The mechanisms driving these changes are unclear. Gap junctions may provide cell–cell adhesion (Elias et al. 2007), although the sparseness of coupling among developing VB neurons (Lee et al. 2010) argues against this. Gap junctions might pass morphologically important signalling molecules, although the non‐Cx36 coupling among KO cells could provide such a role. Cell morphology can also be affected by neural activity (Parrish et al. 2007) and reduced electrical coupling could change firing rates or synchrony.
We found that Cx36 deletion leads to fewer VB neuron pairs with coincident IPSCs (Fig. 7 B), which suggests fewer divergent inputs from TRN. Less complex dendritic trees in KO‐VB neurons would reduce the ability of neighbouring neurons to capture inputs from a common presynaptic axon, and thus could lead to smaller receptive fields in VB. Less divergent input would also tend to reduce the influence of each TRN neuron on the VB neuron population, and therefore produce more specific feedforward and feedback inhibition in the thalamocortical network. Cx36 KO mice exhibit impaired function in cognitive and behavioural tasks (Allen et al. 2011; Zlomuzica et al. 2012), perhaps in part because of abnormal inhibitory synaptic properties. Taken together, our data strongly suggest that Cx36‐dependent electrical coupling is necessary for the normal development of thalamic inhibitory networks.
Regulation of chemical synapse development by electrical synapses
Cx36 is important for regulating synchronized activity in central networks (Gibson et al. 1999; Landisman et al. 2002; Buhl et al. 2003; Long et al. 2004, 2005; Mancilla et al. 2007). Network synchronization during development may contribute to the stabilization of chemical synaptic connections by co‐ordinating the activity of neighbouring synapses (Personius and Balice‐Gordon, 2000). Desynchronization as a result of the loss of Cx36 might lead to more competition and pruning among synapses (Kim and Kandler, 2003). The TRN generates spontaneous activity during development, which has been implicated in chemical synaptic development in the thalamus (Pangratz‐Fuehrer et al. 2007). Our findings suggest that the Cx36 KO has less divergent input from the TRN to VB neurons, perhaps because of desynchronized TRN activity and the consequent pruning of its inputs onto VB neurons. Gap junctions in the early postnatal TRN may contribute to patterns of spontaneous activity that support the subsequent maturation of chemical synapses in the thalamus.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
TAZ and BWC conceived and designed the experiments. TAZ and BWC collected, assembled, analysed and interpreted the data. All experiments were performed in the laboratory of BWC at Brown University. TAZ and BWC drafted the article or revised it critically for important intellectual content. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This research was supported by grants from the NIH: NRSA grant NS060448 (to TAZ), NS050434 (to BWC); and from the NSF: EFRI‐0937848 (to BWC).
Acknowledgements
We thank Saundy Patrick for excellent technical assistance and Omar Ahmed for helping with the Matlab code.
References
- Agmon A & Connors BW (1991). Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41, 365–379. [DOI] [PubMed] [Google Scholar]
- Allen K, Fuchs EC, Jaschonek H, Bannerman DM & Monyer H (2011). Gap junctions between interneurons are required for normal spatial coding in the hippocampus and short‐term spatial memory. J Neurosci 31, 6542–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai Y, Gibson JR, Beierlein M, Patrick SL, Ho AM, Connors BW & Golomb D (2002). The spatial dimensions of electrically coupled networks of interneurons in the neocortex. J Neurosci 22, 4142–4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam H, Liu X, Colombo PJ, Corriveau RA & Belousov AB (2005). NMDA receptors regulate developmental gap junction uncoupling via CREB signaling. Nat Neurosci 8, 1720–1726. [DOI] [PubMed] [Google Scholar]
- Auerbach AA & Bennett MV (1969). A rectifying electrotonic synapse in the central nervous system of a vertebrate. J Gen Physiol 53, 211–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal T, von Krosigk M & McCormick DA (1995). Synaptic and membrane mechanisms underlying synchronized oscillations in the ferret lateral geniculate nucleus in vitro. J Physiol 483 (Pt 3), 641–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrio LC, Suchyna T, Bargiello T, Xu LX, Roginski RS, Bennett MV & Nicholson BJ (1992). Gap junctions formed by connexins 26 and 32 alone and in combination are differently affected by applied voltage. Proc Natl Acad Sci USA 88, 8410–8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluardo N, Mudo G, Trovato‐Salinaro A, Le Gurun S, Charollais A, Serre‐Beinier V, Amato G, Haefliger JA, Meda P & Condorelli DF (2000). Expression of connexin36 in the adult and developing rat brain. Brain Res 865, 121–138. [DOI] [PubMed] [Google Scholar]
- Bennett MV (1966). Physiology of electrotonic junctions. Ann NY Acad Sci 137, 509–539. [DOI] [PubMed] [Google Scholar]
- Bennett MV & Zukin RS (2004). Electrical coupling and neuronal synchronization in the mammalian brain. Neuron 41, 495–511. [DOI] [PubMed] [Google Scholar]
- Blankenship AG, Hamby AM, Firl A, Vyas S, Maxeiner S, Willecke K & Feller MB (2011). The role of neuronal connexins 36 and 45 in shaping spontaneous firing patterns in the developing retina. J Neurosci 31, 9998–10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhl DL, Harris KD, Hormuzdi SG, Monyer H & Buzsaki G (2003). Selective impairment of hippocampal gamma oscillations in connexin‐36 knock‐out mouse in vivo. J Neurosci 23, 1013–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukauskas FF, Angele AB, Verselis VK & Bennett MV (2002). Coupling asymmetry of heterotypic connexin 45/ connexin 43‐EGFP gap junctions: properties of fast and slow gating mechanisms. Proc Natl Acad Sci USA 99, 7113–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukauskas FF, Kreuzberg MM, Rackauskas M, Bukauskiene A, Bennett MV, Verselis VK & Willecke K (2006). Properties of mouse connexin 30.2 and human connexin 31.9 hemichannels: implications for atrioventricular conduction in the heart. Proc Natl Acad Sci USA 103, 9726–9731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnevale NT & Johnston D (1982). Electrophysiological characterization of remote chemical synapses. J Neurophysiol 47, 606–621. [DOI] [PubMed] [Google Scholar]
- Catalano SM & Shatz CJ (1998). Activity‐dependent cortical target selection by thalamic axons. Science 281, 559–562. [DOI] [PubMed] [Google Scholar]
- Connors BW, Benardo LS & Prince DA (1983). Coupling between neurons of the developing rat neocortex. J Neurosci 3, 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors BW & Long MA (2004). Electrical synapses in the mammalian brain. Annu Rev Neurosci 27, 393–418. [DOI] [PubMed] [Google Scholar]
- Dantzker JL & Callaway EM (1998). The development of local, layer‐specific visual cortical axons in the absence of extrinsic influences and intrinsic activity. J Neurosci 18, 4145–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zeeuw CI, Chorev E, Devor A, Manor Y, Van Der Giessen RS, De Jeu MT, Hoogenraad CC, Bijman J, Ruigrok TJ, French P, Jaarsma D, Kistler WM, Meier C, Petrasch‐Parwez E, Dermietzel R, Sohl G, Gueldenagel M, Willecke K & Yarom Y (2003). Deformation of network connectivity in the inferior olive of connexin 36‐deficient mice is compensated by morphological and electrophysiological changes at the single neuron level. J Neurosci 23, 4700–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans MR, Gibson JR, Sellitto C, Connors BW & Paul DL (2001). Synchronous activity of inhibitory networks in neocortex requires electrical synapses containing connexin36. Neuron 31, 477–485. [DOI] [PubMed] [Google Scholar]
- Devor A & Yarom Y (2002). Electrotonic coupling in the inferior olivary nucleus revealed by simultaneous double patch recordings. J Neurophysiol 87, 3048–3058. [DOI] [PubMed] [Google Scholar]
- Elias LA, Wang DD & Kriegstein AR (2007). Gap junction adhesion is necessary for radial migration in the neocortex. Nature 448, 901–907. [DOI] [PubMed] [Google Scholar]
- Fischbach GD (1972). Synapse formation between dissociated nerve and muscle cells in low density cell cultures. Dev Biol 28, 407–429. [DOI] [PubMed] [Google Scholar]
- Froemke RC (2015). Plasticity of cortical excitatory–inhibitory balance. Annu Rev Neurosci 38, 195–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furshpan EJ & Potter DD (1959). Transmission at the giant motor synapses of the crayfish. J Physiol 145, 289–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemel J, Lin X, Collins R, Veenstra RD & Beyer EC (2008). Cx30.2 can form heteromeric gap junction channels with other cardiac connexins. Biochem Biophys Res Commun 369, 388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Beierlein M & Connors BW (1999). Two networks of electrically coupled inhibitory neurons in neocortex. Nature 402, 75–79. [DOI] [PubMed] [Google Scholar]
- Harris AL (2001). Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys 34, 325–472. [DOI] [PubMed] [Google Scholar]
- Haas JS, Zavala B & Landisman CE (2011). Activity‐dependent long‐term depression of electrical synapses. Science 334, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He DS, Jiang JX, Taffet SM & Burt JM (1999). Formation of heteromeric gap junction channels by connexins 40 and 43 in vascular smooth muscle cells. Proc Natl Acad Sci USA 96, 6495–6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK & Fagiolini M (2005). Excitatory–inhibitory balance and critical period plasticity in developing visual cortex. Prog Brain Res 147, 115–124. [DOI] [PubMed] [Google Scholar]
- Hoge GJ, Davidson KG, Yasumura T, Castillo PE, Rash JE & Pereda AE (2011). The extent and strength of electrical coupling between inferior olivary neurons is heterogeneous. J Neurophysiol 105, 1089–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormuzdi SG, Pais I, LeBeau FE, Towers SK, Rozov A, Buhl EH, Whittington MA & Monyer H (2001). Impaired electrical signaling disrupts gamma frequency oscillations in connexin 36‐deficient mice. Neuron 31, 487–495. [DOI] [PubMed] [Google Scholar]
- Huguenard JR & McCormick DA (2007). Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci 30, 350–356. [DOI] [PubMed] [Google Scholar]
- Huntsman MM & Huguenard JR (2000). Nucleus‐specific differences in GABA(A)‐receptor‐mediated inhibition are enhanced during thalamic development. J Neurophysiol 83, 350–358. [DOI] [PubMed] [Google Scholar]
- Jaslove SW & Brink PR (1986). The mechanism of rectification at the electrotonic motor giant synapse of the crayfish. Nature 323(6083), 63–65. [DOI] [PubMed] [Google Scholar]
- Jones EG (2007). The Thalamus, 2nd edn Cambridge: Cambridge University Press. [Google Scholar]
- Kandler K & Katz LC (1995). Neuronal coupling and uncoupling in the developing nervous system. Curr Opin Neurobiol 5, 98–105. [DOI] [PubMed] [Google Scholar]
- Kandler K & Katz LC (1998. a). Coordination of neuronal activity in developing visual cortex by gap junction‐mediated biochemical communication. J Neurosci 18, 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandler K & Katz LC (1998. b). Relationship between dye coupling and spontaneous activity in developing ferret visual cortex. Dev Neurosci 20, 59–64. [DOI] [PubMed] [Google Scholar]
- Kätzel D & Miesenböck G (2014). Experience‐dependent rewiring of specific inhibitory connections in adult neocortex. PLoS Biol 12, e1001798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G & Kandler K (2003). Elimination and strengthening of glycinergic/GABAergic connections during tonotopic map formation. Nat Neurosci 6, 282–290. [DOI] [PubMed] [Google Scholar]
- Koval M, Molina SA & Burt JM (2014). Mix and match: investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett 588, 1193–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzberg MM, Sohl G, Kim JS, Verselis VK, Willecke K & Bukauskas FF (2005). Functional properties of mouse connexin30.2 expressed in the conduction system of the heart. Circ Res 96, 1169–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzberg MM, Deuchars J, Weiss E, Schober A, Sonntag S, Wellershaus K, Draguhn A & Willecke K (2008). Expression of connexin30.2 in interneurons of the central nervous system in the mouse. Mol Cell Neurosci 37, 119–134. [DOI] [PubMed] [Google Scholar]
- Landisman CE, Long MA, Beierlein M, Deans MR, Paul DL & Connors BW (2002). Electrical synapses in the thalamic reticular nucleus. J Neurosci 22, 1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie DJ, Wisden W & Seeburg PH (1992). The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J Neurosci 12, 4151–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Cruikshank SJ & Connors BW (2010). Electrical and chemical synapses between relay neurons in developing thalamus. J Physiol 588, 2403–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Patrick SL, Richardson, KA & Connors BW (2014). Two functionally distinct networks of gap junction‐coupled inhibitory neurons in the thalamic reticular nucleus. J Neurosci 34, 13170–13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Friedberg MH & Ebner FF (1994). The role of GABA‐mediated inhibition in the rat ventral posteriormedial thalamus. I. Assessment of receptive field changes following thalamic reticular nucleus lesions. J Neurophysiol 71, 1702–1715. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu H, Cheng PL, Ge S, Xu H, Shi SH & Dan Y (2012). Clonally related visual cortical neurons show similar stimulus feature selectivity. Nature 486, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XB & Jones EG (2003). Fine structural localization of connexin‐36 immunoreactivity in mouse cerebral cortex and thalamus. J Comp Neurol 466, 457–467. [DOI] [PubMed] [Google Scholar]
- Long MA, Landisman CE & Connors BW (2004). Small clusters of electrically coupled neurons generate synchronous rhythms in the thalamic reticular nucleus. J Neurosci 24, 341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MA, Deans MR, Paul DL & Connors BW (2002). Rhythmicity without synchrony in the electrically uncoupled inferior olive. J Neurosci 22, 10898–10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MA, Jutras MJ, Connors BW & Burwell RD (2005). Electrical synapses coordinate activity in the suprachiasmatic nucleus. Nat Neurosci 8, 61–66. [DOI] [PubMed] [Google Scholar]
- Maher BJ, McGinley MJ & Westbrook GL (2009). Experience‐dependent maturation of the glomerular microcircuit. Proc Natl Acad Sci USA 106, 16865–16870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancilla JG, Lewis TJ, Pinto DJ, Rinzel J & Connors BW (2007). Synchronization of electrically coupled pairs of inhibitory interneurons in neocortex. J Neurosci 27, 2058–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis GZ, Diaz E, Moran LB & Navarrete R (2002). Increased incidence of gap junctional coupling between spinal motoneurones following transient blockade of NMDA receptors in neonatal rats. J Physiol 544, 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mese G, Richard G & White TW (2007). Gap junctions: basic structure and function. J Invest Dermatol 127, 2516–2524. [DOI] [PubMed] [Google Scholar]
- Montoro RJ & Yuste R (2004). Gap junctions in developing neocortex: a review. Brain Res Brain Res Rev 47, 216–226. [DOI] [PubMed] [Google Scholar]
- Moreno AP, Laing JG, Beyer EC & Spray DC (1995). Properties of gap junction channels formed of connexin 45 endogenously expressed in human hepatoma (SKHep1) cells. Am J Physiol Cell Physiol 268, C356–C365. [DOI] [PubMed] [Google Scholar]
- Neunuebel JP & Zoran MJ (2005). Electrical synapse formation disrupts calcium‐dependent exocytosis, but not vesicle mobilization. Synapse 56, 154–165. [DOI] [PubMed] [Google Scholar]
- Niculescu D & Lohmann C (2013). Gap junctions in developing thalamic and neocortical neuronal networks. Cereb Cortex 24, 3097–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangratz‐Fuehrer S, Rudolph U & Huguenard JR (2007). Giant spontaneous depolarizing potentials in the developing thalamic reticular nucleus. J Neurophysiol 97, 2364–2372. [DOI] [PubMed] [Google Scholar]
- Parker PR, Cruikshank SJ & Connors BW (2009). Stability of electrical coupling despite massive developmental changes of intrinsic neuronal physiology. J Neurosci 29, 9761–9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish JZ, Emoto K, Kim MD & Jan YN (2007). Mechanisms that regulate establishment, maintenance, and remodeling of dendritic fields. Annu Rev Neurosci 30, 399–423. [DOI] [PubMed] [Google Scholar]
- Pereda AE (2014). Electrical synapses and their functional interactions with chemical synapses. Nat Rev Neurosci 15, 250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez DESML, Dedek K, Janssen‐Bienhold U, Meyer A, Kreuzberg MM, Lorenz S, Willecke K & Weiler R (2010). Expression and modulation of connexin30.2, a novel gap junction protein in the mouse retina. Vis Neurosci 27, 91–101. [DOI] [PubMed] [Google Scholar]
- Personius KE & Balice‐Gordon RJ (2000). Activity‐dependent editing of neuromuscular synaptic connections. Brain Res Bull 53, 513–522. [DOI] [PubMed] [Google Scholar]
- Personius KE, Chang Q, Mentis GZ, O'Donovan MJ & Balice‐Gordon RJ (2007). Reduced gap junctional coupling leads to uncorrelated motor neuron firing and precocious neuromuscular synapse elimination. Proc Natl Acad Sci USA 104, 11808–11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan P, Goulding LA, Tam JL, Allen MJ, Dawber RJ, Davies JA & Bacon JP (2008). Molecular mechanism of rectification at identified electrical synapses in the Drosophila giant fiber system. Curr Biol 18, 1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D (2004). The thalamic reticular nucleus: structure, function and concept. Brain Res Brain Res Rev 46, 1–31. [DOI] [PubMed] [Google Scholar]
- Prange O & Murphy TH (1999). Correlation of miniature synaptic activity and evoked release probability in cultures of cortical neurons. J Neurosci 19, 6427–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash JE, Curti S, Vanderpool KG, Kamasawa N, Nannapaneni S, Palacios‐Prado N, Flores CE, Yasumura T, O'Brien J, Lynn BD, Bukauskas FF, Nagy JI & Pereda AE (2013). Molecular and functional asymmetry at a vertebrate electrical synapse. Neuron 79, 957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roerig B & Feller MB (2000). Neurotransmitters and gap junctions in developing neural circuits. Brain Res Brain Res Rev 32, 86–114. [DOI] [PubMed] [Google Scholar]
- Sevetson J & Haas JS (2015). Asymmetry and modulation of spike timing in electrically coupled neurons. J Neurophysiol 113, 1743–1751. [DOI] [PubMed] [Google Scholar]
- Sholl DA (1953). Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat 87, 387–406. [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Pangratz‐Fuehrer S, Rudolph U & Huguenard JR (2006). Intrinsic and synaptic dynamics interact to generate emergent patterns of rhythmic bursting in thalamocortical neurons. J Neurosci 26, 4247–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohl G, Maxeiner S & Willecke K (2005). Expression and functions of neuronal gap junctions. Nat Rev Neurosci 6, 191–200. [DOI] [PubMed] [Google Scholar]
- Sutor B & Hagerty T (2005). Involvement of gap junctions in the development of the neocortex. Biochim Biophys Acta 1719, 59–68. [DOI] [PubMed] [Google Scholar]
- Szabo TM & Zoran MJ (2007). Transient electrical coupling regulates formation of neuronal networks. Brain Res 1129, 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo TM, Faber DS & Zoran MJ (2004). Transient electrical coupling delays the onset of chemical neurotransmission at developing synapses. J Neurosci 24, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teubner B, Degen J, Sohl G, Guldenagel M, Bukauskas FF, Trexler EB, Verselis VK, De Zeeuw CI, Lee CG, Kozak CA, Petrasch‐Parwez E, Dermietzel R & Willecke K (2000). Functional expression of the murine connexin 36 gene coding for a neuron‐specific gap junctional protein. J Membr Biol 176, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd KL, Kristan WB, Jr. & French KA (2010). Gap junction expression is required for normal chemical synapse formation. J Neurosci 30, 15277–15285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rijen HV, Wilders R, Van Ginneken AC & Jongsma HJ (1998). Quantitative analysis of dual whole‐cell voltage‐clamp determination of gap junctional conductance. Pflügers Arch 436, 141–151. [DOI] [PubMed] [Google Scholar]
- Warren RA & Jones EG (1997). Maturation of neuronal form and function in a mouse thalamo‐cortical circuit. J Neurosci 17, 277–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Guldenagel M, Deutsch U & Sohl G (2002). Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem 383, 725–737. [DOI] [PubMed] [Google Scholar]
- Yu YC, He S, Chen S, Fu Y, Brown KN, Yao XH, Ma J, Gao KP, Sosinsky GE, Huang K & Shi SH (2012). Preferential electrical coupling regulates neocortical lineage‐dependent microcircuit assembly. Nature 486, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LI & Poo MM (2001). Electrical activity and development of neural circuits. Nat Neurosci 4 (Suppl), 1207–1214. [DOI] [PubMed] [Google Scholar]
- Zlomuzica A, Viggiano D, Degen J, Binder S, Ruocco LA, Sadile AG, Willecke K, Huston JP & Dere E (2012). Behavioral alterations and changes in Ca/calmodulin kinase II levels in the striatum of connexin36 deficient mice. Behav Brain Res 226, 293–300. [DOI] [PubMed] [Google Scholar]