

Abstract
Proteinase 3 (PR3), the autoantigen in granulomatosis with polyangiitis, is expressed at the plasma membrane of resting neutrophils, and this membrane expression increases during both activation and apoptosis. Using surface plasmon resonance and protein-lipid overlay assays, this study demonstrates that PR3 is a phosphatidylserine-binding protein and this interaction is dependent on the hydrophobic patch responsible for membrane anchorage. Molecular simulations suggest that PR3 interacts with phosphatidylserine via a small number of amino acids, which engage in long lasting interactions with the lipid heads. As phosphatidylserine is a major component of microvesicles (MVs), this study also examined the consequences of this interaction on MV production and function. PR3-expressing cells produced significantly fewer MVs during both activation and apoptosis, and this reduction was dependent on the ability of PR3 to associate with the membrane as mutating the hydrophobic patch restored MV production. Functionally, activation-evoked MVs from PR3-expressing cells induced a significantly larger respiratory burst in human neutrophils compared with control MVs. Conversely, MVs generated during apoptosis inhibited the basal respiratory burst in human neutrophils, and those generated from PR3-expressing cells hampered this inhibition. Given that membrane expression of PR3 is increased in patients with granulomatosis with polyangiitis, MVs generated from neutrophils expressing membrane PR3 may potentiate oxidative damage of endothelial cells and promote the systemic inflammation observed in this disease.
Keywords: inflammation, microparticles, molecular modeling, neutrophil, proteinase, serine protease, microvesicles, proteinase 3, vasculitis
Introduction
Proteinase 3 (PR3),7 the autoantigen in granulomatosis with polyangiitis (GPA), is a 29-kDa serine protease expressed exclusively in neutrophils, monocytes, and macrophages (1–3). It belongs to a family of neutrophil-derived proteases including elastase, cathepsin G, and the enzymatically inactive azurocidin and similar to its homologues is mainly stored within azurophilic granules (4). In contrast to other family members, PR3 can also be located at both the plasma membrane of resting neutrophils and within secretory vesicles (5). During neutrophil activation and apoptosis, membrane expression of PR3 increases, and soluble PR3 is also released into the extracellular environment during degranulation (6, 7). In fact, a high percentage of neutrophils bearing membrane PR3 is considered as a risk factor for GPA (8, 9). Using both computational biology and mutation studies, the region of PR3 responsible for membrane anchorage has been identified (10). It is dependent on the hydrophobic patch containing four hydrophobic amino acids, Phe-180, Phe-181, Leu-228, and Phe-229. Mutation of these amino acids to alanine prevented both basal and apoptosis-induced membrane expression. During apoptosis, membrane-bound PR3 serves as a “don't eat me” signal. It is co-externalized with phosphatidylserine (PS) via its association with phospholipid scramblase 1 (PLSCR1), a protein facilitating membrane flip-flop. When PLSCR1 was knocked down using siRNA, not only did it prevent PR3 membrane expression but it also increased the rate of apoptotic cell clearance by macrophages (11). This was linked to the ability of PR3 to associate with calreticulin, a protein involved in apoptotic cell recognition and a major “eat me” signal, thereby inhibiting apoptotic cell clearance by phagocytes (12). In addition to functioning as a don't eat me signal, membrane PR3 can also perpetuate inflammation by increasing the production of proinflammatory cytokines including TNFα, MCP1, and IL-6 (12).
Microvesicles (MVs) are small vesicles generated from the plasma membrane of cells during the budding process following cellular activation or apoptosis (13). Ranging from 100 to 1000 nm in size with most around 300–400 nm in diameter, the majority of MVs contain high levels of PS on their membrane outer leaflet (14) and comprise various molecules from the parent cell including membrane constituents and cytoplasmic content. MVs participate in intercellular communication where they play a role in inflammation, coagulation, and vascular function (15). Although produced at low levels during normal physiological conditions, MV production significantly increases during inflammatory and autoimmune diseases including vasculitis, atherosclerosis, diabetes, and malignancies (16). During active vasculitis, MV levels were significantly increased in the plasma of patients compared with healthy controls, and these originated from platelets, endothelial cells, and leukocytes (17–19). Additionally, levels of endothelial cell-derived MVs correlated with disease activity, possibly serving as a marker of endothelial cell activation or vascular damage (18, 19). Whether MV production during vasculitis is simply a consequence of vascular dysfunction or is actively involved in the process is not yet known. However, anti-neutrophil cytoplasmic antibodies (ANCAs) directed against either PR3 and myeloperoxidase, the autoantigens for GPA and microscopic polyangiitis, respectively, stimulated the production of MVs from human neutrophils primed with TNFα (20). These MVs were able to bind to endothelial cells to induce expression of proinflammatory mediators including IL-6 and IL-8 and up-regulate ICAM1, an adhesion molecule involved in facilitating leukocyte endothelial transmigration (20). Furthermore, neutrophil-derived MVs facilitated endothelial cell damage in vitro. Treatment of human umbilical vein endothelial cells with neutrophil-derived MVs resulted in a loss of membrane integrity and morphological changes characteristic of cell injury (21). Based on these studies, it seems likely that MVs generated during autoimmune vasculitis do indeed play an active role in promoting inflammation and endothelial cell damage. Given that PR3 displays a high affinity for lipids, we investigated whether this protein could bind to PS and whether this interaction affected microvesicle-dependent biological processes known to modulate inflammation.
Experimental Procedures
Cell Culture, Transfection, and Immunoblotting
Rat basophilic leukemia (RBL) cells were transfected with an empty pcDNA plasmid (Invitrogen), pcDNA/PR3, pcDNA/PR3-4H4A, or pcDNA/S204A and cultured as described previously in DMEM (Gibco) supplemented with 10% fetal calf serum and 1 μg/ml Zeocin (Invitrogen) (22). RBL cells were stably transduced with a retroviral vector coding for shRNA/rPLSCR1 targeting rat PLSCR1 as described previously (23). Briefly, the shRNA/rPLSCR1 sequence was inserted between HindIII and BglII sites in the mammalian expression vector pSUPER.retro.puro (OligoEngine), and viruses were generated in HEK293 following transduction. RBL cells were incubated with the HEK293 retrovirus-containing supernatant, and infected cells were selected using puromycin (1 μg/ml) (InvivoGen). Silencing of rat PLSCR1 (rPLSCR1) expression was confirmed by Western blotting as described previously (11). Briefly, RBL cells (1 × 108/ml) were lysed in lysis buffer (50 mm NaCl, 50 mm NaF, 1 mm sodium orthovanadate, 1% Triton, 50 mm HEPES, and antiproteases (4 mm PMSF, 400 μm leupeptin, 400 μm pepstatin, and 2 mm EDTA)) for 15 min at 4 °C. Protein quantification was performed on the soluble fraction, and 20 μg was used for each sample. Proteins were fractionated on SDS-polyacrylamide gels, transferred onto polyvinylidene fluoride (PVDF) membranes (Immobilon-P, Millipore), and probed with an anti-rPLSCR1 antibody (mAb 129.2). The proteins were visualized using a peroxidase-conjugated anti-mouse antibody and a SuperSignal WestPico detection kit (Pierce). Where indicated, apoptosis was induced in RBL cells using 2 μg/ml gliotoxin (Sigma), and cells were collected after 16 h. Cells were stained with Annexin V-phycoerythrin and 7-aminoactinomycin D to quantify apoptosis (24) and exclude necrotic cells (25) as described previously (6). Cells were analyzed using a FACScan flow cytometer and CELLQuest software (BD Biosciences).
Nitrocellulose Membrane Assay
Purified phospholipids including PS, phosphatidylcholine (PC), and phosphatidylethanolamine (PE) (Avanti Polar Lipids, Inc.) were spotted onto a nitrocellulose membrane (Hybond; 0.1 μmol/spot), and membranes were blocked for 3 h with TBS containing 0.1% Tween and 3% human serum albumin. After 3 h, membranes were incubated with either 2 μg/ml purified PR3 or HNE (Athens Research and Technology) for 16 h. PR3 and HNE binding was determined using Western blotting analysis with either an anti-PR3 (clone CLB 12.8, Sanquin) or anti-HNE (Biogenesis) antibody, respectively, followed by HRP-conjugated anti-mouse antibody. Purified HNE was also spotted directly onto the membrane as a positive control, and the membranes were probed as above with anti-HNE antibody. The neutrophil cytosolic fraction obtained by nitrogen cavitation in relaxation buffer (100 mm KCl, 3 mm NaCl, 3.5 mm MgCl2, and 10 mm PIPES, pH 6.8) as described previously (7) was used as a source of Annexin A1. Nitrocellulose membranes containing PS, PC, and PE were incubated with the neutrophil cytosolic fraction, and binding was detected using an anti-Annexin A1 antibody (Zymed Laboratories Inc.). Similarly, lysates generated from RBL/pcDNA, RBL/PR3, and RBL/PR3-4H4A (10) with 1% digitonin (9 mg/ml) were incubated with the above membranes, and PR3 binding was detected using an anti-PR3 4A5 antibody. To ensure the anti-PR3 antibody was able to detect both wild type PR3 and PR3-4H4A mutant equally, lysates from the three different cells lines were spotted directly onto a membrane, and PR3 was detected using the same anti-PR3 4A5 antibody.
Surface Plasmon Resonance Spectroscopy
Analyses were carried out on a BIAcore 3000 (BIAcore, GE Healthcare). For analyses on PR3-coated surface, PR3 was diluted to 50 μg/ml in 10 mm sodium acetate, pH 4.5, and immobilized onto a CM5 sensor chip (GE Healthcare) using the BIAcore amine coupling kit. The running buffer for PR3 immobilization was 145 mm NaCl, 5 mm EDTA, and 10 mm HEPES, pH 7.4. Binding of 06:0 PS (Avanti Polar Lipids, Inc.) to immobilized PR3 (3500 response units) was measured at a flow rate of 20 μl/min in the running buffer (PBS, pH 7.4). Surfaces were regenerated by one injection of 10 μl of 0.05% SDS. The specific binding signal shown was obtained by subtracting the background signal, which was obtained by injection of the sample over an activated-deactivated surface. Data were analyzed by global fitting to a 1:1 Langmuir binding model of the association and dissociation phases for several concentrations of 06:0 PS using BIAevaluation 3.2 software (GE Healthcare) and were obtained with a statistic χ2 value <2. The apparent equilibrium dissociation constants (KD) were calculated from the ratio of the dissociation and association rate constants (koff/kon). For analyses on phospholipid-coated surface, HPA (hydrophobic) sensor chips (GE Healthcare) were used for preparation of PS- and PE-coated chips as described previously (26). PR3 binding was measured over 1500 resonance units of immobilized phospholipids at flow rate of 10 μl/min in the running buffer (PBS, pH 7.4). The specific binding signal shown was obtained by subtracting the background signal obtained by injection of the protein sample over a surface saturated with BSA.
Molecular Modeling Analysis
The whole protocol, going from equilibration of the bilayer to simulation of the membrane-bound protein, was performed as described previously (27) for PR3 and HNE simulations into a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine bilayer. Briefly, a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPS) bilayer neutralized with Na+ counterions but lacking ionic strength was created at an initial surface area of 58.5 Å2 using the membrane builder module of CHARMM-GUI (28). This bilayer alone was then equilibrated for 150 ns using NAMD2.9 (29) and the CHARMM36 force field (30) including the non-bonded corrections for sodium ions (31). During this phase, the surface area remained stable, averaging 59.4 ± 0.8 Å2. Order parameters and hydrogen bond analyses also revealed that both ordering of the aliphatic chains and interactions between headgroups reached satisfying convergence. The protein was inserted into the bilayer using the previously reported orientation (10). To account for the difference of width between the POPS and the implicit bilayer, the protein was translated by 2.5 Å2 from the position obtained with IMM1. To obtain sufficient sampling of the interface, the PR3/POPS system was simulated for 500 ns. The stability of the bilayer was not affected by the presence of the protein, and the surface area remained stable (59.9 ± 0.9 Å2). All simulations were performed at a temperature of 310 K with an integration step of 2 fs.
Interactions between PR3 and the bilayer were investigated using contacts and hydrogen bond analyses, all computed using the COOR facility of CHARMM. The hydrogen bonds used a 2.4-Å-cutoff distance between hydrogen and acceptor and a 130° donor-hydrogen acceptor angle criterion. The donor and acceptor definitions were obtained from the CHARMM force field. Hydrogen bonds bridged by water molecules were computed using the same geometric criteria where a bridging water molecule had to hydrogen bond simultaneously with the protein and the membrane. Hydrophobic contacts were defined using a 3-Å-cutoff distance between aliphatic groups of the lipids and enzymes (CHARMM atom types ca, cb, cg1, cg2, cg2, ha*, hb*, hg, and hg2*; type cg except for Hsd, Hse, Asn, and Asp; type hg1 except for Cys, Thr, and Ser; type cd except for Arg, Gln, and Glu; type cd1; type cd2 except for Hsd and Hse; types ce1, ce2, and cz and associated hydrogens of Phe and Tyr; types cd1, cd2, ce2, ce3, cz2, and cz3 and associated hydrogen of Trp; type cay; and type hy*). Except for hydrophobic contacts, interactions were sorted into the following subcategories: backbone and side chain for amino acids, headgroup, phosphate, and glycerol for lipids. All analyses were carried out over the 250–500-ns window during which the root mean square deviation averaged 1.47 ± 0.12 Å. Occupancy values given below always refer to the percentage of existence of an interaction over that window. As usual for serine proteases, the chymotrypsin numbering convention is used for the amino acids of PR3 throughout (the active site is thus His-57, Asp-102, and Ser-195).
Binding of Purified PR3 to PS Externalized on Apoptotic RBL Cells or Murine Neutrophils
Murine neutrophils were collected from 10–12-week-old male C57Bl6 mice purchased from Charles River Laboratories. Mice were housed in an specific pathogen-free facility under standard conditions and maintained in a 12-h/12-h light/dark cycle at 22 °C in accordance with the European Union directives. All animal studies were performed in accordance with and approved by the Institutional Animal Care and Use Committee at the Cochin Institute. Murine neutrophils were isolated from the peritoneal lavage 5 h after intraperitoneal injection of 1 mg of zymosan and stained with an allophycocyanin-conjugated anti-Ly6G antibody (clone 1A8, BD Biosciences) to determine neutrophil purity. Spontaneous apoptosis was induced by incubating neutrophils in Roswell Park Memorial Institute (RMPI) 1640 medium supplemented with 10% fetal calf serum and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin) for 16 h at 37 °C. For the binding of exogenous PR3 to apoptotic RBL cells or murine neutrophils, 4 × 105 apoptotic cells were incubated with 4 μg of purified PR3 (Athens Research and Technology) for 4 h at 4 °C. After saturation with PBS containing heat-aggregated goat IgG and 5% fetal calf serum, cells were incubated at 4 °C for 30 min with FITC-conjugated mouse anti-PR3 (2 μg/ml; clone CLB 12.8). Binding of PR3 and Annexin V-phycoerythrin to externalized PS was evaluated by flow cytometry (BD Biosciences). In the case of RBL cells, this binding was performed in PBS alone or in PBS containing either 2.5 mm CaCl2, 1 mm EDTA, or both.
Exogenous PR3 Binding to Human Microvesicles Isolated from Plasma
Plasma-derived MVs were isolated from 2.5 ml of acid-citrate-dextrose-anticoagulated human blood from healthy volunteers obtained after written informed consent (Etablissement Français du Sang Bourgogne Franche-Comté, Besançon, France). The platelet-rich plasma was obtained by centrifugation at 100 × g for 15 min. This plasma was spun at 2500 × g for 15 min, the supernatant was collected, and the plasma-derived MVs were pelleted by centrifugation at 15,000 × g for 1 h at 4 °C. Plasma-derived MVs (0.5 × 106) were resuspended in 200 μl of PBS and incubated with either PBS alone or 8 μg of PR3 at 4 °C for 4 h. MVs were then stained with allophycocyanin-conjugated Annexin V, monoclonal FITC-conjugated anti-PR3 antibody (5 ng; clone CLB12.8), or a mouse FITC-conjugated IgG1 isotype control (Jackson ImmunoResearch Laboratories). MV staining was analyzed using a Navios flow cytometer (Beckman Coulter) and fluorescent microbeads (0.5, 0.9, and 3 μm in diameter; Megamix Biocytex).
Microvesicle Generation and Quantification from RBL
RBL/pcDNA, RBL/PR3, RBL/PR3-4H4A, and RBL/PR3-S204A were used to generate MVs. To induce MV production, RBL cells were treated with either calcium ionophore (2 μm for 30 min) or gliotoxin (2 μg/ml for 2 h). Cell culture medium was collected, centrifuged to eliminate cells (600 × g, 5 min, 4 °C), and centrifuged again to remove cell debris (600 × g, 5 min, 4 °C), and the resulting supernatant was then ultracentrifuged (15,000 × g, 90 min, 4 °C) to pellet MVs. MVs were resuspended in 500 μl of 0.1-μm filtered 0.9% NaCl and stored at −80 °C. MVs were labeled with Annexin V-FITC in a calcium-dependent manner and analyzed by flow cytometry (EPICS XL, Beckman Coulter) as described previously (32). Events less than 1 μm in diameter were identified by forward scatter and side scatter in comparison with fluorescent microbeads (0.5, 0.9, and 3 μm in diameter). MVs were defined as events with a size <1 μm and >0.1 μm that stained with Annexin V-FITC, and numbers were quantified by comparison with calibrator Flowcount beads (Beckman Coulter; 10-μm diameter) with a predetermined concentration. MVs were also detected using CellVue® Claret Far Red staining according to the manufacturer's instructions. Briefly, MVs isolated as above were resuspended in 1 ml of PBS (0.1-μm filtered), which was subsequently mixed with 1 ml of diluent C containing 4 μl of CellVue Claret Far Red. MVs were incubated for 4 min at room temperature before staining was stopped with 2 ml of PBS containing 0.5% BSA. MVs were collected using ultracentrifugation (15,000 × g, 90 min, 4 °C), resuspended in 500 μl of 0.1-μm filtered 0.9% NaCl, and stored at −80 °C.
Measurement of Neutrophil Respiratory Burst by Chemiluminescence
Human neutrophils were isolated from whole blood from healthy volunteers obtained after written informed consent (Etablissement Français du Sang, Paris, France) using plasma/Percoll gradients. Production of NADPH oxidase-induced oxidant was measured by chemiluminescence in a single photon luminometer (AutoLumat LB953, Berthold) with the cyclic hydrazide luminol (5-amino-2,3-dihydro-1,4-phthalazinedione). For each test, 1 × 105 purified human neutrophils were resuspended in 100 μl of Hanks' balanced salt solution in polystyrene tubes containing 100 μl of luminol (0.15 mm) and 45 μl of one of the following: 0.9% NaCl or MVs from RBL/pcDNA, RBL/PR3, RBL/PR3-4H4A, or RBL/PR3-S203A. Luminescence activity was measured immediately in duplicate over 40 min and expressed as integrated total counts as described previously (33). For experiments involving co-treatment with phorbol myristate acetate (PMA), a final concentration of 1 μg/ml PMA was added to each tube. Lastly, MVs from RBL/pcDNA or RBL/PR3 were incubated for 24 h at 4 °C with 50 μg/ml IgGs isolated from a GPA patient with anti-PR3 ANCA or from a normal control (5). This mixture was then added to human neutrophils, and the respiratory burst was measured as above.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 6.05 software. Comparisons were made using the Student's t test or one-way analysis of variance (ANOVA) where appropriate. Differences were considered significant for a p value <0.05.
Results
PR3 Is a Phosphatidylserine Binding Partner
A protein-lipid overlay assay was used to determine whether PR3 was a PS-binding protein. A nitrocellulose membrane spotted with phospholipids including PC, PE, and PS was incubated with purified PR3, elastase (HNE), or Annexin A1. Purified PR3 bound specifically to PS, and no binding was observed to either PC or PE (Fig. 1A). Despite sharing 56% sequence homology with PR3, HNE did not interact with any of the phospholipids. As a positive control, the well established PS-binding protein Annexin A1 specifically interacted with PS in this assay. Surface plasmon resonance spectroscopy was used to characterize the interaction between PS and PR3. In these experiments, purified PR3 was captured on a PS-coated surface but not on a surface coated with PE (Fig. 1B, left panel) or PC (data not shown). As the data generated in this configuration (immobilized PS and soluble PR3) are not sufficient to determine the KD value accurately, mainly due to PR3 aggregation when used at high concentration, further investigation of the PR3-PS interaction was conducted in the reverse orientation using a commercial water-soluble derivative (06:0 PS) comprising the polar moiety of PS connected to two six-carbon saturated hydrocarbon chains. In this case, PR3 was coated on a CM5 sensor chip, and 06:0 PS was used as a soluble ligand. As illustrated in Fig. 1B (right panel), surface plasmon resonance analysis at varying 06:0 PS concentrations demonstrated a dose-dependent interaction with immobilized PR3. In this configuration, the KD value determined using the simple 1:1 interaction model was over 6 μm.
FIGURE 1.

PR3 binds to PS through the hydrophobic patch. A, purified PR3, HNE, or Annexin A1 was incubated with nitrocellulose membranes spotted with PS, PE, and PC. Blots were probed with anti-PR3, anti-HNE, or anti-Annexin A1 antibody. B, surface plasmon resonance analysis of PR3 (250 nm) binding to PS- and PE-immobilized surfaces (left panel) and 06:0 PS (0.15, 0.25, 0.3, 0.5, and 1 mm) to immobilized PR3 (right panel). Association and dissociation curves were recorded for 120 s. A dose-response effect was observed when using increasing concentrations of soluble 06:0 PS with a KD of 6.28 × 10−6 m. C, lysates generated from RBL/PR3, RBL/PR3-4H4A, or control RBL/pcDNA were incubated with nitrocellulose membranes spotted with PS, PE, and PC, and the membranes were subjected to immunoblotting (IB) using an anti-PR3 antibody (left panel). As a positive control (right panel), the lysates from the three RBL cell lines were spotted onto a membrane and probed with anti-PR3. Panels display a representative image obtained from three replicates. Resp. Diff., response difference; RU, response units; CT, control.
The Hydrophobic Patch Present in PR3 Is Essential for PS Binding
To determine the domain responsible for the interaction between PR3 and PS, whole cell lysates from RBL cells transfected with a control plasmid (RBL/pcDNA), plasmid coding for wild type PR3 (RBL/PR3), or plasmid coding for PR3 mutated within the hydrophobic patch (RBL/PR3-4H4A) were generated as described previously (10) and used in a protein-lipid overlay assay (Fig. 1C). In agreement with the results presented in Fig. 1A, when phospholipid-spotted membranes were incubated with lysates from RBL/PR3 cells, PR3 specifically interacted with PS and not PC or PE. Importantly, when the hydrophobic patch was mutated (RBL/PR3-4H4A), PR3 no longer bound to PS. To ensure that the PR3 antibody used in these experiments was able to detect both wild type PR3 and PR3-4H4A, a dot blot was conducted, and no difference in the ability of this antibody to recognize either version of the protein was observed (Fig. 1C, right panel). These results clearly demonstrate that PR3 is a PS-binding protein, and this interaction depends on the hydrophobic patch, which is composed of the four amino acids Phe-180, Phe-181, Leu-228, and Phe-229.
Molecular Simulations Identify a PR3 PS-specific Binding Site
Molecular simulations of PR3 bound to a bilayer composed of POPS were performed to gain structural insights into interactions between PR3 and PS lipids. A POPS bilayer was chosen as a model for lipid-coated surfaces (see previous section) and PS-rich domains upon PS externalization (see next section). The simulation showed that PR3 was stably anchored into the POPS bilayer, and the hydrophobic insertion is mostly achieved by residues located on three loops: β8-β9, β9-β10, and β11-β12 (Fig. 2A). The amino acids responsible for the most stable hydrogen bonds with phosphates and headgroups are shown in Fig. 2, B and C, respectively. Six basic residues alone mediated 75% of the total number of hydrogen bonds. In detail, the side chains of residues Lys-99, Arg-177, Arg-186A, Arg-186B, Lys-187, and Arg-222 formed hydrogen bonds with the phosphates groups with occupancies of 85–100%. Interestingly, as a consequence of their proximity within the structure, the side chains of the pairs Lys-99/Arg-177, Arg-186A/Arg-186B, and Lys-187/Arg-222 trapped the phosphate group of the same lipid during 82, 99, and 80% of the simulation trajectory, respectively. Lys-99, Arg-186B, and Lys-187 simultaneously mediated highly stable hydrogen bonds (occupancies between 75 and 90%) with PS headgroups (Fig. 2C). The aliphatic chains of the corresponding lipids mediated only a few contacts with the protein except the one whose headgroup interacts with Lys-99. That particular lipid participated in a stable network of interactions that also involved residues Glu-97 and Arg-177 as well as the headgroup of a second lipid (Fig. 2D). That second lipid interacted with the first lipid through its headgroup, its phosphate connected residues Lys-99 and Arg-177, and finally its aliphatic chain mediated numerous hydrophobic contacts with PR3, which account for a quarter of the total number of contacts between protein and bilayer. Interestingly, it interacted with Phe-166, Leu-223, and Phe-224 of the hydrophobic patch. The interaction network described depended on PS headgroups and involved both the basic (Arg-186A, Arg-186B, Lys-187, and Arg-22) and hydrophobic (Phe-166, Phe-167, Leu-223, and Phe-224) patches of the interfacial binding site reported earlier (4, 10). Therefore, these simulation suggest that these amino acids constitute a specific PS-binding site.
FIGURE 2.

Molecular modeling of the PR3-PS interaction. Insertion of PR3 into a POPS bilayer is shown. The protein is shown in gray. We highlight the membrane binding interface and the position at the interface of the residues mediating the highest number of hydrophobic contacts with the PS lipids (A) and the amino acids with the highest occupancies for hydrogen bonds with phosphates and head groups of PS (B and C, respectively) shown with magenta, blue, and green color gradients, respectively. D, interaction network of the predicted PR3/PS-binding site. The main hydrogen bonds mediated by two lipids are represented with black dashed lines. The amino acids mediating hydrophobic contacts with the central lipids are shown using spheres and colored using a blue to red gradient following the increasing number of contacts.
Soluble PR3 Binds to PS Externalized on Apoptotic Cells
PS is an important component of the cell membrane preferentially located within the inner leaflet, and during apoptosis a dysregulation in the enzymes responsible for membrane integrity leads to the externalization of PS. To determine whether soluble PR3 was able to bind PS expressed on the membrane of apoptotic cells, control RBL cells or RBL cells transduced with scrambled shRNA or shRNA targeting rPLSCR1 were used. PLSCR1 is responsible for the redistribution of all phospholipids between the two leaflets of the plasma membrane. Transduction of rPLSCR1 shRNA markedly decreased rPLSCR1 expression in RBL cells as assessed by Western blotting analysis, whereas rPLSCR1 expression was unchanged in cells transfected with scrambled shRNA (Fig. 3A). Knockdown of rPLSCR1 resulted in a significant decrease in externalized PS during apoptosis as measured by Annexin V staining and flow cytometry (Fig. 3B). Apoptosis was induced in RBL cells overnight, and cells were subsequently incubated with soluble PR3. Using flow cytometry, PR3 was detected on the surface of PS-positive apoptotic cells (Fig. 3C), whereas PR3 could not be detected on cells lacking externalized PS (see representative dot blots in Fig. 3D). Notably, when PS externalization was decreased using rPLSCR1 shRNA, the proportion of cells able to capture soluble PR3 decreased in the same order of magnitude (Fig. 3C). This strongly suggests that PR3 binding to the surface of apoptotic cells is dependent on PS externalization. To determine whether binding of PR3 to PS was dependent on calcium, soluble PR3 was incubated with a mixed population of live and apoptotic cells in the presence of PBS alone or PBS containing either CaCl2, EDTA, or both. In these experiments, there was no difference in the ability of soluble PR3 to bind to cells expressing externalized PS when CaCl2 or EDTA was present (Fig. 3D). To determine whether the same phenomena occurred in primary cells, neutrophils isolated from mice were examined. Murine neutrophils were incubated overnight at 37 °C to induce spontaneous apoptosis and PS externalization. Apoptotic neutrophils were incubated with soluble PR3, and flow cytometry was performed to examine both PS externalization and PR3 binding. Murine neutrophils bound soluble PR3 only when PS was externalized during apoptosis (Fig. 3E).
FIGURE 3.

Soluble PR3 binding to apoptotic cell surface depends on PS expression. A, RBL cells stably transfected with a retroviral vector coding for rPLSCR1 shRNA displayed decreased PLSCR1 expression compared with those transfected with scrambled shRNA. Actin was used as a loading control, and the panel displays a representative Western blot. B, flow cytometry after Annexin V staining was used to detect PS externalization on the above cells during gliotoxin-induced apoptosis. C, apoptotic control or shRNA rPLSCR1 cells were incubated with soluble PR3, and flow cytometry was used to detect binding. D, soluble PR3 binding to control apoptotic cells was also determined in the presence of CaCl2, EDTA, or both, and representative dot blots obtained after anti-PR3-FITC labeling are displayed. Murine apoptotic neutrophils were incubated with soluble PR3 and subsequently stained with anti-PR3-FITC and Annexin V (AnV)-phycoerythrin (PE) (E). All experiments were performed three times, and data are presented as mean ± S.E. (**, p < 0.01; ***, p < 0.001; Student's t test). Error bars represent S.E.
PR3 Is Captured by Plasma-derived Microvesicles
MVs are small vesicles containing high levels of PS on their outer leaflet (14). MVs isolated from the plasma of healthy donors were incubated with soluble PR3, and events corresponding to MVs were gated by flow cytometry with the help of calibration beads and expression of PS. Incubation with soluble PR3 followed by anti-PR3-FITC allowed for the identification of PR3-positive events (Fig. 4A). Soluble PR3 bound to 45.8 ± 10.3% of events isolated from the plasma of healthy donors (Fig. 4B).
FIGURE 4.
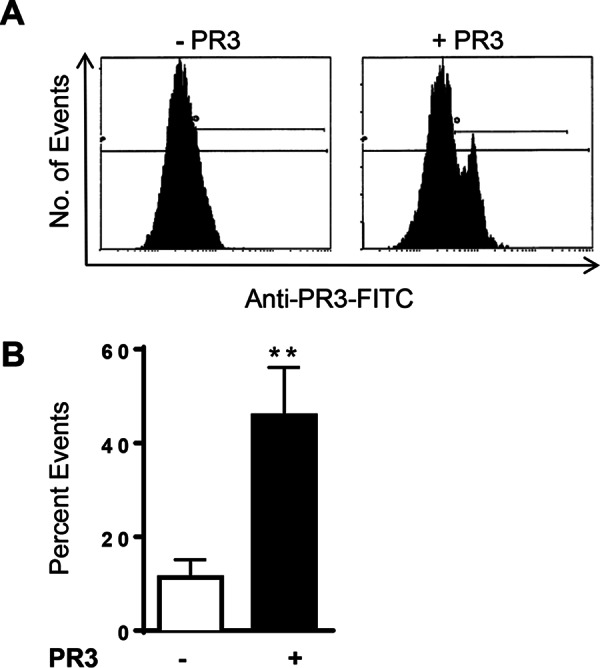
Exogenous PR3 binds plasma-derived microvesicles. MVs isolated from human plasma were incubated with soluble PR3 for 4 h and stained with anti-PR3-FITC. A, representative flow cytometry histograms of PR3 staining events isolated from plasma and incubated with either PBS or soluble PR3. B, percentage of plasma-derived MVs staining positive for PR3. Data are expressed as the mean ± S.E. of six independent experiments (**, p = 0.01; Student's t test). Error bars represent S.E.
Endogenous Membrane PR3 Inhibits Microvesicle Production during Both Activation and Apoptosis
MV generation was assessed by flow cytometry in RBL/pcDNA, RBL/PR3, and RBL/PR3-4H4A following activation with the calcium ionophore A23187. Cells expressing membrane-associated PR3 produced significantly less PS+ MVs compared with cells transfected with the control plasmid (Fig. 5A). Importantly, when PR3 was mutated within the hydrophobic patch and the protein was no longer able to interact with PS, PS+ MV production was restored. To ensure the observed decrease in MV production was not due to PR3 interfering with the binding of Annexin V to PS, an alternative method of detection based on the fluorescent labeling of MVs was used. The same results were obtained when MVs were identified using CellVue staining: PR3 significantly decreased the production of MVs compared with control or PR3-4H4A (Fig. 5B). It should be noted that this latter method detects both PS+ and PS− MVs. PR3 also affected the production of MVs during gliotoxin-induced apoptosis with a significant decrease in PS+ MVs observed when cells expressed membrane PR3 compared with control cells (Fig. 5C). PS+ MV production was restored when PR3 was not expressed at the membrane and lacked the capacity to bind to PS. Again, these results were confirmed using CellVue staining with the same decrease in MV production observed in PR3-expressing cells (Fig. 5D). These results clearly demonstrate that PR3 expressed at the plasma membrane affects the generation of MVs during both activation and apoptosis and that the hydrophobic patch was essential for this effect. To determine whether the serine protease activity of PR3 was required for the decreased MV production, RBL cells were transfected with an enzymatically inactive form of PR3 (PR3/S203A) containing a serine to alanine mutation at position 203 (34). The serine protease activity of PR3 was essential for the decrease in MV production during both activation with calcium ionophore (Fig. 5E) and apoptosis induced by gliotoxin (Fig. 5F) as RBL cells transfected with PR3/S203A produced equivalent numbers of MVs as control RBL cells.
FIGURE 5.

PR3 affects MV production during cell activation and apoptosis. Analysis of MVs following activation with the calcium ionophore A23187 (A, B, and E) or gliotoxin-induced apoptosis (C, D, and F) is shown. MVs isolated from RBL/pcDNA, RBL/PR3, and RBL/4H4A were stained with Annexin V, and flow cytometry was used to determine the number of PS+ MVs (MP)/μl for seven independent MV preparations (A and C). MVs were also detected using CellVue Claret Far Red staining (B and D) where data are displayed as -fold change in MV production compared with RBL/pcDNA from three independent MV preparations. MVs isolated from RBL/pcDNA, RBL/PR3, and RBL/S203A were stained with Annexin V, and flow cytometry was used to determine the number of PS+ MVs/μl for five independent MV preparations (E and F). All data are presented as means ± S.E. (*, p < 0.05; **, p < 0.01; one-way ANOVA). Error bars represent S.E.
MV Derived from Activated PR3-expressing Cells Potentiated Oxidant Production in Neutrophils
As MVs are known to exert both a pro- and anti-inflammatory effect during inflammation (35, 36), the biological function of the MVs generated during activation was examined. Regardless of the parent cell, MVs generated during activation triggered a respiratory response in neutrophils and facilitated the production of oxidants (Fig. 6A), strongly suggesting that activation-induced MVs are proinflammatory. Importantly, a significantly larger respiratory response was recorded in neutrophils exposed to MVs from RBL/PR3 cells compared with controls. No difference in the induction of oxidant production was observed between MVs generated from control cells or those expressing PR3-4H4A, suggesting that the ability of PR3 to associate with PS is key to this proinflammatory effect. MVs generated from RBL/PR3-S203A cells were not able to induce the same increase in oxidant production compared with MVs generated from PR3-expressing cells; rather a respiratory response equivalent to treatment with control MVs was observed. This suggests that the serine protease activity of PR3 is essential in the function of MVs generated during cellular activation (Fig. 6B). The respiratory response observed following co-stimulation with PMA, a compound known to induce the production of oxidants, demonstrated that MVs from PR3-expressing cells again induced a larger respiratory response in human neutrophils compared with those generated from either control or PR3-4H4A cells (Fig. 6C). ANCAs directed against PR3 are known to potentiate the respiratory burst in neutrophils (37, 38). To determine whether ANCA could modulate the respiratory burst triggered by activation-induced MVs, pcDNA or PR3 MVs and either ANCA or isotype control IgG were incubated for 24 h at 4 °C to allow for antibody binding. This mixture was then added to neutrophils, and oxidant production was measured. Addition of ANCA to MVs, regardless of whether MVs were from control or PR3-expressing cells, produced a larger respiratory response in human neutrophils compared with control IgG antibody (Fig. 6D; pcDNA MVs + ANCA versus IgG control, p = 0.0051; PR3 MVs + ANCA versus IgG control, p = 0.0673). This result suggests that ANCA forming immune complexes with any MVs are more inflammatory and that this effect was not dependent on PR3.
FIGURE 6.

MVs from activated cells expressing PR3 induced a larger respiratory burst in human neutrophils. NADPH oxidase activation was evaluated in human neutrophils in response to MV stimulation. Intracellular H2O2 production was measured using luminol-amplified chemiluminescence (CL), and luminescence was recorded for 40 min. Data are means ± S.E. of the integral of the response in cpm. MVs from RBL/pcDNA, RBL/PR3, RBL/4H4A (A), and RBL/S203A (B) were used. The respiratory response was also measured in the presence of PMA (C). MVs from RBL/pcDNA or RBL/PR3 were preincubated with IgGs isolated from either a GPA patient or control individual, and the respiratory response was measured (D). Data in A, C, and D are represented as respiratory burst generated from 1 × 106 MVs; n = 7 independent MV preparations used on neutrophils isolated from different donors (*/#, p < 0.05; **/##, p < 0.01; one-way ANOVA). Data in B represent percent change compared with pcDNA obtained from n = 5 independent MV preparations used on neutrophils isolated from different donors (**, p < 0.01; one-way ANOVA). Error bars represent S.E.
PR3 Hampered the Ability of Apoptosis-induced MVs to Inhibit a Respiratory Burst in Neutrophils
In contrast to those generated during activation, MVs produced during gliotoxin-induced apoptosis impaired the basal respiratory response in neutrophils regardless of the parent cell used to generate them and can therefore be considered anti-inflammatory. Although MVs isolated from PR3-expressing apoptotic cells also down-regulated the production of oxidants in neutrophils, a significantly larger respiratory burst response was observed compared with MVs generated from control apoptotic cells (Fig. 7A). Again, no difference was observed in the induction of oxidant production between MVs generated from control cells and those expressing PR3-4H4A. MVs produced during apoptosis from RBL/PR3-S203A cells were able to inhibit oxidant production more than MVs generated from PR3-expressing cells, and inhibition of the respiratory response was equivalent to results obtained when neutrophils were treated with control MVs. This suggests that the serine protease activity of PR3 is again essential in the function of MVs generated during apoptosis (Fig. 7B). To determine whether apoptosis-induced MVs also hampered the respiratory response following stimulation with a potent activator, neutrophils were co-treated with PMA and MVs from control, PR3, or PR3-4H4A cells. Neutrophils incubated with PMA and MVs from control cells produced a significantly reduced respiratory burst response compared with those stimulated with PMA alone that was consistent with the data presented in Fig. 7A, again suggesting that apoptosis-induced MVs are anti-inflammatory. However, MVs from PR3-expressing cells were no longer able to inhibit the respiratory burst observed following treatment with MVs from both control and PR3-4H4A-expressing cells (Fig. 7C). In fact, the respiratory response observed following co-treatment with PMA and MVs from PR3-expressing cells was equivalent to the response observed following stimulation with PMA alone, indicating that PR3 is able to abolishing the anti-inflammatory effects of MVs generated during apoptosis.
FIGURE 7.

MVs generated from apoptotic cells decreased the respiratory response in human neutrophils, and PR3 hampered this inhibition. NADPH oxidase activation was evaluated in human neutrophils in response to MV stimulation. Intracellular H2O2 production was measured using luminol-amplified chemiluminescence (CL), and luminescence was recorded for 40 min. Data are means ± S.E. of the integral of the response in cpm (A). MVs from RBL/pcDNA, RBL/PR3, RBL/4H4A (A), and RBL/S203A (B) were used. The respiratory response was also measured in the presence of PMA (C). Data in A and C are represented as respiratory burst generated from 1 × 106 MVs; n = 7 independent MV preparations used on neutrophils isolated from different donors (*, p < 0.05; **/##, p < 0.01; ###, p < 0.001; one-way ANOVA). Data in B represent percent change compared with pcDNA obtained from n = 4 independent MV preparations used on neutrophils isolated from different donors (*, p < 0.05; one-way ANOVA). Error bars represent S.E.
Discussion
Here, we report for the first time that PR3, the autoantigen in GPA, is a PS-binding protein, and this interaction affects both the production and function of MVs. The newly identified ability of PR3 to bind to PS is yet another unique biochemical property that distinguishes this protein from other serine proteases including the closely related HNE. Despite sharing a 56% sequence identity, a number of studies have shown that PR3 and HNE have different substrates, localizations, and functions (4). Molecular modeling and surface plasmon resonance demonstrated that PR3 can bind directly and robustly to the plasma membrane as well as lipid vesicles, and this binding occurs with a significantly higher affinity than HNE (27). Previously studies have shown that PR3 is expressed at the membrane of resting neutrophils through its ability to bind to CD177 (39, 40). However, this type of interaction does not appear to play a role in apoptosis-induced membrane expression of PR3, which is independent of CD177 expression (11). Furthermore, we recently demonstrated that membrane expression of PR3 under resting conditions or following TNFα stimulation does not correlate with membrane expression after the induction of apoptosis, strongly suggesting that different pools of PR3 are present within neutrophils that can be mobilized to the cell surface (34). Importantly, regardless of the mechanisms responsible for PR3 externalization, the integrity of the hydrophobic patch is required (10). Extending this further, we found that PR3 was able to directly interact with PS, whereas HNE did not display an affinity for any of the phospholipids. PR3 interacted exclusively with PS and not PC or PE, and using molecular modeling a potential PS-binding site within the basic and hydrophobic patches of PR3 was identified. It has long been established that PR3 is co-externalized with PS during apoptosis (6), and in these experiments inhibiting PS externalization by silencing phospholipid scramblase 1 prevented the expression of PR3 at the plasma membrane (11). PR3 contains many motifs common to other well characterized PS-binding proteins such as annexins including positively charged regions that facilitate electrostatic interactions and hydrophobic residues that act to anchor the protein (41). Molecular modeling and mutation of four hydrophobic amino acids located within the hydrophobic patch demonstrated that this region is indeed critical for the interaction between PR3 and PS (11).
Although these data provide clear evidence demonstrating that PR3 is a PS-binding protein, the question remains what is the significance of this interaction. The most obvious consequence is the modulation of apoptotic cell clearance. PS is externalized upon apoptosis, and extensive research indicates that when expressed at the membrane PS acts as a major eat me signal facilitating the recognition and removal of dying cells (42, 43). By binding to PS, PR3 may again prevent this process and function as a don't eat me signal in line with previous studies (11). PS is not the only eat me signal inhibited by PR3. PR3 also binds calreticulin, a protein expressed at the membrane shown to be important for the efficient phagocytosis of apoptotic cells both in vitro and in vivo (12, 44). Given that PR3 is co-externalized with calreticulin and PS and that the latter is dependent on the activity of phospholipid scramblase 1 (11), we suggest that PR3 may form part of a protein platform expressed at the membrane during apoptosis. However, the ability to inhibit PS-mediated phagocytosis of apoptotic cells may not be the sole consequence of this PR3-PS interaction. PS has multiple binding partners including annexins, PKC, coagulation factors, and Raf-1, and these partners are implicated in a large range of cellular processes (41). For example, outward exposure of PS initiates the binding of several coagulation factors on platelets, which ultimately activates a biochemical reaction cascade that promotes coagulation (45). It is possible that by binding externalized PS on other cells including platelets, neutrophils, and leukocytes PR3 may interfere with natural binding partners and block PS-mediated biological processes.
In this study, we also demonstrate that soluble PR3 is able to bind to PS in a cellular context. When apoptotic neutrophils or MVs containing high levels of PS on their outer leaflet were incubated with soluble PR3, PR3 protein bound to the membrane. Based on this observation, we suggest that PS acts as an anchoring site or receptor for soluble PR3 released during neutrophil degranulation or apoptosis. Although these experiments clearly demonstrate that PR3 binds to PS-positive cells and MVs, we cannot discount the fact that PR3 may also bind to other molecules expressed on the surface such as calreticulin and phospholipid scramblase 1, or there may be other unidentified proteins that can also bind PR3. Given that PR3 expressed at the surface of an apoptotic cell can act as a don't eat me signal (11, 12), it is possible that soluble PR3 released into the extracellular environment may transfer this don't eat me property to any bystander cell expressing membrane PS including activated leukocytes and platelets, thereby preventing these cells from fulfilling their functions. In fact, as PR3 has a number of proinflammatory properties, soluble PR3 binding to any cell type or vesicle expressing PS may modulate the inflammatory process. This could occur at both the local site of inflammation and even distally if PR3 is bound to circulating leukocytes or MVs.
Furthermore, in GPA, soluble PR3 released by activated or apoptotic neutrophils may bind PS expressed on the surface of other cell types including leukocytes, platelets, and endothelial cells or even MVs. For example, PR3 may interact with PS located on the surface of damaged endothelial cells, which may in turn prevent their clearance and further promote inflammation. In fact, defective clearance of apoptotic cells is linked to the development of autoimmune diseases including systemic lupus erythematosus (46). Mouse models also demonstrated that one autoantigen unique to systemic lupus erythematosus, ribonucleoprotein Sm, is associated with apoptotic cell blebs and that apoptotic cells induce an anti-Sm immune response in mice (47). If circulating soluble PR3 is able to bind distant apoptotic cells through PS, it is possible that this may prevent clearance, promote inflammation, or even enhance the anti-PR3 immune responses and facilitate inflammatory processes in GPA. Further work is required to determine whether this occurs in vivo and what implications it may have on the pathogenesis of GPA. It is possible that the autoantibodies in GPA could bind to any cell that has captured soluble PR3 and modulate their normal responses, and this may again promote inflammation and further contribute to endothelial cell damage.
Another biological process known to involve PS is MV formation. MVs contain high levels of PS on their outer membrane and are formed during either cellular activation or apoptosis. During active vasculitis, plasma of patients contains increased numbers of MVs compared with healthy controls (17–19). Using RBL cells expressing enzymatically active PR3, we observed a significant reduction in MV production when PR3 was expressed at the membrane regardless of whether cells were activated or undergoing apoptosis. The fact that the enzymatically inactive form of PR3 is unable to modulate MV production suggests that PR3 cleaves an as yet unidentified protein essential in the formation of these vesicles. A key event in MV formation is an increase in intracellular calcium levels, which activates Ca2+-dependent proteases that in turn facilitate the shedding of MVs (48). Previous experiments examining calcium-mediated degranulation (6) failed to detect any defect in calcium influx or signaling in the presence of PR3. Therefore we suggest that PR3 may affect membrane blebbing or even shedding through its interaction with the phospholipid PS or other unidentified proteins involved with these processes.
Microvesicles generated from PR3-expressing and control cells were then used to assess whether PR3 altered MV function. The first thing to note is the function of the MVs differed dramatically depending on the stimulus used to produce them. MVs produced during cell activation stimulated a significant respiratory burst response in human neutrophils and can be considered as proinflammatory, whereas MVs produced during apoptosis inhibited both the basal respiratory burst and the oxidative burst observed following PMA stimulation and thus acted in an anti-inflammatory manner. The idea that MVs can be either pro- or anti-inflammatory is not a new concept. Although MVs are able to potentiate inflammation by up-regulating the production of cytokines and chemokines and the expression of adhesion molecules as well as activating proinflammatory signaling pathways (49–52), other studies demonstrate an anti-inflammatory effect (53). For example, MVs generated from human neutrophils decreased neutrophil recruitment and facilitated the resolution of inflammation, and they were also capable of entering cartilage and protecting joints during inflammatory arthritis, processes mediated by Annexin A1 (36, 53). MVs also promoted the resolution of inflammation in vivo during sepsis (54). In our current system, we suggest that MVs generated during activation are designed to promote inflammation to ensure the effective elimination of the pathogen or danger signal, whereas apoptotic MVs, much like apoptotic cells themselves, are important for promoting the resolution of inflammation. Regardless of the conditions used to generate them, MVs from PR3-expressing cells were more proinflammatory compared with those generated from control cells. This study found that MVs from PR3-expressing cells were able to activate reactive oxygen species production, eliciting a larger respiratory burst response in human neutrophils. The effect of complexes containing both MVs and ANCA on the respiratory response was also examined, and although the addition of ANCA appeared to increase reactive oxygen species production in human neutrophils, this was not dependent on the type of MVs used. The same increase was observed when ANCAs were preincubated with MVs from control or PR3-expressing cells, indicating that this effect was independent of PR3. Although in this study the effect of MVs generated from PR3-expressing cells on neutrophil responses was assessed, it is highly likely that within the circulatory system or at the local site of inflammation these MVs are also delivered to other immune cells such as monocytes, leukocytes, and macrophages. Therefore future studies examining the effects of these MVs generated during either activation or apoptosis on immune cells such as monocytes and macrophages are essential in understanding how these MVs can modulate the inflammatory process.
As MVs are known to play a role in inflammation and vascular function, we believe that this novel biochemical property of PR3 may have implications not just in inflammation but also in the pathogenesis of GPA. Our results indicate that when PR3 is expressed at the membrane MVs produced during both activation and apoptosis are more inflammatory. Given that GPA patients express increased membrane PR3 compared with healthy donors, it seems very likely that MVs produced from neutrophils of patients could contribute to the vascular inflammation observed in this disease. In fact, the ability of PR3 to be expressed on MVs may provide another mechanism, in addition to soluble PR3, in which the autoantigen is able to disseminate throughout the circulatory system. By doing so, PR3 MVs may contribute to inflammation in distant areas or promote systemic inflammation like that observed in GPA. This is not the first time an autoantigen has been identified on MVs. Studies examining other autoimmune diseases including rheumatoid arthritis and systemic lupus erythematosus have found that autoantigens are expressed on MVs and that these MVs can form potent inflammatory complexes containing autoantibodies (55, 56). This phenomenon may also contribute to inflammation in GPA, although more research is required to determine whether these complexes form when MVs are produced from a PR3-expressing cell.
Author Contributions
K. R. M. performed experiments, drew figures, and wrote the manuscript. C. K.-M. generated the RBL/4H4A mutant, performed protein-lipid overlay assays, and helped draft the manuscript. M. Y. and C. M. B. assisted in characterizing the MVs and helped draft the manuscript. C. G. and N. R. generated the molecular modeling and helped analyze and draft the manuscript. M. P.-R., F. A.-D., A. C., and P. S. assessed the ability of soluble PR3 to bind to PS on cells and MVs and helped draft the manuscript. M. B. provided constructs used to silence rPLSCR1 expression and provided the necessary technical assistance. P. F. performed BIAcore experiments and helped analyze and draft the manuscript. V. W.-S. conceived the project, planned the experiments, analyzed the data, and wrote the manuscript.
Acknowledgments
We greatly acknowledge Prof. Luc Mouthon from the department of Internal Medicine for providing the sera from patients with GPA, Muriel Andrieu and Karine Labroquère of the Cochin Cytometry and Immunobiology facility, the staff of the Animal Care Facilities at the Cochin Institute, the excellent technical assistance of Valérie Gausson for the overlay experiments, and the Etablissement Français du Sang (Saint-Antoine Hospital, Paris, France).
This work was supported in part by the Investissements d'Avenir Program (Grant ANR-11-IDEX-0005-02), Sorbonne Paris Cite, Labex Inflamex, Départements Hospitalo Universitaires Autoimmune and Hormonal Diseases (DHU-AUTHORS), Labex LipSTIC (Grant ANR-11-LABX-0021), and the Chancellerie des Universités de Paris (Legs Poix to V. W.-S.). All authors declare that they have no significant competing financial, professional or personal interests that might have influenced the performance or presentation of the work described in this manuscript.
- PR3
- proteinase 3
- PS
- phosphatidylserine
- MV
- microvesicle
- GPA
- granulomatosis with polyangiitis
- PLSCR1
- phospholipid scramblase 1
- ANCA
- anti-neutrophil cytoplasmic antibody
- RBL
- rat basophilic leukemia
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- HNE
- human neutrophil elastase
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- PMA
- phorbol myristate acetate
- ANOVA
- analysis of variance
- rPLSCR1
- rat PLSCR1
- Hsd
- δ-protonated His
- Hse
- ϵ-protonated His.
References
- 1. Campanelli D., Melchior M., Fu Y., Nakata M., Shuman H., Nathan C., and Gabay J. E. (1990) Cloning of cDNA for proteinase 3: a serine protease, antibiotic, and autoantigen from human neutrophils. J. Exp. Med. 172, 1709–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zimmer M., Medcalf R. L., Fink T. M., Mattmann C., Lichter P., and Jenne D. E. (1992) Three human elastase-like genes coordinately expressed in the myelomonocyte lineage are organized as a single genetic locus on 19pter. Proc. Natl. Acad. Sci. U.S.A. 89, 8215–8219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lepse N., Abdulahad W. H., Kallenberg C. G., and Heeringa P. (2011) Immune regulatory mechanisms in ANCA-associated vasculitides. Autoimmun. Rev. 11, 77–83 [DOI] [PubMed] [Google Scholar]
- 4. Hajjar E., Korkmaz B., and Reuter N. (2007) Differences in the substratebinding sites of murine and human proteinase 3 and neutrophil elastase. FEBS Lett. 581, 5685–5690 [DOI] [PubMed] [Google Scholar]
- 5. Halbwachs-Mecarelli L., Bessou G., Lesavre P., Lopez S., and Witko-Sarsat V. (1995) Bimodal distribution of proteinase 3 (PR3) surface expression reflects a constitutive heterogeneity in the polymorphonuclear neutrophil pool. FEBS Lett. 374, 29–33 [DOI] [PubMed] [Google Scholar]
- 6. Durant S., Pederzoli M., Lepelletier Y., Canteloup S., Nusbaum P., Lesavre P., and Witko-Sarsat V. (2004) Apoptosis-induced proteinase 3 membrane expression is independent from degranulation. J. Leukoc. Biol. 75, 87–98 [DOI] [PubMed] [Google Scholar]
- 7. Witko-Sarsat V., Cramer E. M., Hieblot C., Guichard J., Nusbaum P., Lopez S., Lesavre P., and Halbwachs-Mecarelli L. (1999) Presence of proteinase 3 in secretory vesicles: evidence of a novel, highly mobilizable intracellular pool distinct from azurophil granules. Blood 94, 2487–2496 [PubMed] [Google Scholar]
- 8. Witko-Sarsat V., Lesavre P., Lopez S., Bessou G., Hieblot C., Prum B., Noël L. H., Guillevin L., Ravaud P., Sermet-Gaudelus I., Timsit J., Grünfeld J. P., and Halbwachs-Mecarelli L. (1999) A large subset of neutrophils expressing membrane proteinase 3 is a risk factor for vasculitis and rheumatoid arthritis. J. Am. Soc. Nephrol. 10, 1224–1233 [DOI] [PubMed] [Google Scholar]
- 9. Abdgawad M., Gunnarsson L., Bengtsson A. A., Geborek P., Nilsson L., Segelmark M., and Hellmark T. (2010) Elevated neutrophil membrane expression of proteinase 3 is dependent upon CD177 expression. Clin. Exp. Immunol. 161, 89–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kantari C., Millet A., Gabillet J., Hajjar E., Broemstrup T., Pluta P., Reuter N., and Witko-Sarsat V. (2011) Molecular analysis of the membrane insertion domain of proteinase 3, the Wegener's autoantigen, in RBL cells: implication for its pathogenic activity. J. Leukoc. Biol. 90, 941–950 [DOI] [PubMed] [Google Scholar]
- 11. Kantari C., Pederzoli-Ribeil M., Amir-Moazami O., Gausson-Dorey V., Moura I. C., Lecomte M. C., Benhamou M., and Witko-Sarsat V. (2007) Proteinase 3, the Wegener autoantigen, is externalized during neutrophil apoptosis: evidence for a functional association with phospholipid scramblase 1 and interference with macrophage phagocytosis. Blood 110, 4086–4095 [DOI] [PubMed] [Google Scholar]
- 12. Gabillet J., Millet A., Pederzoli-Ribeil M., Tacnet-Delorme P., Guillevin L., Mouthon L., Frachet P., and Witko-Sarsat V. (2012) Proteinase 3, the autoantigen in granulomatosis with polyangiitis, associates with calreticulin on apoptotic neutrophils, impairs macrophage phagocytosis, and promotes inflammation. J. Immunol. 189, 2574–2583 [DOI] [PubMed] [Google Scholar]
- 13. Rautou P. E., Vion A. C., Amabile N., Chironi G., Simon A., Tedgui A., and Boulanger C. M. (2011) Microparticles, vascular function, and atherothrombosis. Circ. Res. 109, 593–606 [DOI] [PubMed] [Google Scholar]
- 14. Théry C., Zitvogel L., and Amigorena S. (2002) Exosomes: composition, biogenesis and function. Nat. Rev. Immunol. 2, 569–579 [DOI] [PubMed] [Google Scholar]
- 15. VanWijk M. J., VanBavel E., Sturk A., and Nieuwland R. (2003) Microparticles in cardiovascular diseases. Cardiovasc. Res. 59, 277–287 [DOI] [PubMed] [Google Scholar]
- 16. Mause S. F., and Weber C. (2010) Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ. Res. 107, 1047–1057 [DOI] [PubMed] [Google Scholar]
- 17. Daniel L., Fakhouri F., Joly D., Mouthon L., Nusbaum P., Grunfeld J. P., Schifferli J., Guillevin L., Lesavre P., and Halbwachs-Mecarelli L. (2006) Increase of circulating neutrophil and platelet microparticles during acute vasculitis and hemodialysis. Kidney Int. 69, 1416–1423 [DOI] [PubMed] [Google Scholar]
- 18. Erdbruegger U., Grossheim M., Hertel B., Wyss K., Kirsch T., Woywodt A., Haller H., and Haubitz M. (2008) Diagnostic role of endothelial microparticles in vasculitis. Rheumatology 47, 1820–1825 [DOI] [PubMed] [Google Scholar]
- 19. Brogan P. A., Shah V., Brachet C., Harnden A., Mant D., Klein N., and Dillon M. J. (2004) Endothelial and platelet microparticles in vasculitis of the young. Arthritis Rheum. 50, 927–936 [DOI] [PubMed] [Google Scholar]
- 20. Hong Y., Eleftheriou D., Hussain A. A., Price-Kuehne F. E., Savage C. O., Jayne D., Little M. A., Salama A. D., Klein N. J., and Brogan P. A. (2012) Anti-neutrophil cytoplasmic antibodies stimulate release of neutrophil microparticles. J. Am. Soc. Nephrol. 23, 49–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pitanga T. N., de Aragão França L., Rocha V. C., Meirelles T., Borges V. M., Gonçalves M. S., Pontes-de-Carvalho L. C., Noronha-Dutra A. A., and dos-Santos W. L. (2014) Neutrophil-derived microparticles induce myeloperoxidase-mediated damage of vascular endothelial cells. BMC Cell Biol. 15, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Witko-Sarsat V., Canteloup S., Durant S., Desdouets C., Chabernaud R., Lemarchand P., and Descamps-Latscha B. (2002) Cleavage of p21waf1 by proteinase-3, a myeloid-specific serine protease, potentiates cell proliferation. J. Biol. Chem. 277, 47338–47347 [DOI] [PubMed] [Google Scholar]
- 23. Amir-Moazami O., Alexia C., Charles N., Launay P., Monteiro R. C., and Benhamou M. (2008) Phospholipid scramblase 1 modulates a selected set of IgE receptor-mediated mast cell responses through LAT-dependent pathway. J. Biol. Chem. 283, 25514–25523 [DOI] [PubMed] [Google Scholar]
- 24. Koopman G., Reutelingsperger C. P., Kuijten G. A., Keehnen R. M., Pals S. T., and van Oers M. H. (1994) Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 84, 1415–1420 [PubMed] [Google Scholar]
- 25. Schmid I., Uittenbogaart C. H., Keld B., and Giorgi J. V. (1994) A rapid method for measuring apoptosis and dual-color immunofluorescence by single laser flow cytometry. J. Immunol. Methods 170, 145–157 [DOI] [PubMed] [Google Scholar]
- 26. Païdassi H., Tacnet-Delorme P., Garlatti V., Darnault C., Ghebrehiwet B., Gaboriaud C., Arlaud G. J., and Frachet P. (2008) C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J. Immunol. 180, 2329–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schillinger A. S., Grauffel C., Khan H. M., Halskau O., and Reuter N. (2014) Two homologous neutrophil serine proteases bind to POPC vesicles with different affinities: When aromatic amino acids matter. Biochim. Biophys. Acta 1838, 3191–3202 [DOI] [PubMed] [Google Scholar]
- 28. Jo S., Kim T., Iyer V. G., and Im W. (2008) CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 [DOI] [PubMed] [Google Scholar]
- 29. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D. Jr., and Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Venable R. M., Luo Y., Gawrisch K., Roux B., and Pastor R. W. (2013) Simulations of anionic lipid membranes: development of interaction-specific ion parameters and validation using NMR data. J. Phys. Chem. B. 117, 10183–10192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vion A. C., Ramkhelawon B., Loyer X., Chironi G., Devue C., Loirand G., Tedgui A., Lehoux S., and Boulanger C. M. (2013) Shear stress regulates endothelial microparticle release. Circ. Res. 112, 1323–1333 [DOI] [PubMed] [Google Scholar]
- 33. Witko-Sarsat V., Gausson V., Nguyen A. T., Touam M., Drüeke T., Santangelo F., and Descamps-Latscha B. (2003) AOPP-induced activation of human neutrophil and monocyte oxidative metabolism: a potential target for N-acetylcysteine treatment in dialysis patients. Kidney Int. 64, 82–91 [DOI] [PubMed] [Google Scholar]
- 34. Millet A., Martin K. R., Bonnefoy F., Saas P., Mocek J., Alkan M., Terrier B., Kerstein A., Tamassia N., Satyanarayanan S. K., Ariel A., Ribeil J. A., Guillevin L., Cassatella M. A., Mueller A., Thieblemont N., Lamprecht P., Mouthon L., Perruche S., and Witko-Sarsat V. (2015) Proteinase 3 on apoptotic cells disrupts immune silencing in autoimmune vasculitis. J. Clin. Investig. 125, 4107–4121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reid V. L., and Webster N. R. (2012) Role of microparticles in sepsis. Br. J. Anaesth. 109, 503–513 [DOI] [PubMed] [Google Scholar]
- 36. Headland S. E., Jones H. R., Norling L. V., Kim A., Souza P. R., Corsiero E., Gil C. D., Nerviani A., Dell'Accio F., Pitzalis C., Oliani S. M., Jan L. Y., and Perretti M. (2015) Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci. Transl. Med. 7, 315ra190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Falk R. J., Terrell R. S., Charles L. A., and Jennette J. C. (1990) Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc. Natl. Acad. Sci. U.S.A. 87, 4115–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schreiber A., and Kettritz R. (2013) The neutrophil in antineutrophil cytoplasmic autoantibody-associated vasculitis. J. Leukoc. Biol. 94, 623–631 [DOI] [PubMed] [Google Scholar]
- 39. Bauer S., Abdgawad M., Gunnarsson L., Segelmark M., Tapper H., and Hellmark T. (2007) Proteinase 3 and CD177 are expressed on the plasma membrane of the same subset of neutrophils. J. Leukoc. Biol. 81, 458–464 [DOI] [PubMed] [Google Scholar]
- 40. von Vietinghoff S., Tunnemann G., Eulenberg C., Wellner M., Cristina Cardoso M., Luft F. C., and Kettritz R. (2007) NB1 mediates surface expression of the ANCA antigen proteinase 3 on human neutrophils. Blood 109, 4487–4493 [DOI] [PubMed] [Google Scholar]
- 41. Stace C. L., and Ktistakis N. T. (2006) Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim. Biophys. Acta 1761, 913–926 [DOI] [PubMed] [Google Scholar]
- 42. Ravichandran K. S. (2010) Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J. Exp. Med. 207, 1807–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Korkmaz B., Kuhl A., Bayat B., Santoso S., and Jenne D. E. (2008) A hydrophobic patch on proteinase 3, the target of autoantibodies in Wegener granulomatosis, mediates membrane binding via NB1 receptors. J. Biol. Chem. 283, 35976–35982 [DOI] [PubMed] [Google Scholar]
- 44. Gardai S. J., McPhillips K. A., Frasch S. C., Janssen W. J., Starefeldt A., Murphy-Ullrich J. E., Bratton D. L., Oldenborg P. A., Michalak M., and Henson P. M. (2005) Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334 [DOI] [PubMed] [Google Scholar]
- 45. Leventis P. A., and Grinstein S. (2010) The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 39, 407–427 [DOI] [PubMed] [Google Scholar]
- 46. Baumann I., Kolowos W., Voll R. E., Manger B., Gaipl U., Neuhuber W. L., Kirchner T., Kalden J. R., and Herrmann M. (2002) Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 46, 191–201 [DOI] [PubMed] [Google Scholar]
- 47. Qian Y., Wang H., and Clarke S. H. (2004) Impaired clearance of apoptotic cells induces the activation of autoreactive anti-Sm marginal zone and B-1 B cells. J. Immunol. 172, 625–635 [DOI] [PubMed] [Google Scholar]
- 48. Morel O., Jesel L., Freyssinet J. M., and Toti F. (2011) Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler. Thromb. Vasc. Biol. 31, 15–26 [DOI] [PubMed] [Google Scholar]
- 49. Mesri M., and Altieri D. C. (1998) Endothelial cell activation by leukocyte microparticles. J. Immunol. 161, 4382–4387 [PubMed] [Google Scholar]
- 50. Rautou P. E., Leroyer A. S., Ramkhelawon B., Devue C., Duflaut D., Vion A. C., Nalbone G., Castier Y., Leseche G., Lehoux S., Tedgui A., and Boulanger C. M. (2011) Microparticles from human atherosclerotic plaques promote endothelial ICAM-1-dependent monocyte adhesion and transendothelial migration. Circ. Res. 108, 335–343 [DOI] [PubMed] [Google Scholar]
- 51. Scanu A., Molnarfi N., Brandt K. J., Gruaz L., Dayer J. M., and Burger D. (2008) Stimulated T cells generate microparticles, which mimic cellular contact activation of human monocytes: differential regulation of pro- and anti-inflammatory cytokine production by high-density lipoproteins. J. Leukoc. Biol. 83, 921–927 [DOI] [PubMed] [Google Scholar]
- 52. Angelot F., Seillès E., Biichlé S., Berda Y., Gaugler B., Plumas J., Chaperot L., Dignat-George F., Tiberghien P., Saas P., and Garnache-Ottou F. (2009) Endothelial cell-derived microparticles induce plasmacytoid dendritic cell maturation: potential implications in inflammatory diseases. Haematologica 94, 1502–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dalli J., Norling L. V., Renshaw D., Cooper D., Leung K. Y., and Perretti M. (2008) Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood 112, 2512–2519 [DOI] [PubMed] [Google Scholar]
- 54. Dalli J., Norling L. V., Montero-Melendez T., Federici Canova D., Lashin H., Pavlov A. M., Sukhorukov G. B., Hinds C. J., and Perretti M. (2014) Microparticle α2-macroglobulin enhances pro-resolving responses and promotes survival in sepsis. EMBO Mol. Med. 6, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cloutier N., Tan S., Boudreau L. H., Cramb C., Subbaiah R., Lahey L., Albert A., Shnayder R., Gobezie R., Nigrovic P. A., Farndale R. W., Robinson W. H., Brisson A., Lee D. M., and Boilard E. (2013) The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: the microparticle-associated immune complexes. EMBO Mol. Med. 5, 235–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nielsen C. T., Østergaard O., Stener L., Iversen L. V., Truedsson L., Gullstrand B., Jacobsen S., and Heegaard N. H. (2012) Increased IgG on cell-derived plasma microparticles in systemic lupus erythematosus is associated with autoantibodies and complement activation. Arthritis Rheum. 64, 1227–1236 [DOI] [PubMed] [Google Scholar]