

Abstract
Key points
Increases in intracellular Zn2+ concentrations are an early, necessary signal for the modulation of Kv2.1 K+ channel localization and physiological function.
Intracellular Zn2+‐mediated Kv2.1 channel modulation is dependent on calcineurin, a Ca2+‐activated phosphatase.
We show that intracellular Zn2+ induces a significant increase in ryanodine receptor‐dependent cytosolic Ca2+ transients, which leads to a calcineurin‐dependent redistribution of Kv2.1 channels from pre‐existing membrane clusters to diffuse localization. As such, the link between Zn2+ and Ca2+ signalling in this Kv2.1 modulatory pathway is established.
We observe that a sublethal ischaemic preconditioning insult also leads to Kv2.1 redistribution in a ryanodine receptor‐dependent fashion.
We suggest that Zn2+ may be an early and ubiquitous signalling molecule mediating Ca2+ release from the cortical endoplasmic reticulum via ryanodine receptor activation.
Abstract
Sublethal injurious stimuli in neurons induce transient increases in free intracellular Zn2+ that are associated with regulating adaptive responses to subsequent lethal injury, including alterations in the function and localization of the delayed‐rectifier potassium channel, Kv2.1. However, the link between intracellular Zn2+ signalling and the observed changes in Kv2.1 remain undefined. In the present study, utilizing exogenous Zn2+ treatment, along with a selective Zn2+ ionophore, we show that transient elevations in intracellular Zn2+ concentrations are sufficient to induce calcineurin‐dependent Kv2.1 channel dispersal in rat cortical neurons in vitro, which is accompanied by a relatively small but significant hyperpolarizing shift in the voltage‐gated activation kinetics of the channel. Critically, using a molecularly encoded calcium sensor, we found that the calcineurin‐dependent changes in Kv2.1 probably occur as a result of Zn2+‐induced cytosolic Ca2+ release via activation of neuronal ryanodine receptors. Finally, we couple this mechanism with an established model for in vitro ischaemic preconditioning and show that Kv2.1 channel modulation in this process is also ryanodine receptor‐sensitive. Our results strongly suggest that intracellular Zn2+‐initiated signalling may represent an early and possibly widespread component of Ca2+‐dependent processes in neurons.
Key points
Increases in intracellular Zn2+ concentrations are an early, necessary signal for the modulation of Kv2.1 K+ channel localization and physiological function.
Intracellular Zn2+‐mediated Kv2.1 channel modulation is dependent on calcineurin, a Ca2+‐activated phosphatase.
We show that intracellular Zn2+ induces a significant increase in ryanodine receptor‐dependent cytosolic Ca2+ transients, which leads to a calcineurin‐dependent redistribution of Kv2.1 channels from pre‐existing membrane clusters to diffuse localization. As such, the link between Zn2+ and Ca2+ signalling in this Kv2.1 modulatory pathway is established.
We observe that a sublethal ischaemic preconditioning insult also leads to Kv2.1 redistribution in a ryanodine receptor‐dependent fashion.
We suggest that Zn2+ may be an early and ubiquitous signalling molecule mediating Ca2+ release from the cortical endoplasmic reticulum via ryanodine receptor activation.
Abbreviations
- cER
cortical endoplasmic reticulum
- Dan
dantrolene; FK520, ascomycin
- GFP
green fluorescent protein
- MCT
multiple comparison test
- MHB
MEM‐HEPES‐BSA
- RCaMP‐h
red genetically‐encoded calcium indicator protein
- ROI
region of interest
- RyR
ryanodine receptor
- TPEN
N,N,N',N'‐tetrakis(2‐pyridylmethyl)ethane‐1,2‐diamine
- ZnPyr
zinc‐pyrithione
Introduction
Intracellular Zn2+ signalling is a critical regulatory component of both neurotoxic and neuroprotective cell signalling pathways. In neurons, intracellular Zn2+ concentrations transiently increase following exposure to a sublethal ischaemic insult (Frederickson et al. 2005; Aras et al. 2009 b), mediating adaptive responses to subsequent excitotoxic injury. For example, chemical ischaemic preconditioning, where a sublethal ischaemic insult renders neurons resistant to subsequent lethal insults, relies heavily on intracellular Zn2+‐mediated signalling. Indeed, chelation of intracellular Zn2+ during ischaemic preconditioning attenuates the resulting neuroprotective cascade and restores cell susceptibility to excitotoxic death (Aras et al. 2009 b). Conversely, lethal excitotoxic insults in neurons elicit a delayed, sustained increase in free Zn2+ that promotes cell death (Aras et al. 2009 b; Granzotto and Sensi, 2015). Chelation of this delayed free Zn2+ surge supports cell survival. Taken together, these findings implicate intracellular Zn2+ as a regulatory component of both neuroprotective and neurodestructive cellular responses to injury (Sensi et al. 2011).
Injury‐induced adaptive increases in intracellular free Zn2+ are also associated with (and indeed required for) modulation of the neuronal delayed‐rectifying voltage‐gated K+ channel, Kv2.1 (Aras et al. 2009 a), which localizes to highly organized somato‐dendritic cell membrane‐surface clusters under normal conditions (Lim et al. 2000; Misonou et al. 2004; O'Connell et al. 2006; Scannevin et al. 1996; Tamkun et al. 2007). Chemical ischaemic preconditioning and other injurious stimuli have been shown to alter Kv2.1 in three key modalities: (i) they induce Ca2+/calmodulin‐activated calcineurin‐dependent dephosphorylation of the channel at multiple intracellular residues, mostly localized to the C‐terminus; (ii) they induce a hyperpolarizing shift in the channel's voltage‐gated activation kinetics; and (iii) they alter the distribution of Kv2.1 channels from highly localized cell membrane clusters (Lim et al. 2000; Misonou et al. 2004; O'Connell et al. 2006; Scannevin et al. 1996; Tamkun et al. 2007) to a more diffuse distribution throughout the neuronal plasma membrane (Du et al. 2000; Misonou et al. 2004; Misonou et al. 2005 a; Mulholland et al. 2008; Aras et al. 2009 a; Baver and O'Connell, 2012; Shepherd et al. 2012; Shah and Aizenman, 2014). Zn2+ chelation during chemical ischaemic preconditioning blocks Kv2.1 channel declustering and changes in voltage dependency (Aras et al. 2009 a), suggesting a critical role for Zn2+ signalling in Kv2.1 modulation within the context of ischaemic injury.
Intracellular Ca2+ release has also been shown to play a vital role in ischaemic preconditioning‐induced Kv2.1 modulation by activation of calcineurin and subsequent Kv2.1 dephosphorylation, leading to channel declustering (Misonou et al. 2004; Mohapatra and Trimmer, 2006; Mulholland et al. 2008; Aras et al. 2009 a; Baver and O'Connell, 2012; Shepherd et al. 2012; Shah and Aizenman, 2014). However, a mechanistic link between intracellular Zn2+ rises, Ca2+ release, calcineurin activation and Kv2.1 channel modulation has yet to be firmly established. In the present study, we uncover the probable molecular pathway linking these processes, which are intimately associated with mediating neuronal tolerance and homeostatic responses to injury. Briefly, we report that the link between intracellular Zn2+ surges and Ca2+/calcineurin‐dependent modulations of Kv2.1 localization and function comprises activation of the neuronal ryanodine receptors (RyR) by intracellular Zn2+. We also confirm that this RyR‐dependent mechanism of Kv2.1 channel modulation is involved in the established ischaemic preconditioning pathway, which has been shown to result in neuroprotection.
Methods
Ethical approval
To generate the primary cortical neuronal cultures utilized in our experiments, one timed‐pregnant, female Sprague–Dawley rat was killed each week via CO2 inhalation, followed by exsanguination, and neuronal tissue was harvested from embryos. This procedure was carried out in accordance with The University of Pittsburgh Institutional Animal Care and Use Committee and the policies and regulations outlined in ‘Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology’ (Grundy, 2015). The investigators understand the ethical principles under which the journal operates and have complied with these standards.
Cell culture, transfection and drug treatments
All experiments utilized primary cortical neurons cultured from embryonic day 17 Sprague–Dawley rats of either sex (Hartnett et al. 1997). Transfections were performed 21–25 days in vitro using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) (Aras et al. 2009 b). Briefly, neuronal cultures were treated with 1.5 μg of total DNA, 2 μl of Lipofectamine 2000 and 100 μl of Opti‐MEM (Gibco, Grand Island, NY, USA) per well. Cells were utilized for experimentation ∼18–24 h following transfection. Treatment solutions were prepared in modified Minimal Essential Media (Life Technologies, Carlsbad, CA, USA), which contained 25 mm Hepes and 0.01% BSA (MHB). MHB media was utilized for both treatments and imaging to avoid media change‐triggered Kv2.1 declustering (Fox et al. 2015).
Confocal imaging
For analysis of transfected Kv2.1‐GFP channel distribution, neuronal cultures were imaged on a A1+ confocal microscope (Nikon, Tokyo, Japan) at 60× magnification. Between five and 15 optical sections (0.5 μm) were obtained to generate a maximum intensity projection image for visualizing Kv2.1 surface clusters as described earlier (Shah et al. 2014). Utilizing Nikon Instruments Software Elements Advanced Research (NIS‐Elements AR) analysis, the object count feature was customized to analyse metrics on Kv2.1 clusters. Object count parameters defined a Kv2.1 cluster as an area of high intensity green fluorescent protein (GFP) signal (compared to background) measuring 0.05 μm2 or larger (Shah et al. 2014). NIS‐Elements AR was also utilized to measure the somatic area of each neuron to calculate a normalized value of Kv2.1 clusters/μm2 of neuronal soma.
Calcium imaging
To test the capability of intracellular Zn2+ to induce cytosolic Ca2+ release, neuronal cultures were transfected with a GFP‐n1 construct (85% of total DNA) to visualize neurons in their entirety, as well as a genetically‐encoded Ca2+ sensor, red genetically‐encoded calcium indicator protein (RCaMP‐h) (15% of total DNA) to measure intracellular Ca2+ release signals. Upon translation, RCaMP‐h binds intracellular Ca2+ ions with high affinity and emits red fluorescence when excited by 561 nm light (Akerboom et al. 2013). Thus, RCaMP‐h fluorescence measurements provided visual confirmation of intracellular Ca2+ events. Eighteen to 24 h post‐transfection, neuronal cultures on glass coverslips were loaded into an imaging chamber with 0.5 ml of MHB. Neurons were imaged on a A1+ confocal microscope at 60× magnification. Cells were imaged continuously over a total period of ∼20 min, and drug solutions were directly infused into the imaging chamber following baseline fluorescence measurements for ∼10 min. During experiments, both RCaMP‐h and GFP fluorescence measurements from the neuronal soma were recorded with respect to time. Increases in red light emission, relative to GFP fluorescence emission, correlated with RCaMP‐h‐Ca2+ binding, indicating that cytosolic Ca2+ had been liberated near the transfected protein. Traces (e.g. Fig. 4) are shown as the ratio of background‐corrected RCaMP‐h/GFP fluorescence vs. time. To analyse Ca2+ event frequency, the first derivative plot of each RCaMP‐h/GFP trace was obtained using MatLab (MathWorks Inc., Natick, MA, USA), eliminating major background fluctuations in RCaMP‐h fluorescence. A period of baseline quiescence (15–20 s) was defined for the 10 min segments prior to and following drug infusion. A Ca2+ transient was considered as an area on the first derivative plot that spanned 3 SDs above and below the established baseline level, indicating a rise and fall of the RCaMP‐h emission signal slope.
Figure 4. Sublethal surges in intracellular Zn2+ concentration trigger an increase in cytosolic Ca2+ release via neuronal RyR modulation .
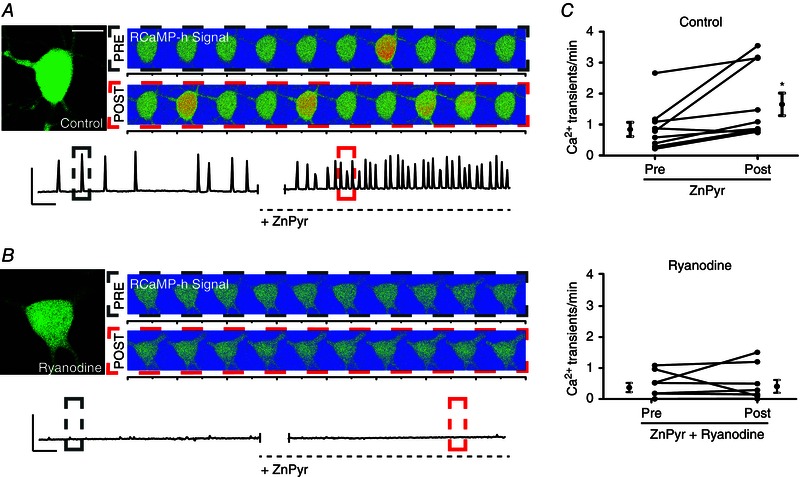
A, cortical neuron transfected with GFP‐n1 and RCaMP‐h plasmid. Left: GFP‐only image; scale bar = 10 μm. Right: two sets of images are shown representative of 40 s segments of RCaMP‐h fluorescence measurements in a representative neuron, correlating with cytosolic Ca2+ binding to expressed intracellular RCaMP‐h protein. Images are shown with a rainbow‐contrast filter to increase visibility of live intracellular Ca2+ release events (red‐orange surges). The top set of images represents Ca2+ events pre‐ZnPyr (30 μm ZnCl2, 300 nm pyrithione) infusion, whereas the bottom set reflects Ca2+ events post‐ZnPyr addition. Below: a trace that reflects background‐subtracted fluorescence measurements of RCaMP‐h normalized to GFP fluorescence vs. time in the given neuronal ROI, both before and after ZnPyr addition. Colour‐coded dotted‐line boxes (grey, PRE; red, POST) on trace segments correlate with the rainbow‐contrast images above. Scale bars: vertical = 0.1 RU; horizontal = 60 s. B, structure is identical to Fig. 4 A; neurons were pre and co‐treated with ryanodine (15 μm) during ZnPyr treatment. C, before and after plots, showing the number of Ca2+ transients min–1 for ∼10 min before and after ZnPyr‐treatment in both control and ryanodine‐supplemented conditions, as well as the pooled mean ± SEM of the data (Pre‐ZnPyr, 0.84 ± 0.23, n = 10; Post‐ZnPyr, 1.65 ± 0.37, n = 10; Pre‐ZnPyr + ryanodine, 0.43 ± 0.15, n = 8; Post‐ZnPyr + ryanodine, 0.47 ± 0.20, n = 8). Each paired data set (pre‐/post‐treatment) is analysed via a paired t test (two‐tailed) (*P < 0.05).
Zinc imaging
To confirm that Zn2+ permeated the neuronal membrane during zinc‐pyrithione (ZnPyr) treatment (30 μm ZnCl2, 300 nm pyrithione), a highly selective, cell‐permeant, Zn2+‐sensitive molecular probe, FluoZin3‐AM (Life Technologies, Carlsbad, CA, USA) was utilized. Neurons were first loaded with FluoZin3‐AM (10 μm for 30 min) prepared in a Hepes buffered salt solution (144 mm NaCl, 3 nm KCl, 10 mm Hepes, 5.5 mm glucose, pH 7.3). Following incubation at 37°C and 5% CO2 with the probe, neuronal cultures were washed with MHB, and immediately transferred to an imaging chamber containing 2.5 ml MHB. Neurons were imaged on a DMIRB microscope (Leica Microsystems, Wetzlar, Germany) at 20× magnification. Neurons within an imaging field (n = 10–20) were excited with 490 nm light every 10 s until baseline fluorescence levels stabilized. ZnPyr treatments were directly infused into the imaging chamber during continuous imaging. Following free Zn2+ increases, a cell‐permeant Zn2+ chelator, N,N,N',N'‐tetrakis(2‐pyridylmethyl)ethane‐1,2‐diamine (TPEN; 20 μm), was utilized to decrease Zn2+ fluorescence signal and confirm its Zn2+‐dependency.
Electrophysiology
Whole‐cell voltage clamp currents were obtained with an Axopatch 200B amplifier and pClamp software (Molecular Devices, LLC., Silicon Valley, CA, USA) using 3–5 MΩ electrodes. Whole‐cell K+ currents were measured immediately before and after application of either ZnPyr (30 μm ZnCl2, 300 nm pyrithione) or ZnPyr + dantrolene (Dan) (30 μm ZnCl2, 300 nm pyrithione, 10 μm Dan) in an extracellular recording solution (115 mm NaCl, 2.5 mm KCl, 2.0 mm MgCl2, 1.0 mm CaCl2, 10 mm Hepes, 10 mm d‐glucose and 0.25 μm tetrodotoxin, pH 7.2). The electrode solution contained 100 mm K‐gluconate, 10 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 2.2 mm Mg2·ATP, 0.33 mm GTP, 11 mm EGTA and 10 mm Hepes (pH 7.2). Series resistance was partially compensated (80%) in all cases. Currents were filtered at 2 kHz and digitized at 10 kHz. K+ currents were evoked with a series of 200 ms voltage steps from a holding potential of −80 to +80 mV in 10 mV increments. Before depolarization, a single 30 ms pre‐pulse to +10 mV was used to inactivate A‐type K+ currents. Conductance (G) was calculated from peak steady‐state current amplitudes (I) using the equation G = I/(V − E K). G/G max was plotted against the membrane potential and fit to a Boltzmann function, G = G max/(1 + exp[−(V − V 1/2)/k]), where V 1/2 is the voltage of half‐maximal activation and k is the slope factor of activation.
Chemical ischaemic preconditioning
Following transfection with a GFP‐tagged Kv2.1 construct (to enable visualization of Kv2.1 localization during microscopy), cortical neurons in vitro were pre‐treated for 20 min with either vehicle (DMSO) or ryanodine [15 μm ryanodol 3‐(1H‐pyrrole‐2‐carboxylate)] solutions, made up in a glucose‐free balanced salt solution (150 mm NaCl, 2.8 mm KCl, 1 mm CaCl2, 10 mm Hepes, pH 7.2). Following pre‐treatment, cells were preconditioned for 90 min with 3 mm KCN (or ddH2O) accompanied by either vehicle (DMSO) or ryanodine co‐treatment, following a protocol similar to that described in Aras et al. (2009 b). After ischaemic preconditioning, coverslips were washed with solutions identical to pre‐treatments. Kv2.1 localization was analysed via confocal microscopy immediately following preconditioning or vehicle‐treatment. Confocal image acquisition followed a protocol similar to that described above.
Statistical analysis
All data are presented as the mean ± SEM. All statistical analyses were performed in PRISM (GraphPad Software Inc., San Diego, CA, USA). When more than two means were compared, a one‐way ANOVA was utilized with Dunnett's multiple comparison test (MCT) vs. vehicle or control‐treated group. Sets of paired observations or experiments only involving two groups were analysed via two‐tailed t tests. α was set at 0.05 for experiments (95% confidence). P < 0.05 was considered statistically significant.
Results
Intracellular Zn2+ increases are sufficient to induce transient Kv2.1 channel declustering
Preconditioning‐induced, sublethal increases in intracellular Zn2+ concentrations are sufficient to induce neuronal tolerance and associated Kv2.1 modulations in neurons (Aras et al. 2009 b; Aras et al. 2009 a). Furthermore, we have previously shown that preconditioning‐induced Kv2.1 channel cluster dispersal is inhibited by chelation of intracellular Zn2+ surges produced by the injurious treatment (Aras et al. 2009 a). Thus, we first examined whether rises in intracellular Zn2+ alone were sufficient to induce Kv2.1 channel declustering, aiming to more closely associate intracellular Zn2+ signalling with induction of the Kv2.1 modulation process. To accomplish this, neurons were transfected with a GFP‐tagged Kv2.1 construct, which produces somatodendritic Kv2.1 channel clusters similar to endogenous channels (Misonou et al. 2004; O'Connell et al. 2006; Shah et al. 2014), to visualize channel distribution. Treatment with exogenous Zn2+ in combination with the selective Zn2+ ionophore, pyrithione (ZnPyr; 30 μm ZnCl2, 300 nm pyrithione), for 2 h facilitated a surge in intracellular Zn2+ (Aras et al. 2009 b). This concentration of Zn2+ has been shown to provide neuroprotection in previous studies and thus serves as an appropriate model for preconditioning‐induced intracellular Zn2+ increases (Aras et al. 2009 b). We found that the ZnPyr treatment yielded significant reduction of Kv2.1 clustering compared to vehicle (300 nm pyrithione)‐treated cells (Vehicle, 0.155 ± 0.011 Kv2.1 clusters/μm2 of neuronal soma, n = 25; ZnPyr 2 h, 0.093 ± 0.015 Kv2.1 clusters/μm2, n = 14) (Fig. 1 A and B).
Figure 1. Sublethal elevation of intracellular Zn2+ concentration induces transient Kv2.1 channel cluster dispersal in neurons .
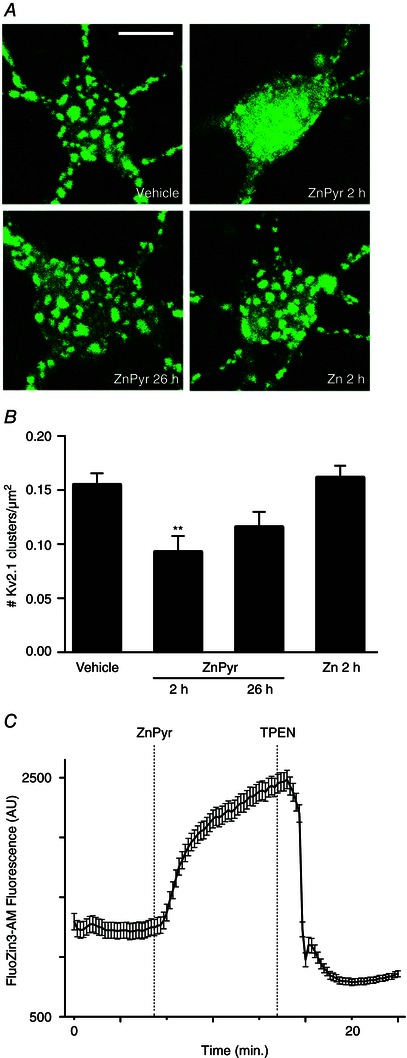
A, confocal images of cortical neurons transfected with a GFP‐tagged Kv2.1 construct, representative of Kv2.1 localization following treatment with either vehicle (ddH2O, 300 nm pyrithione), ZnPyr (30 μm ZnCl2, 300 nm pyrithione) or Zn (30 μm ZnCl2) conditions. ZnPyr‐treated neurons are shown both immediately and 24 h following 2 h treatment. Scale bar = 10 μm. B, showing the mean number of Kv2.1 clusters found in each condition, normalized to neuronal somatic area (number of Kv2.1 clusters μm–2 of neuronal soma). Data are are shown for each condition as the mean ± SEM (Vehicle, 0.155 ± 0.011, n = 25; ZnPyr 2 h, 0.093 ± 0.015, n = 14; ZnPyr 26 h, 0.116 ± 0.014, n = 15; Zn 2 h, 0.162 ± 0.011, n = 16). Analysed via one‐way ANOVA with Dunnett's MCT vs. vehicle‐treated neurons (**P < 0.01). C, trace represents Zn2+ entry into neurons as a function of FluoZin3‐AM (a cell‐permeant, Zn2+‐sensitive probe) fluorescence (AU) vs. time (min). ZnPyr was infused ∼7 min following baseline measurement, whereas TPEN, a cell‐permeant Zn2+ chelator was infused at ∼14 min, to confirm the Zn2+‐dependency of FluoZin3‐AM fluorescence. Data points are shown as the mean ± SEM of fluorescence measurements (AU) for n = 15 cell ROIs measured in the experiment.
In previous studies, Kv2.1 channel clusters returned to their clustered state 24 h following withdrawal of the preconditioning stimulus (Aras et al. 2009 a; Shah et al. 2014). To investigate whether Zn2+‐induced channel declustering was also transient, we imaged neuronal cultures 24 h following the 2 h ZnPyr treatment (Fig. 1 A) and found that Kv2.1 clusters were predominantly restored compared to vehicle‐treated neurons (ZnPyr 26 h, 0.116 ± 0.014 Kv2.1 clusters/μm2, n = 15) (Fig. 1 A and B).
Next, we confirmed that Zn2+ mediated its effect primarily by an intracellular mechanism, and not by binding to the extracellular, ligand‐binding domain of the metabotropic Zn2+ receptor mZnR/GPR39 (Besser et al. 2009), or by mediating some other extracellular signalling‐dependent process. We removed pyrithione from the treatment solution, thereby preventing Zn2+ entry into the neuronal cytosol, and proceeded with Kv2.1 cluster imaging following Zn2+ exposure for 2 h (Zn; 30 μm ZnCl2). No significant level of declustering was displayed in this case (Fig. 1 A), confirming that Zn2+‐dependent Kv2.1 channel declustering effect is indeed mediated by an intracellular, Zn2+‐activated signalling pathway (Zn 2 h, 0.162 ± 0.011 Kv2.1 clusters/μm2, n = 16) (Fig. 1 B).
To confirm the efficacy of pyrithione in facilitating Zn2+ influx during the ZnPyr treatments, especially because our incubating solution contained phosphate, a possible Zn2+‐binding media component, Zn2+ imaging was carried out using the Zn2+‐sensitive cell‐permeant probe, FluoZin3‐AM. ZnPyr treatment during live imaging of neuronal somas (region of interest; ROI) yielded significant increases in FluoZin3‐AM fluorescence (n = 15 cells), which were attenuated by addition of the selective Zn2+ chelator TPEN (20 μm) (Fig. 1 C). Although we had not expected any precipitation of free Zn2+ in our media based on the published solubility product of Zn3(PO4)2 (Yamasaki et al. 2012), this experiment confirms that our experimental conditions are adequate for inducing intracellular increases in free Zn2+.
Zn2+‐induced Kv2.1 channel declustering is critically dependent on calcineurin activation
Similar to increases in intracellular Zn2+, calcineurin‐mediated Kv2.1 channel dephosphorylation is also required for ischaemic preconditioning‐induced channel declustering (Misonou et al. 2005 b; Mohapatra and Trimmer, 2006; Aras et al. 2009 b; Aras et al. 2009 a; Shah et al. 2014). Thus, we hypothesized that calcineurin activation may be critically linked to Zn2+‐induced Kv2.1 channel declustering as well. To test this hypothesis, we co‐treated neurons with ZnPyr in the presence of ascomycin (FK520) (5 μm), a potent inhibitor of calcineurin activity (Lotem et al. 1999; Shah et al. 2014) for 2 h. Indeed, calcineurin antagonism during ZnPyr treatment completely prevented Zn2+‐induced Kv2.1 declustering because neurons presented prominent clusters similar to vehicle‐treated neurons (Fig. 2 A). FK520 alone had no effect on basal Kv2.1 clustering levels (Vehicle, 0.141 ± 0.014 Kv2.1 clusters/μm2, n = 20; ZnPyr 2 h, 0.072 ± 0.008 Kv2.1 clusters/μm2, n = 41; ZnPyr + FK520 2 h, 0.168 ± 0.014 Kv2.1 clusters/μm2, n = 24; FK520 2 h, 0.147 ± 0.020 Kv2.1 clusters/μm2, n = 9) (Fig. 2 B).
Figure 2. Intracellular Zn2+‐induced Kv2.1 channel dispersal is calcineurin‐dependent .
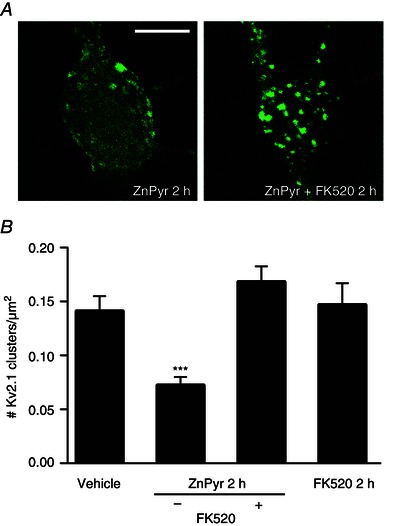
A, confocal images of cortical neurons transfected with a GFP‐tagged Kv2.1 construct, representative of Kv2.1 localization following treatment with either ZnPyr (30 μm ZnCl2, 300 nm pyrithione) or ZnPyr + FK520 (30 μm ZnCl2, 300 nm pyrithione, 5 μm FK520) conditions for 2 h. Control images not relevant to this finding were omitted; both vehicle‐treated and FK520‐treated neurons showed significant Kv2.1 channel clustering. Scale bar = 10 μm. B, showing the mean number of Kv2.1 clusters found in each condition, normalized to neuronal somatic area (number Kv2.1 clusters μm–2 of neuronal soma). Data are shown for each condition as the mean ± SEM (Vehicle, 0.141 ± 0.014, n = 20; ZnPyr 2 h, 0.072 ± 0.008, n = 41; ZnPyr + FK520 2 h, 0.168 ± 0.014, n = 24; FK520 2 h, 0.147 ± 0.020 Kv2.1, n = 9). Analysed via one‐way ANOVA with Dunnett's MCT vs. vehicle‐treated neurons (***P < 0.001).
RyR activity is vital to Zn2+‐induced Kv2.1 channel declustering and functional modulations
In the previous experiments, we confirmed an association between surges in intracellular Zn2+ concentrations and calcineurin activation. Next, we aimed to characterize a possible mechanistic link between free Zn2+ elevations and subsequent cytosolic Ca2+ liberation required for calcineurin activation. We hypothesized that this mechanism may rely on RyRs, large intracellular receptors situated on subsurface cisternae of the cortical endoplasmic reticulum (cER), which allow Ca2+ ion liberation from cER stores when open. Recent studies have noted close apposition between Kv2.1 channel clusters and neuronal RyR clusters of similar morphology, situated on the cER (Mandikian et al. 2014). Furthermore, functional junctions appear to exist between Kv2.1 clusters and the cER, with changes in cER association with the plasma membrane correlating closely with changes in Kv2.1 localization (Fox et al. 2015). Importantly, intracellular Zn2+ has also been noted to directly modulate RyR2 in cardiac myocytes, acting as a primary agonist stimulating Ca2+ release at low micromolar concentrations (Woodier et al. 2015). Moreover, Zn2+ action via RyR or IP3‐mediated pathways may modulate intracellular neuronal Ca2+ levels (Johanssen et al. 2015), although the mechanistic details behind this process have not been directly demonstrated. Given this evidence and our present findings, we hypothesized that intracellular Zn2+‐dependent calcineurin activation and consequent Kv2.1 modulations probably rely on RyR activation‐dependent liberation of cytosolic Ca2+.
To investigate this potentially novel mechanism of intracellular Ca2+ release in neurons, we first examined the effects of RyR inhibition on Zn2+‐induced Kv2.1 channel cluster dispersal. We co‐treated neurons during ZnPyr treatment with the RyR antagonist dantrolene (Dan; 10 μm). In our first set of studies, we found that this concentration of Dan was somewhat toxic to neurons over a 2 h treatment, and thus decreased our exposure to 1 h. We thus needed to confirm that the ZnPyr‐induced Kv2.1 channel declustering still occurred at this time‐point, which was indeed the case (Vehicle, 0.201 ± 0.034 Kv2.1 clusters/μm2, n = 8; ZnPyr 1 h, 0.067 ± 0.011 Kv2.1 clusters/μm2, n = 20) (Fig. 3 A and B). Importantly, the addition of Dan to the ZnPyr treatment significantly attenuated Zn2+‐induced declustering (ZnPyr + Dan 1 h, 0.156 ± 0.026 Kv2.1 clusters/μm2, n = 10) (Fig. 3 A and B), supporting a role for RyR activation in Zn2+‐mediated Kv2.1 channel modulation. Additionally, we performed a similar experiment using ryanodine [15 μm ryanodol 3‐(1H‐pyrrole‐2‐carboxylate)], which inhibits RyR activity at micromolar concentrations (Sutko et al. 1997). Similar to Dan treatment, ryanodine‐mediated inhibition of RyR activity also significantly attenuated Zn2+‐induced Kv2.1 declustering (ZnPyr + ryanodine 1 h, 0.195 ± 0.044 Kv2.1 clusters/μm2, n = 8) (Fig. 3 A and B). Neither ryanodine, nor Dan (with pyrithione vehicle) alone affected basal Kv2.1 channel clustering status (Vehicle + Dan/pyrithione 1 h, 0.173 ± 0.018 Kv2.1 clusters/μm2, n = 9; Vehicle + ryanodine 1 h, 0.161 ± 0.019 Kv2.1 clusters/μm2, n = 6) (Fig. 3 B). These results strongly suggest that the missing link between surges in intracellular Zn2+ and calcineurin activation, resulting in Kv2.1 modulation and changes in localization, may lie within the activation of neuronal RyR to cause Ca2+ release.
Figure 3. Intracellular Zn2+‐induced Kv2.1 channel dispersal is critically reliant on intermediate neuronal RyR activity .
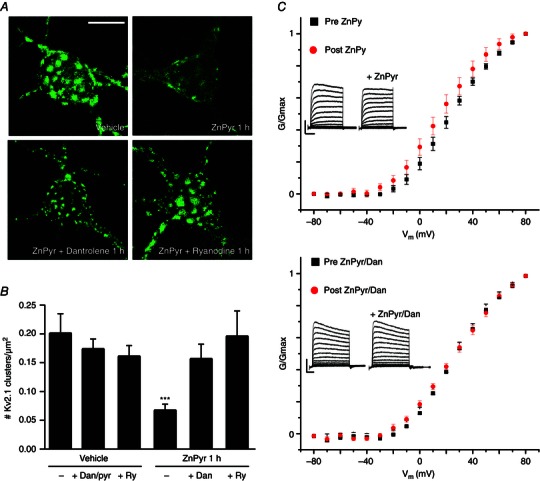
A, confocal images of cortical neurons transfected with a GFP‐tagged Kv2.1 construct, representative of Kv2.1 localization following treatment with either vehicle (ddH2O/DMSO), ZnPyr (30 μm ZnCl2, 300 nm pyrithione), ZnPyr + Dan (30 μm ZnCl2, 300 nm pyrithione, 10 μm Dan) or ZnPyr + ryanodine (30 μm ZnCl2, 300 nm pyrithione, 15 μm ryanodine) conditions for 1 h. Control images not relevant to this finding are not shown; Dan and ryanodine only‐treated neurons showed significant Kv2.1 channel clustering. Scale bar = 10 μm. B, showing the mean number of Kv2.1 clusters found in each condition, normalized to neuronal somatic area (number of Kv2.1 clusters μm–2 of neuronal soma). Data are shown for each condition as the mean ± SEM (Vehicle, 0.201 ± 0.034, n = 8; Vehicle + Dan/pyrithione 1 h, 0.173 ± 0.018, n = 9; Vehicle + ryanodine 1 h, 0.161 ± 0.019, n = 6; ZnPyr + vehicle 1 h, 0.067 ± 0.011 Kv2.1, n = 20; ZnPyr + Dan 1 h, 0.156 ± 0.026, n = 10; ZnPyr + ryanodine 1 h, 0.195 ± 0.044, n = 8). Analysed via one‐way ANOVA with Dunnett's MCT vs. vehicle‐treated neurons (***P < 0.001). C, top: average normalized G–V relationship of peak K+ currents is shown as the mean ± SEM, for all neurons before (black) and after (red) ZnPyr addition. Average half‐maximal activation voltage values (mV) were calculated for statistical analysis (pre‐ZnPyr, 24.15 ± 2.72 mV; post‐ZnPyr, 18.06 ± 3.29 mV; n = 9; P < 0.01); bottom: the same trend before and after ZnPyr + Dan treatment (pre‐ZnPyr + Dan, 19.92 ± 4.21 mV; post‐ZnPyr + Dan, 16.81 ± 5.39 mV; n = 7; (n.s.). Both paired data sets analysed via paired t test (two‐tailed). Below: Inlays representative whole‐cell current traces before and after each treatment condition. Scale bar: vertical = 2000 pA; horizontal = 60 ms.
Activation of calcineurin by intracellular Ca2+ and subsequent dephosphorylation of Kv2.1 are also known to shift the activation voltage of the channel in a hyperpolarizing direction (Park et al. 2006). Although a previous, non‐paired design study conducted in our laboratory failed to observe a measurable shift in the activation voltage of Kv2.1 in ZnPyr‐treated neurons compared to vehicle‐treated cells (Aras et al. 2009 a), we revisited this issue and measured K+ currents in the same neurons before and after the addition of ZnPyr to the culture bath. Under these conditions, we were able to observe a small but statistically significant, hyperpolarizing shift in the half‐maximal activation voltage (pre‐ZnPyr, 24.15 ± 2.72 mV; post‐ZnPyr, 18.06 ± 3.29 mV, n = 9) (Fig. 3 C) during these paired studies. Importantly, Dan inhibition of RyR during ZnPyr treatment was sufficient to abolish the magnitude and significance of the voltage activation shift (pre‐ZnPyr + Dan, 19.92 ± 4.21 mV; post‐ZnPyr + Dan, 16.81 ± 5.39 mV, n = 7) (Fig. 3 C), indicating that the small level of hyperpolarizing shift in Kv2.1 activation kinetics induced by intracellular Zn2+ relies on RyR activation as well.
Intracellular Zn2+ induces a RyR‐dependent increase in cytosolic Ca2+ release event frequency
Because our results suggest Zn2+‐induced Kv2.1 channel modulation to be critically dependent on the neuronal RyR, we investigated the possibility that Zn2+ may be directly modulating these receptors to increase intracellular Ca2+ liberation. Evidence of this phenomenon has been demonstrated in cardiac cells (Woodier et al. 2015) but has not yet been explored as a physiological process in neurons. A demonstration of this process would provide the critical link between Zn2+ and Ca2+/calcineurin dependency of Kv2.1 modulation.
To test this hypothesis, we co‐transfected cortical neurons in vitro with a GFP‐n1 plasmid to visualize neurons, along with a genetically encoded Ca2+‐sensitive fluorophore, RCaMP‐h, aiming to visualize and measure intracellular Ca2+ release in real time, before and after direct ZnPyr infusion into imaging media. In control cells, many neurons displayed spontaneous Ca2+ release events, correlating with an increased fluorescence produced by RCaMP‐h. These release events usually lasted 2–4 s and ranged in magnitude from focal fluorescence in compartmentalized areas of the cell to global release with stronger RCaMP‐h signal production (Fig. 4 A). Following ZnPyr infusion, a significant 96% increase in measurable Ca2+ event frequency occurred, on average (pre‐ZnPyr, 0.84 ± 0.23 Ca2+ transients/min; post‐ZnPyr, 1.65 ± 0.37 Ca2+ transients/min, n = 10) (Fig. 4 C). These results suggest that intracellular Zn2+ can indeed increase intracellular Ca2+ levels, or at least influence the frequency of cytosolic Ca2+ release events.
To test whether Zn2+ was acting through RyR to cause intracellular Ca2+ liberation, we first attempted to treat the cells with Dan (10 μm) both before and during ZnPyr infusion. We found that Dan almost completely quenched the RCaMP‐h signal prior to ZnPyr addition, without affecting GFP fluorescence. Although we have no explanation for this phenomenon, we did identify a study suggesting that calmodulin significantly potentiates the actions of Dan in blocking RyR isoforms (Gomez‐Hurtado et al. 2014). Because the RCaMP‐h protein comes from a family of proteins derived from calmodulin (Akerboom et al. 2013), Dan may have just simply interacted with the fluorescent Ca2+ sensor to abolish its signal. Regardless, under these circumstances, we felt that using Dan in these experiments might yield erroneous conclusions. Accordingly, we turned our attention again to ryanodine itself, which, as mentioned earlier, is known to inhibit RyR at micromolar concentrations (Sutko et al. 1997) and, as shown in the present study, can block ZnPyr‐mediated Kv2.1 cluster dispersal in a fashion similar to Dan (Fig. 3 A). In our experiments described below (n = 8), we found that baseline RCaMP‐h fluorescence was not affected by ryanodine (15 μm), enabling us to use and interpret the results of this experiment (i.e. based on the data generated below, baseline fluorescence measurements appeared to be unaffected upon the addition of ryanodine to the imaging chamber). Importantly, both spontaneous and Zn2+‐induced Ca2+ release events were significantly reduced by ryanodine application (Fig. 4 B and C). Neurons treated with ryanodine showed, on average, just an 8% increase in Ca2+ release events following ZnPyr infusion (pre‐ZnPyr + ryanodine, 0.43 ± 0.15 Ca2+ transients/min; post‐ZnPyr + ryanodine, 0.47 ± 0.20 Ca2+ transients/min, n = 8, ns) (Fig. 3 C) compared to the 96% increase following ZnPyr infusion alone. These results strongly suggest that intracellular Zn2+ causes Ca2+ liberation from the cER and subsequent calcineurin activation via a RyR‐dependent mechanism.
Neuronal RyR inhibition attenuates chemical ischaemic preconditioning‐mediated Kv2.1 channel cluster dispersal
Chemical ischaemic preconditioning with KCN (3 mm) has been shown to induce a rapid, transient increase in intracellular Zn2+ concentration within neurons that results in Kv2.1 channel declustering and concomitant neuronal tolerance to subsequent lethal excitotoxic stimuli (Aras et al. 2009 a). We thus tested whether preconditioning‐induced changes in Kv2.1 were also dependent on RyR activation. Note that, in these experiments, we also utilized ryanodine (15 μm) as our primary RyR antagonist because the combination of KCN and Dan proved to be highly toxic to the cells. We found that RyR antagonism during chemical ischaemic preconditioning treatment significantly attenuated the dispersal of Kv2.1 channel clusters induced by the KCN treatment alone (Vehicle, 0.190 ± 0.014 Kv2.1 clusters/μm2, n = 11; KCN, 0.081 ± 0.025 Kv2.1 clusters/μm2, n = 11; KCN + ryanodine, 0.169 ± 0.032 Kv2.1 clusters/μm2, n = 10) (Fig. 5).
Figure 5. RyR activation by intracellular Zn2+ is critical to Kv2.1 channel cluster dispersal in the chemical ischaemic preconditioning pathway .
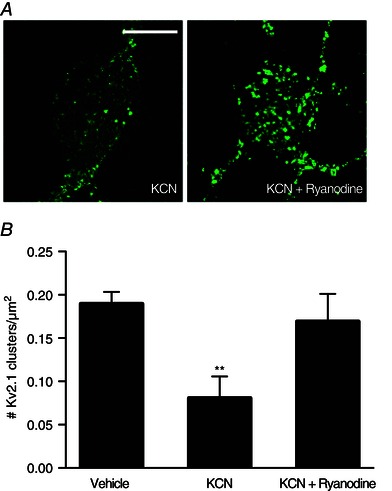
A, confocal images of cortical neurons transfected with a GFP‐tagged Kv2.1 construct, representative of Kv2.1 localization following treatment with either Vehicle (ddH2O, DMSO), KCN (3 mm KCN, DMSO) or KCN + ryanodine (3 mm KCN, 15 μm ryanodine) conditions for 90 min. Prior to treatment, neuronal cultures were incubated with either vehicle or ryanodine‐containing treatment solutions, depending on the treatment condition. Control images not relevant to this finding were omitted; Vehicle‐treated neurons showed significant Kv2.1 channel clustering. Scale bar = 10 μm. B, showing the mean number of Kv2.1 clusters found in each condition, normalized to neuronal somatic area (number of Kv2.1 clusters μm–2 of neuronal soma). Data are shown for each condition as the mean ± SEM (Vehicle, 0.190 ± 0.014, n = 11; KCN, 0.081 ± 0.025, n = 11; KCN + ryanodine, 0.169 ± 0.032, n = 10). Analysed via one‐way ANOVA with Dunnett's MCT vs. vehicle‐treated neurons (**P < 0.01).
Overall, these results suggest that chemical ischaemic preconditioning stimuli mediate changes in Kv2.1 localization and function by a Zn2+/Ca2+ co‐dependent pathway, which is critically reliant on the Ca2+‐releasing activity of neuronal RyRs. These results firmly establish neuronal RyR activation as the missing link between intracellular Zn2+ surges and Ca2+‐dependent processes in Kv2.1 modulation following preconditioning, and possibly by other sublethal injurious processes that result in increases in intracellular Zn2+.
Discussion
Intracellular Zn2+ signalling is a critical step in the modulation of Kv2.1 channel function and the activation of associated neuroprotective mechanisms in neurons (Aras et al. 2009 b; Aras et al. 2009 a). As such, Zn2+ has been shown to be essential to the chemical ischaemic preconditioning process, as well as to the associated alterations in Kv2.1 channel localization and voltage‐gated activation kinetics. Furthermore, Ca2+/calcineurin‐dependent dephosphorylation of the channel is an important step in modulating Kv2.1 in response to a wide range of injurious processes (Misonou et al. 2004; Surmeier and Foehring, 2004; Mulholland et al. 2008; Aras et al. 2009 b; Mohapatra et al. 2009; Baver and O'Connell, 2012; Shepherd et al. 2012; Shah and Aizenman, 2014). However, the link between Zn2+ and Ca2+ in this important adaptive neuronal process has not been established previously.
In the present study, we first confirm our hypothesis that increases in intracellular Zn2+ are alone sufficient for Kv2.1 channel declustering. An acute surge in intracellular Zn2+ concentration induces transient Kv2.1 channel diffusion in the membrane. This localization change is accompanied by a small but significant hyperpolarizing shift in the voltage‐gated activation kinetics of the channel. Because intracellular Zn2+ is an adequate preconditioning stimulus at similar concentrations (Aras et al. 2009 b), capable of promoting neuronal tolerance (i.e. prevention of cell death) in vitro, our results further associate Kv2.1 modulation with the induction of neuronal tolerance. Importantly, we show that Zn2+‐mediated channel declustering is dependent on activation of calcineurin, which is a protein phosphatase known to dephosphorylate Kv2.1 at several intracellular residues (Misonou et al. 2004; Misonou et al. 2005 b; Mulholland et al. 2008; Baver and O'Connell, 2012; Shah et al. 2014). We also found that calcineurin activation in this context is probably promoted by Ca2+ liberation from intracellular stores following direct modulation of neuronal RyRs by Zn2+. As mentioned earlier, a recent study demonstrated direct activation of cardiac isoform RyR in myocytes by Zn2+ (Woodier et al. 2015). The present study indicates that intracellular Zn2+ also probably activates neuronal RyRs, linking intracellular Zn2+ surges to calcineurin activation. Our results therefore suggest that this process of ryanodine receptor activation may be a ubiquitous component of Zn2+ signalling, further solidifying the status of Zn2+ as an important second messenger in a large array of cell types (Yamasaki et al. 2007).
Recent studies have also provided strong evidence indicating that RyR and Kv2.1 are spatially linked. Indeed, accumulated RyR clusters on subsurface cER cisternae are closely apposed to plasma membrane Kv2.1 clusters, even taking the same morphological organization into clusters (Mandikian et al. 2014). Additionally, cER, which is an important source of cytosolic Ca2+ release from RyR, is closely associated with Kv2.1, such that the loss of Kv2.1 cluster‐cER membrane junctions also results in dissociation of surface Kv2.1 channel clusters (Fox et al. 2015). Indeed, Kv2.1 channel clusters may represent the de facto anchor that facilitates the close apposition of the plasma membrane with the cER (Fox et al. 2013), with Kv2.1 surface clusters serving as an important membrane insertion point of new potassium channels, including Kv2.1 (Deutsch et al. 2012). Furthermore, because Kv2.1 surface clusters have been shown to be non‐conducting, with declustered Kv2.1 channels mediating the majority of high threshold K+ currents (O'Connell et al. 2010), these clusters probably serve a function outside of K+ current conduction. Because we have found that Zn2+‐induced Kv2.1 declustering is critically reliant on a signalling mechanism involving neuronal RyR activation and Ca2+ liberation, the close association of RyR‐containing cER to Kv2.1‐containing plasma membrane clusters suggests the existence of a classical signalling microdomain (Hoessli et al. 2000) that enables the channel modulation observed in the present study. In this way, active molecular complexes necessary for Kv2.1 modulations are probably juxtaposed to channel clusters in a confined cytosolic environment (Fig. 6).
Figure 6. Intracellular Zn2+‐induced Kv2.1 channel modulation occurs in compartmentalized signalling domains .
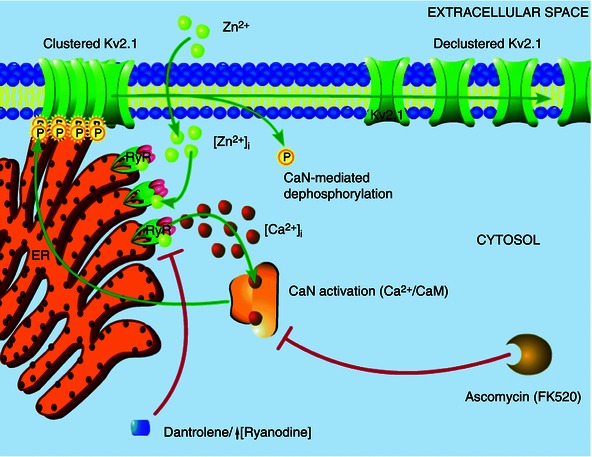
The results of the present study suggest that Zn2+ plays an early, critical role in Kv2.1 modulation. Transient surges in free Zn2+ concentration within the cell (either produced by exogenous ZnPyr treatment, as shown here, or induced by sublethal ischaemic injury), probably modulate neuronal RyR directly, inducing an increased Ca2+ event frequency. These Ca2+ transients, located in close proximity to Kv2.1 channel cluster domains, lead to calcineurin activation and subsequent Kv2.1 dephosphorylation and dispersal of channel localization. This signalling cascade is sensitive to Zn2+ chelation, calcineurin activation and RyR activity, with direct Zn2+‐modulation of RyR being the key link in this process between acute surges in intracellular Zn2+ and calcineurin‐mediated Kv2.1 modulation.
Thus, our model (Fig. 6) suggests that intracellular Zn2+ transiently alters Kv2.1 channel function and localization by direct modulation of the neuronal RyR in a highly compartmentalized mechanism. By associating with closely apposed RyR clusters, intracellular Zn2+ ions mediate an increased frequency of Ca2+ release events from the cER in contact with Kv2.1 channel clusters on the somatic membrane. This cytosolic Ca2+ then rapidly binds a closely associated calcineurin‐calmodulin complex, engaging protein phosphatase activity. In turn, active calcineurin dephosphorylates Kv2.1, leading, in the case reported in the present study, to a small hyperpolarizing shift in the channel's activation kinetics, perhaps aiding neurons with homeostatic adaptation to increased excitation, such as that occurring during excitotoxic injury (Aras et al. 2009 b; Aras et al. 2009 a; Mohapatra et al. 2009; Shepherd et al. 2013). Moreover, this profound, Zn2+‐dependent change in Kv2.1 may also serve as an adaptive response to injury by preventing the insertion of additional Kv2.1 channels, a phenomenon that has been tightly linked to neuronal cell death (Pal et al. 2003; Pal et al. 2006). Notably, we also demonstrate Zn2+dependent activation of RyR is an important component of ischaemic preconditioning‐induced changes in Kv2.1 localization, which also causes a similar increase in free cytosolic Zn2+. It is important to note, overall, that this process probably proceeds in a concerted and spatially linked mechanism where Kv2.1 clusters, subsurface cER cisternae, RyR‐mediated Ca2+ transients and calcineurin are closely associated in a signalling microdomain, allowing a kinetically fast interaction to occur. Interestingly, the calcineurin‐mediated hyperpolarizing shift that we observed in the activation kinetics of Kv2.1 also exists in the presence of 11 mm EGTA in whole‐cell patch pipettes; thus, this is probably not a process dependent on global Ca2+ release or a kinetically slow signalling mechanism. Indeed, the diminished magnitude of this hyperpolarizing shift, when compared with previously reported values closer to 20 mV (Misonou et al. 2005 b), may be partially explained by the presence of this slow Ca2+ chelator.
In sum, intracellular Zn2+ has been strongly indicated as a transient, early and crucial signalling ion active in response to cellular injury, preceding cytosolic Ca2+ liberation. Although Zn2+‐dependent Ca2+ increases have been previously shown to occur in neurons (Medvedeva et al. 2009; Johanssen et al. 2015), the mechanism behind this process had not been defined. Specifically, the link between Zn2+ and Ca2+ had not been clarified. The present study provides a critical, mechanistic link between intracellular Zn2+ signalling and RyR‐dependent cytosolic Ca2+ liberation leading to Kv2.1 declustering and cellular adaptive responses. As such, Zn2+ may well be a ubiquitous signalling ion in the central nervous system, critically regulating neuronal function and cellular homeostasis in response to a wide range of environmental insults.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
AJS, NHS, JAJ, RDM, ZPW and EA designed the research. AJS, NHS, JAJ, RDM and ZPW collected data. AJS, NHS, JAJ, RDM, ZPW and EA analysed and interpreted data. AJS, NHS, JAJ and EA wrote the manuscript. EA provided financial support. ZPW provided some reagents and materials. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by NIH grant NS043277 to EA. NHS was supported by the American Heart Association Pre‐doctoral Fellowship 12PRE11070001. JAJ is supported by NIH T32 NS086749.
Acknowledgements
We thank D. P. Mohapatra, Washington University, St Louis, for GFP‐Kv2.1‐expressing plasmid; C. A. Anderson for helpful suggestions; and K. A. Hartnett for technical support.
References
- Akerboom J, Calderón NC, Tian L, Wabnig S, Prigge M, Tolö J, Gordus A, Orger MB, Severi KE & Macklin JJ (2013). Genetically encoded calcium indicators for multi‐color neural activity imaging and combination with optogenetics. Front Mol Neurosci 6, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aras MA, Saadi RA & Aizenman E (2009. a). Zn2+ regulates Kv2. 1 voltage‐dependent gating and localization following ischemia. Eur J Neurosci 30, 2250–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aras MA, Hara H, Hartnett KA, Kandler K & Aizenman E (2009. b). Protein kinase C regulation of neuronal zinc signaling mediates survival during preconditioning. J Neurochem 110, 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baver SB & O'Connell KM (2012). The C‐terminus of neuronal Kv2. 1 channels is required for channel localization and targeting but not for NMDA‐receptor‐mediated regulation of channel function. Neuroscience 217, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besser L, Chorin E, Sekler I, Silverman WF, Atkin S, Russell JT & Hershfinkel M (2009). Synaptically released zinc triggers metabotropic signaling via a zinc‐sensing receptor in the hippocampus. J Neurosci 29, 2890–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E, Weigel AV, Akin EJ, Fox P, Hansen G, Haberkorn CJ, Loftus R, Krapf D & Tamkun MM (2012). Kv2. 1 cell surface clusters are insertion platforms for ion channel delivery to the plasma membrane. Mol Biol Cell 23, 2917–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Haak LL, Phillips‐Tansey E, Russell JT & McBain CJ (2000). Frequency‐dependent regulation of rat hippocampal somato‐dendritic excitability by the K+ channel subunit Kv2. 1. J Physiol 522, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PD, Loftus RJ & Tamkun MM (2013). Regulation of Kv2. 1 K+ conductance by cell surface channel density. J Neurosci 33, 1259–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PD, Haberkorn CJ, Akin EJ, Seel PJ, Krapf D & Tamkun MM (2015). Induction of stable endoplasmic reticulum/plasma membrane junctions by Kv2. 1 potassium channels. J Cell Sci 128, 2096–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson CJ, Koh J‐Y & Bush AI (2005). The neurobiology of zinc in health and disease. Nat Rev Neurosci 6, 449–462. [DOI] [PubMed] [Google Scholar]
- Gomez‐Hurtado N, Oo YW, Laver D & Knollmann B (2014). Presence of calmodulin potentiates block of ryanodine receptor calcium release channels by dantrolene and flecainide. Circulation 130, A18157–A18157. [Google Scholar]
- Granzotto A & Sensi SL (2015). Intracellular zinc is a critical intermediate in the excitotoxic cascade. Neurobiol Dis 81, 25–37. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . Exp Physiol 100, 755–758. [DOI] [PubMed] [Google Scholar]
- Hartnett K, Stout A, Rajdev S, Rosenberg P, Reynolds I & Aizenman E (1997). NMDA Receptor‐mediated neurotoxicity: a paradoxical requirement for extracellular Mg2+ in Na+/Ca2+‐free solutions in rat cortical neurons in vitro. J Neurochem 68, 1836–1845. [DOI] [PubMed] [Google Scholar]
- Hoessli DC, Ilangumaran S, Soltermann A, Robinson PJ & Borisch B (2000). Signaling through sphingolipid microdomains of the plasma membrane: the concept of signaling platform. Glycoconj J 17, 191–197. [DOI] [PubMed] [Google Scholar]
- Johanssen T, Suphantarida N, Donnelly PS, Liu XM, Petrou S, Hill AF & Barnham KJ (2015). PBT2 inhibits glutamate‐induced excitotoxicity in neurons through metal‐mediated preconditioning. Neurobiol Dis 81, 176–185. [DOI] [PubMed] [Google Scholar]
- Lim ST, Antonucci DE, Scannevin RH & Trimmer JS (2000). A novel targeting signal for proximal clustering of the Kv2. 1 K+ channel in hippocampal neurons. Neuron 25, 385–397. [DOI] [PubMed] [Google Scholar]
- Lotem J, Kama R & Sachs L (1999). Suppression or induction of apoptosis by opposing pathways downstream from calcium‐activated calcineurin. Proc Natl Acad Sci USA 96, 12016–12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandikian D, Bocksteins E, Parajuli LK, Bishop HI, Cerda O, Shigemoto R & Trimmer JS (2014). Cell type‐specific spatial and functional coupling between mammalian brain Kv2. 1 K+ channels and ryanodine receptors. J Comp Neurol 522, 3555–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedeva YV, Lin B, Shuttleworth CW & Weiss JH (2009). Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization, and cell death in an acute slice oxygen‐glucose deprivation model of ischemia. J Neurosci 29, 1105–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP & Trimmer JS (2005. a). Kv2. 1: a voltage‐gated K+ channel critical to dynamic control of neuronal excitability. Neurotoxicology 26, 743–752. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Menegola M & Trimmer JS (2005. b). Calcium‐ and metabolic state‐dependent modulation of the voltage‐dependent Kv2. 1 channel regulates neuronal excitability in response to ischemia. J Neurosci 25, 11184–11193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE & Trimmer JS (2004). Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci 7, 711–718. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP & Trimmer JS (2006). The Kv2. 1 C terminus can autonomously transfer Kv2. 1‐like phosphorylation‐dependent localization, voltage‐dependent gating, and muscarinic modulation to diverse Kv channels. J Neurosci 26, 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Misonou H, Sheng‐Jun P, Held JE, Surmeier DJ & Trimmer JS (2009). Regulation of intrinsic excitability in hippocampal neurons by activity‐dependent modulation of the KV2. 1 potassium channel. Channels 3, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland PJ, Carpenter‐Hyland EP, Hearing MC, Becker HC, Woodward JJ & Chandler LJ (2008). Glutamate transporters regulate extrasynaptic NMDA receptor modulation of Kv2. 1 potassium channels. J Neurosci 28, 8801–8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell KM, Loftus R & Tamkun MM (2010). Localization‐dependent activity of the Kv2. 1 delayed‐rectifier K+ channel. Proc Natl Acad Sci USA 107, 12351–12356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell KM, Rolig AS, Whitesell JD & Tamkun MM (2006). Kv2. 1 potassium channels are retained within dynamic cell surface microdomains that are defined by a perimeter fence. J Neurosci 26, 9609–9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Takimoto K, Aizenman E & Levitan E (2006). Apoptotic surface delivery of K+ channels. Cell Death Differ 13, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Hartnett KA, Nerbonne JM, Levitan ES & Aizenman E (2003). Mediation of neuronal apoptosis by Kv2. 1‐encoded potassium channels. J Neurosci 23, 4798–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K‐S, Mohapatra DP, Misonou H & Trimmer JS (2006). Graded regulation of the Kv2. 1 potassium channel by variable phosphorylation. Science 313, 976–979. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Murakoshi H, Rhodes KJ & Trimmer JS (1996). Identification of a cytoplasmic domain important in the polarized expression and clustering of the Kv2. 1 K+ channel. J Cell Biol 135, 1619–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Paoletti P, Koh J‐Y, Aizenman E, Bush AI & Hershfinkel M (2011). The neurophysiology and pathology of brain zinc. J Neurosci 31, 16076–16085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NH & Aizenman E (2014). Voltage‐gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl Stroke Res 5, 38–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NH, Schulien AJ, Clemens K, Aizenman TD, Hageman TM, Wills ZP & Aizenman E (2014). Cyclin E1 regulates Kv2. 1 channel phosphorylation and localization in neuronal ischemia. J Neurosci 34, 4326–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AJ, Loo L & Mohapatra DP (2013). Chemokine co‐receptor CCR5/CXCR4‐dependent modulation of Kv2. 1 channel confers acute neuroprotection to HIV‐1 glycoprotein gp120 exposure. PloS ONE 8, e76698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AJ, Loo L, Gupte RP, Mickle AD & Mohapatra DP (2012). Distinct modifications in Kv2. 1 channel via chemokine receptor CXCR4 regulate neuronal survival‐death dynamics. J Neurosci 32, 17725–17739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ & Foehring R (2004). A mechanism for homeostatic plasticity. Nat Neurosci 7, 691–692. [DOI] [PubMed] [Google Scholar]
- Sutko JL, Airey JA, Welch W & Ruest L (1997). The pharmacology of ryanodine and related compounds. Pharmacol Rev 49, 53–98. [PubMed] [Google Scholar]
- Tamkun MM, O'Connell KM & Rolig AS (2007). A cytoskeletal‐based perimeter fence selectively corrals a sub‐population of cell surface Kv2. 1 channels. J Cell Sci 120, 2413–2423. [DOI] [PubMed] [Google Scholar]
- Woodier J, Rainbow RD, Stewart AJ & Pitt SJ (2015). Intracellular zinc modulates cardiac ryanodine receptor‐mediated calcium release. J Biol Chem 290, 17599–17610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki K, Kigawa T, Watanabe S, Inoue M, Yamasaki T, Seki M, Shinozaki K & Yokoyama S (2012). Structural basis for sequence‐specific DNA recognition by an Arabidopsis WRKY transcription factor. J Biol Chem 287, 7683–7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki S, Sakata‐Sogawa K, Hasegawa A, Suzuki T, Kabu K, Sato E, Kurosaki T, Yamashita S, Tokunaga M & Nishida K (2007). Zinc is a novel intracellular second messenger. J Cell Biol 177, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]