

Abstract
The human-restricted pathogen Neisseria gonorrhoeae encodes a single N-acetylmuramyl-l-alanine amidase involved in cell separation (AmiC), as compared with three largely redundant cell separation amidases found in Escherichia coli (AmiA, AmiB, and AmiC). Deletion of amiC from N. gonorrhoeae results in severely impaired cell separation and altered peptidoglycan (PG) fragment release, but little else is known about how AmiC functions in gonococci. Here, we demonstrated that gonococcal AmiC can act on macromolecular PG to liberate cross-linked and non-cross-linked peptides indicative of amidase activity, and we provided the first evidence that a cell separation amidase can utilize a small synthetic PG fragment as substrate (GlcNAc-MurNAc(pentapeptide)-GlcNAc-MurNAc(pentapeptide)). An investigation of two residues in the active site of AmiC revealed that Glu-229 is critical for both normal cell separation and the release of PG fragments by gonococci during growth. In contrast, Gln-316 has an autoinhibitory role, and its mutation to lysine resulted in an AmiC with increased enzymatic activity on macromolecular PG and on the synthetic PG derivative. Curiously, the same Q316K mutation that increased AmiC activity also resulted in cell separation and PG fragment release defects, indicating that activation state is not the only factor determining normal AmiC activity. In addition to displaying high basal activity on PG, gonococcal AmiC can utilize metal ions other than the zinc cofactor typically used by cell separation amidases, potentially protecting its ability to function in zinc-limiting environments. Thus gonococcal AmiC has distinct differences from related enzymes, and these studies revealed parameters for how AmiC functions in cell separation and PG fragment release.
Keywords: cell wall, Gram-negative bacteria, infectious disease, NOD-like receptor (NLR), pathogen-associated molecular pattern (PAMP), peptidoglycan, Neisseria gonorrhoeae, amidase, cell separation
Introduction
Peptidoglycan (PG)2 is a critical structural macromolecule that makes up the cell wall surrounding the cytoplasmic membrane of nearly all bacteria. This cage-like structure is made up of long polysaccharide strands that are composed of repeating units of N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) with short peptide stems (3–5 amino acids) covalently attached to the MurNAc moiety (1). Depending on the organism, some proportion of peptide stems are cross-linked to peptides on adjacent strands, whereas others remain non-cross-linked. Cross-linking provides structural integrity for the macromolecule and contributes to the determination of bacterial shape.
Neisseria gonorrhoeae (the gonococcus or GC) is a Gram-negative bacterium, obligate human pathogen, and etiologic agent of the sexually transmitted infection gonorrhea. Gonorrhea is the second most common reportable bacterial infection in the United States with an estimated 820,000 cases annually (2). A robust inflammatory response is the cause of much of the damage observed during GC infection and is triggered by the release of lipo-oligosaccharide, porin proteins, and fragments of PG (3–5). In particular, N. gonorrhoeae release a greater abundance of PG fragments than Escherichia coli during normal growth (6, 7). Furthermore, rather than the peptides and disaccharides released by E. coli, GC release a larger proportion of N-acetylglucosaminyl-1,6-anhydromuramyl-tripeptide and -tetrapeptide fragments (disaccharide-peptides or PG monomers), as well as stem peptides, GlcNAc-anhMurNAc disaccharides, and some larger fragments (8–11). The abundant release of PG fragments is analogous to the situation in Bordetella pertussis, where the peptidoglycan fragment tracheal cytotoxin (TCT; N-acetylglucosaminyl-1,6-anhydromuramyl-tetrapeptide) is released by the bacterium and acts as a major virulence factor responsible for inflammatory pathology and the death of ciliated epithelial cells in the host (12).
Although thought of as a rigid scaffold, PG is a dynamic structure, remodeled throughout growth by a variety of specialized enzymes and ultimately requiring some coordinated disassembly at the septum during cell division and separation (13). Two components known to be critical for cleaving PG at the division septum include PG hydrolases of the LytC type (i.e. N-acetylmuramyl-l-alanine amidases) and LytM domain-containing metalloendopeptidase-like factors. Periplasmic N-acetylmuramyl-l-alanine amidases hydrolyze the amide bond between the lactyl moiety of MurNAc and the l-alanine of the peptide stem in sacculi and have variable specificity for soluble versus insoluble PG and cross-linked versus non-cross-linked peptide stems (14–16). E. coli encodes three periplasmic cell separation amidases (AmiA, AmiB, and AmiC) that are all zinc-dependent metallopeptidases with a common fold type and partially redundant functions (17). Deleting amiA, amiB, or amiC genes alone does not impact cell separation, but double mutants or mutations in the TAT pathway of export (which exports both AmiA and AmiC) result in cell chaining (AmiB in E. coli is exported via the Sec pathway) (14, 18, 19). Although AmiA and AmiC travel similar paths to the periplasm, it is AmiB and AmiC that contain N-terminal extensions targeting them to the E. coli septum, whereas AmiA remains diffusely distributed in the periplasm (20). The structure of this N-terminal extension (AMIN domain) has been solved and shown to bind peptidoglycan independently of the C-terminal catalytic domain (21). The AMIN domain is important in the septal localization of AmiC paralogs, even in filamentous bacteria with contiguous PG sacculi, indicating a high level of functional conservation across diverse Gram-negative bacteria (22–24). The recruitment of amidases to the division site is part of a coordinated assembly of cell separation factors, with AmiB recruitment dependent on the presence of FtsZ and FtsN and the assembly of septal PG (20). The septal targeting of amidases in E. coli occurs after, but independently of, the recruitment of the LytM domain-containing proteins (20).
Proteins with LytM domains (EnvC and/or NlpD in several species) participate in controlling the activity of amidases through direct protein-protein interactions (25–27). Proteins with LytM domains are active metalloendopeptidases in Gram-positive bacteria, cleaving cross-links between opposing peptide chains to separate glycan strands (28). In E. coli, the LytM homologs EnvC and NlpD lack two of the four predicted metalloendopeptidase active-site residues (25), display no detectable enzymatic activity, and have been shown to act in a regulatory role, enhancing the activity of amidases in specific cognate pairs (AmiA and AmiB with EnvC and AmiC with NlpD) (26, 29). The specific mechanism by which amidase activity is regulated has been determined for the EnvC-AmiB pair in E. coli, where an amphipathic α-helix occludes the active-site cleft and is anchored by a glutamic acid (Glu-303) that coordinates the active-site zinc (21, 27). This helix is then displaced during interaction with a cognate activator (25). The importance of the anchoring residue also applies to E. coli AmiA (Glu-167) and Vibrio cholerae AmiB (Glu-286), demonstrating the broader applicability of this structure-function relationship (27). The crystal structure of E. coli AmiC reveals an α-helix with a glutamine (Gln-299) in a similar position, predicting a comparable relationship between AmiC and NlpD, which have been shown to bind to each other (21). Along with an AmiC homolog, gonococci contain an NlpD homolog that has an important role in normal cell separation, enhances the activity of AmiC, and binds PG, all without any apparent enzymatic activity (30).
In N. gonorrhoeae, AmiC is critical for proper cell separation, is necessary for normal PG fragment release, and functions as an autolysin (31). Several aspects of AmiC activity remain uncharacterized, however, including the nature of its substrates and products as well as how the active site functions. Our goals were to probe the function of two active-site residues, comparing their functional contribution to other characterized cell separation amidases, and to determine how those residues contribute to both the characteristic PG fragment release profile of gonococci and the ability of cells to separate normally. By purifying GC-AmiC, we were also able to define the products of GC-AmiC activity on whole GC sacculi and synthetic PG GlcNAc-MurNAc(pentapeptide)-GlcNAc-MurNAc(pentapeptide) substrate.
Experimental Procedures
Bacterial Growth
MS11 was used as the wild-type N. gonorrhoeae strain in all assays and as the parent strain for strains generated in this study. Piliated variants of MS11 were used for all transformations, and nonpiliated variants were used in all other assays. Gonococcal strains were grown at 37 °C with 5% CO2 on gonococcal base (GCB) agar plates (Difco) containing Kellogg's supplement (32, 33). Liquid cultures were started from overnight plates and grown in gonococcal base liquid medium (GCBL) containing Kellogg's supplement and 0.042% NaHCO3 (complete GCBL or cGCBL) as described previously (34). Chloramphenicol was used at 5–10 μg/ml in GCB plates for selection of the insertion of inducible constructs at the aspC-lctP chromosomal site. Gene expression was induced in gonococci (for complementation or overexpression) with 0.1 mm isopropyl β-d-thiogalactopyranoside (IPTG) (31). E. coli strains were grown on Luria agar plates or in Luria broth (LB) at 37 °C. Antibiotics were used at the following concentrations for E. coli: ampicillin, 50–100 μg/ml; kanamycin, 40 μg/ml; erythromycin, 500 μg/ml; and chloramphenicol, 25 μg/ml. Gene expression was induced in E. coli with anhydrotetracycline (aTc) or IPTG at concentrations noted below.
Bioinformatics
The sequences of amiB and amiC from E. coli were retrieved from NCBI, and the sequence for N. gonorrhoeae strain MS11 amiC was retrieved from the Broad Institute website. To predict catalytic site residues in GC-AmiC, the protein sequence was aligned with E. coli amidases using MUSCLE. To determine whether the predicted zinc-binding residues were likely to be spatially oriented in GC-AmiC similarly to other characterized amidases, the GC-AmiC sequence was submitted to Phyre2 V2.0 (protein homology/analogy recognition engine) for modeling (35). The resulting model was visualized and aligned separately with the crystal structures of N-acetylmuramoyl-l-alanine amidase (AmiB) from Bartonella henselae (Protein Data Bank ID: 3NE8) (27) and AmiC from E. coli (Protein Data Bank ID: 4BIN) using PyMOL. The predicted location of zinc-coordinating residues in GC-AmiC was confirmed to match the placement of the same conserved residues in B. henselae and E. coli.
Construction of Plasmids
To generate point mutations in the gonococcal chromosome, plasmids were first made with the desired target mutations using E. coli TAM1 cells. To generate a construct with the E229D substitution (pKX2), primers with the introduced changes in the amiC sequence were used to amplify areas upstream and downstream of the desired mutation site. A portion of amiC downstream, including the base change, was amplified with the primers mamiC3R (5′-GGCTCGAGGCACCAGTTTGCCGAGTTTG-3′) and amiCE229DF (5′-GCGGTCTGCAGGATAAACACGTCGTCCTC-3′), where the underlined bases indicate changes. A portion of amiC upstream, including the same base change, was amplified with the primers mamiC3F (5′-CCGAATTCAAAGTTATGGCGGACGACCC-3′) and amiCE229DR (5′-TGTTTATCCTGCAGACCGCCCGG-3′), where the underlined bases indicate changes. The upstream and downstream portions were then joined by overlap extension PCR using mamiC3F and mamiC3R. The final construct contains an E229D change and introduces a silent mutation that creates a PstI restriction site for screening. The amiCE229D insert was digested with EcoRI and XhoI and cloned into pIDN3 to generate pKX2. The Q316K substitution construct (pJDL45) was made in a similar fashion with the upstream portion amplified using the primers amiC2kbF (31) and JLDNA37 (5′-CGCGTCGGCATTATTTTTGGTTT-3′) and the downstream portion amplified with the primers JLDNA36 (5′-CCAAATTCCTCGAGCAAACCAAAAATAA-3′) and amiC2kbKpnIR (31). The underlined bases indicate differences from the WT sequence that result in the Q316K change and introduction of a silent XhoI site. The upstream and downstream portions were then joined by overlap extension PCR using amiC2kbF and amiC2kbKpnIR. The amiCQ316K insert was digested with EcoRI and KpnI and cloned into pIDN3 to generate pJDL45.
To generate constructs for protein purification, the amiC gene lacking its predicted signal sequence was amplified from MS11 chromosomal DNA (WT) or KX503 chromosomal DNA (amiCE229D) using primers JLDNA35 (5′-CTAGCTAGCAAAACGGTACGCGCCCCG-3′) and JLDNA4 (5′-GGAATTCTATGCCTGCCTTCATCCGACA-3′), which introduce NheI and EcoRI sites, underlined above. amiCQ316K was amplified using the overlap extension protocol above by first amplifying a 5′ portion with JLDNA35 and JLDNA37 and a 3′ portion with JLDNA36 and JLDNA4 and then joining these 5′ and 3′ portions with JLDNA35 and JLDNA4 to form a single insert. Each of the above inserts (amiC, amiCE229D, and amiCQ316K) was digested with NheI and EcoRI and cloned into pTEV5 (36) to generate pJDL37, pJDL38, and pJDL41, respectively. The resulting plasmids were transformed into chemically competent TAM1 E. coli, Ampr colonies were selected, plasmids prepared, and inserts sequenced to confirm proper in-frame assembly. Prior to protein purification, the prepared plasmids were transformed into BL21 Star(DE3) cells (Invitrogen).
Cloning of His6-tagged NlpD for protein purification was performed as described elsewhere (30). Briefly, nlpD lacking its predicted signal sequence was amplified from FA1090 chromosomal DNA by PCR using primers NlpD-10 and NlpD-11 and then cloned into pCR-BLUNT. The nlpD insert was excised from pCR-BLUNT with a NheI/BamHI digest and cloned into NheI/BamHI-digested pET28a overexpression vector. When not otherwise indicated above, plasmids were purified using the QIAprep Spin MiniPrep kit (Qiagen) according to the manufacturer's directions. All plasmid inserts and ligation junctions were sequenced to ensure maintenance of the reading frame and to check for point mutations.
To generate constructs for E. coli lysis assays, C-terminally hemagglutinin (HA)-tagged versions of amiC were amplified from MS11 chromosomal DNA (WT) or KX503 chromosomal DNA (amiCE229D) using primers JLDNA14 (5′-CGAGCTCATGCCTGCTGACCGCCCATA-3′) (SacI site underlined and start codon bolded) and JLDNA12 (5′-GACTAGTTCAAGCGTAATCTGGAACATCGTATGGGTATCCACCACCCCGCTTCAATACGGATG-3′) (SpeI site underlined and stop codon bolded). The HA tag with a diglycine spacer introduced on JLDNA12 is indicated with italics. amiCQ316K was amplified using overlap extension by first amplifying a 5′ portion of amiC with JLDNA14 and JLDNA37, and a 3′ portion of amiC with JLDNA36 and JLDNA12 and then joining an insert with JLDNA14 and JLDNA12. Each of the above inserts (amiC, amiCE229D, and amiCQ316K) was digested with SacI and SpeI and cloned into pMR68 (37) to generate pJDL7, pJDL39, and pJDL40, respectively. The resulting plasmids were transformed into chemically competent TAM1 E. coli; Kanr colonies were selected, plasmids prepared, and inserts sequenced to confirm proper in-frame assembly and the addition of the C-terminal HA tag.
For constructs used to purify protein for metal dependence assays, DNA corresponding to residues 199–432 of AmiC (AmiC-CTD) was PCR-amplified from a vector containing full-length AmiC using the forward primer 5′-CCCCCGGATCCGCACGGGGCAAAAACGGGCGCAGA-3′ and reverse primer 5′-AGACTCGAGTCAACCCCGCCTTCAATACGGATGTATTGAT-3′. BamHI and XhoI sites are underlined in the forward and reverse primers, respectively, and the stop codon is indicated in bold in the reverse primer. PCR products were digested with BamHI and XhoI and ligated into pGEX-6P-3 (GE Healthcare) and then later subcloned into pT7Htb by digesting both vectors with the restriction enzymes BamHI and NotI. Following ligation, the completed plasmid was transformed into E. coli DH5α chemically competent cells (Invitrogen).
Construction of Gonococcal Mutants
Point mutations of amiC were made by introducing pIDN3-based plasmids into GC strain MS11 for double-crossover recombination (38). Point mutation and deletion strains were generated by transforming MS11 with pKX2 (to generate the amiCE229D strain KX503) or pJDL45 (to generate the amiCQ316K strain JL535). For transformations, 2 μg of plasmid was linearized by overnight digestion with PciI, and the digestion was spotted onto a GCB agar plate. Piliated GC were spread across the area containing absorbed plasmid DNA. Transformations were incubated overnight as described above, and the resulting growth was plated for isolation on GCB. For point mutations, amiC was amplified from isolated colonies by PCR, and the resulting product was screened by digestion with either PstI (for amiCE229D) or XhoI (for amiCQ316K), where sensitivity to the enzyme indicated the introduction of a silent restriction site at or near the desired mutation. PCR product was then sequenced to confirm the desired mutation. All complementation strains were generated by introducing pKH37-based plasmids into GC essentially as described above, except that following spot transformation insertions were selected on GCBcam. pDG016 was transformed into the amiCE229D strain (KX503) to generate the complemented strain JL537 and into the amiCQ316K strain (JL535) to generate the complemented strain JL536. Following selection, camr colonies were screened by PCR for insertion of plasmids at the aspC/lctP complementation site.
Radiolabeling and PG Fragment Release
Gonococcal peptidoglycan was labeled metabolically with [6-3H]glucosamine or [3H]rac-2,6-diaminopimelic acid ([3H]DAP), and fragments were assessed quantitatively, essentially as described previously (39, 40). At the conclusion of labeling and washing, cells were suspended in cGCBL (glucose-supplemented), and the amount of incorporated label in samples was measured by liquid scintillation counting. Based on these counts, the culture volumes were adjusted accordingly, and growth was continued for 2.5 h at 37 °C. At the conclusion of this chase period, cells were pelleted by centrifugation (4000 × g for 5 min). The supernatant was removed, passed through a 0.2-μm filter, and then saved at −20 °C. Supernatants from quantitative PG fragment release were separated on tandem size exclusion chromatography columns, and fractions were measured by liquid scintillation counting.
Peptidoglycan Isolation
[3H]glucosamine-labeled PG from MS11 and KH530 was purified by the boiling SDS method, essentially as described previously (39, 41). Following initial purification, PG pellets were suspended in 200 μl of phosphate buffer (PB) with 20 μl of 2 mg/ml Pronase (Sigma) and incubated for 2 h at 37 °C. At the conclusion of Pronase digestion, 200 μl of 8% SDS and 800 μl of PB were added to the digests, and the pellets were boiled for 30 min. Pellets were again cooled, centrifuged at 45,000 × g for 30 min, and washed a minimum of three times with PB prior to suspension in 100–200 μl of sodium phosphate buffer. A sample from each pellet was measured by liquid scintillation counting to determine input for sacculi digest experiments.
Unlabeled PG was isolated from 1 liter of log-phase cultures of MS11 and KH530 using similar methodology. Cells were harvested by centrifugation (8000 × g for 10 min at 4 °C), washed with PB, suspended in 10 ml of PB, and added dropwise to 10 ml of boiling 8% SDS in an Oak Ridge tube. Samples were boiled for 1 h, cooled, centrifuged (45,000 × g for 30 min at 15 °C), and subjected to another 1-h round of SDS boiling. Samples were then cooled and centrifuged as before, and pellets were washed a minimum of four times with 10–20 ml of PB to remove SDS. Following the final wash and centrifugation, pellets were suspended in 500 μl of PB, transferred to a 1.5-ml tube with 50 μl of 2 mg/ml Pronase, and incubated overnight at 37 °C with constant agitation. The following day, PG was again added to 10 ml of boiling 8% SDS and boiled for 2 h followed by a minimum of four washes as described above. The concentration of PG was measured with o-phthalaldehyde reagent solution (Sigma), and MS11 and KH530 PG levels were normalized to each other in phosphate buffer.
Thin-section Electron Microscopy
Scanning electron microscopy services were provided by the University of Wisconsin-Madison Medical School Electron Microscopy Facility. For each experiment, gonococci were cultured as described above on GCB plates at 37 °C with 5% CO2 overnight followed by growth for 3 h in cGCBL liquid culture (including 0.1 mm IPTG induction where necessary). At the conclusion of growth, cells were gently centrifuged (4000 × g for 5 min), washed once in PBS, centrifuged again, and suspended in fixative. The fixed cells were then processed for thin-section electron microscopy, essentially as described previously (42). To quantify cellular clustering in thin-section electron microscopy, 5–6 fields of the wild-type, amiCQ316K, or amiCQ316K+amiC strains, taken at a magnification of ×5600, were counted to determine the number of cells present as individuals (1), diplococci (2), clusters of three (3), or clusters of four (4). Each field contained on average 178 ± 20 cells, and between 877 and 1180 cells were counted per condition (>3000 total). Differences were assessed using one-way analysis of variance with a Bonferroni post-test to compare all conditions.
Protein Purification
Overexpression and purification of His6-tagged AmiC and NlpD were performed as described elsewhere (30), and AmiCQ316K was purified in essentially the same manner as wild-type AmiC. To overexpress a C-terminal version of AmiC used for metal dependence assays, cells harboring the pT7Htb-AmiC plasmid were incubated at 37 °C in LBkan until an A600 of ∼0.6–0.8 was reached. Protein expression was induced by the addition of IPTG to 0.4 mm, and growth was continued overnight at 19 °C at 225 rpm. Cells were harvested by centrifugation and suspended in TNG buffer, pH 8.0 (20 mm Tris, 0.5 m NaCl, 10% glycerol, and HCl to adjust pH) containing 2 mm EDTA, 0.1 mm benzamidine, and 0.1 mm PMSF. Cells were lysed by three flash freeze/thaw cycles in liquid nitrogen followed by sonication at 60% amplitude (Model 500 Sonic Dismembrator, Fisher Scientific). Cell lysate was clarified by centrifugation at 10,000 × g (Beckman Coulter J2-21) and an ammonium sulfate (∼50% v/v) precipitation. The protein pellet was resuspended in TNG and filtered through a 0.22-μm syringe filter unit (Millipore). Purification of His6-AmiC-CTD was achieved by passage over a HiTrap nickel affinity column (GE Healthcare) and eluted with a 0–500 mm imidazole linear gradient in TNG buffer using the ÄKTA FPLC system (GE Healthcare). The fusion protein was dialyzed against TNG and digested with 0.1 mg of His6-TEV protease/5 mg of fusion protein overnight at 4 °C. AmiC-CTD was separated from tobacco etch virus protease and the purification tag by a second passage over the HiTrap nickel column as described above, except the tobacco etch virus, purification tag, and undigested protein, all of which contain His6, were trapped on the column and AmiC-CTD was eluted in the flow-through or low-concentration imidazole wash. The average yield was 10 mg protein/liter of culture. Protein samples were stored at −80 °C in 20 mm Tris, pH 8.0, 0.5 m NaCl, and 10% glycerol.
Solubilization of PG Sacculi
The testing of enzymes for their ability to solubilize whole gonococcal sacculi was performed essentially as described previously (43). Reactions were prepared with 100 nm His-AmiC or His-AmiCQ316K, 20 mm sodium phosphate, pH 6, 2 mm ZnCl2, and ∼200,000 cpm of [3H]glucosamine-labeled peptidoglycan prepared from either MS11 or KH530 in a total reaction volume of 600 μl. A control reaction lacking any enzyme was set up in parallel for both of the sacculi substrates (MS11 and KH530), and all reactions were conducted in duplicate. Reactions were incubated at 37 °C, and 100-μl samples were removed at various time points. Samples were added to 500 μl of 20% trichloroacetic acid (TCA) with 20 μl of unlabeled PG (matched to the labeled strain) added as carrier. Samples were allowed to precipitate for 30 min on ice prior to centrifugation (45,000 × g, 30 min at 4 °C). Following centrifugation, 500 μl of the supernatant was removed, and the presence of soluble labeled PG was measured by liquid scintillation counting. The activity of His-AmiC versus His-AmiCQ316K was compared at each time point using Student's t test.
E. coli Lysis Assay
Chemically competent TAM1 E. coli (Active Motif) cells were transformed with vectors containing aTc-inducible copies of the arginase argJ (pMR90) (37), C-terminally HA-tagged wild-type amidase amiC (pJDL7), C-terminally HA-tagged amiC with a E299D mutation (pJDL39), or C-terminally HA-tagged amiC with a Q316K mutation (pJDL40). E. coli strains were struck from frozen stocks to isolation on LBkan plates, and then single colonies were cultured overnight at 37 °C in LBkan. In the morning, each strain was subcultured to an A600 of 0.05 in LBkan to seed 6 wells/strain at 1 ml/well in a 24-well plate (Costar). Strains were then grown for 3 h with moderate shaking at 37 °C in an automated plate reader (BioTek Synergy), at which time 3 wells/strain were treated with 20 ng/ml aTc. Growth was continued for a total of 8 h. The blank-adjusted A600 at each time point was averaged from the replicate wells of each condition and graphed using GraphPad Prism 4.0c (displayed as average ± S.D.).
To confirm that wild-type and altered copies of amiC produced protein at comparable levels, the above conditions were used to grow the same strains, and samples were taken from each well preinduction and then at 30 min postinduction. Samples from replicate wells were pooled and bacteria harvested by centrifugation (13,000 × g for 1 min). Bacteria were washed once in LB and resuspended in 200 μl of ddH2O prior to disruption by sonication. Protein levels in samples were determined by BCA assay (Pierce), and each sample was normalized to 200 ng in a 20-μl total volume. 5 μl of Laemmli buffer was added to each sample, and all were boiled for 5 min prior to separation by SDS-PAGE. Proteins were transferred to PVDF membrane (Bio-Rad) for 1 h at 100 V, and the membrane was blocked in TBS with Tween-20 (TBST) and 5% milk overnight at 4 °C. For detection of the HA tag, the membrane was incubated with a 1:10,000 dilution of mouse monoclonal anti-HA antibody (Sigma) in 5% milk/TBST for 1 h, washed three times for 10 min with TBST, incubated with a 1:50,000 dilution of HRP-conjugated goat anti-mouse antibody (Santa Cruz Biotechnology) in TBST, washed three times for 5 min with TBST, developed using Clarity ECL Western substrate (Bio-Rad), and exposed to film.
HPLC and Liquid Chromatography-Mass Spectrometry (LC/MS) Analysis of Sacculi and Synthetic PG Dimer Digests
Digestions of whole gonococcal sacculi were performed with 100 μl of purified unacetylated KH530 sacculi (MS11 pacAH329Q), 100 mm sodium phosphate buffer, pH 6, 2 mm ZnCl2, and 500 nm (each) purified enzyme in a total reaction volume of 200 μl. Reactions were incubated overnight at 37 °C and stopped by boiling for 10 min. Reactions were then centrifuged (13,000 × g for 10 min) to remove the remaining insoluble PG, and soluble portions were filtered through a 10,000 MWCO (molecular weight cut-off) Centricon filter by centrifugation (13,000 × g for 30 min). Digestions of synthetic PG dimer contained 10 μg of dimer substrate, 100 mm sodium phosphate buffer, pH 6, 2 mm ZnCl2, and 500 nm (each) purified enzyme in a total reaction volume of 100 μl. Reactions were incubated for 4 h at 37 °C, stopped by boiling for 5 min, and filtered through a 10,000 MWCO Centricon filter as described above. For the HPLC analysis of amidase digestions of the synthetic PG dimer, the filtered reaction products were separated by reverse-phase HPLC on a Prevail C18 column (5 μm, 250 × 4.6 mm) (Grace Vydac) operating at room temperature. Runs were performed at 0.5 ml/min in a mobile phase (A) of 0.05% trifluoroacetic acid (TFA) in water and an eluent (B) of 25% acetonitrile and 0.05% TFA using a linear gradient of 0–15% B over 60 min. Elution of the PG products was monitored at 206 nm.
For determinion of the reaction products following amidase digestion of both whole sacculi and synthetic PG dimer, LC-MS analysis was carried out by the University of Wisconsin-Madison Biotechnology Center Mass Spectrometry/Proteomics Facility. For analysis of whole sacculi digests, 10 μl of filtered digests were diluted 1:1 in water with 0.1% formic acid and separated on a Zorbax SB-C18 column (1.8 μm, 2.1 × 50 mm) (Agilent Technologies) operating at 40 °C. Runs were performed at 0.2 ml/min in a mobile phase (A) of 0.1% formic acid in water and an eluent (B) of 0.1% formic acid in acetonitrile using linear gradients of 2–40% B over 48 min, 40–60% B over 7 min, and 60–95% B over 5 min. Products were then analyzed with an Agilent 1200 series LC/MSD TOF using electrospray ionization in positive mode, acquiring the mass range of 50 to 3200 m/z. For analysis of PG dimer digests, the same instrumentation was used as described above with the same operating parameters, using two linear gradients: 0–40% B over 26 min followed by 40–95% B over 12 min. In both cases, the top 300 peaks from LC (by intensity) were considered for analysis. Synthetic PG GlcNAc-MurNAc(pentapeptide)-GlcNAc-MurNAc(pentapeptide) was prepared as reported previously (44).
LC-MS/MS analysis product 2 of reaction of synthetic PG dimer digest with AmiC was carried out at the University of Notre Dame Mass Spectrometry and Proteomics Facility. Product 2 was analyzed with a Bruker MicrOTOF-Q II quadrupole time-of-flight hybrid mass spectrometer using electrospray ionization in positive mode. Fragmentation of the protonated molecule was evaluated under single collision conditions with argon gas at collision energy levels of 10 to 15 eV.
Metal Dependence and Chelator Assays
For quantitative analysis of PG degradation in the metal and metal chelator assays, enzyme activity was measured using fluorescein-labeled peptidoglycan (EnzChek® lysozyme assay kit, Invitrogen) according to the manufacturer's instructions. Reactions included 5 μm enzyme and 250 μg of labeled peptidoglycan in a 100-μl reaction buffer (0.1 m sodium phosphate, pH 7.5, 0.1 m NaCl) and were incubated at room temperature for 30–60 min. Fluorescence was detected by excitation at 485 nm, and its emission was monitored at 595 nm in a microplate reading fluorimeter (PerkinElmer Victor3 1420 multilabel counter, Wallac 1420 Manager software). Reactions were carried out in flat-bottom solid black 96-well plates (Corning Costar), and all activity was compared with a standard curve of lysozyme provided in the kit. To measure the dependence of catalytic activity on the presence of zinc (or other metal), the purified C-terminal domain of AmiC was dialyzed overnight at 4 °C against 10 mm metal chelators (EDTA, EGTA, CDTA, and 1,10-phenanthroline) in a 0.1 m Tris, pH 8.5, 0.5 m NaCl buffer. Excess chelator was removed by dialysis against the same buffer without chelators. A range of concentrations (1–10 mm) of various metals including zinc chloride, magnesium chloride, manganese chloride, nickel chloride, cobalt chloride, calcium chloride, and iron chloride were added to the dialyzed protein. The activity of untreated protein (no chelator and no additional metal) was used as a positive control, and the activity of the chelator-treated protein without the addition of metal was used as a negative control.
HEK-Blue Human NOD1 (hNOD1) Reporter Assay
To generate supernatants for assessing hNOD1 activation by released products of gonococci, three independent cultures of nonpiliated MS11, DG005, KX503, and JL537 were grown in liquid culture (cGCBL) on three separate days, and supernatants were prepared from these strains by removing whole cells by low-speed centrifugation (4,000 × g for 5 min) and passing the supernatants through a 0.22-μm syringe filter. Growth of the strains was determined by total protein accumulation in cells as measured by a BCA assay (Pierce), and supernatants were normalized to cellular growth using blank growth media (cGCBL). HEK-Blue hNOD1 and Null1 cells (InvivoGen) were used to measure the NF-κB and AP-1 activation through hNOD1, essentially as described previously (45).
Cells were grown at 37 °C with 5% CO2 in Dulbecco's modified Eagle's medium with 4.5 g/liter glucose and 2 mm l-glutamine supplemented with 10% heat-inactivated fetal bovine serum, 100 μg/ml Normocin, 100 μg/ml Zeocin, and 30 μg/ml blasticidin (hNOD1 cells only). Prior to use in any assay, cells were passaged according to the manufacturer's recommendations. For each assay, HEK-Blue cells were washed with warm PBS, gently dislodged from the flasks, and resuspended in the above media (without selective antibiotics Zeocin or blasticidin) at a density of 2.8 × 105 cells/ml. Cells were plated in 180 μl volumes in the wells of a 96-well plate that contained 20-μl volumes of either 1) bacterial supernatants, 2) 10 μg/ml Tri-DAP positive control (InvivoGen), 3) 10 μg/ml muramyl-dipeptide negative control (InvivoGen), or 4) blank media (cGCBL). All of the conditions listed above were assayed in triplicate wells on both the hNOD1 and Null1 cell lines. Following incubation at 37 °C with 5% CO2 for 16–20 h, 20 μl was removed from each well to a new 96-well assay plate and mixed with 180 μl of QUANTI-Blue medium (InvivoGen) for the detection of alkaline phosphatase. After 1 h incubation, absorbance was read at 650 nm in a plate reader. The A560 value from the hNOD1 cells was subtracted from the background measurement of all the samples assayed in parallel on the matching Null1 cell line. Using this subtracted value, triplicate technical replicates of control wells were averaged (± S.D.), and the technical replicate averages from the three biological replicates (supernatant from each strain) were averaged ± S.E. Data were graphed and analyzed in GraphPad Prism 4.0c.
Results
An amiCE229D Mutation Alters PG Fragment Release
In E. coli AmiC, the zinc ion in the catalytic site is coordinated by five residues (His-196, Glu-211, His-265, Asp-267, and Gln-299) (21). Based on sequence alignment of GC-AmiC to both E. coli AmiB and AmiC, we predicted the glutamate at position 229 to be the zinc-coordinating residue responsible for the catalytic activity of AmiC in N. gonorrhoeae and therefore introduced a mutation that changes the glutamate to aspartate (E229D) in the gonococcal chromosome. Quantitative [3H]glucosamine labeling of WT (MS11), amiCE229D (KX503), and a complemented amiCE229D strain with inducible WT amiC expression from a distant site (JL537) revealed a similar phenotype for the amiCE229D strain to that seen previously with an amiC deletion strain (31). In both an amiC deletion strain and a strain carrying an amiCE229D mutation, there is an increase in the release of PG fragments with two disaccharide units and two peptide stems (compound family 1), whereas the release of disaccharide (compound 4) is eliminated (Fig. 1, A and B). IPTG induction of amiC in the complementation strain reduces the release of compound family 1, increases the release of tetrasaccharide-peptide (compound family 2), and restores the release of disaccharide (compound 4) (Fig. 1C). The compounds found in peaks 1–4 are represented by the structures shown in Fig. 1D.
FIGURE 1.

An amiCE229D mutant releases larger [3H]glucosamine-labeled peptidoglycan fragments relative to the wild type and no disaccharide. A–C, N. gonorrhoeae strains wild type (MS11) (A), amiCE229D (KX503) (B), and amiCE229D+amiC (JL537) (C) were metabolically pulse-chase-labeled with [3H]glucosamine, and the released peptidoglycan fragments were fractionated by size-exclusion chromatography. Traces are representative of two independent labeling and chromatography analyses. D, chemical structures of potential variants for released PG fragments found within the numbered peaks in A–C and heretofore referenced as compound families 1–4. Pictured chain lengths for compound families 1 and 2 are based upon their presence in whole sacculi, but the released proportions of compounds 1A–1G and 2A–2D are unknown. Compounds 3A and 3B are released at an ∼1:3 ratio.
Because glucosamine labeling only allows detection of PG fragments containing the sugar moiety, and because amidases cleave between the sugar backbone and peptide chain, we also employed metabolic labeling with [3H]DAP to specifically label the meso-DAP residue, which is unique to the peptide stem of PG. Quantitative PG fragment release following [3H]DAP labeling reveals that the E229D mutation causes a loss of peptide stem release (compound family 5) (Fig. 2, A and B), which can be complemented by amiC overexpression (Fig. 2C). Loss of peptide release is also seen in the amiC deletion strain (DG005) (Fig. 2D). Peptide stems in peak 5 are represented by the structures shown in Fig. 2E. Together, these changes in PG fragment release indicate that Glu-229 is a critical residue necessary for the enzymatic activity of AmiC on N. gonorrhoeae PG and that the predicted amidase activity of AmiC is responsible for generating fragments that are ultimately released from N. gonorrhoeae.
FIGURE 2.

An amiCE229D mutant does not release some [3H]DAP-labeled peptide fragments, similar to a ΔamiC strain. N. gonorrhoeae strains wild type (MS11) (A), amiCE229D (KX503) (B), amiCE229D+amiC (JL537) (C), and ΔamiC (DG005) (D) were metabolically pulse-chase-labeled with [3H]DAP, and the released peptidoglycan fragments were fractionated by size-exclusion chromatography. Structures for the compounds corresponding to the indicated fragment peaks are found in Fig. 1D (compound families 1-4) or shown here (E) (compound family 5). Traces are representative of two independent labeling and chromatography analyses.
An amiCE229D Mutation Causes a Defect in Cell Separation
In E. coli, deletion of the multiple genes for redundant cell separation amidases (amiA, amiB, and amiC) or disruption of the TAT-dependent export pathway causes cells to form chains and fail to separate properly (18, 26). In N. gonorrhoeae, which has only one cell separation amidase (AmiC) and divides in alternating planes, deletion of amiC results in cells that grow in clusters and have formed septa but fail to separate (31). This phenotype is similar to that observed in N. gonorrhoeae lacking the lytic transglycosylase LtgC (46), implying that LtgC may act in concert with AmiC. Because more than one single-gene deletion can cause cell separation defects in N. gonorrhoeae, we wanted to investigate whether amiC mutants are defective in cell separation because they act in a complex with other proteins responsible for proper cell separation or because of the catalytic action of AmiC. Using thin-section transmission electron microscopy, we observed that MS11 amiCE229D (KX503) has a defect in cell separation, which can be complemented by expression of wild-type amiC (Fig. 3). This result shows that the catalytic activity of amiC is critical for proper cell separation in N. gonorrhoeae.
FIGURE 3.

amiCE229D mutation results in a defect in cell separation. Thin-section electron microscopy of wild-type (MS11), amiCE229D (KX503), and amiCE229D+amiC (JL537) strains are shown. Images are representative of multiple fields taken from each sample at each magnification, and each strain was grown, processed, and imaged a minimum of two independent times. Scale bar, 1 μm at ×15,000 and 2 μm at ×5,600.
An amiCQ316K Mutation Causes Increased Activity on Whole Sacculi and Lysis of E. coli
AmiC from N. gonorrhoeae was designated as an N-acetylmuramyl-l-alanine amidase: 1) by homology with E. coli cell separation amidases; and 2) because deletion of amiC results in an alteration in PG fragment release, indicative of loss of amidase activity (31). To investigate the biochemical activity of N. gonorrhoeae AmiC in more detail, we purified N-terminally His6-tagged AmiC. When tested for its ability to digest [3H]glucosamine-radiolabeled PG from N. gonorrhoeae, His6-AmiC was able to facilitate the release of radioactivity from intact purified sacculi regardless of the PG acetylation state (41), presumably through the cleavage of peptides to liberate labeled glycan strands (Fig. 4, A and B).
FIGURE 4.

amiCQ316K mutation has increased enzymatic and lytic activity, whereas amiCE229D mutation protects E. coli from lytic activity. A and B, [3H]glucosamine-labeled whole sacculi from N. gonorrhoeae (A) or an isogenic pacA mutant that does not acetylate its peptidoglycan (B) were digested with purified His-AmiC or His-AmiCQ316K at 37 °C, and samples were taken at 0, 1, 2, and 4 h. Error bars represent S.D. from duplicate reactions. Differences between His-AmiC and His-AmiCQ316K were determined by Student's t test (*, p < 0.05). Assays in A and B are representative of five independent experiments. C, protein sequence alignment of AmiB and AmiC from E. coli and AmiC from N. gonorrhoeae using MUSCLE (partial sequence displayed). Underlines indicate an α-helix known to block activity in E. coli AmiB; δ symbol indicates site of Glu-229 and κ indicates site of Gln-316 in N. gonorrhoeae; *, indicates other putative Zn-coordinating active-site residues. D, E. coli TAM1 with aTc-inducible, C-terminally HA-tagged versions of AmiC (WT), AmiCE229D, AmiCQ316K, or an untagged metabolic enzyme (argJ) were grown to mid-log phase and induced with aTc, and lysis was monitored for 5 h. Error bars represent mean ± S.D. of triplicate wells. The assay is representative of three independent experiments. E, immunoblot against the HA tag showing that AmiC and all variants are made in E. coli during lysis assays. E. coli were grown as described in D except that samples were removed for immunoblot analysis just prior to and 30 min following aTc addition. Immunoblot is representative of two independent growth experiments.
In E. coli, the cell separation amidases AmiA, AmiB, and AmiC are only partially active without a cognate activator to displace an α-helix occluding the active site (27). Sequence alignment of GC AmiC with E. coli AmiB and AmiC indicates the likely presence of a similar inhibitory α-helix from residues 307–321 (Fig. 4C). The charged residue that anchors this amphipathic helix in E. coli AmiB is Glu-303, and mutating this residue can increase basal AmiB activity (27). In both E. coli and N. gonorrhoeae AmiC, this residue is a glutamine. To determine whether AmiC activity can be increased by changing this residue to lysine, akin to the E303K substitution in E. coli AmiB, we changed glutamine 316 to lysine (Q316K) in N. gonorrhoeae AmiC. This change increased the activity of His-AmiC on intact sacculi, again regardless of PG acetylation state (Fig. 4, A and B).
Because AmiCQ316K appears to have more in vitro peptidoglycanase activity compared with wild-type AmiC, we also wanted to determine whether this change translated into increased lytic ability in vivo, in this case using E. coli as a surrogate. For these experiments, E. coli strains carried aTc-inducible, C-terminally HA-tagged versions of N. gonorrhoeae amiC or arginase (argJ) as a negative control (37). Along with wild-type amiC and amiCQ316K, we included amiCE229D as an additional negative control, as the cell separation and fragment release defects caused by this mutation (Figs. 1–3) indicated that it should have impaired activity. Strains were grown in triplicate for 3 h prior to induction with 20 ng/ml aTc. Uninduced cultures continued to grow normally, whereas induced cultures producing the ArgJ control or AmiCE229D-HA exhibited slightly slower growth (Fig. 4D). Expression of wild-type AmiC-HA, however, caused a more severe growth attenuation and lysis, while AmiCQ316K-HA caused the most severe growth attenuation and lysis (Fig. 4D). In an identical growth experiment, uninduced and induced cultures were sampled at 30 min postinduction, and AmiC production was assessed by immunoblotting for the HA tag. Each version of AmiC was detected postinduction, indicating that the differences observed were not due to differences in production or to degradation of AmiCE229D (Fig. 4E). Taken together, the above results indicate that purified AmiC acts on intact sacculi from GC and that AmiC is capable of lysing E. coli. The E229D mutation eliminates lysis activity, whereas lysis is enhanced by the Q316K mutation.
An amiCQ316K Mutation Has Only Intermediate Effects on PG Fragment Release and Causes a Slight Defect in Cell Separation
Based upon our observations that AmiCQ316K has increased ability to digest PG and has more lytic activity compared with wild-type AmiC when expressed in E. coli, we hypothesized that making the Q316K mutation in N. gonorrhoeae would lead to greater PG fragment release. To investigate this hypothesis, we used quantitative [3H]glucosamine pulse-chase labeling to compare a parental strain to an isogenic amiCQ316K strain (JL535) and a complemented mutant (JL536). Contrary to our hypothesis, however, we observed a phenotype similar to, but less severe than, that of an amiCE229D mutation. Specifically, we observed an increase in the release of tetrasaccharide with two peptide stems (compound 1) and a decrease in the release of disaccharide (compound 4) (Fig. 5, A and B). Complementation with wild-type amiC increased disaccharide and tetrasaccharide-peptide (compound 2) release (Fig. 5C), similar to what was seen in the amiCE229D complementation (Fig. 1C). Because the amiCQ316K mutation generated a fragment release profile indicating a defect in PG fragment release activity, we wanted to determine how this mutation impacts cell separation. Using thin-section transmission electron microscopy, we observed that the amiCQ316K mutation causes a slight defect in cell separation (Fig. 5, D and E). This defect appeared to be less severe than that observed with the inactivating amiCE229D mutation (Fig. 3). When quantified, the Q316K change does result in a significant decrease in the presence of individual cells (monococci), a significant increase in the presence of diplococci (especially those that have completed septation but failed to separate), and an increase in three- and four-cell clusters (Fig. 5F). Complementation with an inducible amiC largely restores wild-type cell separation (Fig. 5F). From these data, we concluded that in addition to displacement of the α-helix blocking the active site, other factors are involved in modulating the activity of AmiC in N. gonorrhoeae. In other characterized amidases, an N-terminal targeting domain (AMIN domain) is responsible for the binding of amidase to PG (21), and binding at the division site is coordinated with the sequential recruitment of other divisome components (20). An N-terminal AMIN domain is also present in GC-AmiC. In the folded structure of E. coli amidases, this domain is located distantly from the catalytic site (21), and active-site mutations have not been shown to impact its function. The observed PG fragment release and cell separation phenotypes caused by the autoactivating Q316K mutation are therefore likely to be due to disruption of an important temporal or spatial interaction with a cognate activator (NlpD) or accessory protein.
FIGURE 5.

amiCQ316K mutation causes intermediate PG fragment release and cell separation phenotypes. A–C, N. gonorrhoeae strains wild type (MS11) (A), amiCQ316K (JL535) (B), and amiCQ316K+amiC (JL536) (C) were metabolically pulse-chase-labeled with [3H]glucosamine, and the released peptidoglycan fragments were fractionated by size-exclusion chromatography. Structures for the compounds corresponding to the indicated fragment peaks are found in the legend for Fig. 1D (compound families 1–4). D and E, thin-section electron microscopy of amiCQ316K (JL535) (D) and amiCQ316K+amiC (JL536) (E) strains. Images are representative of multiple fields taken for each sample at each magnification. Scale bar = 2 μm. F, quantification of thin-section electron microscopy. Cells presenting as individuals (1), diplococci (2), clusters of three (3), or clusters of four (4) were enumerated from a minimum of five fields taken of the wild-type, amiCQ316K, and amiCQ316K+amiC strains (total of >3000 cells). For each group, bars represent the mean ± S.D. of the percentage of cells in a given group within a field. Differences were determined by one-way analysis of variance (***, p < 0.001; **, p < 0.01).
AmiC Generates Peptides from Sacculi
The amiCQ316K mutation results in an enzyme with more in vitro activity but an in vivo disruption in fragment release and cell separation. To understand whether the observed increase in enzyme activity is from an increase in fragment production or from an alteration of enzyme specificity (resulting in products different from the wild type), we determined which PG fragments were made when sacculi were digested with purified His-AmiC or His-AmiCQ316K. Because the Q316K mutation was intended to mimic the activation provided by the LytM domain activator protein NlpD, we also tested a mixture of purified His-AmiC with His6-NlpD to determine: 1) whether NlpD augments AmiC activity in N. gonorrhoeae and 2) whether the Q316K mutation and/or the addition of NlpD facilitate production of the same products. NlpD in N. gonorrhoeae contains a predicted M23B peptidase domain but lacks two of the four residues predicted to be necessary for the enzymatic function seen in Gram-positive bacteria (30). Residues Asn-304 and Gln-383 in NlpD of N. gonorrhoeae strain MS11 are the same two residues that are altered from the consensus active site in E. coli, leading to a loss of enzyme activity in E. coli EnvC (25) and NlpD. However, the N. gonorrhoeae peptidoglycanase NG1686, which has a complete M23B peptidase active site, has been shown to retain activity even with the homologous M23B active-site residues mutated (43). NlpD may therefore have an independent or complementary activity that should be detectable by assessing the products of macromolecular PG digestion by LC/MS.
For these experiments, we prepared unlabeled macromolecular PG from MS11 pacAH329Q (KH530) and subjected it to digestion with 500 nm His-AmiC alone (1× in Table 1), 500 nm His-AmiCQ316K alone, 2.5 μm His-AmiC alone (5× in Table 1), 2.5 μm NlpD alone, or a reaction with 500 nm His-AmiC and 500 nm His-NlpD. We then removed insoluble (undigested) PG and identified the major soluble products by LC/MS (Table 1). Digesting gonococcal PG with AmiC resulted in the recovery of soluble fragments of predicted masses matching tripeptide stem (l-Ala-γ-d-Glu-mDAP), tetrapeptide stem (l-Ala-γ-d-Glu-mDAP-d-Ala), pentapeptide stem (l-Ala-γ-d-Glu-mDAP-d-Ala-d-Ala), cross-linked tetrapeptide-tripeptide (4-3), cross-linked tetrapeptide-tetrapeptide (4-4), and cross-linked tetrapeptide-tetrapeptide-tetrapeptide (4-4-4). The observed peptide products are those that would be predicted from amidase activity. The LC analysis indicated that the most abundant product is a cross-linked tetrapeptide-tetrapeptide (4-4) followed by tetrapeptide stem, cross-linked tetrapeptide-tripeptide (4-3), pentapeptide stem, tetrapeptide-tetrapeptide-tetrapeptide (4-4-4), and tripeptide stem. Very small amounts of product with GlcNAc-anhMurNAc attached to either 4-3, 4-4, or 3-3 cross-linked peptide stems were also observed (<0.25% of the total detector counts for each run). These are known products of partial amidase digestion (16). These results are consistent with a highly cross-linked sacculus, noted previously as being unusual among Gram-negatives (47), and a sacculus composed primarily of tetrapeptide stems (9).
TABLE 1.
PG fragments identified in LC-MS of sacculi digests
—, not observed.
| PG peptide fragment | Calculated exact mass | Ion | AmiC (1×) |
AmiC (5×) |
NlpD (5×) |
AmiC + NlpD |
AmiCQ316K |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Observed m/z | Observed mass | Observed m/z | Observed mass | Observed m/z | Observed mass | Observed m/z | Observed mass | Observed m/z | Observed mass | |||
| Tetra-tetra-tetra | 1347.62 | [M + 2H]2+ | 674.82 | 1347.62 | 674.82 | 1347.62 | — | — | 674.82 | 1347.62 | 674.82 | 1347.62 |
| Tetra-tetra | 904.41 | [M + H]+ | 905.42 | 904.41 | 905.42 | 904.42 | — | — | 905.42 | 904.41 | 905.42 | 904.41 |
| [M + 2H]2+ | 453.22 | 904.42 | — | — | — | — | — | — | — | — | ||
| Tetra-tri | 833.38 | [M + H]+ | 834.38 | 833.38 | 834.38 | 833.38 | — | — | 834.38 | 833.38 | 834.38 | 833.38 |
| [M + 2H]2+ | 417.70 | 833.39 | — | — | — | — | 417.70 | 833.38 | 417.70 | 833.38 | ||
| Penta | 532.25 | [M + H]+ | 533.26 | 532.25 | 533.26 | 532.25 | — | — | 533.26 | 532.25 | 533.26 | 532.25 |
| Tetra | 461.21 | [M + H]+ | 462.22 | 461.21 | 462.22 | 461.21 | — | — | 462.22 | 461.21 | 462.22 | 461.21 |
| [M + 2H]2+ | — | — | 231.61 | 461.21 | — | — | — | — | — | — | ||
| Tri | 390.18 | [M + H]+ | 391.18 | 390.18 | 391.18 | 390.17 | — | — | 391.18 | 390.18 | 391.18 | 390.18 |
In all the reactions containing AmiC or AmiCQ316K, the above products were observed; however, none of these products was observed among the top 300 most abundant masses in the reaction with NlpD alone, indicating no detectable peptidoglycanase activity for NlpD. Neither the addition of NlpD nor the Q316K mutation in AmiC appeared to change the substrate specificity or the released products. These data indicate that the AmiCQ316K mutation acts to derepress amidase activity in vitro in a manner comparable with the addition of NlpD and that AmiC and NlpD in GC likely exist in a similar amidase-activator relationship to that seen in other organisms.
AmiC Can Cleave One Peptide Stem from Synthetic PG Dimer
The cell separation amidases are known to act on macromolecular PG but are unable to target the muramyl-l-alanine linkage in fragments such as UDP-MurNAc-pentapeptide, disaccharide-peptide (compound family 3), and tetrasaccharide peptide (compound family 2) (48). This has left open the question of the nature of the minimal substrate for AmiC. When we overexpressed amiC during complementation of our active-site point mutants, we noticed not only an increase in disaccharide (compound 4) release (Fig. 1C), but an increase in released tetrasaccharide-peptide (compound family 2) (Figs. 1C and 5C) (9). Although this result implies that amidases participate in the generation of tetrasaccharide-peptide fragments, it is unclear whether the amidase acts on peptide stems within the cell wall (prior to liberation by lytic transglycosylases) or on liberated tetrasaccharide fragments containing two peptide stems (compounds 1A–1D). To test whether amidases can target tetrasaccharide fragments with two peptide stems, we employed a synthetic PG fragment (GlcNAc-MurNAc(pentapeptide)-GlcNAc-MurNAc(pentapeptide)) (44). This synthetic PG fragment does not have a reducing end but terminates in a 1β-methoxy moiety as a mimetic of the continuing backbone of sugars. A 10-μg portion of the synthetic PG compound was used as substrate in reactions with 500 nm of His-tagged AmiC, NlpD, AmiCQ316K, or both AmiC and NlpD (along with a no enzyme control), and the resulting products were examined by LC/MS. The predicted reaction was either partial digestion (removal of one peptide stem, P1 or P2) or complete digestion (removal of both peptide stems, P3) (Fig. 6A).
FIGURE 6.

AmiC is capable of cleaving a synthetic PG substrate. A, structure of synthetic PG substrate and four possible products of reaction with AmiC (P1–P4). Partial amidase reaction would give a tetrasaccharide-pentapeptide P1 and/or P2 (predicted mass of 1502.62) along with pentapeptide P4 (predicted mass of 532.25). The complete amidase reaction would give tetrasaccharide P3 and pentapeptide P4. B, collision-induced spectrum of AmiC tetrasaccharide-peptide product and fragmentation. The m/z values in bold (682, 822, and 1025) are unique fragment ions and can only be formed in P1, not P2. C, reverse-phase HPLC analysis of digestions of PG dimer using His-AmiC, His-NlpD, His-AmiCQ316K, or a combination of enzymes. S, synthetic dimer substrate; P1, tetrasaccharide-pentapeptide; P4, pentapeptide. Products were confirmed by mass spectrometry. HPLC analysis is representative of two independent experiments on a synthetic dimer under identical conditions used for independent LC-MS analysis of reactions (Table 2).
Within the reactions containing AmiC, AmiC + NlpD, and AmiCQ316K, the two identifiable major products have masses of 532.25 and 1502.62, matching the predicted size of pentapeptide P4 and tetrasaccharide-pentapeptide (P1 or P2), respectively (Fig. 6A and Table 2). None of the reactions contained a mass corresponding to the tetrasaccharide P3, GlcNAc-MurNAc-GlcNAc-MurNAc-OCH3 (988.39), leading us to conclude that AmiC is able to recognize tetrasaccharide PG as a substrate but can differentiate between the two muramyl-l-alanine amide linkages. Subsequent LC-MS/MS analysis of the tetrasaccharide-peptide product indicated that the pentapeptide chain closer to the methoxy end was the one retained (P1, Fig. 6B). Hence, there appears to be a substrate preference that limits the catalytic action to only one of the two peptides. HPLC analysis of digestion reactions showed that AmiC activity is enhanced by the presence of NlpD or by the Q316K substitution (Fig. 6C). NlpD alone does not have enzymatic activity on this substrate, as shown both by the lack of product observed on HPLC (Fig. 6C) and by the lack of recognizable products in the LC-MS results (data not shown). Thus, it is likely that NlpD plays a role as a cognate activator of AmiC in N. gonorrhoeae, although AmiC in GC does display a higher basal activity than seen in other species (Figs. 6C and 4, A and B) (21, 26). The ability of AmiC to cut one peptide stem, but not both, for each tetrasaccharide unit may also inform as to how AmiC works on macromolecular PG. Hydrolytically cutting potentially every second stem peptide provides a model for how the subsequent action of lytic transglycosylases, which digest PG into fragments containing the 1,6-anhydromuramyl units, would produce a mix of the observed disaccharide-peptides (compound family 3), disaccharide (compound 4), and disaccharides with cross-linked peptides (compounds 1E–1G) rather than only the disaccharide and peptides that are observed in E. coli (10, 11).
TABLE 2.
PG fragments identified in LC-MS of synthetic PG substrate digests
S, synthetic dimer substrate; P1, tetrasaccharide-pentapeptide; P4, pentapeptide; —, not observed.
| PG fragment | Calculated exact mass | Ion | AmiC |
AmiC + NlpD |
NlpD |
AmiCQ316K |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| Observed m/z | Observed mass | Observed m/z | Observed mass | Observed m/z | Observed mass | Observed m/z | Observed mass | |||
| S | 2016.86 | [M + 2H]2+ | 1009.44 | 2016.86 | 1009.94 | 2016.86 | 1009.43 | 2016.85 | 1009.94 | 2016.86 |
| [M + 3H]3+ | — | — | 673.29 | 2016.86 | — | — | 673.29 | 2016.86 | ||
| P1 | 1502.62 | [M + H]+ | — | — | — | — | — | — | 1503.63 | 1502.62 |
| [M + 2H]2+ | 752.32 | 1502.63 | 752.32 | 1502.62 | — | — | 752.32 | 1502.63 | ||
| [M + 3H]3+ | 501.88 | 1502.63 | 501.88 | 1502.63 | — | — | — | — | ||
| P4 | 532.25 | [M + H]+ | 533.26 | 532.25 | 533.26 | 532.25 | — | — | 533.26 | 532.25 |
| [M + 2H]2+ | — | — | — | — | — | — | 267.13 | 532.25 | ||
AmiC Can Utilize Several Divalent Metal Cations for Activity
The N-acetylmuramyl-l-alanine amidases of E. coli have been classified as zinc-dependent, although other divalent metal cations have been shown to facilitate amidase function in the cell separation amidases of Bacillus (49). To determine the metal dependence of GC-AmiC, the purified C-terminal domain of AmiC, dialyzed overnight against the metal chelators EDTA, CDTA, and EGTA and the zinc-specific chelator 1,10-phenanthroline, was then used to digest fluorescein-labeled PG. All of these metal chelators produced a significant decrease in activity compared with untreated AmiC. Treatment with 1,10-phenanthroline resulted in complete loss of AmiC activity, whereas EGTA and CDTA resulted in a ∼90% reduction in activity and EDTA in a 70% reduction (Fig. 7A). The response to all of these chelators was dose-dependent, and analysis of chelator-treated proteins by circular dichroism confirmed that the proteins remained folded (data not shown).
FIGURE 7.

AmiC requires metal cations for activity. A, treatment of AmiC-CTD with metal chelators results in nearly complete ablation of activity. The most marked effect was with the zinc-specific chelator 1,10-phenanthroline. B, addition of metal ions rescue AmiC-CTD activity. Following treatment of AmiC-CTD with 1,10-phenanthroline, the chelator was removed by dialysis, and 10 mm metal salts were added to the protein. The addition of ZnCl2, MgCl2, and CaCl2 resulted in 70–80% restoration of protein activity. Error bars represent mean ± S.D. of triplicate reactions. (**, p < 0.01; *, p < 0.05).
Because the loss of a metal cofactor in reactions resulted in a loss of activity, we explored the range of metals capable of rescuing AmiC activity. Following treatment with 10 mm CDTA, we dialyzed out this chelator and reintroduced physiologically relevant metal ions by the addition of 10 mm ZnCl2, MgCl2, CaCl2, CoCl2, or NiCl2. Some restoration of activity was observed with all of the metal salts. The addition of ZnCl2, MgCl2, and CaCl2 was the most effective, restoring 70–80% of the activity, with NiCl2 restoring ∼50% and CoCl2 the least effective at only ∼25% (Fig. 7B). Some of these variations in recovery of activity may have been due to differences among these metal ions for their preferred coordination chemistry. In general, it is reasonable to characterize AmiC as a zinc-dependent amidase, but our data suggest that the requirement for the divalent metal ion by this protein is not strict. It is therefore possible that other metal ions could substitute for a zinc ion in AmiC as necessary, thus preserving the activity of this critical cell separation enzyme, even when zinc ion concentration could be limiting.
Loss of Released Peptides Does Not Impact NOD-dependent NF-κB Activation by GC
The NOD-like receptor family of eukaryotic intracellular PG sensors includes NOD1, which recognizes PG-derived ligands that contain mesod-diaminopimelic acid (mDAP) (50, 51). This C-carboxylated lysine is unique to the stem peptides of PG and is found throughout Gram-negative bacteria, as well as in some Gram-positive bacteria. N. gonorrhoeae releases several PG fragments that contain mDAP, including peptide stems (compound family 5), disaccharide-peptides (compound family 3), tetrasaccharide-peptides (compound family 2), and tetrasaccharide with peptide stems on each MurNAc (compound family 1) (8, 9). Human NOD1 (hNOD1) specifically recognizes PG fragments terminating in mDAP, whereas tracheal cytotoxin and tetrapeptide stems are poor hNOD1 agonists (52). Previously, it was shown that the GC enzymes LtgA and LtgD are responsible for the production of PG fragments through their lytic transglycosylase activity, and eliminating these enzymes abolishes disaccharide-peptide PG fragment release (53). Although disaccharide-peptide fragments are a large proportion of the potential hNOD1 agonist PG released by N. gonorrhoeae, the loss of these fragments does not eliminate the ability of gonococci to signal through hNOD1.3 Because amiC mutants still release disaccharide-peptide but do not release peptide stems (Fig. 2, B and D), we were interested to see whether eliminating the peptides released by AmiC activity (Table 1) reduces hNOD1 activation by gonococci. To test this hypothesis, we harvested conditioned supernatants following the growth of wild-type, ΔamiC, amiCE229D, and amiCE229D + amiC strains and normalized the supernatant to the total cellular protein.
GC supernatant alone has been shown to contain measurable hNOD1 agonist (45, 54). Supernatants were used for treatment of HEK-293 cells overexpressing human NOD1 and carrying a secreted embryonic alkaline phosphatase reporter (HEK-BLUE hNOD1 cells). To avoid saturating the reporter system, we tested the supernatants at two different dilutions and included the NOD1 agonist l-Ala-γ-d-Glu-mDAP (or TriDAP) and the NOD2 agonist muramyl dipeptide as the positive and negative controls, respectively. Eliminating the activity of amiC did not result in an appreciable decrease in hNOD1-dependent NF-κB activation (Fig. 8). This result indicates that the release of either disaccharide-peptide PG fragments or PG peptides from gonococci is sufficient to activate hNOD1.
FIGURE 8.
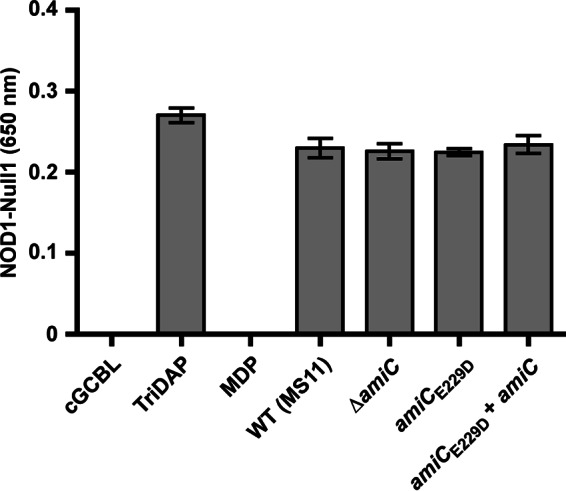
Reducing the peptide and disaccharide release does not reduce signaling through hNOD1. HEK-Blue cells overexpressing hNOD1 and carrying a reporter driven by NF-κB and AP-1 were exposed to supernatant from wild-type (MS11), amiCE229D (KX503), amiCE229D+amiC (JL537), and ΔamiC (DG005) strains. For each assay, each strain was grown three independent times, and supernatants from each of these replicates were assayed in triplicate wells alongside positive and negative controls on the same passage of HEK-Blue cells. All exposures on hNOD1 cells were mirrored on the parental Null1 strain grown in parallel, and values from Null1 cells were subtracted as background from the values obtained for hNOD1 cells. Error bars of controls represent the mean ± S.D. of triplicate technical replicates, and error bars of experimental samples represent the means ± S.E. of the value obtained for the three biological replicates after averaging the triplicate technical replicate wells for each. TriDAP, l-Ala-γ-d-Glu-mDAP (hNOD1 agonist, 10 μg/ml); MDP, muramyl dipeptide (hNOD2 agonist, 10 μg/ml).
Discussion
N. gonorrhoeae encodes a single periplasmic N-acetylmuramyl-l-alanine amidase, AmiC, involved in cell separation. We have demonstrated that this enzyme indeed possesses the amidase activity that was predicted in previous studies (31), with the enzymatic activity responsible both for its role in cell separation and for the release of fragments of PG during growth. N. gonorrhoeae with an amiCE229D mutation no longer releases PG-derived GlcNAc-anhMurNAc disaccharide (compound 4) or stem peptides (compound family 5) during growth; instead it releases more of the larger tetrasaccharide fragments with attached peptide stems (compound family 1) (Fig. 1). This mutation alone disrupts proper cell separation in N. gonorrhoeae, suggesting that the amidase activity of AmiC cannot be substituted with other peptidoglycanases during cell separation. Likewise, the E229D change abolishes the lytic activity of amidase overexpression in E. coli, consistent with a loss of enzymatic activity. Although we noted that AmiC can be activated by the LytM-domain-containing protein NlpD, similar to the amidase-activator arrangement seen in E. coli (26), our studies have revealed that GC AmiC is able to digest PG largely without a necessity for NlpD. The presence of NlpD does enhance AmiC activity, as does a mutation in a putative autoinhibitory α-helical structure, Q316K.
Mimicking an autoactivating E303K mutation from E. coli (27), the Q316K mutation in N. gonorrhoeae increases amidase activity in vitro (and E. coli lysis); but unexpectedly this results in a slight defect in cell separation and PG fragment release, which resembles a milder version of the inactivating E229D mutation. It was not previously known how autoactivating mutations like those made in E. coli AmiB affect phenotypes other than by increasing PG hydrolase activity and decreasing cell growth (27). We showed here that lowering the autoinhibition of AmiC impairs its normal function in cell separation. This result suggests that it is not only important for cell separation amidases to be derepressed by cognate activators but that this activating interaction may also be dependent on spatiotemporal factors, or it may require additional accessory factors to achieve coordinated cell separation activity (20). In E. coli, AmiB and its cognate activator EnvC, as well as AmiC and its cognate activator NlpD, both arrive at the divisome independently, although the amidases and activators have different requirements for the presence of the septal ring component FtsN and the timing of their interactions (18, 20). In V. cholerae, which has only a single amidase but two activators, the localization of the amidase is activator-dependent (29). In GC, it would be of interest to determine in what order AmiC and/or NlpD localizes to the division site and which (if either) has a localization that depends on other components of the divisome. One candidate for a co-localizing divisome component in GC is the lytic transglycosylase LtgC, which also has a single-deletion cell separation phenotype comparable with an amiC deletion and has a role in the release of GlcNAc-anhMurNAc disaccharide (46). In considering other potential candidates for protein-protein interactions with AmiC in gonococci, it should be noted that GC lacks homologs of the high-molecular-weight E. coli PBPs 1b and 2, as well as the low-molecular-weight PBPs 5, 6, and 6b. This shortened list of PG synthesis components still includes PBP3, a homolog of E. coli PBP4/DacB, and PBP4, a homolog of E. coli PBP7 (55, 56). Deletions of PBP4 and/or PBP7 in E. coli exacerbate the cell separation defects of an amiC mutant and are implicated as accessory factors in E. coli cell separation (57). A strain of N. gonorrhoeae with PBP3 and PBP4 deleted also displays aberrant morphology with variations in cell size (56).
Prior to our work, it was unknown which PG fragments were generated by AmiC digestion of intact N. gonorrhoeae sacculi or whether there was a size limit for the digestion of smaller PG fragments. Even in E. coli, the substrate specificity of amidases, and how this dictates their functions in cell division, remains somewhat unclear (57). We have demonstrated that AmiC from GC is capable of liberating peptide chains of all lengths, as well as cross-linked peptides from whole macromolecular PG, without the action of other enzymes. GC-AmiC is capable of this biochemical activity even though only the non-cross-linked peptides are observed as naturally released by GC (8). The abundance of peptide fragments solubilized by AmiC also corresponds to the known proportions of these chains in GC sacculi (which have mostly tetrapeptide stems) and reflects the known high degree of cross-linking (8, 47). AmiC has no apparent substrate preference, based on peptide chain length or configuration, and is therefore more similar to E. coli AmiC than to the more substrate-restricted AmiA and AmiB (14, 57). In E. coli, AmiA is more active on intact sacculi than AmiC, as AmiA is present throughout the periplasm and capable of remodeling activity throughout the sacculi, whereas AmiC is septally restricted and shows a preference for septal PG over sidewall PG (14, 18). GC is coccal in shape and lacks components of the PG elongation machinery (58, 59), making it unlikely to have sidewall PG. It may be that a single broad specificity, cell separation amidase is sufficient when there is no need for multiple amidases that differentiate between septal and sidewall PG.
The ability of AmiC to act on a synthetic PG dimer fragment informs how we consider the order of enzyme reactions during cell separation and the mechanism by which released PG fragments are generated in N. gonorrhoeae. By treating synthetic PG dimer with amidase, we showed that AmiC can recognize a substrate as small as a tetrasaccharide (two GlcNAc-MurNAc disaccharide units) but cuts only one of the two peptide stems. This represents the smallest substrate yet identified for a cell separation amidase (48). Because not every peptide stem in the sacculus is cross-linked, it is possible that amidases do not need to work on every stem to facilitate the separation of glycan strands during cell separation. This result suggests a role for endopeptidases working in concert with AmiC to trim the remaining cross-links. Without that activity, disaccharides joined by peptide cross-links would be a larger proportion of naturally released fragments, given the degree of cross-linking in the GC sacculi. Possible candidates for this activity include PBP3, which has endopeptidase and d-Ala-d-Ala carboxypeptidase activity, and PBP4, which has dd-endopeptidase activity (55, 56). In GC the deletion of either PBP3 or PBP4 reduces the release of disaccharide and impacts autolysis (60), phenotypes also associated with AmiC activity. Tetrasaccharide fragments (with either one or two peptide stems) are a minority of the released products, making it unlikely that AmiC works exclusively on liberated tetrasaccharide fragments. It is more likely that it operates processively on glycan strands, which are later degraded by lytic transglycosylases.
The release of PG fragments during the growth of gonococci has been shown to contribute to host damage, specifically the death and sloughing of ciliated cells present in the fallopian tube (5). This situation is analogous to the release of tracheal cytotoxin, the major PG fragment released by B. pertussis, which causes ciliated cell death in the hamster trachea (12). PG fragments of the composition released by GC are potent agonists of the human intracellular PG sensor NOD1, and conditioned media from GC can activate NOD1 (54). We demonstrated that cross-linked and non-cross-linked peptides can be generated from GC sacculi by the activity of AmiC, including the known agonist l-Ala-γ-d-Glu-mDAP (50, 51). We assumed that both the disaccharide-peptide fragments and the released stem peptides terminating in mDAP were capable of activating NF-κB through hNOD1, as we observed that eliminating the release of disaccharide-peptides by deleting the lytic transglycosylases ΔltgA and ΔltgD (53) did not affect hNOD1 signaling.3 Thus, in this work, we sought to determine whether eliminating the release of stem peptides by AmiC could reduce hNOD1 activation by gonococcal supernatant. We considered two possible scenarios: 1) small mDAP-containing peptides are more efficient agonists and contribute most of the hNOD1 signaling; or 2) disaccharide-peptide fragments and peptide stems contribute equally and are both sufficient for hNOD1 signaling. Our data indicate that as long as either mDAP-containing product is released, soluble PG from GC is capable of being detected by NOD1. Thus, multiple soluble PG fragments released by gonococci may contribute to hNOD1-dependent inflammatory responses and cell death.
As the only cell separation amidase in gonococci, AmiC is critical for normal cell separation. Although AmiC homologs in E. coli have low background amidase activity (21), we see significant activity from purified gonococcal AmiC and increased PG fragment release (indicative of more amidase activity) when wild-type AmiC is overexpressed. In a system without redundancy, the flexibility of gonococcal AmiC to favor an “on” state as opposed to a baseline “off” state may prevent a block in cell division and may also shed light on what makes GC so notably autolytic (61). The ability of GC AmiC to utilize several different metal cations for activity (although clearly preferring zinc) may also be a property that increases biological flexibility. A similar ability to utilize other divalent metal cations for activity was demonstrated for the amidase Rv3717 from Mycobacterium tuberculosis, which can use copper, cobalt, and magnesium in place of zinc (62). However, this enzyme is more similar in function to AmpD, which is used in PG recycling (63, 64), making AmiC the first cell separation amidase shown to have promiscuous metal specificity. The ability of AmiC to act without explicit activation and to utilize other metal ions when necessary may be advantageous in a zinc-limiting environment during host infection.
PG has long been a target of antimicrobial activity, from phage autolysins (many of which are amidases) to β-lactams that target PG synthesis. The emergence of multi-drug-resistant gonococci in the community and the potential future losses of effective antibiotics against GC (65) should turn our focus to the development of antimicrobials that attack this pathogen in new ways. The approach of disrupting division or cell separation has shown promise, with several division inhibitors targeting FtsZ entering preclinical evaluation (66). In the gonococcus, the non-redundant, seemingly non-essential enzymes that are important in cell separation (like AmiC or LtgC) should be considered as antimicrobial targets. These enzymes undertake a critical cellular process tied to bacterial lysis; inhibiting them can lower the MIC for existing antibiotics, and disrupting them may aid the host by slowing bacterial growth and presenting a more attractive target for immune defenses.
Author Contributions
J. D. L. and J. P. D. designed and conceived this study, and J. D. L. wrote the paper. J. D. L. designed, performed, and analyzed all experiments not specifically noted below. E. A. S. purified NlpD. K. T. H. and K. X. designed and constructed vectors and made the E229D mutant in Neisseria gonorrhoeae. K. F. made the initial observations that led to the work in Figs. 3 and 4B. M. L. and D. H. synthesized the synthetic PG dimer used in Fig. 6 and Table 2. M. L. performed and analyzed the experiment in Fig. 6C. R. M. R. designed, performed, and analyzed the experiments shown in Fig. 7. H. S. S., S. M., C. D., and J. P. D. provided technical assistance and aided in the preparation of this manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Randall Massey of the University of Wisconsin-Madison Medical School Electron Microscopy Facility for assistance with electron micrographs. We also thank Grzegorz Sabat and Leigh Anson of the University of Wisconsin-Madison Biotechnology Center Mass Spectrometry/Proteomics Facility for assistance with LC-MS.
This work was supported by National Institutes of Health Grants R01 AI097157 and R21 AI099539 (to J. P. D.), T32 AI055397 and F32 AI115911 (to J. D. L.), R37 AI033493 and R01 AI044239 (to E. A. S. and H. S. S.), R01 GM066861 (to C. D.), and R01 AI090348 and R01 GM061629 (to S. M.). The authors declare that they have no conflict of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
K. T. Hackett and J. P. Dillard, unpublished data.
- PG
- peptidoglycan
- GC
- gonococcus (Neisseria gonorrhoeae)
- GCB
- gonococcal base
- GCBL
- gonococcal base liquid medium
- IPTG
- isopropyl β-d-thiogalactopyranoside
- aTc
- anhydrotetracycline
- DAP
- diaminopimelic acid
- mDAP
- meso-diaminopimelic acid
- CTD
- C-terminal domain
- PB
- phosphate buffer.
References
- 1. Vollmer W. (2008) Structural variation in the glycan strands of bacterial peptidoglycan. FEMS Microbiol. Rev. 32, 287–306 [DOI] [PubMed] [Google Scholar]
- 2. Centers for Disease Control and Prevention (2013) Antibiotic Resistance Threats in the United States, 2013, http://www.cdc.gov/drugresistance/threat-report-2013/
- 3. Gregg C. R., Melly M. A., Hellerqvist C. G., Coniglio J. G., and McGee Z. A. (1981) Toxic activity of purified lipopolysaccharide of Neisseria gonorrhoeae for human fallopian tube mucosa. J. Infect. Dis. 143, 432–439 [DOI] [PubMed] [Google Scholar]
- 4. Massari P., Henneke P., Ho Y., Latz E., Golenbock D. T., and Wetzler L. M. (2002) Cutting edge: immune stimulation by neisserial porins is toll-like receptor 2 and MyD88 dependent. J. Immunol. 168, 1533–1537 [DOI] [PubMed] [Google Scholar]
- 5. Melly M. A., McGee Z. A., and Rosenthal R. S. (1984) Ability of monomeric peptidoglycan fragments from Neisseria gonorrhoeae to damage human fallopian tube mucosa. J. Infect. Dis. 149, 378–386 [DOI] [PubMed] [Google Scholar]
- 6. Boneca I. G. (2005) The role of peptidoglycan in pathogenesis. Curr. Opin. Microbiol. 8, 46–53 [DOI] [PubMed] [Google Scholar]
- 7. Hebeler B. H., and Young F. E. (1976) Chemical composition and turnover of peptidoglycan in Neisseria gonorrhoeae. J. Bacteriol. 126, 1180–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rosenthal R. S. (1979) Release of soluble peptidoglycan from growing gonococci: hexaminidase and amidase activities. Infect. Immun. 24, 869–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sinha R. K., and Rosenthal R. S. (1980) Release of soluble peptidoglycan from growing gonococci: demonstration of anhydro-muramyl-containing fragments. Infect. Immun. 29, 914–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goodell E. W., and Schwarz U. (1985) Release of cell wall peptides into culture medium by exponentially growing Escherichia coli. J. Bacteriol. 162, 391–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Uehara T., and Park J. T. (2007) An anhydro-N-acetylmuramyl-l-alanine amidase with broad specificity tethered to the outer membrane of Escherichia coli. J. Bacteriol. 189, 5634–5641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goldman W. E., and Cookson B. T. (1988) Structure and functions of the Bordetella tracheal cytotoxin. Tokai J. Exp. Clin. Med. 13, (suppl.) 187–191 [PubMed] [Google Scholar]
- 13. Frirdich E., and Gaynor E. C. (2013) Peptidoglycan hydrolases, bacterial shape, and pathogenesis. Curr. Opin. Microbiol. 16, 767–778 [DOI] [PubMed] [Google Scholar]
- 14. Heidrich C., Templin M. F., Ursinus A., Merdanovic M., Berger J., Schwarz H., de Pedro M. A., and Höltje J. V. (2001) Involvement of N-acetylmuramyl-l-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol. Microbiol. 41, 167–178 [DOI] [PubMed] [Google Scholar]
- 15. Lee M., Artola-Recolons C., Carrasco-López C., Martínez-Caballero S., Hesek D., Spink E., Lastochkin E., Zhang W., Hellman L. M., Boggess B., Hermoso J. A., and Mobashery S. (2013) Cell-wall remodeling by the zinc-protease AmpDh3 from Pseudomonas aeruginosa. J. Am. Chem. Soc. 135, 12604–12607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martínez-Caballero S., Lee M., Artola-Recolons C., Carrasco-López C., Hesek D., Spink E., Lastochkin E., Zhang W., Hellman L. M., Boggess B., Mobashery S., and Hermoso J. A. (2013) Reaction products and the x-ray structure of AmpDh2, a virulence determinant of Pseudomonas aeruginosa. J. Am. Chem. Soc. 135, 10318–10321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Firczuk M., and Bochtler M. (2007) Folds and activities of peptidoglycan amidases. FEMS Microbiol. Rev. 31, 676–691 [DOI] [PubMed] [Google Scholar]
- 18. Bernhardt T. G., and de Boer P. A. (2003) The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol. Microbiol. 48, 1171–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Craig M., Sadik A. Y., Golubeva Y. A., Tidhar A., and Slauch J. M. (2013) Twin-arginine translocation system (TAT) mutants of Salmonella are attenuated due to envelope defects, not respiratory defects. Mol. Microbiol. 89, 887–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peters N. T., Dinh T., and Bernhardt T. G. (2011) A fail-safe mechanism in the septal ring assembly pathway generated by the sequential recruitment of cell separation amidases and their activators. J. Bacteriol. 193, 4973–4983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rocaboy M., Herman R., Sauvage E., Remaut H., Moonens K., Terrak M., Charlier P., and Kerff F. (2013) The crystal structure of the cell division amidase AmiC reveals the fold of the AMIN domain, a new peptidoglycan binding domain. Mol. Microbiol. 90, 267–277 [DOI] [PubMed] [Google Scholar]
- 22. Berendt S., Lehner J., Zhang Y. V., Rasse T. M., Forchhammer K., and Maldener I. (2012) Cell wall amidase AmiC1 is required for cellular communication and heterocyst development in the cyanobacterium Anabaena PCC 7120 but not for filament integrity. J. Bacteriol. 194, 5218–5227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Büttner F. M., Faulhaber K., Forchhammer K., Maldener I., and Stehle T. (2016) Enabling cell-cell communication via nanopore formation: structure, function, and localization of the unique cell wall amidase, AmiC2 of Nostoc punctiforme. FEBS J. 10.1111/febs.13673 [DOI] [PubMed] [Google Scholar]
- 24. Lehner J., Zhang Y., Berendt S., Rasse T. M., Forchhammer K., and Maldener I. (2011) The morphogene AmiC2 is pivotal for multicellular development in the cyanobacterium Nostoc punctiforme. Mol. Microbiol. 79, 1655–1669 [DOI] [PubMed] [Google Scholar]
- 25. Peters N. T., Morlot C., Yang D. C., Uehara T., Vernet T., and Bernhardt T. G. (2013) Structure-function analysis of the LytM domain of EnvC, an activator of cell wall remodelling at the Escherichia coli division site. Mol. Microbiol. 89, 690–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uehara T., Parzych K. R., Dinh T., and Bernhardt T. G. (2010) Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis. EMBO J. 29, 1412–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang D. C., Tan K., Joachimiak A., and Bernhardt T. G. (2012) A conformational switch controls cell wall-remodelling enzymes required for bacterial cell division. Mol. Microbiol. 85, 768–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Firczuk M., Mucha A., and Bochtler M. (2005) Crystal structures of active LytM. J. Mol. Biol. 354, 578–590 [DOI] [PubMed] [Google Scholar]
- 29. Möll A., Dörr T., Alvarez L., Chao M. C., Davis B. M., Cava F., and Waldor M. K. (2014) Cell separation in Vibrio cholerae is mediated by a single amidase whose action is modulated by two nonredundant activators. J. Bacteriol. 196, 3937–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stohl E. A., Lenz J. D., Dillard J. P., and Seifert H. S. (2015) The gonococcal NlpD protein facilitates cell separation by activating peptidoglycan cleavage by AmiC. J. Bacteriol. 198, 615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garcia D. L., and Dillard J. P. (2006) AmiC functions as an N-acetylmuramyl-l-alanine amidase necessary for cell separation and can promote autolysis in Neisseria gonorrhoeae. J. Bacteriol. 188, 7211–7221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kellogg D. S. Jr., Peacock W. L. Jr., Deacon W. E., Brown L., and Pirkle D. I. (1963) Neisseria gonorrhoeae. I. Virulence genetically linked to clonal variation. J. Bacteriol. 85, 1274–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morse S. A., and Bartenstein L. (1974) Factors affecting autolysis of Neisseria gonorrhoeae. Proc. Soc. Exp. Biol. Med. 145, 1418–1421 [DOI] [PubMed] [Google Scholar]
- 34. Dillard J. P. (2011) Genetic manipulation of Neisseria gonorrhoeae. Curr. Protoc. Microbiol. 23, A:4A.2:4A.1.1–4A.1.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction, and analysis. Nat. Protoc. 10, 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rocco C. J., Dennison K. L., Klenchin V. A., Rayment I., and Escalante-Semerena J. C. (2008) Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid 59, 231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramsey M. E., Hackett K. T., Kotha C., and Dillard J. P. (2012) New complementation constructs for inducible and constitutive gene expression in Neisseria gonorrhoeae and Neisseria meningitidis. Appl. Environ. Microbiol. 78, 3068–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hamilton H. L., Schwartz K. J., and Dillard J. P. (2001) Insertion-duplication mutagenesis of Neisseria: use in characterization of DNA transfer genes in the gonococcal genetic island. J. Bacteriol. 183, 4718–4726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosenthal R. S., and Dziarski R. (1994) Isolation of peptidoglycan and soluble peptidoglycan fragments. Methods Enzymol. 235, 253–285 [DOI] [PubMed] [Google Scholar]
- 40. Cloud K. A., and Dillard J. P. (2002) A lytic transglycosylase of Neisseria gonorrhoeae is involved in peptidoglycan-derived cytotoxin production. Infect. Immun. 70, 2752–2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dillard J. P., and Hackett K. T. (2005) Mutations affecting peptidoglycan acetylation in Neisseria gonorrhoeae and Neisseria meningitidis. Infect. Immun. 73, 5697–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mehr I. J., Long C. D., Serkin C. D., and Seifert H. S. (2000) A homologue of the recombination-dependent growth gene, rdgC, is involved in gonococcal pilin antigenic variation. Genetics 154, 523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stohl E. A., Chan Y. A., Hackett K. T., Kohler P. L., Dillard J. P., and Seifert H. S. (2012) Neisseria gonorrhoeae virulence factor NG1686 is a bifunctional M23B family metallopeptidase that influences resistance to hydrogen peroxide and colony morphology. J. Biol. Chem. 287, 11222–11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee M., Hesek D., Shah I. M., Oliver A. G., Dworkin J., and Mobashery S. (2010) Synthetic peptidoglycan motifs for germination of bacterial spores. Chembiochem 11, 2525–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Woodhams K. L., Chan J. M., Lenz J. D., Hackett K. T., and Dillard J. P. (2013) Peptidoglycan fragment release from Neisseria meningitidis. Infect. Immun. 81, 3490–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cloud K. A., and Dillard J. P. (2004) Mutation of a single lytic transglycosylase causes aberrant septation and inhibits cell separation of Neisseria gonorrhoeae. J. Bacteriol. 186, 7811–7814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosenthal R. S., Wright R. M., and Sinha R. K. (1980) Extent of peptide cross-linking in the peptidoglycan of Neisseria gonorrhoeae. Infect. Immun. 28, 867–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jensen S. E., and Campbell J. N. (1976) Amidase activity involved in peptidoglycan biosynthesis in membranes of Micrococcus luteus (sodonensis). J. Bacteriol. 127, 319–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shida T., Hattori H., Ise F., and Sekiguchi J. (2001) Mutational analysis of catalytic sites of the cell wall lytic N-acetylmuramoyl-l-alanine amidases CwlC and CwlV. J. Biol. Chem. 276, 28140–28146 [DOI] [PubMed] [Google Scholar]
- 50. Girardin S. E., Travassos L. H., Hervé M., Blanot D., Boneca I. G., Philpott D. J., Sansonetti P. J., and Mengin-Lecreulx D. (2003) Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J. Biol. Chem. 278, 41702–41708 [DOI] [PubMed] [Google Scholar]
- 51. Lee J., Tattoli I., Wojtal K. A., Vavricka S. R., Philpott D. J., and Girardin S. E. (2009) pH-dependent internalization of muramyl peptides from early endosomes enables Nod1 and Nod2 signaling. J. Biol. Chem. 284, 23818–23829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Magalhaes J. G., Philpott D. J., Nahori M. A., Jéhanno M., Fritz J., Le Bourhis L., Viala J., Hugot J. P., Giovannini M., Bertin J., Lepoivre M., Mengin-Lecreulx D., Sansonetti P. J., and Girardin S. E. (2005) Murine Nod1 but not its human orthologue mediates innate immune detection of tracheal cytotoxin. EMBO Rep. 6, 1201–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cloud-Hansen K. A., Hackett K. T., Garcia D. L., and Dillard J. P. (2008) Neisseria gonorrhoeae uses two lytic transglycosylases to produce cytotoxic peptidoglycan monomers. J. Bacteriol. 190, 5989–5994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mavrogiorgos N., Mekasha S., Yang Y., Kelliher M. A., and Ingalls R. R. (2014) Activation of NOD receptors by Neisseria gonorrhoeae modulates the innate immune response. Innate Immun. 20, 377–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barbour A. G. (1981) Properties of penicillin-binding proteins in Neisseria gonorrhoeae. Antimicrob. Agents Chemother. 19, 316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stefanova M. E., Tomberg J., Olesky M., Höltje J. V., Gutheil W. G., and Nicholas R. A. (2003) Neisseria gonorrhoeae penicillin-binding protein 3 exhibits exceptionally high carboxypeptidase and β-lactam binding activities. Biochemistry 42, 14614–14625 [DOI] [PubMed] [Google Scholar]
- 57. Priyadarshini R., Popham D. L., and Young K. D. (2006) Daughter cell separation by penicillin-binding proteins and peptidoglycan amidases in Escherichia coli. J. Bacteriol. 188, 5345–5355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sauvage E., Kerff F., Terrak M., Ayala J. A., and Charlier P. (2008) The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32, 234–258 [DOI] [PubMed] [Google Scholar]
- 59. Veyrier F. J., Biais N., Morales P., Belkacem N., Guilhen C., Ranjeva S., Sismeiro O., Péhau-Arnaudet G., Rocha E. P., Werts C., Taha M. K., and Boneca I. G. (2015) Common cell shape evolution of two nasopharyngeal pathogens. PLoS Genet. 11, e1005338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Garcia D. L. (2007) Liberation of Pro-inflammatory Peptidoglycan Fragments from the Gonococcal Cell. Ph.D. thesis, University of Wisconsin, Madison [Google Scholar]
- 61. Hebeler B. H., and Young F. E. (1976) Mechanism of autolysis of Neisseria gonorrhoeae. J. Bacteriol. 126, 1186–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kumar A., Kumar S., Kumar D., Mishra A., Dewangan R. P., Shrivastava P., Ramachandran S., and Taneja B. (2013) The structure of Rv3717 reveals a novel amidase from Mycobacterium tuberculosis. Acta Crystallogr. D Biol. Crystallogr. 69, 2543–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Prigozhin D. M., Mavrici D., Huizar J. P., Vansell H. J., and Alber T. (2013) Structural and biochemical analyses of Mycobacterium tuberculosis N-acetylmuramyl-l-alanine amidase Rv3717 point to a role in peptidoglycan fragment recycling. J. Biol. Chem. 288, 31549–31555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Carrasco-López C., Rojas-Altuve A., Zhang W., Hesek D., Lee M., Barbe S., André I., Ferrer P., Silva-Martin N., Castro G. R., Martínez-Ripoll M., Mobashery S., and Hermoso J. A. (2011) Crystal structures of bacterial peptidoglycan amidase AmpD and an unprecedented activation mechanism. J. Biol. Chem. 286, 31714–31722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Barbee L. A. (2014) Preparing for an era of untreatable gonorrhea. Curr. Opin. Infect. Dis. 27, 282–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. den Blaauwen T., Andreu J. M., and Monasterio O. (2014) Bacterial cell division proteins as antibiotic targets. Bioorg. Chem. 55, 27–38 [DOI] [PubMed] [Google Scholar]