

Abstract
Children with cancer are increasingly benefiting from novel therapeutic strategies and advances in supportive care, as reflected in improvements in both their survival and quality of life. However, the continuous emergence of new oncology drugs and supportive care agents has also increased the possibility of deleterious drug interactions and healthcare providers need to practice extreme caution when combining medications. In this review, we discuss the most common interactions of chemotherapeutic agents with supportive care drugs such as anticonvulsants, antiemetics, uric acid–lowering agents, acid suppressants, antimicrobials, and pain management medications in pediatric oncology patients. As chemotherapy agents interact not only with medications but also with foods and herbal supplements that patients receive during the course of their treatment, we also briefly review such interactions and provide recommendations to avoid unwanted and potentially fatal interactions in children with cancer.
Keywords: Drug interactions, Cancer therapy toxicity, Adverse side effects, CYP450 enzyme system, Pediatric oncology
INTRODUCTION
Cancer treatment is based on the concept of drug interaction. Rational combination of drugs with additive or synergistic effects and non-overlapping toxicities has significantly contributed to improving the five-year survival rate of children with cancer from less than 50% in the 1960s to 80% in 2008.1–3 The parallel advancement in supportive care has been pivotal in alleviating the toxicity of cancer therapy, allowing patients a better quality of life and a superior survival. As more drugs are being used in the care of patients with cancer, healthcare providers need to take measures to avoid undesired drug interactions. Adult patients on polypharmacy for preexisting comorbidities before their cancer diagnosis are at high risk of being exposed to an unwanted drug interaction.4 In pediatric oncology, chemotherapy drug interactions occur mostly with supportive care agents prescribed during treatment. These interactions are complicated further by individual variabilities in drug disposition arising from factors such as age and genetic profile.5,6 The advent of the pharmacogenetic era will allow a safer individualized administration and combinations of drugs.7
This article reviews the major and most common drug-drug interactions between chemotherapy and supportive care agents in pediatric oncology with the intent of helping healthcare professionals recognize and reduce the risk of harmful drug interactions in their practice.
GENERAL CONCEPTS OF DRUG INTERACTIONS
Drug interactions are usually pharmacokinetic, pharmacodynamic or pharmacogenetic in nature. Pharmacokinetic interactions occur when one drug affects the absorption, distribution, metabolism or elimination of another medication.8 Most antineoplastic agents are excreted from the body after being metabolized into less lipid-soluble byproducts. The liver is the major organ that metabolizes drugs through phase I (oxidation, reduction, or hydrolysis) or phase II reactions. Phase I oxidative metabolism largely occurs through the cytochrome P450 (CYP450) enzyme system, which consists of approximately 100 isoenzymes such as CYP3A4, 2D6, 2C19, and 2C9. CYP3A4 is the most abundant isoenzyme in the human liver and accounts for approximately 50% of all CYP450 drug metabolisms.9,10 In addition to being metabolized by the CYP450 system, some medications can also induce or inhibit the effects of the isoenzymes (Figure 1). Administration of chemotherapy agents that are catabolized by CYP450 with inducers of these isoenzymes can facilitate chemotherapy clearance, potentially resulting in decreased efficacy and in turn to an increased risk of relapse.11 On the other hand, administration of chemotherapy agents with inhibitors of CYP450 can result in slow clearance and increased toxicity.12 The effect of inhibiting or inducing metabolism is the opposite with agents that are administered as prodrugs (e.g., cyclophosphamide, mercaptopurine).13 It is important to note the timing required to achieve cytochrome induction and inhibition: Induction increases the de novo synthesis of a CYP isoform and necessitates up to weeks for the full effect of the interaction to take place. Once the offending agent is discontinued, it can take days to weeks for the isoform’s activity to return to baseline. The effect of inhibiting a CYP isoform depends on the type of inhibition. A competitive inhibition (which occurs when two drugs are substrates of the same CYP isoform) occurs in a dose dependent manner and becomes significant when the isoform is saturated. In the case of a non-competitive inhibition, the CYP isoform is destroyed by the offending medication. Once the offending agent is discontinued, the inhibition is rapidly reversed in the case of a competitive inhibition. In the case of a non-competitive inhibition, it can take days to weeks for the isoform’s activity to return to baseline because de novo synthesis is required.4 Phase II reactions are mostly catabolic in nature and usually involve conjugation, sulfation, glucuronidation, acetylation, methylation, or glutathione conjugation of a drug into a more hydrophilic moiety thus allowing the metabolites to be excreted primarily through the kidneys. Most drugs undergoing phase II biotransformations undergo glucuronidation.13 It is also important to remember that genetic polymorphisms of hepatic drug-metabolizing enzyme systems can contribute to significant inter-individual variability in drug disposition.14,15
Figure 1.
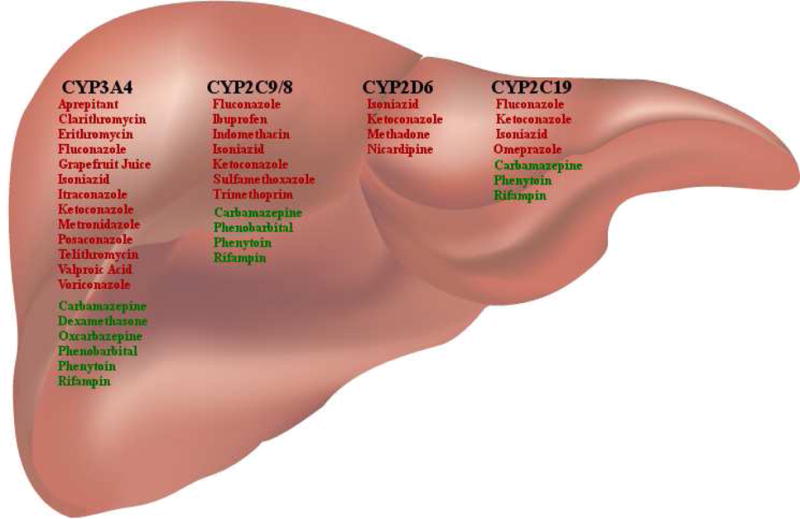
Major cytochrome P450 isoform inducers and inhibitors used in pediatric oncology patients. Medications listed in red are inhibitors and those listed in green are inducers of the CYP450 enzyme system.
Protein binding and affinity to plasma proteins is another mechanism of pharmacokinetic interactions. Drugs that are highly bound to plasma proteins (such as phenytoin) can be displaced by agents that have a higher affinity to the protein itself. This could result in a higher concentration of free (active) drug leading to increased drug toxicity. This interaction is especially important in oncology because chemotherapy agents have a narrow therapeutic index.4 Pharmacokinetic interactions can also be due to transport proteins located on the cell wall such as P-glycoprotein (P-gp), an active transport protein located on the cell membrane of many tissues in the body, including the biliary tract, brain, proximal tubule cells of the kidney, and epithelial cells of the intestine.16 It serves as an efflux transporter to remove chemical agents from the cell and transport them back into the circulation for elimination. P-glycoprotein therefore does not metabolize the drugs with which it interacts but merely transports them to a new location, thereby presenting them to metabolic machinery such as the CYP450 enzyme system.16
Pharmacodynamic interactions occur when two or more drugs have additive or antagonistic effects on each other. Excessive response and increased toxicity is observed if the interaction is additive as would be the case when using two drugs that have the potential to prolong the QTc interval or when combining leucovorin with 5-fluorouracil.17 A reduction in the response with decreased efficacy of medications occurs when drugs have antagonistic effects. This type of drug interaction has served as the basis for chemotherapy drug combination.17 It is also important to keep in mind that combining nephrotoxic agents can lead to mild or severe renal dysfunction. Additive kidney injuries caused by combining multiple nephrotoxic medications can delay the elimination of chemotherapy agents that are cleared through the kidney and thus increase their toxicity potential.18
SEARCH STRATEGY AND SELECTION CRITERIA
References for this review were identified through a search of PubMed, using the terms “chemotherapy”, “drug interaction”, “methotrexate”, “platinum chemotherapy”, “topoisomerase inhibitor”, “anthracycline”, “alkylating agent”, “mercaptopurine”, “vinca alkaloid”, “epipodophyllotoxin”, and “tyrosine kinase inhibitor”. Further articles were identified from the reference list of the retrieved publications and by searching cited references. Papers published in English and French published between January 1966, and October 2009 were included. Only published articles, and no abstracts, were taken into consideration.
ANTICONVULSANTS
Seizures are common complications of cancer and its therapy. Most of the old-generation anticonvulsants (phenytoin, carbamazepine, phenobarbital, and valproic acid) are CYP450 enzyme inducers or inhibitors. Concomitant administration of anticonvulsants and chemotherapy drugs that are metabolized through the CYP450 system can decrease the efficacy or potentiate the toxicity of these drugs. Phenobarbital, frequently used to prevent seizure recurrence in infants, is an inducer of the CYP3A4, 2C9/8, 2B6, and 1A2/6 enzymes.10 Concomitant administration of cyclophosphamide and phenobarbital in mice was found to decrease the concentration of phosphoramide mustard (the active metabolite of cyclophosphamide) by 26%.19 This interaction can be generalized to all barbiturates as they are all enzyme inducing agents. Such effects have also been observed with other chemotherapy agents metabolized by CYP3A4 (Panel 1).
Panel 1.
Substrates of hepatic cytochrome P450 isoforms involved in the catabolism or anabolism of chemotherapeutic agents used in pediatric oncology
| CYP3A4 substrates | ||
|
| ||
| Bortezomib | Etoposide | Paclitaxel |
| Busulfan | Everolimus | Prednisone |
| Cyclophosphamide | Gefitinib | Prednisolone |
| Dasatinib | Ifosfamide | Sirolimus |
| Cyclosporine | Irinotecan | Sorafenib |
| Dexamethasone | Lapatinib | Sunitinib |
| Docetaxel | Methylpresnisolone | Tacrolimus |
| Doxorubicin | Nilotinib | Tamoxifen |
| Erlotinib | Temsirolimus | |
|
| ||
| CYP2D6 substrates | ||
|
| ||
| Bortezomib | Lomustine | Vinblastine |
| Doxorubicin | Tamoxifen | Vinorelbine |
| Imatinib | ||
|
| ||
| CYP2C9 substrates | ||
|
| ||
| Bortezomib | Imatinib | Tamoxifen |
| Cyclophosphamide | Paclitaxel | Tretinoin |
| Ifosfamide | ||
|
| ||
| CYP2C19 substrates | ||
|
| ||
| Bortezomib | Ifosfamide | |
| Cyclophosphamide | Imatinib | |
Note: Drugs are listed in alphabetical order
Phenytoin, an inducer of the CYP3A4, 2B6, 2C9, and 2C19 enzymes can also decrease the effectiveness of chemotherapy agents that are extensively catabolized by the liver.10 The topoisomerase I inhibitor irinotecan is anabolized by carboxylesterases to the active metabolite SN-38, and it also undergoes oxidative catabolism by CYP3A4 to the inactive metabolite 7-ethyl-10-[4-N-[(5-aminopentanoic acid)-1-piperidino]-carbonyloxy-camptothecin (APC). The concomitant administration of irinotecan with the CYP450 inducing agent phenytoin has been found to cause a marked decrease in the systemic exposure to irinotecan and SN-38 as well as an increase in APC in pediatric patients.20 In another study, the use of phenytoin or phenobarbital caused a 1·5-fold increase the clearance of etoposide (23·7 and 13·4 mL/min/m2 with and without anticonvulsants, respectively, p < 0·01); however, correlation analysis with remission or cure rates was not conducted.21
Carbamazepine induces the CYP3A4, 1A2, 2B6, 2C9/8, and 2C19 enzymes.10 Adult patients with brain tumor who received carbamazepine had a higher vincristine clearance (mean clearance 925 mL/min) than those not receiving it (mean clearance 569 mL/min, p=0.004).22 Other investigators have also reported similar effects of increased clearance of the chemotherapeutic agent, but the impact of this interaction on clinical outcome was first demonstrated in children with acute lymphoblastic leukemia (ALL).11 In a study by Relling and colleagues, 40 (5·6%) of 716 children received long-term treatment with phenytoin, phenobarbital, carbamazepine, or their combination simultaneously with antileukemic therapy.11 After adjusting for age and ploidy, anticonvulsant therapy was found to be significantly associated with a worse event free survival and a higher rate of hematological and CNS relapse in the 566 patients with precursor B-cell ALL. Anticonvulsants could also increase the clearance of anticancer drugs through other mechanisms, such as induction of enzymes and proteins involved in conjugative drug metabolism and in drug excretion and transport.23
Valproic acid, an antiepileptic increasingly used in patients with cancer for its antitumor and antiangiogenic activities via inhibition of histone deacetylase, is an inhibitor of the CYP3A4, 2D6, 2C9/8, and 2C19 enzymes.10 In a study of 70 adult patients receiving a cisplatin-based regimen for high-grade gliomas, the incidence of hematologic toxicity (thrombocytopenia, neutropenia, or both) was three times higher in the 24 patients who concomitantly received valproic acid.24
Whenever possible, enzyme-inducing anticonvulsants and valproic acid should be avoided as maintenance agents for seizure control in patients receiving chemotherapy. However, if a patient cannot be weaned off the antiepileptic drug before the start of therapy, chemotherapy should be initiated without any delay and the weaning of the anticonvulsant should occur after therapy has begun. Agents that are less likely to interfere with drug-metabolizing enzymes (e.g., levetiracetam, gabapentin, topiramate) should be considered as alternative antiepileptic drugs whenever possible. Most oncology phase I and II clinical trials preclude the concomitant use of enzyme-inducing and enzyme-inhibiting drugs in order to avoid the potential effect of such drugs on study results, such as determination of the maximum tolerated dose of the investigational agent under evaluation.
ANTIEMETICS
Aprepitant is the first neurokinin 1 inhibitor approved to prevent the acute and delayed nausea and vomiting associated with highly emetogenic cancer chemotherapy.25 The use of aprepitant in pediatric oncology has been limited because it can potentially interact with chemotherapeutic agents and because data on its dosing in children is limited. Aprepitant is a substrate and an inhibitor of CYP3A4, and can also inhibit 2C9/8 and 2C19.10 Because many chemotherapy agents are metabolized through the CYP3A4 pathway, there is a potential risk of increased toxicity with the use of aprepitant. Case reports in adult patients have implicated aprepitant in the increased toxicity of ifosfamide and in the inhibition of cyclophosphamide bioactivation and thiotepa metabolism.26,27 However, aprepitant was reported not to affect the pharmacokinetics of vinorelbine in adult patients with metastatic solid tumors.28
Another agent used to prevent nausea and vomiting in patients receiving highly emetogenic chemotherapy is dexamethasone. Studies have shown a 2·2-fold increase in the area under the curve (AUC) of dexamethasone when it is combined with aprepitant.29 The American Society of Clinical Oncology as well as the aprepitant package insert recommend decreasing the dexamethasone dose by half when it is combined with aprepitant for antiemetic use in cancer patients.30
In addition to serving as a CYP3A4 substrate, dexamethasone (and other glucocorticoids) is a CYP3A4 inducer and a P-gp modulator.10 Its use as an antiemetic could potentially increase the clearance of chemotherapy agents that are substrates of the CYP3A4 enzyme and P-gp. The addition of prednisone to etoposide causes an increased clearance of etoposide (from a median of 29·2 mL/min/m2 to a median of 47·4 mL/min/m2, p<0·0001) in a study of pediatric patients with ALL.31
The antiemetic ondansetron has been shown to affect the pharmacokinetics of chemotherapy agents. A study conducted in adult bone marrow transplant patients, demonstrated that the AUC of cyclophosphamide was lower when patients received ondansetron compared to prochlorperazine (76·6 mg/ml/min and 90·6 mg/ml/min respectively, p=0.001).32 The same findings applied to cisplatin (525 mcg/ml/min and 648 mcg/ml/min respectively, p=0.001) however these differences in AUC were not correlated with long-term survival.
URIC ACID–LOWERING AGENTS
The xanthine analog allopurinol is commonly used in the management of hyperuricemia associated with tumor lysis syndrome. Allopurinol competitively inhibits xanthine oxidase thus blocking the conversion of the purine metabolites xanthine and hypoxanthine to uric acid.33 Allopurinol also reduces the clearance of other purine analogs, such as mercaptopurine and azathioprine; leading to increased toxicity and myelosuppression requiring dose reductions of mercaptopurine by up to 75%.34,35 The recombinant urate oxidase rasburicase is a very effective uricolytic agent with no known drug interactions, but significantly higher cost.36
ACID SUPPRESSANTS
Proton pump inhibitors (PPIs) are frequently used to manage reflux and gastritis in children with cancer. Omeprazole has been reported to delay the elimination of high-dose methotrexate in a child with osteosarcoma.37 A recent study has shown that the concurrent administration of PPIs with high-dose methotrexate is associated with delayed excretion of methotrexate in a cohort that consisted mostly of adult patients.38 Although the exact mechanism of this interaction remains to be determined, the delayed excretion can be explained by competition for the efflux protein transporters. It has been suggested that patients with CYP2C19 polymorphisms (an essential enzyme in the metabolism of PPIs) are at high risk of toxicity as they have high levels of the PPIs in the blood.38 In such patients, it is recommended that the use of PPIs be discontinued at least a day before administering high-dose methotrexate and restarted when the methotrexate has cleared. If acid suppression is required during chemotherapy, sucralfate or a type-2 histamine-receptor blocker can be considered. A summary of the drug interactions that occur with methotrexate is available in table 1.
Table 1.
Summary of the most commonly observed drug interactions with intravenous high-dose methotrexate therapy
| Offending Agent | Mechanism of Interaction | Effect of Interaction |
|---|---|---|
| Amphotericin B18 | Additive nephrotoxicity | Renal failure |
| Cisplatin39 | Additive nephrotoxicity | Renal failure |
| Folic Acid and multivitamins containing folic acid40 | Folic acid overcomes the dihydrofolate reductase inhibition of MTX | At high doses, decreased anticancer activity of MTX* |
| High dose Acyclovir41 | Additive nephrotoxicity | Acute renal failure |
| NSAIDs and bismuth subsalicylate42 | Salicylates decrease renal tubular secretion of MTX | Increased MTX toxicity |
| Penicillins43 | Penicillins reduce renal clearance of MTX | Increased MTX toxicity |
| Probenecid44 | Probenecid decreases renal tubular secretion of MTX | Increased MTX toxicity |
| Proton pump inhibitors (PPI)37 | PPIs decrease active secretion of MTX by the kidneys (probable mechanism) | Increased MTX toxicity |
| p-glycoprotein inducers45 | Increase efflux of MTX from cell | MTX resistance: decrease effectiveness of MTX |
| p-glycoprotein inhibitors46 | Decreased efflux of MTX from cell | Increased MTX toxicity |
Abbreviations: NSAIDs, non-steroidal anti-inflammatory drugs; MTX, Methotrexate
Leucovorin rescue is routinely used in patients receiving high dose MTX therapy, this interaction is due to the excess folic acid administered
Proton pump inhibitors as well as antacids increase gastric pH, leading to decreased absorption of tyrosine kinase inhibitors (TKIs).47 It is recommended to avoid the use of antacids while patients are receiving TKIs (with the exception of imatinib). However, if both drugs need to be used concomitantly, there should be at least a 4-hour gap between administration of TKIs and the antacids.
ANTIMICROBIALS
Antifungals
Azole antifungals are responsible for most of the interactions in this class. Azoles such as fluconazole, voriconazole, and posaconazole are used both as prophylactic and treatment agents against fungal infections. Azole antifungals were developed because they could inhibit sterol synthesis by the CYP450 family in fungi. These enzymes have varying cross-reactivity with the human CYP450 enzymes. All three agents are inhibitors of CYP3A4 and P-gp: Fluconazole and voriconazole are inhibitors of CYP2C9 and 2C19, posaconazole has no significant inhibiting activity on these two isoforms.10 The use of itraconazole, a CYP3A4 and P-gp inhibitor, has now greatly decreased because of its high incidence of causing hepatotoxicity and multiple drug interactions.
Increased neurotoxicity, including paralytic ileus, paralysis of extremities, and even seizures, have been reported when azole antifungals were administered with vinca alkaloids (vincristine, vinblastine, and vinorelbine).12,48,49 This increased toxicity occurs due to an increase in the plasma concentration of vinca alkaloids. Some centers hold the azole antifungal for 24 hours before chemotherapy, and restart them 24 hours after administration of a vinca alkaloid. If antifungal coverage is required during this time, an echinocandin antifungal (e.g., micafungin, caspofungin, anidulafungin) may be substituted for the azole agent.
The use of the calcineurin inhibitors cyclosporine and tacrolimus (both CYP3A4 substrates) concomitantly with azole antifungals (that inhibit their metabolism) is occurring more frequently in patients undergoing allogeneic hematopoietic stem cell transplant.50 When combined with voriconazole, a 1·7-fold increase in the AUC of cyclosporine and a 2·48-fold rise in the serum trough concentration of cyclosporine was observed.50 Increased serum concentrations of cyclosporine and tacrolimus have also been reported with fluconazole and ketoconazole.51–53 The symptoms of toxicity vary from tingling and redness of the palms and soles to hyperkalemia, neurotoxicity, and acute renal failure.
In clinical practice, and to avoid toxicity, the dose of cyclosporine and tacrolimus can be preemptively decreased by 30–50% on the day the azole antifungal is instituted. The dose of the calcineurin inhibitor should be subsequently adjusted depending on the serum concentration of the drug.50 This type of interaction is used favorably in infants and young children, who tend to have higher systemic clearances of cyclosporine and tacrolimus, therefore resulting in an inability to maintain adequate serum trough concentrations to prevent graft rejection.54,55 Because patients undergoing hematopoietic stem cell transplantation routinely receive antifungal prophylaxis, using an azole antifungal instead of an echinocandin can help increase the serum concentration of the calcineurin inhibitor to the desired range to prevent graft rejection in young patients. Combining these drugs in order to gain benefit from the drug interaction should be done with extreme caution and with very tight therapeutic drug monitoring to avoid toxicities. Amphotericin B in all its formulations is widely known for its nephrotoxic potential. It should not be combined with chemotherapy agents that are known to be nephrotoxic, such as cisplatin and high-dose methotrexate. The combination of cisplatin and amphotericin B has been found to be lethal in rat glioma models: all rats had massive hematuria within 24 hours of administering the two agents.56 Echinocandins or azole antifungals (except for intravenous voriconazole) are preferable agents for antifungal coverage in patients receiving nephrotoxic chemotherapy.
Antibiotics
Penicillins interfere with the proximal tubular secretion of methotrexate thus delaying the excretion of MTX and resulting in severe renal, hepatic, and hematologic toxicities.43,57 This interaction may be more pronounced in patients receiving high-dose methotrexate therapy, but it can potentially result in myelosuppression and mucositis even in patients receiving low-dose methotrexate (e.g., during the maintenance phase of ALL therapy).43
Due to the ototoxic and nephrotoxic side effect of aminoglycosides, it is preferable not to combine this class of antibiotics with cisplatin or high-dose methotrexate.58 The same recommendation applies to vancomycin. If vancomycin or aminoglycosides are clinically required, serum concentrations should be monitored closely and doses adjusted accordingly to prevent toxicity.
Trimethoprim-sulfamethoxazole (TMP/SMX) may slightly increase methotrexate toxicity. Both TMP and methotrexate block the reduction of dihydrofolate to tetrahydrofolate by inhibiting dihydrofolate reductase, resulting in additive methotrexate toxicity.59 Also, sulfamethoxazole (and all sulfa drugs) can displace methotrexate from its plasma protein binding site and inhibit its renal tubular secretion. Mean plasma concentrations of methotrexate can be 66% higher when administered concurrently with TMP/SMX.60 Although some pediatric oncology centers hold TMP/SMX when patients are receiving high-dose methotrexate, other institutions continue administering TMP/SMX if it is prescribed at the Pneumocystis jiroveci pneumonia prophylaxis dose.18
The macrolide antibiotics (clarithromycin, telithromycin, azithromycin, and erythromycin) are CYP3A4 and P-glycoprotein inhibitors.10 If the use of a macrolide is necessary, azithromycin is recommended as it is the least likely to interact with other drugs. The use of this class of antibiotics is restricted in many phase I and II trials.
The prevalence of cancer with tuberculosis infection is rare in developed countries but not uncommon in the developing world. Tuberculosis medications are strong inhibitors of the CYP450 isoenzyme system. Isoniazid is an inhibitor of CYP3A4, 2C9, and 2C19, whereas rifampin is an inducer of the same CYP450 enzymes as well as P-glycoprotein.10 Caution should be exercised when combining these drugs with chemotherapy agents that are metabolized through the CYP450 system (Table 2).61
Table 2.
Summary of the common drug interactions between supportive care and anticancer agents in pediatric oncology and recommendations for alternative agents
| Supportive Care Agent | Chemotherapy Agent | Outcome and Mechanism of Interaction | Alternatives | Ref |
|---|---|---|---|---|
| Phenytoin, Carbamazepine, Phenobarbital, | Anthracyclines, Cyclophosphamide, Ifosfamide, Epipodophyllotoxins, Tyrosine kinase inhibitors, Vinca alkaloids | Decreased effectiveness of chemotherapy agent through induction of CYP450 that catabolize anticancer agents | Use non-enzyme inducing anticonvulsants | 19–22 |
| Valproic acid (VPA) | Cisplatin | Increased cisplatin hematologic toxicity | - Use non-enzyme inhibiting anticonvulsants | 23,24 |
| Methotrexate | Decreased absorption of VPA | - Monitor valproic acid serum concentration | 23 | |
| Aprepitant | Anthracyclines, Epipodophyllotoxins, Cyclophosphamide, Ifosfamide, Dexamethasone, Tyrosine kinase inhibitors,Thiotepa | Increased toxicity of the chemotherapy agent through CYP450 inhibition | Use alternative antiemetic | 26,27,29 |
| Proton pump inhibitors | Methotrexate | Decreased methotrexate excretion via transporter inhibition | - Use ranitidine or sucralfate | 37 |
| Tyrosine kinase inhibitors (except imatinib) | Decreased absorption of the chemotherapy agent by decreasing gastric pH | - Separate drug administration by 3–4 hours | 47 | |
| Azole antifungals | Anthracyclines, Busulfan, Calcineurin inhibitors, Epipodophyllotoxins, Tyrosine kinase inhibitors, Vinca alkaloids | Increased toxicity through inhibition of CYP450 catabolism of active anticancer drugs | Use echinocandin antifungals or amphotericin B formulations | 12,48–51,62 |
| Amphotericin B | Cisplatin, High-dose methotrexate | Decreased excretion of renally cleared anticancer drug due to amphotericin nephrotoxicity | Use echinocandin antifungals | 56 |
| Penicillins | Methotrexate | Interference with tubular excretion of methotrexate | Consider an alternative non-penicillin antibiotic | 43,57 |
| TMP/SMX | Methotrexate | Delayed clearance of methotrexate | Some institutions hold TMP/SMX; others do not | 59,60 |
| NSAIDs | Methotrexate | Decreased excretion of methotrexate | Avoid NSAIDs, use opioids for pain if needed | 42,63,64 |
| Acyclovir (high dose) | Cisplatin, High-dose methotrexate | Additive nephrotoxicity | Do not combine if possible | 41 |
| Sargramostim, filgrastim | Vinca alkaloids | Severe atypical neuropathy | Some institutions hold factor support; others do | 65 |
| St. John’s Wort | Anthracyclines, Calcineurin inhibitors, Epipodophyllotoxins, Tyrosine kinase inhibitors, Vinca alkaloids | Decreased effectiveness of chemotherapy agent through induction of CYP450 induction | Avoid the use of St. John’s Wort during chemotherapy | 66 |
Abbreviations: VPA, valproic acid; trimethoprim-sulfamethoxazole (TMP/SMX); NSAIDs, non-steroidal anti-inflammatory drugs.
Antivirals
Most antivirals do not have any clinically significant interactions with chemotherapy. However, when used intravenously, acyclovir can induce acute renal failure in patients who are volume depleted.41 As recommended for other nephrotoxic agents, it is preferable not to use high-dose intravenous acyclovir in combination with antineoplastics that cause renal toxicity (e.g., cisplatin and high-dose methotrexate).
PAIN MANAGEMENT
Non-steroidal anti-inflammatory drugs
Non-steroidal anti-inflammatory drugs (NSAIDs) inhibit the formation of prostaglandins, leading to decreased renal perfusion and decreased methotrexate clearance when both agents are combined.42,63,64 Although the most common culprits tend to be indomethacin, ibuprofen, and naproxen, it is assumed that this is a class effect and that NSAIDs should be avoided when patients receive high-dose methotrexate.42 If pain management is required, opioids could be prescribed as alternative to NSAIDs. It is preferable to avoid using NSAIDs altogether in cancer patients as their effect on platelet aggregation could increase the risk of bleeding, and their antipyretic effect could mask a fever in neutropenic patients. Salicylates also increase MTX toxicity because they inhibit its active tubular secretion in the kidneys.
Acetaminophen should also be avoided because of its antipyretic effect and its potential to aggravate the hepatotoxicity of certain drugs such as imatinib.66
GRANULOCYTE COLONY STIMULATING FACTORS
In one study, the use of hematopoietic growth factors in patients with lymphoma was associated with severe atypical neuropathy of the lower extremities when combined with the vinca alkaloid vincristine.65 Symptoms of neuropathy included foot drop, decreased muscle strength, and constant sharp burning pain in the feet; with sparing of the upper extremities. Of the 28 patients who received factor support in this study, 11 (39%) developed severe neuropathy compared with only one (4%) of the 26 patients who did not receive any colony stimulating agent (p=0·0024).65 It was hypothesized that this neuropathy occurs because of an interference with nerve growth factors that are involved in nerve end repair. This finding has not been confirmed in other studies, and many pediatric oncology centers do not hold or restrict the use of the granulocyte stimulating growth factor analog filgrastim in patients receiving vincristine. However, prescribers should keep this interaction in mind if a patient develops severe neuropathy after receiving vincristine while on hematopoietic growth factors.
FOOD INTERACTIONS
Grapefruit and grapefruit juice
Grapefruit contains various furanocoumarins that inhibit CYP3A4.10 If given concomitantly with drugs that are metabolized by CYP3A4 in the intestinal wall prior to absorption, it results in increased drug levels. Interactions are most pronounced for drugs with low oral bioavailability that normally undergo significant pre-systemic metabolism. Grapefruit also weakly inhibits intestinal cell P-gp, decreasing the efflux of some absorbed drugs back into the gut lumen. Organic anion transporting polypeptide (OATP) is another transporter system affected by grapefruit. Unlike with CYP3A4 and P-gp, drugs handled by OATP may have decreased absorption when taken with grapefruit, possibly leading to decreased efficacy.
Patients who took cyclosporine concurrently with grapefruit juice had higher trough concentrations (87 ng/mL) than those who took cyclosporine with water (40 ng/mL, p=0·004).(38)67 On the other hand, the bioavailability of etoposide decreased from 73·2% to 52·4% when patients took grapefruit juice.68 Since grapefruit can alter the pharmacokinetics parameters of a number of drugs through its effects on drug transport and metabolism, patients and their parents should be counseled about not consuming grapefruit-containing products with chemotherapy agents that are substrates of CYP3A4 and P-gp (Panel 1).
Milk and dairy products
Mercaptopurine is metabolized by xanthine oxidase into inactive metabolites. Ingestion of foods that contain xanthine oxidase, such as milk and dairy products, can decrease the effectiveness of mercaptopurine.69 Many case reports have shown that the bioavailability of mercaptopurine is decreased when it is administered with milk (AUC 143 μmol/L·min without milk vs. 105 μmol/L·min with milk, p<0·01).69,70 Mercaptopurine is best absorbed when administered on an empty stomach, preferably at night.70 There should be a two-hour gap between the administration of mercaptopurine and milk and other dairy products.
HERBAL PRODUCT INTERACTIONS
The most common herbal product chemotherapy agents interact with is St. John’s Wort (Hypericum perforatum). The use of this agent to manage symptoms of depression is more widespread in adults than it is in children. St John’s Wort is a CYP3A4, 2D6, 2C9, and 1A2 inducer and can therefore increase the clearance of chemotherapy agents metabolized through any of these cytochromes (Panel 1).71 The use of St. John’s Wort with chemotherapy agents such as dasatinib, cyclophosphamide, irinotecan, and temsirolimus can decrease the activity of chemotherapy agents.72 For a complete review of interactions between herbal products and chemotherapy drugs, see the article by Sparreboom and colleagues.73
Many cancer patients and their families resort to nutritional supplements and holistic medicine. Although good nutrition should be encouraged and spiritual beliefs respected, it is important to discuss any multivitamin or supplement intake with patients. Multivitamins containing folic acid should be avoided in patients receiving methotrexate, as they can decrease its antineoplastic activity. The exact ingredients of several over-the-counter nutritional supplements may not be listed, and listed ingredients are seldom studied and can have unknown adverse effects or interactions. It is therefore crucial for healthcare providers to review all supplements that patients receive and to inform them of the potential undesired effects.
CONCLUSION
The last decade has witnessed the development of several novel agents that allow oncologists unprecedented flexibility in treating their patients. A thorough review of all the medications a patient receives should be conducted at every patient visit in order to document compliance, prevent drug duplication, discontinue medications that are no longer needed, and avoid undesirable drug interactions. This review provides a summary of the most common drug interactions in pediatric oncology. However, because of the constant release of new medications on the market, it is important for healthcare providers to check the most updated references before making recommendations about drug interactions. The most accessible way to check for a drug interaction with a single chemotherapeutic agent is to refer to the drug’s package inset (or summary of product characteristics). Online drug interaction softwares such as Lexi-Interact™ and Micromedex (available only through paid subscriptions) are increasingly being used by prescribers to identify clinically significant drug interactions especially since they allow the user to check for interactions between multiple agents at the same time. Prescribers can also refer to other online aids such as the P450 drug interaction table by the University of Indiana (http://medicine.iupui.edu/clinpharm/ddis/table.asp).
Finally, although it is beyond the scope of this article. prescribers should also remember that antineoplastic agents also interact with each other and can have additive toxicities or synergistic effects. Although uncommon in children, healthcare professionals should also check drug interactions between chemotherapy agents and agents that prolong the QT interval. A comprehensive list of medications that have the potential to prolong the QT interval can be found on the University of Arizona’s website (http://www.azcert.org/medical-pros/drug-lists/drug-lists.cfm).
Acknowledgments
This work was supported in part by a Cancer Center Support Grant (CA21765) from the National Cancer Institute and by the American Lebanese Syrian Associated Charities (ALSAC).
The funding source supports the author’s salary and technical support in manuscript formatting (figures, tables, etc).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Cyrine Haidar and Sima Jeha report no conflict of interest relevant to this manuscript.
Reference List
- 1.Balis FM, Holcenberg JS, Zimm S, Tubergen D, Collins JM, Murphy RF, et al. The effect of methotrexate on the bioavailability of oral 6-mercaptopurine. Clin Pharmacol Ther. 1987;41(4):384–7. doi: 10.1038/clpt.1987.45. [DOI] [PubMed] [Google Scholar]
- 2.Furman WL, Navid F, Daw NC, McCarville MB, McGregor LM, Spunt SL, et al. Tyrosine kinase inhibitor enhances the bioavailability of oral irinotecan in pediatric patients with refractory solid tumors. J Clin Oncol. 2009;27:4599–604. doi: 10.1200/JCO.2008.19.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.National Cancer Institute. A snapshot of Pediatric Cancer. 2008 Sep 30; http://planning.cancer.gov/disease/snapshots.shtml. [Accessed August 2009]
- 4.Blower P, de WR, Goodin S, Aapro M. Drug-drug interactions in oncology: why are they important and can they be minimized? Crit Rev Oncol Hematol. 2005;55:117–42. doi: 10.1016/j.critrevonc.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Kearns GL, bdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology–drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349:1157–67. doi: 10.1056/NEJMra035092. [DOI] [PubMed] [Google Scholar]
- 6.McLeod HL, Krynetski EY, Relling MV, Evans WE. Genetic polymorphism of thiopurine methyltransferase and its clinical relevance for childhood acute lymphoblastic leukemia. Leukemia. 2000;14:567–72. doi: 10.1038/sj.leu.2401723. [DOI] [PubMed] [Google Scholar]
- 7.Evans WE, Relling MV. Moving towards individualized medicine with pharmacogenomics. Nature. 2004;429:464–8. doi: 10.1038/nature02626. [DOI] [PubMed] [Google Scholar]
- 8.Beijnen JH, Schellens JHM. Drug interactions in oncology. Lancet Oncol. 2004;5:489–96. doi: 10.1016/S1470-2045(04)01528-1. [DOI] [PubMed] [Google Scholar]
- 9.Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286:487–91. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 10.Cavallari LM, Ellingrod VL, Kolesar JM. Pharmaco-Genomics Handbook. 2nd. Lexi-Comp, Inc; 2009. [Google Scholar]
- 11.Relling MV, Pui CH, Sandlund JT, Rivera GK, Hancock ML, Boyett JM, et al. Adverse effect of anticonvulsants on efficacy of chemotherapy for acute lymphoblastic leukaemia. Lancet. 2000;356:285–90. doi: 10.1016/S0140-6736(00)02503-4. [DOI] [PubMed] [Google Scholar]
- 12.Bermudez M, Fuster JL, Llinares E, Galera A, Gonzalez C. Itraconazole-related increased vincristine neurotoxicity: case report and review of literature. J Pediatr Hematol Oncol. 2005;27:389–92. doi: 10.1097/01.mph.0000172751.06286.5b. [DOI] [PubMed] [Google Scholar]
- 13.Kashuba AD. Drug metabolism, transport, and the influence of hepatic disease. In: Burton ME, Shaw LM, Schentag JJ, Evans WE, editors. Applied pharmacokinetics and pharmacodynamics. 4th. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 121–64. [Google Scholar]
- 14.Madadi P, Koren G. Pharmacogenetic insights into codeine analgesia: implications to pediatric codeine use. Pharmacogenomics. 2008;9:1267–84. doi: 10.2217/14622416.9.9.1267. [DOI] [PubMed] [Google Scholar]
- 15.Iyer L, King CD, Whitington PF, et al. Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J Clin Invest. 1998;101:847–54. doi: 10.1172/JCI915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho RH, Kim RB. Transporters and drug therapy: implications for drug disposition and disease. Clin Pharmacol Ther. 2005;78:260–77. doi: 10.1016/j.clpt.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 17.McLeod HL. Clinically relevant drug-drug interactions in oncology. Br J Clin Pharmacol. 1998;45:539–44. doi: 10.1046/j.1365-2125.1998.00719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Relling MV, Fairclough D, Ayers D, Crom WR, Rodman JH, Pui CH, et al. Patient characteristics associated with high-risk methotrexate concentrations and toxicity. J Clin Oncol. 1994;12:1667–72. doi: 10.1200/JCO.1994.12.8.1667. [DOI] [PubMed] [Google Scholar]
- 19.Alberts DS, Peng YM, Chen HS, Struck RF. Effect of phenobarbital on plasma levels of cyclophosphamide and its metabolites in the mouse. Br J Cancer. 1978;38:316–24. doi: 10.1038/bjc.1978.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murry DJ, Cherrick I, Salama V, et al. Influence of phenytoin on the disposition of irinotecan: a case report. J Pediatr Hematol Oncol. 2002;24:130–3. doi: 10.1097/00043426-200202000-00014. [DOI] [PubMed] [Google Scholar]
- 21.Rodman JH, Murry DJ, Madden T, Santana VM. Altered etoposide pharmacokinetics and time to engraftment in pediatric patients undergoing autologous bone marrow transplantation. J Clin Oncol. 1994;12:2390–7. doi: 10.1200/JCO.1994.12.11.2390. [DOI] [PubMed] [Google Scholar]
- 22.Villikka K, Kivisto KT, Maenpaa H, Joensuu H, Neuvonen PJ. Cytochrome P450-inducing antiepileptics increase the clearance of vincristine in patients with brain tumors. Clin Pharmacol Ther. 1999;66:589–93. doi: 10.1053/cp.1999.v66.103403001. [DOI] [PubMed] [Google Scholar]
- 23.Yap KY, Chui WK, Chan A. Drug interactions between chemotherapeutic regimens and antiepileptics. Clin Ther. 2008;30:1385–407. doi: 10.1016/j.clinthera.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 24.Bourg V, Lebrun C, Chichmanian RM, Thomas P, Frenay M. Nitroso-urea-cisplatin-based chemotherapy associated with valproate: increase of haematologic toxicity. Ann Oncol. 2001;12:217–9. doi: 10.1023/a:1008331708395. [DOI] [PubMed] [Google Scholar]
- 25.Curran MP, Robinson DM. Aprepitant: a review of its use in the prevention of nausea and vomiting. Drugs. 2009;69:1853–78. doi: 10.2165/11203680-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 26.Jarkowski A., III Possible contribution of aprepitant to ifosfamide-induced neurotoxicity. Am J Health Syst Pharm. 2008;65:2229–31. doi: 10.2146/ajhp080069. [DOI] [PubMed] [Google Scholar]
- 27.deJonge ME, Huitema AD, Holtkamp MJ, van Dam SM, Beijnen JH, Rodenhuis S. Aprepitant inhibits cyclophosphamide bioactivation and thiotepa metabolism. Cancer Chemother Pharmacol. 2005;56:370–8. doi: 10.1007/s00280-005-1005-4. [DOI] [PubMed] [Google Scholar]
- 28.Loos WJ, de Wit R, Freedman SJ, et al. Aprepitant when added to a standard antiemetic regimen consisting of ondansetron and dexamethasone does not affect vinorelbine pharmacokinetics in cancer patients. Cancer Chemother Pharmacol. 2007;59:407–12. doi: 10.1007/s00280-006-0359-6. [DOI] [PubMed] [Google Scholar]
- 29.McCrea JB, Majumdar AK, Goldberg MR, et al. Effects of the neurokinin1 receptor antagonist aprepitant on the pharmacokinetics of dexamethasone and methylprednisolone. Clin Pharmacol Ther. 2003;74:17–24. doi: 10.1016/S0009-9236(03)00066-3. [DOI] [PubMed] [Google Scholar]
- 30.Kris MG, Hesketh PJ, Somerfield MR, et al. American Society of Clinical Oncology guideline for antiemetics in oncology: update 2006. J Clin Oncol. 2006;24:2932–47. doi: 10.1200/JCO.2006.06.9591. [DOI] [PubMed] [Google Scholar]
- 31.Kishi S, Yang W, Boureau B, et al. Effects of prednisone and genetic polymorphisms on etoposide disposition in children with acute lymphoblastic leukemia. Blood. 2004;103:67–72. doi: 10.1182/blood-2003-06-2105. [DOI] [PubMed] [Google Scholar]
- 32.Cagnoni PJ, Matthes S, Day TC, Bearman SI, Shpall EJ, Jones RB. Modification of the pharmacokinetics of high-dose cyclophosphamide and cisplatin by antiemetics. Bone Marrow Transplant. 1999;24:1–4. doi: 10.1038/sj.bmt.1701832. [DOI] [PubMed] [Google Scholar]
- 33.Spector T. Inhibition of urate production by allopurinol. Biochem Pharmacol. 1977;26:355–8. doi: 10.1016/0006-2952(77)90191-5. [DOI] [PubMed] [Google Scholar]
- 34.Coffey JJ, White CA, Lesk AB, Rogers WI, Serpick AA. Effect of allopurinol on the pharmacokinetics of 6-mercaptopurine (NSC 755) in cancer patients. Cancer Res. 1972;32:1283–9. [PubMed] [Google Scholar]
- 35.Zimm S, Collins JM, O’Neill D, Chabner BA, Poplack DG. Inhibition of first-pass metabolism in cancer chemotherapy: interaction of 6-mercaptopurine and allopurinol. Clin Pharmacol Ther. 1983;34:810–7. doi: 10.1038/clpt.1983.254. [DOI] [PubMed] [Google Scholar]
- 36.Jeha S, Kantarjian H, Irwin D, et al. Efficacy and safety of rasburicase, a recombinant urate oxidase (Elitek), in the management of malignancy-associated hyperuricemia in pediatric and adult patients: final results of a multicenter compassionate use trial. Leukemia. 2005;19:34–8. doi: 10.1038/sj.leu.2403566. [DOI] [PubMed] [Google Scholar]
- 37.Beorlegui B, Aldaz A, Ortega A, Aquerreta I, Sierrasesumega L, Giraldez J. Potential interaction between methotrexate and omeprazole. Ann Pharmacother. 2000;34:1024–7. doi: 10.1345/aph.19094. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki K, Doki K, Homma M, et al. Co-administration of proton pump inhibitors delays elimination of plasma methotrexate in high-dose methotrexate therapy. Br J Clin Pharmacol. 2009;67:44–9. doi: 10.1111/j.1365-2125.2008.03303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crom WR, Pratt CB, Green AA, Champion JE, Crom DB, Stewart CF, et al. The effect of prior cisplatin therapy on the pharmacokinetics of high-dose methotrexate. J Clin Oncol. 1984;2:655–61. doi: 10.1200/JCO.1984.2.6.655. [DOI] [PubMed] [Google Scholar]
- 40.Salim A, Tan E, Ilchyshyn A, Berth-Jones J. Folic acid supplementation during treatment of psoriasis with methotrexate: a randomized, double-blind, placebo-controlled trial. Br J Dermatol. 2006;154:1169–74. doi: 10.1111/j.1365-2133.2006.07289.x. [DOI] [PubMed] [Google Scholar]
- 41.Izzedine H, Launay-Vacher V, Deray G. Antiviral drug-induced nephrotoxicity. Am J Kidney Dis. 2005;45:804–17. doi: 10.1053/j.ajkd.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 42.Tracy TS, Krohn K, Jones DR, Bradley JD, Hall SD, Brater DC. The effects of a salicylate, ibuprofen, and naproxen on the disposition of methotrexate in patients with rheumatoid arthritis. Eur J Clin Pharmacol. 1992;42:121–5. doi: 10.1007/BF00278469. [DOI] [PubMed] [Google Scholar]
- 43.Williams WM, Chen TS, Huang KC. Effect of penicillin on the renal tubular secretion of methotrexate in the monkey. Cancer Res. 1984;44:1913–7. [PubMed] [Google Scholar]
- 44.Basin KS, Escalante A, Beardmore TD. Severe pancytopenia in a patient taking low dose methotrexate and probenecid. J Rheumatol. 1991;18:609–10. [PubMed] [Google Scholar]
- 45.Callaghan R, Crowley E, Potter S, Kerr ID. P-glycoprotein: so many ways to turn it on. J Clin Pharmacol. 2008;48:365–78. doi: 10.1177/0091270007311568. [DOI] [PubMed] [Google Scholar]
- 46.Kim RB. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev. 2002;34:47–54. doi: 10.1081/dmr-120001389. [DOI] [PubMed] [Google Scholar]
- 47.Egorin MJ, Shah DD, Christner SM, et al. Effect of a proton pump inhibitor on the pharmacokinetics of imatinib. Br J Clin Pharmacol. 2009;68:370–4. doi: 10.1111/j.1365-2125.2009.03466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eiden C, Palenzuela G, Hillaire-Buys D, et al. Posaconazole-increased vincristine neurotoxicity in a child: a case report. J Pediatr Hematol Oncol. 2009;31:292–5. doi: 10.1097/MPH.0b013e31819b9d01. [DOI] [PubMed] [Google Scholar]
- 49.Kamaluddin M, McNally P, Breatnach F, et al. Potentiation of vincristine toxicity by itraconazole in children with lymphoid malignancies. Acta Paediatr. 2001;90:1204–7. doi: 10.1080/080352501317061675. [DOI] [PubMed] [Google Scholar]
- 50.Romero AJ, Le PP, Nilsson LG, Wood N. Effect of voriconazole on the pharmacokinetics of cyclosporine in renal transplant patients. Clin Pharmacol Ther. 2002;71:226–34. doi: 10.1067/mcp.2002.121911. [DOI] [PubMed] [Google Scholar]
- 51.Gomez DY, Wacher VJ, Tomlanovich SJ, Hebert MF, Benet LZ. The effects of ketoconazole on the intestinal metabolism and bioavailability of cyclosporine. Clin Pharmacol Ther. 1995;58:15–9. doi: 10.1016/0009-9236(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 52.Sud K, Singh B, Krishna VS, et al. Unpredictable cyclosporine–fluconazole interaction in renal transplant recipients. Nephrol Dial Transplant. 1999;14:169–703. doi: 10.1093/ndt/14.7.1698. [DOI] [PubMed] [Google Scholar]
- 53.Pai MP, Allen S. Voriconazole inhibition of tacrolimus metabolism. Clin Infect Dis. 2003;36:1089–91. doi: 10.1086/374252. [DOI] [PubMed] [Google Scholar]
- 54.Fanta S, Jonsson S, Backman JT, Karlsson MO, Hoppu K. Developmental pharmacokinetics of ciclosporin–a population pharmacokinetic study in paediatric renal transplant candidates. Br J Clin Pharmacol. 2007;64:772–84. doi: 10.1111/j.1365-2125.2007.03003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fukudo M, Yano I, Masuda S, et al. Population pharmacokinetic and pharmacogenomic analysis of tacrolimus in pediatric living-donor liver transplant recipients. Clin Pharmacol Ther. 2006;80:331–45. doi: 10.1016/j.clpt.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 56.Bergstrom P, Johnsson A, Cavallin-Stahl E, Bergenheim T, Henriksson R. Effects of cisplatin and amphotericin B on DNA adduct formation and toxicity in malignant glioma and normal tissues in rat. Eur J Cancer. 1997;33:153–9. doi: 10.1016/s0959-8049(96)00339-5. [DOI] [PubMed] [Google Scholar]
- 57.Titier K, Lagrange F, Pehourcq F, Moore N, Molimard M. Pharmacokinetic interaction between high-dose methotrexate and oxacillin. Ther Drug Monit. 2002;24:570–2. doi: 10.1097/00007691-200208000-00018. [DOI] [PubMed] [Google Scholar]
- 58.Haas A, Anderson L, Lad T. The influence of aminoglycosides on the nephrotoxicity of cis-diamminedichloroplatinum in cancer patients. J Infect Dis. 1983;147:363. doi: 10.1093/infdis/147.2.363. [DOI] [PubMed] [Google Scholar]
- 59.Govert JA, Patton S, Fine RL. Pancytopenia from using trimethoprim and methotrexate. Ann Intern Med. 1992;117:877–8. doi: 10.7326/0003-4819-117-10-877_2. [DOI] [PubMed] [Google Scholar]
- 60.Ferrazzini G, Klein J, Sulh H, Chung D, Griesbrecht E, Koren G. Interaction between trimethoprim-sulfamethoxazole and methotrexate in children with leukemia. J Pediatr. 1990;117:823–6. doi: 10.1016/s0022-3476(05)83351-7. [DOI] [PubMed] [Google Scholar]
- 61.Fardel O, Lecureur V, Loyer P, Guillouzo A. Rifampicin enhances anti-cancer drug accumulation and activity in multidrug-resistant cells. Biochem Pharmacol. 1995;49:1255–60. doi: 10.1016/0006-2952(95)00045-2. [DOI] [PubMed] [Google Scholar]
- 62.Mori T, Aisa Y, Kato J, Nakamura Y, Ikeda Y, Okamoto S. Drug interaction between voriconazole and calcineurin inhibitors in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transplant. 2009;44:371–4. doi: 10.1038/bmt.2009.38. [DOI] [PubMed] [Google Scholar]
- 63.Cassano WF. Serious methotrexate toxicity caused by interaction with ibuprofen. Am J Pediatr Hematol Oncol. 1989;11:481–2. [PubMed] [Google Scholar]
- 64.Frenia ML, Long KS. Methotrexate and nonsteroidal antiinflammatory drug interactions. Ann Pharmacother. 1992;26:234–7. doi: 10.1177/106002809202600219. [DOI] [PubMed] [Google Scholar]
- 65.Weintraub M, Adde MA, Venzon DJ, et al. Severe atypical neuropathy associated with administration of hematopoietic colony-stimulating factors and vincristine. J Clin Oncol. 1996;14:935–40. doi: 10.1200/JCO.1996.14.3.935. [DOI] [PubMed] [Google Scholar]
- 66.Ridruejo E, Cacchione R, Villamil AG, Marciano S, Gadano AC, Mando OG. Imatinib-induced fatal acute liver failure. World J Gastroenterol. 2007;13:6608–111. doi: 10.3748/wjg.v13.i48.6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brunner LJ, Pai KS, Munar MY, Lande MB, Olyaei AJ, Mowry JA. Effect of grapefruit juice on cyclosporin A pharmacokinetics in pediatric renal transplant patients. Pediatr Transplant. 2000;4:313–21. doi: 10.1034/j.1399-3046.2000.00136.x. [DOI] [PubMed] [Google Scholar]
- 68.Reif S, Nicolson MC, Bisset D, Reid M, Kloft C, Jaehde U, et al. Effect of grapefruit juice intake on etoposide bioavailability. Eur J Clin Pharmacol. 2002;58:491–4. doi: 10.1007/s00228-002-0495-9. [DOI] [PubMed] [Google Scholar]
- 69.de Lemos ML, Hamata L, Jennings S, Leduc T. Interaction between mercaptopurine and milk. J Oncol Pharm Pract. 2007;13:237–40. doi: 10.1177/1078155207080802. [DOI] [PubMed] [Google Scholar]
- 70.Riccardi R, Balis FM, Ferrara P, Lasorella A, Poplack DG, Mastrangelo R. Influence of food intake on bioavailability of oral 6-mercaptopurine in children with acute lymphoblastic leukemia. Pediatr Hematol Oncol. 1986;3:319–24. doi: 10.3109/08880018609031233. [DOI] [PubMed] [Google Scholar]
- 71.Gurley BJ, Gardner SF, Hubbard MA, et al. Cytochrome P450 phenotypic ratios for predicting herb-drug interactions in humans. Clin Pharmacol Ther. 2002;72:276–87. doi: 10.1067/mcp.2002.126913. [DOI] [PubMed] [Google Scholar]
- 72.Mathijssen RH, Verweij J, de BP, Loos WJ, Sparreboom A. Effects of St. John’s wort on irinotecan metabolism. J Natl Cancer Inst. 2002;94:1247–9. doi: 10.1093/jnci/94.16.1247. [DOI] [PubMed] [Google Scholar]
- 73.Sparreboom A, Cox MC, Acharya MR, Figg WD. Herbal remedies in the United States: potential adverse interactions with anticancer agents. J Clin Oncol. 2004;22:2489–503. doi: 10.1200/JCO.2004.08.182. [DOI] [PubMed] [Google Scholar]