

Abstract
The carbohydrate-response element-binding protein (ChREBP) is a glucose-responsive transcription factor that plays an essential role in converting excess carbohydrate to fat storage in the liver. In response to glucose levels, ChREBP is regulated by nuclear/cytosol trafficking via interaction with 14-3-3 proteins, CRM-1 (exportin-1 or XPO-1), or importins. Nuclear localization of ChREBP was rapidly inhibited when incubated in branched-chain α-ketoacids, saturated and unsaturated fatty acids, or 5-aminoimidazole-4-carboxamide ribonucleotide. Here, we discovered that protein-free extracts of high fat-fed livers contained, in addition to ketone bodies, a new metabolite, identified as AMP, which specifically activates the interaction between ChREBP and 14-3-3. The crystal structure showed that AMP binds directly to the N terminus of ChREBP-α2 helix. Our results suggest that AMP inhibits the nuclear localization of ChREBP through an allosteric activation of ChREBP/14-3-3 interactions and not by activation of AMPK. AMP and ketone bodies together can therefore inhibit lipogenesis by restricting localization of ChREBP to the cytoplasm during periods of ketosis.
Keywords: allosteric regulation, AMP, glucose metabolism, hepatocyte, lipogenesis, ChREBP, ketogenesis, glucose sensing, nuclear localization
Introduction
Carbohydrate-response element-binding protein (ChREBP)2 is a glucose-responsive transcription factor that increases transcription of multiple genes containing glucose-responsive elements in promoters (1). ChREBP is expressed at very high concentrations in liver and stimulates gene expression of glycolytic enzymes and all of the lipogenic enzymes, resulting in the conversion of excess carbohydrate into storage fat (2, 3). The action of ChREBP in the liver is independent of insulin effects and accounts for more than half of de novo fat synthesis in the liver.
ChREBP is a large transcription factor of nearly 100 kDa containing functional domains, including two nuclear export signals, NES1 and NES2, and a nuclear import signal (NLS) in the N-terminal region (1–250 amino acids) (Fig. 1A). The nuclear transport factors bind to the NES1, NES2, and NLS within the sequences of the ChREBP to affect their nuclear export and import, respectively. ChREBP functions at two levels, nuclear localization and DNA binding. The N-terminal region binds 14-3-3 proteins and importin α and importin β and is responsible for regulating subcellular localization in response to changing glucose levels. The C-terminal region contains a DNA-binding basic helix-loop-helix/Zip domain and is responsible for transcriptional activity by forming a heterodimer with Max-like protein (Mlx), which binds to E boxes in the promoters of target genes. Phosphorylation/dephosphorylation is the primary mechanism by which ChREBP activity is regulated in response to the glucose level (4, 5). When glucose availability is low, Ser196 of ChREBP is phosphorylated by PKA, and an inactive pool of ChREBP(P)·14-3-3 complex is localized in the cytosol. Phosphorylation by AMPK also inhibits glycolysis and lipogenesis through inactivation of ChREBP. As the glucose level rises, the concentration of the pentose shunt intermediate xylulose 5-P increases, activating a specific protein phosphatase (PP2A-ABδC), which leads to dephosphorylation of ChREBP, nuclear localization by binding to importins, and activation of transcriptional activity of ChREBP (Fig. 1B) (6, 7). In addition to phosphorylation, acetylation and GlcNAc modification of ChREBP have also been reported (8–10) to be involved in activation of ChREBP.
FIGURE 1.

A, schematic overview of ChREBP and N-terminal (81–196) regulatory region (11) and representation of important sites, including the nuclear export signals NES1, NES2, and NLS. B, proposed nuclear-cytosol trafficking pathway of ChREBP. In response to high glucose, phosphorylated inactive ChREBP is dephosphorylated by xylulose 5-P (Xu5P)-activated PP2A and translocates into the nucleus through interactions with importin α and importin β. ChREBP is further dephosphorylated in the nucleus and induces transcription of lipogenic genes. A decrease in glucose levels results in inactivation of ChREBP by phosphorylation by PKA and complex formation with 14-3-3 and CRM-1 to the cytosol (6). bHLH, basic helix-loop-helix.
Contrastingly, 14-3-3 proteins and importin α play important roles in nuclear/cytosol trafficking of ChREBP. Previously, we have shown that 14-3-3 proteins bind to two α-helices of ChREBP, a primary binding site on α2 helix (residues 125–135) and secondary binding site on an extended α3 helix and phosphorylated Ser196 (6). Importin α binds to an extended bidentate NLS site (residues 158–190) that overlaps with the α3 helix and helps explain our previous finding that 14-3-3 has a dual regulatory function; one function is to stimulate export of inactive and phosphorylated ChREBP out of nucleus, while at the same time blocking importin α from binding to the NLS site, thus restricting ChREBP to the cytoplasm under low glucose concentrations (11, 12). As the glucose levels rise after a meal, the inactive Ser(P)196-ChREBP is dephosphorylated to form active ChREBP, leading to the dissociation of 14-3-3 from the NLS site. This dissociation permits importin α to bind to ChREBP at the NLS site, allowing ChREBP to then be localized in the nucleus.
In addition to the regulation of ChREBP activity by reversible phosphorylation, we showed recently that metabolites play allosteric regulatory roles by altering affinities of the protein/protein interaction between ChREBP-14-3-3 and ChREBP-importin α. We have demonstrated that β-hydroxybutyrate (β-HB) and acetoacetate (AcAc) produced in the livers of starved rats inhibit nuclear localization of ChREBP not only by activation of ChREBP and 14-3-3 interaction and stabilization of the complex in cytoplasm, but also by inhibiting importin α binding to the NLS site of ChREBP in starved livers (12).
We presented evidence previously that AMP generated during fatty acid metabolism inhibits the transcriptional activity of ChREBP by AMPK-mediated phosphorylation of Ser568, located near the BHLH region (5). Later, Dentin et al. (13) claimed that only polyunsaturated fatty acids specifically inhibit ChREBP mainly by decreasing nuclear localization. They measured the nuclear localization by separating the nucleus and the cytoplasm from the hepatocytes incubated in the fatty acids for 24 h. As far as we are aware, it is not possible to isolate the nucleus or to separate the nucleus from cytoplasm from the primary culture of hepatocytes after a few hours. Moreover, hepatocytes are unstable and undergo slow lysis in the presence of polyunsaturated fatty acids after only a few hours. However, fatty acid ingestion leads to the AMP inhibition of ChREBP, but the mechanism of the AMP inhibition is still unclear. As we proposed before, one possibility is AMP activation of AMPK, but the mechanism appears more complex because of the following reason. By using AMPK-deficient mice, it was determined that metformin and other small molecular weight compounds such as A769662, a known activator of AMPK, were not effective in inhibiting lipogenesis completely in hepatocytes (14). Metformin is the most widely used drug to treat type 2 diabetes, but the molecular mechanism of its action is unknown.
We therefore extended the study of metabolite regulation of ChREBP nuclear localization by focusing on the metabolism of branched-chain α-ketoacids (BCKA) and fatty acids in rat hepatocytes. Branched-chain amino acids (BCAAs), including leucine, isoleucine, and valine, are converted to BCKAs in the muscle, and the α-ketoacids are transported to liver where they are oxidized by BCKA dehydrogenase to acyl-CoA. The acyl-CoA is further metabolized to form ketone bodies. Additionally, some of the α-ketoisocaproate (KIC) is oxidized by dioxygenase to form 3-hydroxy-3-methylbutyrate (HMB), which is converted to HMB-CoA, generating AMP. HMB-CoA is ultimately metabolized to AcAc, and mevalonate is converted to cholesterol (15, 16). Under starvation or prolonged exercise, acetyl-CoA formed during oxidation of fatty acids can either enter the citric acid cycle for energy production or undergo conversion to ketone bodies, including AcAc and β-HB for export to other tissues. In this communication, we report that AMP inhibits ChREBP nuclear localization activity by a new mechanism, which is not dependent on the phosphorylation by AMPK.
Experimental Procedures
Chemicals and Vectors
All chemicals were purchased from Sigma unless otherwise indicated. AMPK activators KM-2-100 and LW-V-152 are two structurally distinct experimental small molecules that activate AMPK in vitro via an unknown mechanism and were discovered and synthesized by Jef K. De Brabander (University of Texas Southwestern Medical Center, Dallas). The catalytic subunit of cAMP-dependent protein kinase (PKA) and AMPK were purchased from Promega (Madison, WI). Bacterial expression vector for the human 14-3-3β was a gift from Dr. Steven L. McKnight (University of Texas Southwestern Medical Center) and that for GST-tagged importin α was a gift from Dr. Yoshihiro Yoneda (Osaka University, Osaka, Japan). Both 14-3-3β protein and importin α were purified as described previously (12). FLAG-tagged and GFP fusion ChREBP and Duet ChREBP-14-3-3β proteins were used for each experiment (6, 12, 17).
Interaction of ChREBP and 14-3-3β or ChREBP and Importin
The following three methods were used.
Assay A, Pulldown Assay
FLAG-tagged ChREBP was expressed in HEK293 cells and purified from cell lysate by binding to anti-FLAG antibody beads in the reaction mixture containing 20 mm HEPES-KOH, pH 7.3, 0.5 mm EGTA, 60 mm KAc, 2 mm MgAc, 5 mm NaAc, 0.01% Nonidet P-40 and rocked for 1 h at 4 °C. Homogeneous 14-3-3β or importin α was added to the beads bound with ChREBP and incubated for 1.5 h at 4 °C with gentle rocking. Bound FLAG-ChREBP and 14-3-3 β were eluted with SDS-PAGE sample buffer and separated by SDS-PAGE (10%) and subjected to gel electrophoresis followed by immunoblotting.
Assay B, Fluorescence Polarization
Fluorescence polarization experiments were carried out in a 384-well black microplate (Corning Glass) in a total volume of 50 μl in each well. Rhodamine-tagged ChREBP peptides (residues 117–140 as “α2 helix peptide” or residues 151–190 as “NLS peptide,” 30 nm as final concentration) were mixed with various concentrations of His-tagged 14-3-3 proteins or GST-tagged importin α proteins, or without proteins as control, in Tris-HCl (50 mm, pH 7.5) containing EDTA (0.2 mm) in the plate, then covered, and incubated at room temperature for 1 h. Fluorescence intensity measurements were performed with a SpectraMax i3 plate reader (Molecular Devices, Sunnyvale, CA) with a rhodamine FP cartridge (λex = 535 nm, λem = 595 nm). Fluorescence polarization values were calculated from vertical fluorescence intensity (S-channel) and horizontal fluorescence intensity (P-channel) and expressed as milli-polarization units.
Assay C, Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) experiments were carried out using a VP-ITC microcalorimeter (MicroCal) as described previously (12).
Animals
Male 5–10-week-old Sprague-Dawley rats were reared in temperature-controlled facilities with a 12-h light/dark cycle and fed with standard rodent chow (National Institutes of Health) (Harlan-Teklad Mouse/Rat Diet 7002; Harlan-Teklad Premier laboratory Diets). High sucrose (HS) and HF diets were described previously (18). All animals were cared for in accordance with the Institutional Animal Care and Use Research Advisory Committee of the Veterans Affairs Medical Center.
Animal Rights
Animal manipulations were approved by the Animal Welfare Committee of Veterans Affairs Medical Center.
Preparation of Protein-free Extracts of Livers and HPLC Separation of Metabolites
To isolate metabolites in liver, protein-free extract of rat liver was prepared according to the methods described previously (12, 19). Briefly, freeze-clamped liver of rats fed with different diets was homogenized in 3 volumes of ice-cold 0.6 n HClO4 using a Polytron homogenizer at 4 °C. After centrifugation, the supernatant solution was neutralized with KOH, and the precipitate was removed by centrifugation. The supernatant solution was used for isolation of metabolites by HPLC as described previously (12). Metabolite extracts (0.2 ml) were applied on a CarboPac PA1 column (4 × 250 mm) (Dionex, Sunnyvale, CA) and eluted with a linear gradient of ammonium acetate (0–1 m, pH 5.0) at a flow rate of 1 ml/min. Eluted fractions were collected, concentrated by lyophilization, and then used for identification of the metabolites with MS/MS and for analysis of binding activity of ChREBP and for pulldown assays with 14-3-3.
Primary Cultures of Rat Hepatocytes
Primary hepatocytes were isolated from male Sprague-Dawley rats using the collagenase perfusion method (5, 20). The isolated cells were used for the following three assays.
Luciferase Reporter Activity of ChREBP
The isolated cells were plated in collagen-coated 6-well tissue culture plates (BD Biosciences) at a density of 5 × 105 cells/well in Dulbecco's modified Eagle's medium supplemented with 100 nm dexamethasone, 10 nm insulin, 100 units/ml penicillin, 100 μg/ml streptomycin, 10% dialyzed fetal bovine serum, and 27.5 mm glucose (HG medium). Following attachment for 4 h, the cells were subjected to transfection with pGL-LPK, pRL-TK, and FLAG-ChREBP using Lipofectamine 2000 (Life Technologies, Inc.) according to the manufactured procedure and incubated for 4 h. The medium containing the liposome·DNA complex was removed and replaced with low glucose medium or HG medium in the absence or presence of BCKAs, fatty acids, β-HB, and AMPK activators for 20 h. Luciferase reporter activity was measured sequentially using a Dual-Luciferase reporter assay system (Promega, Fitchburg, WI) using a model TD-20E Luminometer (Turner Design, Sunnyvale, CA), as described previously (6).
Subcellular Localization of ChREBP in Hepatocytes
The isolated cells were plated in collagen-coated 35-mm glass bottom tissue culture plates (MatTek Corp., Ashland, MA) at a density of 1 × 106 cells/well in HG medium. Following attachment for 5 h, the cells were subjected to transfection with pEGFP-ChREBP using Lipofectamine LTX (Life Technologies, Inc.) according to the manufacturer's instruction and incubated for 4 h. The medium containing the liposome·DNA complex was removed and replaced with HG medium for 12 h (expression period). The cells were incubated with low glucose or high glucose medium in the presence of BCKAs, fatty acids, BCAAs, β-HB, and AMPK activators. Subcellular localization of GFP-fused ChREBP was determined using a confocal laser microscope as described before (4).
Preparation of Nuclear and Cytosolic Extracts
A piece of liver (50 mg) from a rat fed either high fat (HF) or HS diet was homogenized in 500 μl of Cytosolic Extraction Reagent buffer-1 (“CER-1,” Thermo Fisher Scientific) as described previously (1). CER-2 buffer (20 μl) was added to the homogenate, and the homogenate was centrifuged at 16,000 × g for 5 min, and the supernatant solution was stored at −80 °C as cytosolic fraction. The pellet fraction was resuspended in 200 μl of NER buffer and centrifuged at 16,000 × g for 10 min. The supernatant solution was stored at −80 °C as nuclear fraction. The extracts of the nuclear and cytosolic fractions were size-fractionated on an SDS-12% polyacrylamide gel and transferred onto a Trans-Blot nitrocellulose membrane, and the ChREBP band was visualized with an ECL Western blotting detection system. The densitometry was performed using ImageJ to evaluate the ChREBP expression levels.
Metabolite Measurements
The measurement of a number of metabolites is required to estimate the free cytosolic ADP and AMP and the ΔG′ of ATP hydrolysis (21). Stable isotope dilution GC-MS analyses for the metabolites, di-hydroxyacetone phosphate, 3-phosphoglycerate, lactate, and pyruvate was performed (22) to estimate the free cytosolic ADP concentrations from the ratios of these concentrations. The free cytosolic AMP and the ΔG′ of ATP hydrolysis were determined as described previously (22). Free intracellular [Mg2+] was estimated by determining the concentrations and the ratio of [citrate]/[isocitrate] because the Keq of the GAPDH-3-phosphoglycerate kinase reaction is dependent upon free [Mg+2] (23) as is the Keq of the myokinase reaction. Finally, an estimate of the intracellular pH is required. Total ATP and AMP were determined by capillary electrophoresis mass spectrometry as described (21, 22).
Phosphorylation Assay
Pure AMPK and the catalytic subunit of PKA were purchased from Promega. (N)-ChREBP-14-3-3β protein was expressed and used as substrate (1–12 μg) for AMPK (15 ng) or PKA (5 ng) at 30 °C for 30 min. The reaction mixture was subjected to SDS-PAGE and immunoblotting using phospho-AMPK substrate (Cell Signaling Technology, Danvers, MA) or PKA substrate antibody (catalog no. 9624; Cell Signaling Technology). As a positive control of these AMPK phosphorylation assays, two other AMPK substrates were used, glycogen synthase purchased from Novus Biologicals (Littleton, CO) and acetyl-CoA carboxylase 2 (ACC2), a gift from Dr. Jay Horton (University of Texas Southwestern Medical School).
Protein Expression and Purification
The procedure for the bacterial expression and purification of the heterodimer protein containing the full-length mouse 14-3-3β and the N-terminal region of ChREBP (residues 1–250,“N-ChREBP”) was described previously (17), with the addition of 20 mm AMP at the wash and elution steps.
Crystallization, Data Collection, and Structure Determination
Crystals were prepared under similar conditions as described previously (17) with slight modification. The N-ChREBP-14-3-3 heterodimer (15 mg/ml) was mixed with 100 mm AMP and incubated on ice for 30 min. To the mixture (2 μl) was added 2 μl of well solution containing 100 mm sodium citrate, pH 6.5, 150 mm sodium/potassium tartrate, 2.2 m ammonium sulfate, and 10 mm tris(2-carboxyethyl)phosphine-HCl and incubated in the hanging-drop plate at 20 °C. Crystals grew to a diffraction quality in 10 days and were harvested, cryo-protected with 2.0 m lithium sulfate, and flash-frozen in the liquid nitrogen. Diffraction data were collected at beamline 19-ID, Advanced Photon Source, Argonne National Laboratory, Argonne, IL. Data were processed using HKL3000 (24). Initial phases were obtained by the molecular replacement using MOLREP (25) with apo-14-3-3β and ChREBP structure (Protein Data Bank code 4GNT) as a search model. The adenine ring and the phosphate molecules were manually built into the density using COOT (26), and the structure was further refined using phenix.refine (27). The coordinates of 14-3-3β-ChREBP complex bound with AMP have been deposited in the Protein Data Bank (PDB access code 5F74).
Statistics
Results were expressed as mean ± S.E. Comparisons among groups were made by factorial analysis of variance or repeated-measure analysis of variance using Tukey-Kramer. Differences were considered statistically significant if the p value was smaller than 0.05. Significance was indicated as p < 0.05 and p < 0.01.
Results
Subcellular Localization of ChREBP in Rat Livers Fed a High Fat or a High Sucrose Diet
Rats were fed HF and HS diets. The cellular distribution of ChREBP in the nucleus and the cytosol was determined by differential centrifugation (Fig. 2). As expected, the cellular distribution of ChREBP in the livers was opposite with these diets; more ChREBP was localized in the nucleus than the cytosol under HS, although its localization was less in the nucleus under the HF diet. These in vivo results confirmed the cellular distribution of ChREBP extensively studied using primary hepatocytes (5, 12).
FIGURE 2.
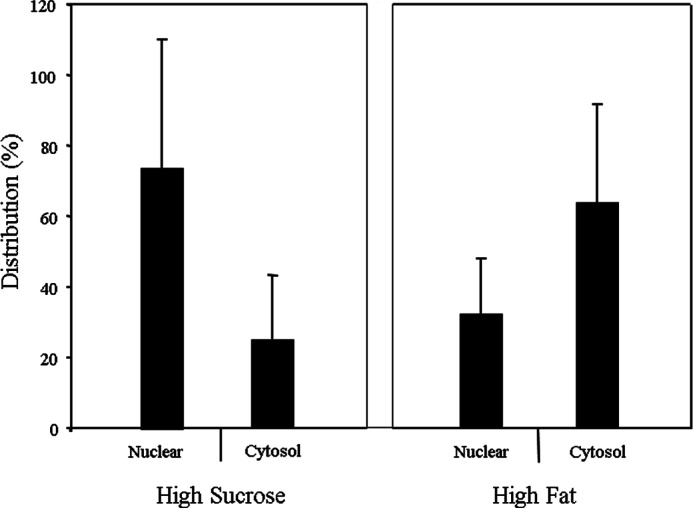
Subcellular localization of ChREBP in rat livers fed a high fat or a high sucrose diet. The nuclear and cytosol extracts from rat liver were prepared using NE-PERTM nuclear and cytoplasmic extraction reagents according to the manufacturer's instructions. The nuclear and cytosol fractions (50 μg) were analyzed by SDS-PAGE and immunoblotting using antibodies specific for ChREBP as described under “Experimental Procedures.” The values presented are the mean ± S.E. of three livers.
Metabolites in High Fat-fed Liver-activated ChREBP and 14-3-3 Interaction
Previously, we have shown that ketone bodies produced in starved rat livers and HF diet-fed rat livers inhibit the nuclear localization as well as the transcriptional activities of ChREBP (12). This inhibition is attributed to the ability of ketone bodies to activate protein/protein interaction between ChREBP and 14-3-3, as demonstrated by pulldown assays (12). The HPLC fractions of the metabolite extract from the starved liver contained only one fraction (Fraction 10 in Fig. 3B) (12), which activated the ChREBP/14-3-3 interaction. In contrast, the HPLC fractions of the metabolite extract of HF-fed livers contained two fractions (fractions 10 and 16), which were shown to activate the interaction (Fig. 3). Similarly to the starved liver, the metabolites in fraction 10 of the HF-fed liver were identified as β-HB and AcAc by GC/MS (12). The metabolite in the HPLC fraction 16 was identified as AMP based on its UV absorption, MS/MS analysis, and co-elution with standard AMP using HPLC (see under “Experimental Procedures”).
FIGURE 3.
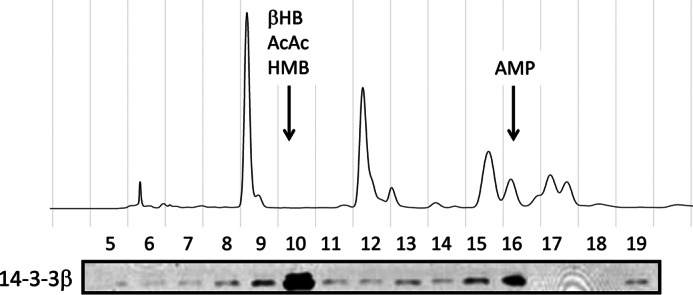
Effects of high fat liver extract on the binding of 14-3-3β to ChREBP. To identify the metabolite stabilizer of the ChREBP and 14-3-3 interaction, protein-free metabolite extract of livers from rats fed with HF diet for 3 days was subjected to HPLC analysis using a CarboPac PA1 column (4 × 250 mm). The elution of UV-absorbing compounds (A254 nm) are shown (top panel). The eluted fractions were assayed using pulldown assay for the interaction of ChREBP and 14-3-3β (bottom panel).
A closer examination of fraction 10 of the HPLC also revealed the presence of an additional metabolite, which co-eluted with ketone bodies in the same fraction. This metabolite was recently identified as HMB by GC/MS and HPLC. HMB is a known metabolite of KIC produced by KIC dioxygenase (11, 28). The concentration of HMB in the extract of HF liver was 95 μm, which was 50% of the AcAc level (180 μm) and 20% of β-HB (760 μm) in the extract (data not shown). It is not surprising that HMB also activates the interaction of ChREBP and 14-3-3 in the pulldown assay, but this activation was ∼30% that by β-HB.
Because the ketone bodies in the HF extract were less than 30% of those in the starved liver extract, but the ChREBP/14-3-3 interaction activation activity of the extract was >50% of the starved liver extract, ketone bodies alone could not account for all the activity in the HF extract and seemed to require the presence of AMP. These results suggest that both ketone bodies and AMP together are responsible for promoting the ChREBP-14-3-3 heterodimer formation and stabilization, which results in the inhibition of ChREBP nuclear localization.
Nuclear Localization of ChREBP Is Inhibited by Metabolism of α-Ketoacids in Hepatocytes
The HF diet also contained casein representing ∼10% of total calories, and the casein contained BCAAs including leucine, isoleucine, and valine. As discussed in the Introduction, these amino acids are converted to α-ketoacids in muscle and metabolized further in the liver to yield ketone bodies and AMP. Because similar metabolites were produced, one expects that the α-ketoacid metabolites have same inhibitory effects as fatty acid metabolism on the ChREBP nuclear localization and transcriptional activity.
When primary cultures of hepatocytes were incubated in high glucose (27.5 mm) containing 2 mm of each KIC, KIV, or α-keto-β-methylvalerate (KMV), the nuclear localization of GFP-ChREBP was inhibited by all these α-ketoacids (Fig. 4A). However, the rates of inhibition were different; the inhibition by KIC and KMV was rapid during the 1st h, and it continued more slowly beyond 6 h (Fig. 4A). The inhibition by KIV proceeded more slowly than the other ketoacids for 6 h. The ChREBP nuclear localization was inhibited more strongly by unsaturated fatty acids, oleate and linoleate, than saturated fatty acids, palmitate (Fig. 4B), confirming our previous results (12). To ensure AMP and ketone bodies were responsible for the inhibition, we examined the effects of AICAR and β-HB. AICAR is phosphorylated to form ZMP in vivo, and it serves as an activator of AMPK. The inhibition by AICAR and β-HB, in contrast to α-ketoacids, was more rapid and reached the maximum in less than 3 h, but the extent of the nuclear localization of ChREBP were less than half of those by fatty acids (Fig. 4C). The observation that the inhibition by the α-ketoacids was greater than by β-HB alone strongly suggests that AMP and ketone bodies together contribute to the inhibition of ChREBP nuclear localization.
FIGURE 4.

BCKAs and unsaturated fatty acids suppress nuclear localization and inhibit transcription of ChREBP. A–C, primary cultures of rat hepatocytes were transfected with GFP-ChREBP in DMEM containing low glucose (5.5 mm) for 4 h and then the medium was changed to DMEM containing high glucose (27.5 mm) for 12 h. The cells were incubated in high glucose in the presence of BCKAs (KIC, KIV, and KMV) (A), fatty acids (oleate, linoleate, and palmitate) (B), and β-HB or AICAR (C). The cells incubated in low and high glucose alone served as the controls. At the indicated time, the cells were fixed with 4% paraformaldehyde, and subcellular localization of GFP-ChREBP in the nucleus and cytoplasm was determined using a confocal microscopy. The values are presented as the mean ± S.E. of triplicate cultures. *, p < 0.05 (versus high glucose incubation). D–F, BCKAs and unsaturated fatty acids suppress transcription activity of ChREBP. Primary cultures of rat hepatocytes were co-transfected with pGL-LPK, pRL-TK, and FLAG-ChREBP. After transfection, cells were incubated in DMEM containing low glucose (5.5 mm) for 4 h and then incubated in high glucose (27.5 mm) for 12 h with medium containing high glucose. The cells were incubated in low glucose or high glucose with or without 2 mm each BCKA (KIC, KIV, and KMV) (D), 2 mm fatty acids (oleate, linoleate, and palmitate) (E), and 2 mm HMB, 2 mm β-HB, or 0.2 mm AICAR (F) for 20 h. Luciferase reporter activity was determined by luciferase reporter system as described previously (12). The values are presented as the mean ± S.E. of three independent experiments. ns, not significant. *, p < 0.05, and **, p < 0.01 (versus high glucose incubation).
Using isotope dilution mass spectrometry, the concentrations of total ATP and AMP in these hepatocytes after 1 h of incubation in low glucose, HG control, KIC in HG, or oleate in HG was determined (Table 1). The concentrations of free ADP and AMP were determined as described previously (21). Although there were no differences in total AMP concentrations between the HG group and other groups, there was a significant elevation of free AMP (3.3-fold) in the KIC group compared with the control in the presence of high glucose (Table 1). In contrast to the free AMP, the measured total AMPs produced by KIC or oleate are approximately the same. Also, the calculated phosphorylation potential or the energy states of these cells decreased only 0.4 and 0.1 kcal/mol in KIC and oleate, respectively, compared with that in HG, but these were not significant.
TABLE 1.
Free and total adenine nucleotides, phosphorylation potential, and free energy of ATP hydrolysis (ΔG′ATP)
The abbreviations used are as follows: LG, low glucose; oleate, oleic acid; AMPfree, calculated from the phosphorylation potential; free AMP, as calculated from free ADP, as determined from the ratio of the glycolytic intermediates (see under “Experimental Procedures”) and total AMP as determined by mass spectrometry.
| Substrate | AMPfree | Phosphorylation potential | ΔG′ATP | ATP | ADP | AMPtotal |
|---|---|---|---|---|---|---|
| μm | kcal/mol | mm | μm | μm | ||
| LG | 5.64 ± 0.01 | 7433 ± 379 | −13.4 ± 0.14 | 1.78 ± 0.22 | 220 ± 23 | 157 ± 27 |
| HG | 4.27 ± 0.05 | 10,276 ± 4692 | −13.6 ± 0.12 | 1.33 ± 0.07 | 191 ± 8 | 119 ± 12 |
| KIC | 14.30 ± 0.03a | 4442 ± 1650 | −13.2 ± 0.03 | 1.16 ± 0.08 | 341 ± 14 | 104 ± 17 |
| Oleate | 5.36 ± 0.01 | 4731 ± 1869 | −13.5 ± 0.2 | 1.06 ± 0.17 | 141 ± 23 | 97 ± 23 |
a Significantly different from HG (control) using analysis of variance with post hoc Bonferroni correction.
The transcriptional activity of ChREBP was inhibited ∼20–30% by KIC, KMV, and KIV in the presence of high glucose (Fig. 3D), similar to the nuclear localization of ChREBP. In contrast, unsaturated fatty acids, oleate and linoleate, inhibited ChREBP transcription activity completely, whereas the saturated fatty acid, palmitate, showed only a small inhibitory effect (10%) under these conditions (Fig. 3E). The ChREBP transcriptional activity of hepatocytes was also inhibited ∼30, 15, and 55%, respectively, in the presence of 2 mm HMB, 2 mm β-HB, and 0.2 mm AICAR in hepatocytes in 20 h (Fig. 4F). The observation that AICAR showed a nearly 2-fold greater inhibition of the ChREBP transcriptional activity than β-HB suggests that AICAR inhibits both the nuclear localization and the transcription activities of ChREBP in hepatocytes, whereas the ketone bodies affect only the nuclear import. These different degrees of inhibition produced by BCKA, unsaturated fatty acids, and ketone bodies appear to result partly from the differences in the mechanisms used by AMP whether allosteric binding or AMPK affecting two different activities of ChREBP.
Mechanism of AMP Inhibition of ChREBP Nuclear Localization
Two possible mechanisms were considered for AMP inhibition of the ChREBP nuclear localization as follows: (a) the activation of AMPK resulting in phosphorylation/inactivation, and (b) the allosteric activation of ChREBP/14-3-3 interaction and stabilization of the heterodimer in cytoplasm, similar to ketone bodies.
To distinguish between these possibilities, we examined the effect of dorsomorphin (compound C), which is a known AMPK inhibitor, on the initial rates of the ChREBP nuclear localization. Compound C in the high glucose conditions did not affect the ChREBP nuclear localization activity (Fig. 5A). In contrast, AICAR (0.2 mm) showed inhibition of the nuclear localization in the presence or absence of compound C. The lack of the effects of the AMPK inhibitor on the nuclear localization rules out a possible involvement of AMPK and favors the AMP action by an AMPK-independent mechanism.
FIGURE 5.

Effects of AMPK activators and AMPK inhibitor on nuclear localization and transcription activity of ChREBP. A, effects of AMPK inhibitor, compound C (1 μm) with or without 200 μm AICAR, on nuclear localization were determined as described in Fig. 2. B, inhibition of transcription activities of ChREBP by AICAR, KIC, and β-HB was recovered by the addition of AMPK inhibitor, compound C (1 μm). C, AMPK activators, 10 μm KM-2-100-1.3, 20 μm LW-V-152, 0.5 mm metformin, and 1 mm A769662 had little effect on the nuclear localization of ChREBP. D, effect of AMPK activators (10 or 20 μm KM-2-100-1.3, 10 or 20 μm LW-V-152, 1 mm A769662, 0.1–1 mm metformin, and 200 μm AICAR) on the transcription activities of ChREBP was determined as described in Fig. 2. The values are presented as the mean ± S.E. of three rats. ns, not significant. *, p < 0.05, and **, p < 0.01 (versus high glucose incubation).
Because we have shown previously that the transcriptional activity of ChREBP is inhibited by AMPK by phosphorylation of the DNA binding domain (5), we investigated the effects of compound C on the transcriptional activity of ChREBP in the presence of KIC, β-HB, and AICAR in hepatocytes under high glucose conditions. The results in Fig. 5B show that KIC and β-HB inhibited ∼50 and 25%, respectively, that of high glucose-induced activation of the ChREBP transcriptional activity. Compound C completely reversed the inhibition. The inhibition by AICAR was significantly larger than KIC, and compound C was able to partially overcome the inhibition. These results are consistent with a mechanism in which the AMP inhibition of ChREBP nuclear localization is AMPK-independent, but its effect on the transcriptional activity is mediated by AMPK.
Effects of Activators of AMPK in Hepatocytes
Next, we investigated the effects of compounds that are known to activate specifically AMPK, including A769662, metformin,“KM-2-100-1.3,” and “LW-V-152,” on the ChREBP activities under high glucose in hepatocytes. A769662 is known to mimic the effect of AMP on AMPK by binding to the allosteric site and activates AMPK, independently of AMP. Metformin is a known activator of AMPK, but its exact mechanism of action is unclear. KM-2-100-1.3 and LW-V-152 are experimental small molecules discovered in the De Brabander laboratory (University of Texas Southwestern Medical Center, Dallas, TX) that strongly activate AMPK in vitro. The exact mechanism of action remains unclear. The results demonstrated that 1 mm A769662, 0.5 mm metformin, 10 μm KM-2-100-1.3, and 20 μm LW-V-152 showed little effect on the nuclear localization of ChREBP under high glucose concentrations (Fig. 5C). However, the transcriptional activity was completely inhibited by these drugs (Fig. 5D). The concentrations of A769662 required to inactivate the ChREBP transcriptional activity in the hepatocytes were 10-fold higher than those reported in the literature (Fig. 4B). The reason for the difference is unclear. These results support the above conclusion that the nuclear localization of ChREBP is not mediated by AMPK, but the transcriptional activity was inhibited by these drugs, known activators of AMPK, as expected. The results suggest that AMP employs two different mechanisms to regulate the ChREBP activities as follows: the nuclear localization by AMP-induced conformational changes leading to allosteric inhibition, and the transcriptional activity by AMPK-mediated phosphorylation. Interestingly, metformin only inhibits the transcriptional activity of ChREBP probably by AMPK activation without affecting the nuclear localization in hepatocytes in the high glucose conditions employed here.
(N)-ChREBP Is Not Phosphorylated by AMPK
To make certain that there is no AMPK-dependent phosphorylation site on the N-terminal region of ChREBP, we prepared a homogeneous (N)-ChREBP (residues 44–196)-14-3-3 heterodimer (17). Several attempts were made to phosphorylate the (N)-ChREBP-14-3-3 heterodimer, with pure AMPK (Promega) in vitro, under similar conditions to which a peptide substrate (S-adenosylmethionine, Promega) of AMPK, glycogen phosphorylase, and acetyl-CoA carboxylase were phosphorylated (data not shown). The same heterodimer of (N)-ChREBP-14-3-3 also contains a cAMP-dependent protein kinase (PKA) site at Ser196 (4), which was phosphorylated by the protein kinase under the same conditions. However, we could not rule out the possibility that an AMPK phosphorylation site may exist in the N-terminal region containing amino acid residues 1–43, but it is unlikely based on the other evidence presented above.
Altogether, these results strongly suggest that the AMP inhibition of the ChREBP nuclear localization is not mediated by ChREBP phosphorylation by AMPK, instead it favors an AMP-induced allosteric mechanism.
AMP Binds to (N)-ChREBP-14-3-3 Heterodimer
Next, we investigated an alternative mechanism in which AMP binds directly to ChREBP and allosterically induces conformational changes resulting in activation of (N)-ChREBP/14-3-3 interaction and inhibition of the nuclear localization. Using isothermal titration calorimetry (ITC, assay B), we determined that AMP binds directly to the (N)-ChREBP-14-3-3 heterodimer with a Kd value of 7.6 ± 0.7 × 10−6 m (Fig. 6A). However, neither (N)-ChREBP nor 14-3-3 alone bound AMP under the same conditions. Moreover, AMP had little effect on the importin α binding to the NLS peptide of (N)-ChREBP, suggesting that 14-3-3 binds weakly to the NLS site.
FIGURE 6.

AMP binds directly to ChREBP and increases the affinity for 14-3-3β. A, ITC experiments were carried out using a VP-ITC microcalorimeter (MicroCal). n = 3. B, activation of the interaction between FLAG-ChREBP and 14-3-3β by varying concentrations of AMP was determined by fluorescence polarization assay. n = 7. The values are presented as the mean ± S.E. of three independent experiments. C, reciprocal effects of AMP on the interaction FLAG-ChREBP-14-3-3β and FLAG-ChREBP-importin α as determined by pulldown assay. AMP activates ChREBP/14-3-3 interaction and inhibits ChREBP-importin α binding. n = 4. Data shown are representative of three separate experiments. D, effects of ZMP on the interaction of FLAG-ChREBP and 14-3-3 (top) and FLAG-ChREBP and importin α (bottom). ZMP inhibited only ChREBP-14-3-3 but no effect on ChREBP-importin. n = 2. Data shown are representative of three separate experiments.
Using fluorescence polarization technique (assay C), we also determined that the presence of 50 μm AMP increased the affinity of the α2 peptide (amino acid residues 117–140) to 14-3-3β 5-fold (Kd = 0.03 μm in the presence versus 0.16 μm in the absence of AMP) (Fig. 6B). This peptide was chosen because we have shown previously that 14-3-3 binds to the α helix consisting of amino acids residues 117–137 of ChREBP in the crystal structure of the ChREBP-14-3-3 heterodimer (17). However, AMP again failed to bind to either the ChREBP peptide or 14-3-3 alone, under similar conditions. These results support a mechanism in which AMP binds directly to the ChREBP in the (N)-ChREBP-14-3-3 heterodimer and induces conformational changes resulting in the increased affinity of ChREBP for 14-3-3 and stabilization of the heterodimer. The large differences in those Kd values between ITC and fluorescence polarization methods are due to the differences in 14-3-3 binding to two different sites on the ChREBP molecule. We have shown that ChREBP has two 14-3-3-binding sites, the primary α2 helix and the secondary α3 sites (6, 11). In the ITC measurement the primary site of the (N)-ChREBP-14-3-3 heterodimer was already occupied by 14-3-3β; consequently; the exogenously added 14-3-3β is binding to the secondary site, although in the fluorescence polarization measurements 14-3-3β is binding specifically to the primary α2 helix peptide. In addition, the differences could be due to the binding of the peptide (N)-ChREBP versus the ChREBP protein in the (N)-ChREBP-14-3-3 heterodimer. Nevertheless, it is important to note that those results showed the direct binding of AMP to (N)-ChREBP-14-3-3.
Effects of AMP and ZMP on ChREBP/14-3-3 and ChREBP/Importin α Interaction
Previously we reported that 14-3-3 inhibits importin α binding to the NLS of ChREBP (6, 11) and that ketone bodies in the starved rat liver extract enhanced the reciprocal effects on the 14-3-3 and importin α bindings to ChREBP (12). It was of interest to determine whether AMP binding affects the ChREBP interaction with those proteins in a reciprocal manner, similar to that of ketone bodies. We employed the pulldown assay to determine the effects of AMP and ZMP on ChREBP-14-3-3 and ChREBP-importin α binding. The results demonstrated that AMP at ranges of 10 μm to 1 mm activated the 14-3-3β/ChREBP interactions while inhibiting the importin/α-ChREBP interactions (Fig. 6, C and D). In contrast, ZMP, a metabolite of AICAR, also activated 14-3-3 binding to ChREBP at 10-fold lower concentrations than AMP, but ZMP showed little effect on the interaction between ChREBP and importin α under these conditions. These results provide additional evidence in support for the direct binding of AMP to the N-terminal region of the ChREBP/14-3-3 heterodimer, independently of AMPK. It should be noted that the AMP concentrations employed in the pulldown assay are within the physiological ranges of free AMP in hepatocytes (Table 1) and in rat liver (21).
Crystal Structure of AMP Bound to (N)ChREBP/14-3-3 Heterodimer
We have also investigated the binding of AMP to the ChREBP/14-3-3 heterodimer by elucidating the crystal structure (Fig. 7A and Table 2). The interface between ChREBP and 14-3-3 has a large space toward the solvent access and a smaller space that is surrounded by the basic residues from both proteins. This basic cavity accommodates the phosphate group from the AMP perfectly, and the major binding energy for AMP to the ChREBP·14-3-3 complex was through the phosphate group of AMP. The orientation of the phosphate molecule is in a manner that both nitrogens of all the surrounding arginine residues (Arg58 and Arg129 of 14-3-3 and Arg128 of ChREBP) can interact with two oxygen atoms of the phosphate (Fig. 7B). In the apo-ChREBP/14-3-3 structure, Arg128 of ChREBP has a different orientation, and only one nitrogen atom has an interaction with the sulfate molecule, which was found in the same site that the phosphate occupies in our structure. The distances between oxygen atoms of phosphate and these amino acid residues range from 2.74 to 3.30 Å. Trp127 of ChREBP has an interaction with the phosphate oxygen with the distance of 3.54 Å, and Lys51 of 14-3-3 also has an interaction with the phosphate oxygen with the distance of 2.58 Å. These same basic residues also participate in the binding with a sulfate molecule in our previous structure (17). There were a few interactions found with the adenine ring of AMP. In the absence of AMP as a control, there is no density for the adenine ring. The most noticeable interaction with the adenine is between Ser47 of 14-3-3 and the 6-amino group of adenine ring with the distance of 2.86 Å (Fig. 7C). There are more residues from 14-3-3 that come close to the adenine ring such as Lys122 and Asn175 of 14-3-3 and the distances between adenine ring and those residues are 3.73 and 3.92 Å, respectively. The electron density of the ribose ring is extremely low, and only the adenine ring and the phosphate were modeled in the electron density.
FIGURE 7.

Binding of AMP at the interface of ChREBP and 14-3-3β. A, overview of AMP binding with ChREBP (plum)-14-3-3β (gray) revealed that the phosphate group of AMP buried deep in the basic cavity at the interface of ChREBP-14-3-3β. B, detailed view of the interaction of the phosphate of AMP and ChREBP-14-3-3β showed that the phosphate group of AMP has interactions with Arg58, Arg129, and Tyr130 of 14-3-3β and Trp127 and Arg128 of ChREBP. C, detailed view of the interaction of the adenine ring of AMP and ChREBP/14-3-3β. The adenine ring has no significant interaction with ChREBP, and all the interactions were found with 14-3-3β, including the interactions with Ser47, Phe119, Lys122, and Asn175 of 14-3-3β. Possible interactions are shown in dotted lines.
TABLE 2.
Summary of crystallographic data collection and refinement statistics
| ChREBP:14-3-3:AMP | |
|---|---|
| Data collection | |
| Space group | P4222 |
| Unit cell dimension | 115.62, 115.62, 59.37 (Å) |
| 90.00, 90.00, 90.00(o) | |
| Resolution (Å) | 50.0-2.35 (2.39-2.35) |
| Rsym | 0.041 (0.628) |
| I/σ | 23.0 (1.3) |
| Completeness (%) | 100.0 (100.0) |
| Redundancy | 6.5 (4.9) |
| Refinement | |
| No. of reflections | 17314 |
| Rwork/Rfree | 19.5/25.2 |
| No. of atoms | |
| Protein | 2058 |
| Ligand/ion | 21 |
| Water | 38 |
| B factor | |
| Protein | 57.7 |
| Ligand/ion | 74.3 |
| Water | 50.0 |
| Root mean square deviation | |
| Length | 0.008 |
| Angle | 1.072 |
As in the previously reported structure (17), additional stabilization from Trp127, Trp130, and Tyr134 of ChREBP and Arg62 of the 14-3-3 αC helix was also found in our AMP-bound structure. These four side chains intercalate one another and give extra stabilization through van der Waals interactions.
This AMP-bound structure of ChREBP-14-3-3 provides the most direct evidence in support for the idea for an AMP-induced conformational change resulting in the formation and stabilization of the ChREBP/14-3-3 heterodimer, although the density of ribose was missing in the data.
Mutagenesis Analysis of the AMP-binding Site
Previously, we made mutation of those same residues on ChREBP as well as the residues on 14-3-3 involved in the interaction resulted in complete loss of the protein/protein interaction as shown using the pulldown assay (17). To evaluate the observed AMP-binding site on ChREBP, we made site-directed mutations on ChREBP, including W127A and R128A, to interrupt its effects on transcriptional activity of ChREBP. The ChREBP mutants were overexpressed in primary hepatocytes, and the cells were incubated in the presence of KIC, oleate, or AICAR in high glucose. One expects the mutation of these amino acids to result in decreased AMP binding leading to activation of the nuclear localization. However, the mutagenesis effects on the transcriptional activity are more complex because the nuclear localization is expected to increase, but AMP activation of AMPK results in inactivation of the ChREBP transcriptional activity, and the net effect depends on the strength of these two opposing reactions. The results demonstrated that the transcriptional activity of ChREBP-W127A was activated in high glucose compared with the WT-ChREBP, but the effects of these substrates and AICAR-generating AMP were negligible, which suggests that the Trp127 residue of ChREBP is not as important in the AMP binding (Fig. 8). However, ChREBP-R128A lost the transcriptional activity completely in high glucose even in the absence of the AMP-generating substrates KIC, oleate, or AICAR. The results suggest that Arg128 plays an essential role in AMP binding. The complete loss of the ChREBP activity of ChREBP-R128A is not surprising because it is well known that 14-3-3 is essential in the stabilization of ChREBP as a ChREBP/14-3-3 heterodimer in hepatocytes, and ChREBP by itself undergoes rapid proteolysis when unable to bind to the 14-3-3 protein (17).
FIGURE 8.

Effects of mutagenesis of W127A and R128A of ChREBP on the nuclear localization and transcriptional activities in hepatocytes. Primary cultured rat hepatocytes were co-transfected with pGL-3 Basic LPK reporter and ChREBP-WT, ChREBP-W127A, ChREBP-R128A, or ChREBP-w127/R128A. After transfection, cells were cultured in 5.5 (open bar) and 27.5 (filled bars) mm glucose, as in Fig. 5 legend. The values presented are the mean ± S.D. of three replicate cultures from a single experiment representative of more than three independent experiments.* versus HG, p < 0.05.
Together, these results show that AMP inhibits ChREBP activities with two different mechanisms as follows: (a) the transcriptional activity by activation of AMPK leading to the phosphorylation of Ser568 near the basic helix-loop-helix/ZIP in the C-terminal region (5), and (b) the nuclear localization by binding directly to the N-terminal region of ChREBP (Fig. 9). The latter is a new mechanism for the action of AMP, independently of AMPK, which involves ligand-induced conformational changes consistent with allosterically induced inhibition.
FIGURE 9.

Role of AMP as an inhibitor of ChREBP activity. In response to low glucose, BCKA metabolized from amino acids such as leucine in the skeletal muscle and FA produced from triacylglycerol (TG) in adipocytes are used for energy source. AMP as well as ketone bodies are produced from the metabolism of BCKA and free fatty acid (FA) in liver. AMP inhibits ChREBP activity by 1) the increase in the allosteric activation of the interaction ChREBP-14-3-3 through direct binding, resulting in stabilizing cytosolic localization of ChREBP, and 2) the promotion of the phosphorylation of ChREBP through AMPK activation, resulting in suppression of ChREBP transcriptional activity. These regulatory mechanisms will contribute to the inhibition of de novo fat synthesis and saving the glucose during starvation and prolonged exercise.
Discussion
To elucidate the mechanism(s) by which changes in glucose concentration regulate the subcellular localization of ChREBP, we have focused the past several years on the functions of the N-terminal region of ChREBP and demonstrated that the N-terminal ChREBP regulatory region (1–251) is necessary and sufficient for glucose-responsive ChREBP and nuclear import and export (11, 17). We have identified three α helices in the N terminus which are essential for the nuclear/cytosol trafficking of ChREBP (6). These α-helices and their functions include the following: (a) the “α1” helix as NES2 together with NES1 on the N terminus for a CRM-1 binding; (b) “α2” helix for 14-3-3 binding; and (c) extended and disordered “α3” helix as NLS for importin α binding (Fig. 1). We also determined that the 14-3-3 plays dual regulatory roles in the nuclear/cytoplasm trafficking of ChREBP-14-3-3 heterodimer by not only activating the export of inactive P-ChREBP out of nucleus but also inhibiting nuclear import by blocking importin α from binding to NLS of ChREBP under low glucose conditions (12).
In addition to phosphorylation/dephosphorylation, especially under starved conditions, the ChREBP activity is also regulated by metabolites when fed a high fat diet, which resulted in inhibition of the nuclear localization of ChREBP in liver, in contrast to high sucrose (Fig. 2). This decreased nuclear localization could be explained by AMPK-dependent phosphorylation (5). However, the phosphorylation site for AMPK is on the C-terminal region of ChREBP and not on the N terminus, where the cellular localization is regulated. We hypothesized that an alternative explanation could be an allosteric regulation metabolite. We have previously demonstrated nuclear localization of ChREBP in hepatocytes is inhibited by metabolites β-HB and AcAc (12) from starved livers. These ketone bodies are produced in high concentrations by β-oxidation of fatty acids in liver and serve as fuels for other tissues under starved conditions.
We demonstrate herein that the BCKAs, including KIC, KIV, and KMV produced from leucine, isoleucine, and valine, also inhibit the ChREBP nuclear localization in hepatocytes. BCAAs, similarly to fatty acids, produce ketone bodies, and these metabolites contribute to the inhibition of ChREBP. However, the amounts of ketone bodies produced by the metabolism of ketoacids or fatty acids are much lower than under starvation and account for only a part of the inhibition (12). Importantly, we show here that AMP also inhibited the ChREBP nuclear localization, and the inhibition by both AMP and ketones together as in the extract of rat liver fed the HF diet appears to account for the inhibition of ChREBP. AMP is produced by the metabolism of α-ketoacids and fatty acids in the acyl-CoA synthetase reaction and serves cellular signaling roles.
A widely accepted mechanism of AMP in nutrient sensing is that AMP activates AMPK, which is responsive to the lower cellular energy status resulting from hydrolysis of ATP to ADP/AMP (29). To maintain energy homeostasis, the cells respond by increasing metabolism by AMP activation of AMPK, which in turn stimulates catabolic pathways, including glycolysis and fatty acid oxidation, and inhibits anabolic pathways such as glycogen synthesis, protein synthesis, etc. We present here a new alternative mechanism of AMP in the regulation of ChREBP. AMP binds directly to ChREBP as an allosteric ligand and stimulates a protein/protein interaction between ChREBP and 14-3-3. Thus AMP binding stabilizes the heterodimer in the cytoplasm, which leads to inhibition of nuclear localization of ChREBP. The crystal structure of AMP bound to the N terminus of α2 helix in the (N)-ChREBP-14-3-3 heterodimer provides the most direct evidence in support of the mechanism in which AMP serves as an allosteric regulatory ligand, instead of stimulating the AMPK-dependent phosphorylation. Although we have not yet obtained a three-dimensional structure for β-HB and AcAc binding to the heterodimer, we assume that the ketone bodies function as allosteric ligands to activate/stabilize the ChREBP·14-3-3 complex by a similar mechanism. Besides being a source of energy during fasting or exercise, β-HB has been shown recently to have important roles in signaling mechanisms, including regulation of transcription by inhibiting histone deacetylases (30) and inflammasome activation (31).
We note that the cellular energy states and the AMP concentrations reported in the literature related to AMPK are inaccurate. Guynn and Veech (32) and Veech et al. (21) have pointed out that for understanding energy state of cells under physiological conditions it is critical to determine “free metabolite” instead of “total metabolite” concentrations in cells. As demonstrated by the authors, the amounts of AMP in a tissue extract as measured by HPLC and enzymatic assay represent the total AMP, but most of the AMP is bound to enzymes or localized within mitochondria and is unavailable for cytoplasmic reactions in cells. In contrast, free AMP and free ADP represent those nucleotides that are available for enzymes and cellular proteins in vivo, and the concentrations of the free nucleotides can be calculated based on phosphorylation potentials. The authors determined that the actual free energy “(ΔG′) of ATP hydrolysis” or “available energy” in cells calculated on the basis of the concentrations of ATP and free ADP and phosphate was ΔG′ = −13.3 kcal/mol, compared with that using the total adenine nucleotides was −0.557 kcal/mol (Table 5 in Ref. 21). If one converts these ΔG′ values to the free energy of hydrolysis of ATP under the standard conditions, the ΔG0 would be −7.60 kcal/mol, based on the free metabolites, although the value is −0.243 kcal/mol based on the total concentrations, which is obviously incorrect (32). This large difference (30-fold) clearly points to the importance of knowing free ATP, ADP, and AMP concentrations to estimate the true energy states of cells.
We have determined here that the free AMP increased 4-fold in KIC, oleate, and low glucose in 1 h in the hepatocytes compared with HG (Table 1). The energy states (ΔG′ATP) of the cells in the low glucose, KIC, and oleate decreased by ∼0.1 to 0.4 kcal/mol or less than 3% compared with that in HG in the hepatocytes. Moreover, the energy content of the hepatocytes in HG compared with those in KIC and oleate was higher (Table 1), yet the ChREBP transcription activity was lower, which also indicates the AMP inhibition was unrelated to the energy states of the cell. It is difficult to assess the physiological significance of the magnitude of the energy differences. However, if one compares the large (3-fold) increase in the free [AMP] and the small change (<3%) in the energy status, it appears more likely that the hepatocytes respond more strongly to the changes in the concentration of AMP than the cellular energy states under these conditions. In general, allosteric regulatory metabolites are extremely effective because its signal is amplified severalfold. Therefore, the large increase of AMP is highly significant toward its effect on inhibition of ChREBP nuclear localization activity and must be physiologically very important to ensure fat synthesis is down-regulated during the up-regulated fatty acid oxidation to generate cellular energy.
These findings as well as the results of the previous study (5) suggest that AMP inhibits ChREBP by two mechanisms as follows: (a) the transcriptional activity by AMP activation of AMPK-dependent phosphorylation near the basic helix-loop-helix/Zip in the C-terminal region, which inhibits the DNA binding and the transcription; and (b) the nuclear localization by AMP binding directly to the ChREBP-14-3-3 heterodimer and induced allosteric activation and stabilization of ChREBP-14-3-3 in the cytoplasm. AMP serves as an allosteric inhibitory ligand of ChREBP nuclear localization. Therefore, AMP-mediated inhibition may be an important alternative mechanism to the PKA-mediated phosphorylation and regulation of ChREBP, especially under high fat or high protein diet.
Based on these results, we conclude that the metabolites of BCKA and fatty acid metabolism, AMP, and ketone bodies coordinately play allosteric regulatory roles in lipid homeostasis, by inhibiting ChREBP and lipogenesis during ketosis in livers with a drop in circulating glucose concentrations. These results suggest a use of ketogenic diets in treating diabetes, obesity, and over-weight individuals. Furthermore, the crystal structure of AMP bound to the unique interface between ChREBP and 14-3-3 also provides a potential target to design drugs to increase this protein/protein interaction to prevent the nuclear translocation of ChREBP.
Author Contributions
S. S., H. J., W. R. L., H. S., and K. U. planned and carried out the project and wrote the manuscript. S. S., W. R. L., and T. N. executed molecular biology and animal experiments. H. J. performed cloning, protein purification, crystallization, and data processing. T. N. and T. T. performed HPLC and T. T., S. L., B. T., and J. B. M. carried out metabolite identification with LC/MS and MS/MS. T. T. and R. M. W. performed fluorescence polarization and ITC measurements. R. P. and R. L. V. analyzed metabolites by GC/MS and calculated the phosphorylation potentials. J. K. D. B. synthesized AMPK activator compounds.
Acknowledgments
We thank Dr. Hong Zhang, the Biochemistry and Biophysics Department, University of Texas Southwestern Medical Center for use of the crystallization chamber, and Dr. Jay Horton, Internal Medicine, University of Texas Southwestern Medical Center, for acetyl-CoA carboxylase. We also thank Dr. Martin Kemper, the National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD, for assistance in calculation of phosphorylation potentials. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by University of Chicago Argonne, LLC, for the United States Department of Energy, Office of Biological and Environmental Research under Contract DE-AC02-06CH11357.
This work was supported the Department of Veterans Affairs and the Robert A. Welch Foundation Grants I-1720 and I-1422. This work was authored, in whole or in part, by National Institutes of Health staff. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5F74) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ChREBP
- carbohydrate-response element-binding protein
- NLS
- nuclear localization signal
- AMPK
- AMP-activated protein kinase
- β-HB
- β-hydroxybutyrate
- AcAc
- acetoacetate
- KIV
- α-ketoisovalerate
- KIC
- α-ketoisocaproate
- KMV
- α-keto-β-methylvalerate
- BCKA
- branched-chain α-ketoacid
- BCAA
- branched-chain amino acid
- HMB
- 3-hydroxy-3-methylbutyrate
- HG
- high glucose
- HF
- high fat
- ITC
- isothermal titration calorimetry
- HS
- high sucrose
- AICAR
- 5-aminoimidazole-4-carboxamide ribonucleotide.
References
- 1. Yamashita H., Takenoshita M., Sakurai M., Bruick R. K., Henzel W. J., Shillinglaw W., Arnot D., and Uyeda K. (2001) A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. U.S.A. 98, 9116–9121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ishii S., Iizuka K., Miller B. C., and Uyeda K. (2004) Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. U.S.A. 101, 15597–15602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Iizuka K., Bruick R. K., Liang G., Horton J. D., and Uyeda K. (2004) Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. U.S.A. 101, 7281–7286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kawaguchi T., Takenoshita M., Kabashima T., and Uyeda K. (2001) Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. U.S.A. 98, 13710–13715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kawaguchi T., Osatomi K., Yamashita H., Kabashima T., and Uyeda K. (2002) Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J. Biol. Chem. 277, 3829–3835 [DOI] [PubMed] [Google Scholar]
- 6. Sakiyama H., Wynn R. M., Lee W. R., Fukasawa M., Mizuguchi H., Gardner K. H., Repa J. J., and Uyeda K. (2008) Regulation of nuclear import/export of ChREBP; interaction of an α-helix of ChREBP with the 14-3-3 proteins and regulation by phosphorylation. J. Biol. Chem. 283, 24899–24908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kabashima T., Kawaguchi T., Wadzinski B. E., and Uyeda K. (2003) Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl. Acad. Sci. U.S.A. 100, 5107–5112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bricambert J., Miranda J., Benhamed F., Girard J., Postic C., and Dentin R. (2010) Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Invest. 120, 4316–4331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sakiyama H., Fujiwara N., Noguchi T., Eguchi H., Yoshihara D., Uyeda K., and Suzuki K. (2010) The role of O-linked GlcNAc modification on the glucose response of ChREBP. Biochem. Biophys. Res. Commun. 402, 784–789 [DOI] [PubMed] [Google Scholar]
- 10. Guinez C., Filhoulaud G., Rayah-Benhamed F., Marmier S., Dubuquoy C., Dentin R., Moldes M., Burnol A. F., Yang X., Lefebvre T., Girard J., and Postic C. (2011) O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 60, 1399–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ge Q., Nakagawa T., Wynn R. M., Chook Y. M., Miller B. C., and Uyeda K. (2011) Importin-α protein binding to a nuclear localization signal of carbohydrate response element-binding protein (ChREBP). J. Biol. Chem. 286, 28119–28127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakagawa T., Ge Q., Pawlosky R., Wynn R. M., Veech R. L., and Uyeda K. (2013) Metabolite regulation of nucleo-cytosolic trafficking of carbohydrate response element-binding protein (ChREBP): role of ketone bodies. J. Biol. Chem. 288, 28358–28367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dentin R., Benhamed F., Pégorier J. P., Foufelle F., Viollet B., Vaulont S., Girard J., and Postic C. (2005) Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J. Clin. Invest. 115, 2843–2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hunter R. W., Foretz M., Bultot L., Fullerton M. D., Deak M., Ross F. A., Hawley S. A., Shpiro N., Viollet B., Barron D., Kemp B. E., Steinberg G. R., Hardie D. G., and Sakamoto K. (2014) Mechanism of action of compound-13: an α1-selective small molecule activator of AMPK. Chem. Biol. 21, 866–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harper A. E., Miller R. H., and Block K. P. (1984) Branched-chain amino acid metabolism. Annu. Rev. Nutr. 4, 409–454 [DOI] [PubMed] [Google Scholar]
- 16. Wong R. H., and Sul H. S. (2010) Insulin signaling in fatty acid and fat synthesis: a transcriptional perspective. Curr. Opin. Pharmacol. 10, 684–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ge Q., Huang N., Wynn R. M., Li Y., Du X., Miller B., Zhang H., and Uyeda K. (2012) Structural characterization of a unique interface between carbohydrate response element-binding protein (ChREBP) and 14-3-3β protein. J. Biol. Chem. 287, 41914–41921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iizuka K., Miller B., and Uyeda K. (2006) Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am. J. Physiol. Endocrinol. Metab. 291, E358–E364 [DOI] [PubMed] [Google Scholar]
- 19. Newton R. P., Salih S. G., Salvage B. J., and Kingston E. E. (1984) Extraction, purification and identification of cytidine 3′,5′-cyclic monophosphate from rat tissues. Biochem. J. 221, 665–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berry M. N., and Friend D. S. (1969) High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J. Cell Biol. 43, 506–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Veech R. L., Lawson J. W., Cornell N. W., and Krebs H. A. (1979) Cytosolic phosphorylation potential. J. Biol. Chem. 254, 6538–6547 [PubMed] [Google Scholar]
- 22. Srivastava S., Kashiwaya Y., Chen X., Geiger J. D., Pawlosky R., and Veech R. L. (2012) Microwave irradiation decreases ATP, increases free [Mg2+], and alters in vivo intracellular reactions in rat brain. J. Neurochem. 123, 668–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Veloso D., Guynn R. W., Oskarsson M., and Veech R. L. (1973) The concentrations of free and bound magnesium in rat tissues. Relative constancy of free Mg2+ concentrations. J. Biol. Chem. 248, 4811–4819 [PubMed] [Google Scholar]
- 24. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 25. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 26. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gohda T., Makita Y., Shike T., Kobayashi M., Funabiki K., Haneda M., Kikkawa R., Watanabe T., Baba T., Yoshida H., and Tomino Y. (2001) Association of the DD genotype and development of Japanese type 2 diabetic nephropathy. Clin. Nephrol. 56, 475–480 [PubMed] [Google Scholar]
- 29. Efeyan A., Comb W. C., and Sabatini D. M. (2015) Nutrient-sensing mechanisms and pathways. Nature 517, 302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shimazu T., Hirschey M. D., Newman J., He W., Shirakawa K., Le Moan N., Grueter C. A., Lim H., Saunders L. R., Stevens R. D., Newgard C. B., Farese R. V. Jr., de Cabo R., Ulrich S., Akassoglou K., and Verdin E. (2013) Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Youm Y. H., Nguyen K. Y., Grant R. W., Goldberg E. L., Bodogai M., Kim D., D'Agostino D., Planavsky N., Lupfer C., Kanneganti T. D., Kang S., Horvath T. L., Fahmy T. M., Crawford P. A., Biragyn A., et al. (2015) The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guynn R. W., and Veech R. L. (1973) The equilibrium constants of the adenosine triphosphate hydrolysis and the adenosine triphosphate-citrate lyase reactions. J. Biol. Chem. 248, 6966–6972 [PubMed] [Google Scholar]