

Abstract
Our recently discovered, selective, on-resin route to N(τ)-alkylated imidazolium-containing histidine residues affords new strategies for peptide mimetic design. In our current work, we demonstrate the use of this chemistry to prepare a series of macrocyclic phosphopeptides, in which imidazolium groups serve as ring-forming junctions. Interestingly, these cationic moieties subsequently serve to charge-mask the phosphoamino acid group that directed their formation. Neighbor-directed histidine N(τ)-alkylation opens the door to new families of phosphopeptidomimetics for use in a range of chemical biology contexts.
Keywords: Plk1, polo kinase, polo-box domain, crystal structure, cationic dialkyl histidine, peptide macrocycles, phosphopeptides
INTRODUCTION
Modification of amino acid residues in biologically active peptides is a powerful tool for peptidomimetic design. These efforts can be greatly facilitated by transforming reactions that are selective and can be performed on solid-phase resin. Histidine is unique among the coded amino acids in having an imidazole ring with its two associated unsubstituted nitrogen atoms. Yet, exploiting histidine’s full potential for constructing peptidomimetics has been limited by the challenges of differentially functionalizing its N(π)- and N(τ)-positions. We have recently discovered the unanticipated ability of phosphoamino acids to direct the N(τ)-alkylation of proximal histidine residues already bearing an N(π)-substituent.1,2 These reactions are highly selective and can be conducted on fully protected peptides bound to solid-phase resin using Mitsunobu chemistries. The facile access to N(π), N(τ)-derivatized histidine residues affords as yet unexplored access to new families of peptidomimetics.
In the first practical application of this technology, we turned our attention to the sequence, “Ac-Pro-Leu-His-Ser-pThr-amide” (1, Figure 1), which is based on the C-proximal residues of the polo-box interacting protein 1 (PBIP1).3 The polo-like family of serine/threonine kinases (Plks) plays critical roles in cell cycle regulation, and antagonists of Plk1 are being developed as potential anticancer agents. The Plks are characterized as having polo-box domains (PBDs), which recognize phosphothreonine (pThr) and phosphoserine (pSer)-containing sequences. In designing Plk1 PBD-binding antagonists, we had originally started with peptide 1 and found that adding long-chain alkylphenyl groups to the N(π) position of the His residue significantly enhances PBD-binding affinity.4 It was in subsequent studies with peptides of the form, “Ac-Pro-Leu-His*-Ser-pThr-amide” (2, Figure 1), where “His*” indicates the presence of an N(τ)–(CH2)8Ph group, that we first observed the preferential alkylation at the His N(τ)-position under Mitsunobu conditions.1 The formation of N(π), N(τ) bis-alkyl-His species was providential, since these peptides showed little or no diminution of PBD-binding affinity in in vitro assays, yet cellular potencies increased. This latter fact points to greater membrane transit made possible by intramolecular “charge-masking” of one anionic pThr phosphoryl hydroxyl by the bis-alkyl-His imidazolium cation.5
Figure 1.
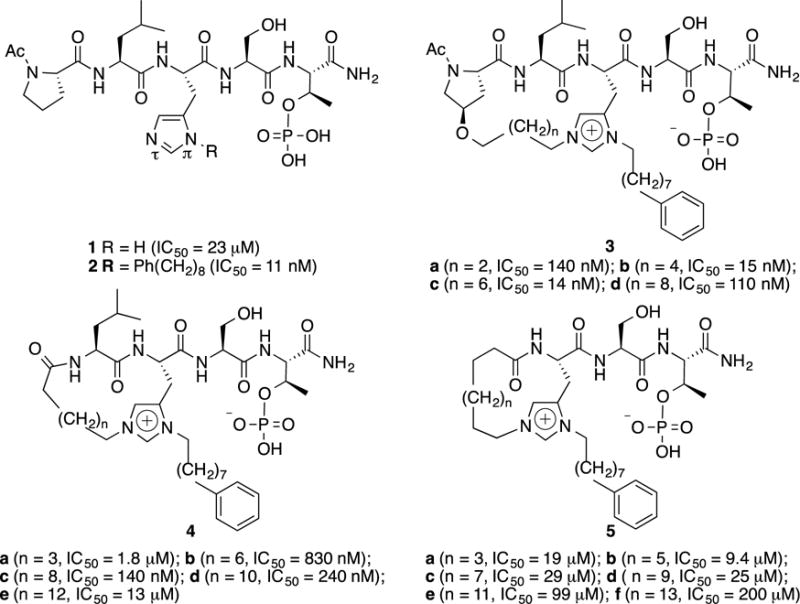
Structures of target PBD-binding peptides with Plk1 PBD-binding affinities.
We anticipated that N(τ)–alkylation could serve as a specific means of introducing structural diversification into the His imidazole side chain,2 and that such species could be employed for the construction of new classes of macrocyles. The importance of macrocyclization for constraining flexible ligands is widely recognized,6–9 and using the bifurcated His residue for ring closure could provide advantages in the design of macrocyclic PBD-binding peptides relative to more standard methodologies.10
RESULTS AND DISCUSSION
In order to examine the application of phospho-dependent His N(τ)–alkylation for macrocyclization, we designed three families of cyclized variants based on the parent sequence 1 (3 – 5, Figure 1). We incorporated the requisite His* residues under standard Fmoc conditions using our recently reported reagent.11 Macrocyclization was accomplished using ring-closing metathesis (RCM),12 which required preparing the appropriate open-chain bis-alkenyl precursors. Amino-terminal alkenyl chains were appended either as ethers of R-4-hydroxy-Pro13 (for peptides 3) or as N-alkenoyl amides of Leu (peptides 4) and His* (peptides 5) (Figure 1).
Synthetic Approaches to Macrocycles 3, 4 and 5
Macrocycles 3a – 3d
Type 3 macrocycles were prepared in 18, 20, 22 and 24-member ring sizes (3a–3d, respectively, Figure 1). Resin-bound peptides having terminal Pro residues containing either an R-4-(4-pentenyloxy) or R-4-(2-propenyloxy) group (peptides 6a and 6b, respectively) were subjected to pThr-directed Mitsunobu alkylations of their His* N(τ)–nitrogens using a series of alkenyl alcohols.1,2 This afforded the requisite resin-bound bis-alkyl-His-containing peptides (7a–7d, Figure 2). On-resin microwave-assisted RCM ring closure using a common Pro residue having a 3-(4-pentenyloxy) group and the appropriate length N(τ)–alkenyl group was possible for the 20, 22 and 24-member macrocycles (8b–8d, respectively, Figure 2). Microwave irradiation can decrease reaction times, enhance catalyst performance and improve overall yields of RCM reactions.14–19 Typically, these reactions are conducted in a solvent combination of CH2Cl2: dimethylformamide (DMF) (10:1) using 2nd Generation Grubbs20 or Hoveyda-Grubbs21 catalyst. The DMF facilitates peptide solubility and absorbs microwave radiation, while CH2Cl2 swells the resin.
Figure 2. Reagents and Conditions.

i) ROH (10 eq.), PPh3 (10.0 eq.), DEAD (10.0 eq.), CH2Cl2, 4 h; ii) Hoveyda-Grubbs 2nd generation cat. (0.3 eq.), CH2Cl2/DMF (10:1, degassed), microwave, 120 °C, 30 min; iii) NBSH (20 eq.), Et3N (40 eq.), CH2Cl2, microwave, 50 °C, 30min; iv) TFA/H2O/TIS = 95: 2.5: 2.5, 4 h; v) 1% TFA in CH2Cl2; vi)) Hoveyda-Grubbs 2nd generation cat. (0.5 eq.), CH2Cl2/DMF (10:1, degassed), room temp., 24 h (two-times); vii) H2, 10% Pd•C, THF/DMF (10:1), overnight.
During initial attempts to prepare the shorter 18-member macrocycle (3a) using 2-propenyl groups on both the 3-oxy-Pro and His*- N(τ) positions, it was found that on resin microwave-assisted RCM macrocyclization resulted in the loss of the Pro 3-alkoxy group, as was also the case when a Pro 3-butenyloxy group was employed. We rationalized that these losses could reflect Ru-catalyzed isomerization to a more labile 2-butenyl species.22 We circumvented these difficulties by using a 1-cinnamoyl group at the N(τ)–position (7a), which served as a 2-propenyl surrogate.23 In this fashion we obtained the desired 18-membered macrocycle (11) by solution-phase RCM of the precursor peptide (10) at room temperature following cleavage from the resin (Figure 2).
Because cyclization under RCM conditions typically yields mixtures of E/Z products,24,25 we eliminated geometric isomers by reducing the ring-closing double bonds. We examined diimine reduction, since this is compatible with both solution and solid-support protocols.26,27 We found that on-resin reduction of the 20-member macrocycle (8b) using 2-nitrobenzenesulfonohydrazine (NBSH) and NEt326,27 resulted in poor conversion to the desired saturated macrocycle (9b). However, for the larger macrocycle ring sizes (22 and 24 member; 8c and 8d, respectively), we achieved good conversion to the desired products (9c and 9d, respectively) using NBSH (20 equiv) and NEt3 (40 equiv) in CH2Cl2 with microwave irradiation (50 °C, 30 min) Subsequently, we found that cleavage of resin-bound 8b to give 11b and hydrogenation in solution using H2 over 10% Pd•C in tetrahydrofuran (THF):DMF (10:1) at room temperature (overnight) gave very high-yield conversion to the saturated product (3b). Ring closure required for the synthesis of the shorter 18-member macrocycle (3a) could not be performed on-resin, due to lability of the Pro allyloxy group under the microwave conditions needed for the on-resin RCM reaction. However, RCM cyclization could be achieved in solution at room temperature on 10 following cleavage of 7a from the resin. This yielded the unsaturated macrocycle (11a), which was hydrogenated (H2 over 10% Pd•C) to provide the desired final macrocycle 3a (Figure 2).
Macrocycles 4a – 4e
Type 4 macrocycles, based on the “Leu-His*-Ser-pThr” motif, were prepared similar to that described above for type 3 macrocycles. Precursor open-chain peptides for 18, 20, 22 and 24-member macrocyles (4b–4e, respectively) were assembled on-resin by initial N-terminal acylation with either hex-5-enoic acid (12a) or oct-7-enoic acid (12b). The peptides were subjected to pThr-directed Mitsunobu N(τ)–alkylation using the appropriate alkenyl alcohols, pent-4-ene-1-ol (for 13b and 13c); hept-6-ene-1-ol (for 13d) and none-8-ene-1-ol (for 13e, Figure 3). On-resin RCM ring closures to the corresponding peptides 14b–14e were carried out as indicated above, using Hoveyda-Grubbs 2nd generation catalyst in CH2Cl2:DMF (10:1) with microwave irradiation (120 °C, 30 min). Diimine-mediated on-resin reduction of the resulting ring-forming double bonds yielded 15b–15e [(20 and 40 equiv of NBSH and Et3N, respectively in CH2Cl2 with microwave irradiation (50 °C, 30 min)]. The final macrocyclic peptides (4b–4e) were obtained by resin cleavage and high performance liquid chromatography (HPLC) purification (Figure 3). We found in the attempted synthesis of the 16-member type 4 macrocycle using N-terminal acylation with but-3-enoic acid, that on-resin microwave-assisted RCM macrocylization resulted in ring-contraction (loss of –CH2–). This may have occurred by isomerization of the but-3-enoyl amide to the conjugated but-2-enoyl species prior to ring closure. Subsequent on-resin diimine reduction of the smaller, 15-member macrocycle also failed. However, cleavage of the peptide from the resin and followed by solution-phase reduction (H2 over 10% Pd•C) gave the 15-member macrocycle (4a).
Figure 3. Reagents and Conditions.
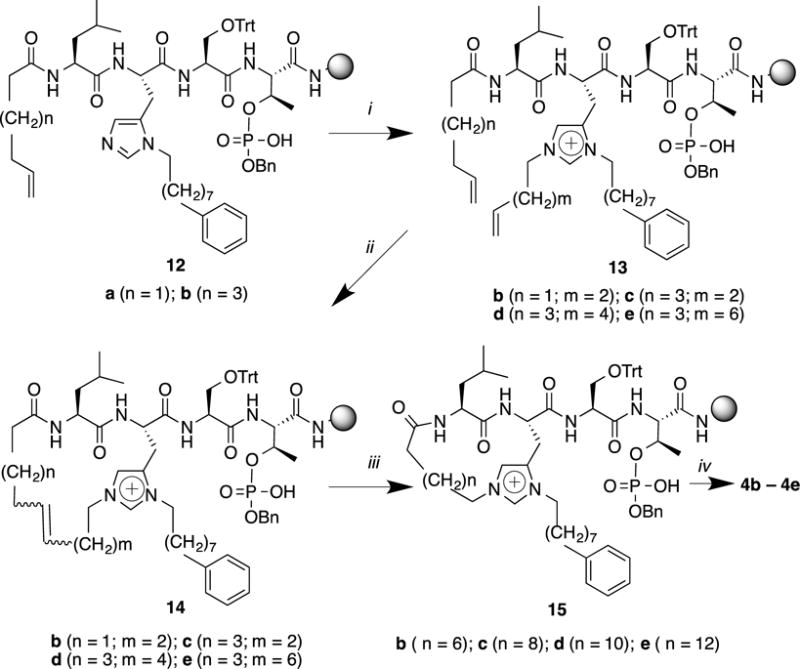
i) ROH (10 eq.), PPh3 (10.0 eq.), DEAD (10.0 eq.), CH2Cl2, 4 h; ii) Hoveyda-Grubbs 2nd generation cat. (0.3 eq.), CH2Cl2/DMF (10:1, degassed), microwave, 120 °C, 30 min; iii) NBSH (20 eq.), Et3N (40 eq.), CH2Cl2, microwave, 50 °C, 30 min; iv) TFA/H2O/TIS = 95: 2.5: 2.5, 4 h.
Macrocycles 5a – 5f
Syntheses of the “His*-Ser-pThr”–based type 5 macrocycles were achieved entirely on solid-phase. Ring sizes were determined by on–resin pThr-directed N(τ)–alkylation of peptides (16a–16c) with appropriate-length alcohols to give the corresponding series of bis-alkenyl-containing peptides (17a–17f) (Figure 4). Microwave irradiation was employed both for RCM macrocyclization (120 °C, 30 min) to yield the corresponding unsaturated macrocycles (18a–18f), and for the subsequent diimine double bond reduction (50 °C, 30 min). Cleavage of the saturated macrocycles (19a–19f) gave the desired final products 5a–5f, having ring sizes of 14-, 16-, 18-, 20-, 22- and 24-members, respectively (Figure 4).
Figure 4. Reagents and Conditions.

i) ROH (10 eq.), PPh3 (10.0 eq.), DEAD (10.0 eq.), CH2Cl2, 4 h; ii) Hoveyda-Grubbs 2nd generation cat. (0.3 eq.), CH2Cl2/DMF (10:1, degassed), microwave, 120 °C, 30 min; iii) NBSH (20 eq.), Et3N (40 eq.), CH2Cl2, microwave, 50 °C, 30 min; iv) TFA/H2O/TIS = 95: 2.5: 2.5, 4 h.
Mass Spectral Characterization
We had previously verified the structures of on-resin Mitsunobu His* N(τ)-alkylation products by high-resolution liquid chromatography – tandem mass spectrometry (LC/MS/MS) analyses using electrospray ionization (ESI).2 We performed similar analyses for peptides of the current series. For all macrocyclic final products, both the protonated peptide (MH+) and the double-protonated peptide (M+2H)2+ ions were observed and had masses consistent with the expected elemental composition (see Supporting Information). Interestingly, the (M+2H)2+ ions of these macrocycles were of consistently lower relative abundance than those observed in the ESI mass spectra of their linear analogues reported earlier.2 The increased spatial constraint of these molecules may make multiple charging less energetically favored than with the linear pThr-containing pentapeptide derivatives. LC/MS/MS analysis also confirmed the presence of a free phosphate moiety and the absence of any macrocyclic derivatives resulting from RCM through the isomeric phosphate ester.
Typically, the primary low energy MS/MS fragmentation observed for pSer- and pThr-containing peptides is loss of neutral phosphoric acid (H3PO4) or HPO3, with a much reduced fragmentation of the amide bond peptide backbone to generate b and y sequence-indicating ions.28 The abundance of these ions depends on the charge-state of the precursor ion, the structure of the peptide and the nature of the collision-induced dissociation (CID).29,30 For the macrocyclic phosphopeptide 3c, which contains a ring-constrained, cationic bis-alkyl-His and a free phosphate, a predominant and diagnostic CID-mediated neutral loss should be that of H3PO4 (98 da), resulting in a product ion at m/z 891.5702. Other prominent neutral losses are expected to be those that are related, such as MH+-HPO3 and MH+-(H3PO4 + NH3), as well as MH+-NH3 from the C-terminal amide. These product ions are all observed in the MS/MS spectrum resulting from CID of the MH+ of 3c (Figure 5 and Supporting Information). The bn and yn sequence-indicating ions for the macrocyclic pThr-containing peptide derivatives are also expected to be somewhat different from those of the linear analogues. Thus in the case of 3c, the bn ions that are seen are those that incorporate the cyclic His*-imidazolium cation, namely b’3 and b’4 (Figure 5). (Note that the b’n ions observed for this and other macrocycles are designated by a prime to indicate a localized charge and numbered on the basis of amino acid residues in the cyclic His*-imidazolium cation.) Furthermore, because the macrocycle is formed through His*, no y3 ions are seen for 3c, since cleavage at the Leu-His* amide bond no longer results in a distinct yn product ion. Thus, taken all together, the MS/MS spectrum of 3c (Figure 5) and those of the other macrocyclic derivatives (Supporting Information) support structures containing a free phosphate and a macrocyclic ring formed through a His*-imidazolium cation.
Figure 5.
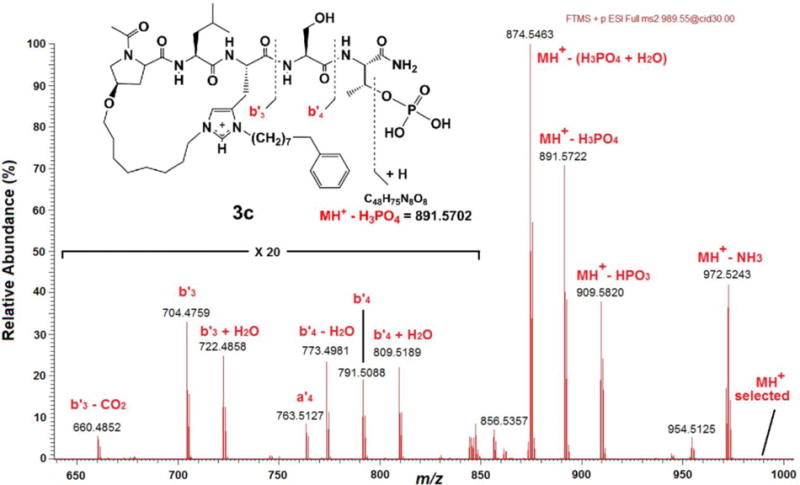
Partial, high resolution, CID MS/MS mass spectrum of the MH+ of 3c. The presence of a free phosphate moiety is indicated by the product ions corresponding to MH+-HPO3, MH+-H3PO4, and MH+-(H3PO4 + NH3). Insert: Structure and expected CID fragmentation of the MH+ (m/z 989.5471) of 3c.
Determination of Plk1 PBD-binding Affinities
PBIP1 is a physiological substrate of Plk1, that once phosphorylated at Thr78, forms a Plk1–PBIP1 complex through the interaction between the PBD of Plk1 and the pThr78 of PBIP1.31 Peptide 1 represents residues 74–78 of PBIP1. In order to determine their Plk1 PBD-binding affinities, the synthetic macrocyclic peptides were evaluated in an enzyme-linked immunosorbent assay (ELISA)-based assay that measured their ability to compete with an immobilized pThr78-derived peptide for binding to human influenza hemagglutinin-green fluorescent protein (HA-GFP)- fused Plk1 expressed in HEK 293A cells. Consistent with our previous findings, introducing the N(π)–(CH2)8Ph group (peptide 2, IC50 = 12 nM) significantly increased binding affinity relative to the parent peptide (1, IC50 = 23 μM) (Table 1).4 In the type 3 macrocycles, peptides having 20– and 22–membered ring sizes (3b, IC50 = 15 nM and 3c, IC50 = 14 nM, respectively) bound with greater affinities than those having 18– and 24–member ring sizes (3a, IC50 = 140 nM and 3d, IC50 = 110 nM, respectively). For the type 4 macrocycles, affinities increased sequentially in going from the 15–membered to the 20–membered ring sizes (from 4a, IC50 = 1.8 μM to 4c, IC50 = 140 nM) and then decreased progressively as the ring size increased to 24–member (4e, IC50 = 13 μM). None of the type 5 macrocycles bound with good affinity (Table 1).
Table 1.
Plk1 PBD IC50 values measured in an ELISA competition assay.a
| No. | Ring Size | IC50 (μM) |
|---|---|---|
| 1 | – | 29 |
| 2 | – | 0.011 |
| 3a | 18 | 0.14 |
| 3b | 20 | 0.015 |
| 3c | 22 | 0.014 |
| 3d | 24 | 0.11 |
| 4a | 15 | 1.8 |
| 4b | 18 | 0.83 |
| 4c | 20 | 0.14 |
| 4d | 22 | 0.24 |
| 4e | 24 | 13 |
| 5a | 14 | 19 |
| 5b | 16 | 9.4 |
| 5c | 18 | 29 |
| 5d | 20 | 25 |
| 5e | 22 | 99 |
| 5f | 24 | 200 |
Values were obtained in an ELISA-based assay that measured the ability of peptides to compete with an immobilized pT78 peptide for binding to HEK 293A cell lysate-derived GFP-HA-fused Plk1.
X-Ray Co-crystal Studies
To understand the effects of macrocyclization on PBD binding modes, we obtained the Plk1 PBD co-crystal structures of the highest affinity macrocycles (3b, 3c, and 4c) and compared these with the previously reported structure of the PBD-bound open-chain parent peptide 2 [Protein Data Bank (PDB) accession code 3RQ7].4 By superimposing all structures using standard protocols included in MolSoft ICM Pro modeling software,32,33 we found that folding of the flexible ring-closing polymethylene chains resulted in striking correspondence with 2 in the binding orientations of the peptide portions of all macrocycles (Figure 6). Interactions were preserved within the pThr-binding pocket with the key phosphoryl-binding residues His538 and Lys540.3,34 Consistent with previous reports that a hydrophobic region formed by the Trp414 and Phe535 residues are important in ligand binding,3,34,35 we observed that for the current complexes, the largest contact interactions occurred with the Trp414 residue. The contact surface areas for 3b and 3c were larger than with the parent peptide 2. Increased contact between the ligand Pro carbonyl and the Arg516 was also observed for 3b. Contact with the Phe535 residue was increased for 3c, where interactions with the ligand Pro pyrrolidine C4 and C5 methylenes as well as proximal components of the ring-closing chain were possible. In contrast, a dramatic loss of contact with Phe535 was incurred for 4c due to the absence of a ligand Pro residue.
Figure 6.

X-ray crystal structures of Plk1 PBD-bound ligands. (A) Protein ribbon rendition of PBD-bound 2 (PDB: 3RQ7) with key interacting protein residues shown and the structures of PBD-bound macrocycles 3b, 3c and 4c superimposed. (B) Enlargement of overlaid PBD-bound peptides shown in (A). For both (A) and (B) ligand coloring is as indicated.
MATERIALS AND METHODS
Materials
Fmoc-Leu-OH, Fmoc-Ser(Trt)-OH and Fmoc-Thr(PO(OBzl)OH)-OH, NovaSyn®TGR amide resin and NovaSyn® TG Siber resin were purchased from Novabiochem. Allyl alcohol, 3-butenoic acid, 3-buten-1-ol, cinnamyl alcohol, 9-decenoic acid, 9-decen-1-ol, 5-hexenoic acid, 5-hexen-1-ol, 7-octenoic acid and 4-penten-1-ol were purchased from Sigma-Aldrich. 6-Hepten-1-ol, 7-octen-1-ol and 8-nonen-1-ol were purchased from Tokyo Chemical Industry Co., Ltd. (TCI). Fmoc-His(N(π)-(CH2)8Ph)-OH,11 N-Fmoc (2S,4R)-4-(allyloxy)pyrrolidine-2-carboxylic acid,36 N-Fmoc (2S,4R)-4-(pent-4-en-1-yloxy)pyrrolidine-2-carboxylic acid36 and NBSH26 were prepared according to the indicated literature procedures.
Solid-phase Peptide Synthesis
Resin-bound open chain peptides 6a and 6b (Figure 2) were synthesized on NovaSyn® TG Siber resin, while peptides 12a and 12b (Figure 3) and 16(a – c) (Figure 4) were synthesized on NovaSyn®TGR resin following standard Fmoc-based solid-phase protocols using N-methyl-2-pyrrolidone (NMP) as solvent. 1-O-Benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate (HBTU) (5.0 equiv), hydroxybenzotriazole (HOBT) (5.0 equiv) and N,N-diisopropylethylamine (DIPEA) (10.0 equiv) were used as coupling reagents. Amino terminal acetylation was achieved using 1-acetylimidazole (10 equiv) in DMF (4h) (for 6a and 6b), and appropriate alkenoic acids (5 equiv), HBTU (5.0 equiv), HOBt (5.0 equiv) and DIPEA (10 equiv) in NMP (2 h) [for 12a, 12b and 16 (a-c)].
Detailed Solid-phase Protocol (Synthesis of Peptide 6a)
NovaSyn® TG Siber resin (0.21 mmol/g, 476 mg, 0.1 mmol) was swelled (CH2Cl2, 5 mL, 1 h) and then washed thoroughly with DMF (5 mL × 5). The N-Fmoc group was removed by treating with 20% piepridine in DMF (4 mL, 20 minutes) and the resin was then washed with DMF (5 mL × 6). A solution of Fmoc-Thr(PO(OBzl)OH)-OH (256 mg, 0.5 mmol), HBTU (195 mg, 0.5 mmol), HOBt (68 mg, 0.5 mmol) and DIPEA (0.174 mL, 1.0 mmol) in NMP (5 mL) was added and the resin was mixed by gentle rotation (2 h). The sequence of Fmoc-deprotection and coupling was completed with appropriate protected amino acids as specified in Materials. Following deprotection of the final residue, acetylation of the terminal amine was achieved using 1-acetylimidazole (110 mg, 1.0 mmol) in DMF (5 mL) (4 h). The resin was then washed with DMF (5 mL × 5); MeOH (5 mL × 5); CH2Cl2 (5 mL × 5) and diethyl ether and the resin was dried under high vacuum to afford resin-bound peptide 6a (0.1 mmol). The syntheses of resin-bound peptides 6b, 12a, 12b, 16a, 16b and 16c were achieved similarly using appropriate amino acids as indicated in Materials.
Resin Cleavage
NovaSyn® TGR Resin: Resin-bound peptide was cleaved by treatment with trifluoroacetic acid (TFA): triispropylsilane (TIS) : H2O (95 : 2.5 : 2.5) at room temperature (4 h). The resin was removed by filtration and the filtrate was concentrated under vacuum. NovaSyn® TG Siber Resin: Resin bound peptide was cleaved by treatment with 1% TFA in CH2Cl2 at room temperature (5 min) and the resin was filtered and the filtrate collected. This procedure was repeated (5 x) and the combined filtrates were concentrated under vacuum.
HPLC Purification
Crude peptides (from 0.1 mmol resin) were dissolved in 50% aqueous CH3CN (5 mL) and purified by reverse phase preparative HPLC using a Phenomenex C18 column (21 mm dia × 250 mm, cat. no: 00G-4436-P0) with a linear gradient from 30% aqueous CH3CN (0.1% TFA) to 100% CH3CN (0.1% TFA) over 30 min at a flow rate of 10.0 mL/min. Lyophilization gave the desired products as white amorphous solids.
On-resin Mistunobu N(τ)-His Alkylation
Resin-bound peptides 6a and 6b (Figure 2); 12a and 12b (Figure 3) and 16(a – c) (Figure 4) (0.1 mmol) were treated with triphenylphosphine (PPh3) (262 mg, 1.0 mmol), diethyl azidodicarboxylate (DEAD) (0.46 mL, 40% solution in toluene, 1.0 mol) and appropriate alcohols (1.0 mmol) in dry CH2Cl2 (4 mL) at room temperature (4 h). The resulting resin-bound peptides [7(a – d) (Figure 2); 13(b – e) (Figure 3) or 17(a – f) (Figure 4)] were washed sequentially with DMF, MeOH, CH2Cl2 and Et2O and dried under vacuum.
On-resin Ring-closing Metathesis
To resin-bound peptide [7(b – d) (Figure 2); 13(b – e) (Figure 3) or 17(a – f) (Figure 4)] (0.1 mmol) under helium was added degassed CH2Cl2:DMF (1:1; 4 mL) and the resin was allowed to swell (30 min). A solution of Hoveyda-Grubbs 2nd generation catalyst (Aldrich cat. No. 569755) (18.8 mg, 0.03 mmol) in degassed CH2Cl2:DMF (10:1; 0.2 mL) was added and the mixture was stirred under microwave irradiation (120 °C; 30 min). The resulting resins [8(b – d), Figure 2; 14(b – e), Figure 3 or 18(a – f), Figure 4] were washed sequentially with DMF, MeOH, CH2Cl2 and Et2O and dried under vacuum.
Solution Phase Ring-closing Metathesis
Resin-bound open-chain peptide 7a was cleaved from the resin and purified by HPLC as indicated above to yield peptide 10 (Figure 2). To the peptide under helium was added a solution of Hoveyda-Grubbs 2nd generation catalyst (0.5 equiv) 1 mM in degassed CH2Cl2:DMF (10:1) and the mixture was stirred at room temperature (24 h). Addition of catalyst (0.5 equiv) was repeated and the mixture was stirred at room temperature (24 h) and then concentrated under vacuum and purified by HPLC to yield 11a.
On-resin Diimine Double Bond Reduction
To a solution of NBSH26 (0.44 g, 1 mmol) in anhydrous CH2Cl2 (10 mL) was added Et3N (0.56 mL, 2 mmol) and this was immediately added to resins [8(b – d), Figure 2; 14(b – e), Figure 3 or 18(a – f), Figure 4] (0.05 mmol) under helium. The mixture was subjected to microwave irradiation (50 °C; 30 min). The resulting resins [9(b – d), 15(b – e) and 19(a – f), respectively] were washed sequentially with DMF, MeOH, CH2Cl2 and Et2O and dried under vacuum. Resin cleavage and HPLC purification as indicated above provided the title peptides [3c and 3d; 4(b – e) and 5(a – f), respectively].
Solution-Phase Double Bond Reduction (Hydrogenation)
A solution of alkene-containing peptide (11a and 11b), 1 mM in DMF:THF (10:1) was hydrogenated over 10% Pd•C under H2 at room temperature (overnight). The mixture was filtered and the filtrate was concentrated under vacuum and purified by HPLC to yield the title peptides 3a and 3b, respectively.
Mass Spectral Characterization
Low resolution, positive ion, electrospray ionization (ESI) mass spectra were obtained by LC/MS analysis on an Agilent LC/MSD single quadrupole system that was also equipped with an in-line diode-array ultraviolet (UV) detector. A narrow-bore (100 × 2.1 mm), small-particle (3.5-μm), Zorbax Rapid-Resolution reversed-phase C18 column coupled with a C18 guard column (12.5 × 2.1 mm) was eluted with a 5–90% gradient of MeOH:H2O containing 0.1% AcOH at a flow rate of 300 μL/min to separate the components of crude reaction isolates and purified final products. Background-subtracted mass spectra and the appropriate compound-indicating extracted ion chromatograms were generated using the LC/MSD ChemStation software (version B.04.02 SP1).
High-resolution LC/MS and LC/MS/MS analyses were conducted on a Thermo-Fisher LTQ-XL Orbitrap hybrid mass spectrometer system operated under Xcalibur (version 2.1.0 SP1) control for data acquisition and qualitative analysis. A similar narrow-bore, small particle (3.5 μm) Zorbax Rapid-Resolution reversed-phase C18 column (100 × 2.1 mm) - guard column (12.5 × 2.1 mm) combination was eluted with CH3CN:H2O containing 0.1% AcOH at 250 μL/min. An initial linear gradient of 2–90% CH3CN:H2O over 15 min was followed by a 5 min isocratic hold at the final conditions before LC reset and equilibration (30 mi. cycle). Primary mass spectra (MS1) for accurate mass measurement of a molecular species ([M+H]+, [M+Na]+ or [M+2H]+2) were obtained at a resolution of 30,0000 (full width at half maximum), while MS/MS (MS2) studies employing collision-induced dissociation (CID) or higher energy CID (HCD) were conducted at a resolution of 15,000. For these MS2 studies, [M+H]+ or [M+2H]+2 precursor ions were selected using a mass window that encompassed the isotopic profile of the ion of interest. For the compounds in question, CID and HCD were carried out at an optimized energy setting of 30 and 27.5, respectively, in order to give the highest intensity product ion mass spectrum. Product ion spectra containing multiply charged ions (i.e., those from [M+2H]+2 precursors) were deconvoluted to a single charge state using the Xtract software module of Xcalibur in order to facilitate structural assignment and analysis. In the case of selected open-chain macrocyclic precursors (e.g., compound 7c), MS2 high-resolution extracted ion chromatograms (HR-XICs) were generated post-analysis using a 20 or 30 mDa mass window centered on the observed accurate mass of the appropriate diagnostic ions. These HR-XICs were then used to locate and measure the relative amounts of isomeric bis-alkyl-His-containing phosphopeptide and esterified histidyl phosphopeptide present in the crude precyclized isolates. MS/MS structural assignments are as indicated in Figure 2, which is the MS/MS product ion mass spectrum resulting from the CID fragmentation of the [M+H]+ ion of 3b. A summary of the high resolution MS1 and MS2 data for each compound can be found in the Supporting Information.
X-ray Crystallography
Crystallization of Macrocycle-PBD Complexes
Plk1 PBD protein (residues 371–603) was obtained from Dr. Dan Lim who purified it as previously described.3 Frozen stocks of protein at 37 mg/mL in 10 mM Tris pH 8, 0.5 M NaCl, 10 mM dithiothreitol (DTT)were thawed and diluted to 10 mg/mL with the same buffer. Complexes with each of three macrocycle compounds (3b, 3c and 4b) were prepared by adding 100 mM stocks of the macrocycle in dimethylsulfoxide (DMSO) directly to the diluted protein to achieve a final concentration of 1 mM. Crystals were grown by hanging drop vapor diffusion, with drops made by mixing equal volumes of protein-macrocycle complex and well solution containing 2–6% polyethylene glycol (PEG)-3350. Crystals were cryo-protected by quickly dipping in a solution of 37.5% ethylene glycol in well solution and frozen in liquid nitrogen.
Structure Solution and Refinement
X-ray diffraction data were collected at the Advance Proton Source (APS) using the Northeastern Collaborative Access Team (NE-CAT) 24-ID-C beam line. Data were indexed, integrated and scaled using HKL2000.37 All crystals belonged to the space group P21 with nearly identical unit cell parameters. The structures were solved by molecular replacement using PHASER38 with PDB entry 4DFW (minus solvent and bound ligand) as a search model. The models were built with COOT39 and refined using PHENIX.40 Refinement constraints for the bound macrocycle ligands were generated by the GRADE web server (http://grade.globalphasing.org). Data collection and refinement statistics are in the Supporting Information.
ELISA-based PBD-binding Inhibition Assays
Similar to previously reported procedures,3,4 biotinylated p-T78 peptide was first diluted with 1 × coating solution (KPL Inc., Gaithersburg, MD) to the final concentration of 0.3 μM, and then 100 μL of the resulting solution was immobilized onto a 96-well streptavidin-coated plate (Nalgene Nunc, Rochester, NY). The wells were washed once with phosphate buffered saline (PBS) plus 0.05% Tween20 (PBST), and incubated with 200 μL of PBS plus 1% bovine serum albumin (BSA) (blocking buffer) for 1 h to prevent non-specific binding. Mitotic 293A lysates expressing HA-EGFP-Plk1 were prepared in TBSN buffer [20mM Tris-Cl (pH 8.0), 150 mM NaCl, 0.5% Nonidet P-40, 5 mM EGTA, 1.5 mM EDTA, 20 mM p-nitrophenyl phosphate, and protease inhibitor mixture (Roche)] (~ 60 μg total lysates in 100 μl buffer), mixed with the indicated amount of the competitors (p-T78 peptide and its derivative compounds), provided immediately onto the biotinylated peptide-coated ELISA wells, and then incubated with constant rocking for 1 h at 25 °C. Following the incubation, ELISA plates were washed 4 × with PBST. To detect bound HA-EGFP-Plk1, the plates were probed for 2 h with 100 μL/well of anti-HA antibody at a concentration of 0.5 μg/mL in blocking buffer and then washed 5 ×. The plates were further probed for 1 h with100 μL/well of horse radish peroxidase (HRP)-conjugated secondary antibody (GE Healthcare, Piscataway, NJ) at a 1:1,000 dilution in blocking buffer. The plates were washed 5 × with PBST and incubated with 100 μL/well of 3,3′,5,5′-tetramethylbenzidine (TMB) solution (Sigma, St. Louis, MO) as a substrate until a desired absorbance was reached. The reactions were stopped by the addition of 100 μL/well of stop solution (Cell Signaling Technology, Danvers, MA). The optical density (O.D.) was measured at 450 nm by using an ELISA plate reader (Molecular Devices, Sunnyvale, CA). Calculated IC50 values are listed in Figure 1 and Table 1.
CONCLUSION
The ability of neighboring acidic residues to facilitate Mitsunobu N(τ)–alkylation of His residues bearing an N(π)–alkyl group provides a useful means of specific structural modification that opens a range of applications. Our use of the bis-alkyl-His imidazolium ring for the synthesis of charge-masked macrocyclic phosphopeptides provides one example of an application to biologically important protein-protein interactions.
Supplementary Material
Acknowledgments
This work was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick and the National Cancer Institute, National Institutes of Health (W.-J. Q., J.-E. P., C. C. L., J. A. K., K. S. L. and T. R. B.), an NCI Director’s Innovation Career Development Award (W.-J. Q.) and National Institutes of Health grants ES015339 and GM104047 (M. B. Y.). This work is based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by a grant from the National Institute of General Medical Sciences (P41 GM103403) from the National Institutes of Health. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Appreciation is expressed to Wei Dai, New York University School of Medicine, NY for reagents and to Dan Lim of the David H. Koch Institute for Integrative Cancer Research for providing purified Plk-1 PBD.
Footnotes
SUPPORTING INFORMATION
Supporting Information includes mass spectral characterization of product macrocycles and selected precursors and X-ray crystallography refinement data and SigmaA weighted 2Fo–Fc electron density maps for Plk1-bound 3b, 3c and 4c.
References
- 1.Qian W-J, Park J-E, Lim D, Lai CC, Kelley JA, Park S-Y, Lee KW, Yaffe MB, Lee KS, Burke TR. Biopolymers Pept Sci. 2014;102:444–455. doi: 10.1002/bip.22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qian W-J, Burke TR., Jr Org Biomol Chem. 2015;13:4221–4225. doi: 10.1039/c5ob00171d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yun S-M, Moulaei T, Lim D, Bang JK, Park J-E, Shenoy SR, Liu F, Kang YH, Liao C, Soung N-K, Lee S, Yoon D-Y, Lim Y, Lee D-H, Otaka A, Appella E, McMahon JB, Nicklaus MC, Burke TR, Jr, Yaffe MB, Wlodawer A, Lee KS. Nat Struct Mol Biol. 2009;16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu F, Park J-E, Qian W-J, Lim D, Graber M, Berg T, Yaffe MB, Lee KS, Burke TR., Jr Nat Chem Biol. 2011;7:595–601. doi: 10.1038/nchembio.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allentoff AJ, Mandiyan S, Liang H, Yuryev A, Vlattas I, Duelfer T, Sytwu I-I, Wennogle LP. Cell Biochem Biophys. 1999;31:129–140. doi: 10.1007/BF02738168. [DOI] [PubMed] [Google Scholar]
- 6.Davies JS. J Pept Sci. 2003;9:471–501. doi: 10.1002/psc.491. [DOI] [PubMed] [Google Scholar]
- 7.Jiang S, Li Z, Ding K, Roller PP. Curr Org Chem. 2008;12:1502–1542. [Google Scholar]
- 8.White CJ, Yudin AK. Nat Chem. 2011;3:509–524. doi: 10.1038/nchem.1062. [DOI] [PubMed] [Google Scholar]
- 9.Dharanipragada R. Future Med Chem. 2013;5:831–849. doi: 10.4155/fmc.13.25. [DOI] [PubMed] [Google Scholar]
- 10.Murugan RN, Park J-E, Lim D, Ahn M, Cheong C, Kwon T, Nam K-Y, Choi SH, Kim BY, Yoon D-Y, Yaffe MB, Yu D-Y, Lee KS, Bang JK. Bioorg Med Chem. 2013;21:2623–2634. doi: 10.1016/j.bmc.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian W, Liu F, Burke TR., Jr J Org Chem. 2011;76:8885–8890. doi: 10.1021/jo201599c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blackwell HE, Sadowsky JD, Howard RJ, Sampson JN, Chao JA, Steinmetz WE, O’Leary DJ, Grubbs RH. J Org Chem. 2001;66:5291–5302. doi: 10.1021/jo015533k. [DOI] [PubMed] [Google Scholar]
- 13.Liu F, Giubellino A, Simister PC, Qian W, Giano MC, Feller SM, Bottaro DP, Burke TR., Jr Biopolymers. 2011;96:780–788. doi: 10.1002/bip.21692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chapman RN, Arora PS. Org Lett. 2006;8:5825–5828. doi: 10.1021/ol062443z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson AJ, Elaridi J, Van Lierop BJ, Mujcinovic S, Jackson WR. J Pept Sci. 2007;13:280–285. doi: 10.1002/psc.840. [DOI] [PubMed] [Google Scholar]
- 16.Robinson AJ, van Lierop BJ, Garland RD, Teoh E, Elaridi J, Illesinghe JP, Jackson WR. Chem Commun. 2009:4293–4295. doi: 10.1039/b909056h. [DOI] [PubMed] [Google Scholar]
- 17.García-Aranda MI, Marrero P, Gautier B, Martín-Martínez M, Inguimbert N, Vidal M, García-López MT, Jiménez MA, González-Muñiz R, Vega MJPD. Bioorg Med Chem. 2011;19:1978–1986. doi: 10.1016/j.bmc.2011.01.056. [DOI] [PubMed] [Google Scholar]
- 18.Mulder MPC, Kruijtzer JAW, Breukink EJ, Kemmink J, Pieters RJ, Liskamp RM. J Bioorg Med Chem. 2011;19:6505–6517. doi: 10.1016/j.bmc.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 19.Heapy AM, Williams GM, Fraser JD, Brimble MA. Org Lett. 2012;14:878–881. doi: 10.1021/ol203407z. [DOI] [PubMed] [Google Scholar]
- 20.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 21.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179. [Google Scholar]
- 22.Michalak M, Wicha J. Org Biomol Chem. 2011;9:3439–3446. doi: 10.1039/c1ob05086a. [DOI] [PubMed] [Google Scholar]
- 23.Stoianova DS, Hanson PR. Org Lett. 2000;2:1769–1772. doi: 10.1021/ol005952o. [DOI] [PubMed] [Google Scholar]
- 24.Heckrodt TJ, Singh R. Synth Commun. 2012;42:2854–2865. [Google Scholar]
- 25.Maysuya Y, Takayanagi S-I, Nemoto H. Chem - Eur J. 2008;14:5275–5281. doi: 10.1002/chem.200800249. [DOI] [PubMed] [Google Scholar]
- 26.Buszek KR, Brown NJ. Org Chem. 2007;72:3125–3128. doi: 10.1021/jo0622173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.García-Aranda MI, González-Muñiz R, García-López MT, Pérez de Vega MJ. Eur J Org Chem. 2009;2009:4149–4157. [Google Scholar]
- 28.Palumbo AM, Tepe JJ, Reid GE. J Proteome Res. 2008;7:771–779. doi: 10.1021/pr0705136. [DOI] [PubMed] [Google Scholar]
- 29.Boersema PJ, Mohammed S, Heck AJR. J Mass Spectrom. 2009;44:861–878. doi: 10.1002/jms.1599. [DOI] [PubMed] [Google Scholar]
- 30.Bythell BJ, Suhai S, Somogyi Á, Paizs B. J Am Chem Soc. 2009;131:14057–14065. doi: 10.1021/ja903883z. [DOI] [PubMed] [Google Scholar]
- 31.Kang YH, Park J-E, Yu L-R, Soung N-K, Yun S-M, Bang JK, Seong Y-S, Yu H, Garfield S, Veenstra TD, Lee KS. Mol Cell. 2006;24:409–422. doi: 10.1016/j.molcel.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 32.ICM Pro Software v3.8-0/MacOSX. MolSoft LLC; La Jolla, CA: http://www.molsoft.com. [Google Scholar]
- 33.Abagyan R, Orry A, Raush E, Totrov M. ICM-Chemist-Pro User Guide v38. http://wwwmolsoftcom/chemistpro/
- 34.Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. Cell. 2003;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 35.Murugan RN, Ahn M, Lee WC, Kim H-Y, Song JH, Cheong C, Hwang E, Seo J-H, Shin SY, Choi SH, Park J-E, Bang JK. PLoS One. 2013;8:e80043. doi: 10.1371/journal.pone.0080043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peters C, Bacher M, Buenemann CL, Kricek F, Rondeau J-M, Weigand K. Chem Bio Chem. 2007;8:1785–1789. doi: 10.1002/cbic.200700362. [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. In: Macromolecular Crystallography, Part A. Carter CW Jr, Sweet RM, editors. Academic Press; New York, NY: 1997. pp. 307–326. [Google Scholar]
- 38.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.