

Abstract
The molecular mechanism underlying PKA-mediated regulation of Na+,K+-ATPase was explored in mutagenesis studies of the potential PKA site at Ser-938 and surrounding charged residues. The phosphomimetic mutations S938D/E interfered with Na+ binding from the intracellular side of the membrane, whereas Na+ binding from the extracellular side was unaffected. The reduction of Na+ affinity is within the range expected for physiological regulation of the intracellular Na+ concentration, thus supporting the hypothesis that PKA-mediated phosphorylation of Ser-938 regulates Na+,K+-ATPase activity in vivo. Ser-938 is located in the intracellular loop between transmembrane segments M8 and M9. An extended bonding network connects this loop with M10, the C terminus, and the Na+ binding region. Charged residues Asp-997, Glu-998, Arg-1000, and Lys-1001 in M10, participating in this bonding network, are crucial to Na+ interaction. Replacement of Arg-1005, also located in the vicinity of Ser-938, with alanine, lysine, methionine, or serine resulted in wild type-like Na+ and K+ affinities and catalytic turnover rate. However, when combined with the phosphomimetic mutation S938E only lysine substitution of Arg-1005 was compatible with Na+,K+-ATPase function, and the Na+ affinity of this double mutant was reduced even more than in single mutant S938E. This result indicates that the positive side chain of Arg-1005 or the lysine substituent plays a mechanistic role as interaction partner of phosphorylated Ser-938, transducing the phosphorylation signal into a reduced affinity of Na+ site III. Electrostatic interaction of Glu-998 is of minor importance for the reduction of Na+ affinity by phosphomimetic S938E as revealed by combining S938E with E998A.
Keywords: membrane transport, Na+/K+-ATPase, protein kinase A (PKA), site-directed mutagenesis, sodium transport, P-type ATPase, PKA site, post-translational regulation, sodium binding
Introduction
Na+,K+-ATPase is a plasma membrane-associated ion pump protein that actively extrudes three Na+ ions from the cell while importing two K+ ions for each ATP being hydrolyzed (1–4). It is found in all animal cells where its main function is to establish and maintain transmembrane Na+ and K+ gradients that are fundamental to a variety of physiological processes ranging from electrical excitability of neurons in the brain to Na+ reabsorption in the kidney. The Na+,K+-ATPase belongs to the superfamily of P-type ATPases and is a hetero-oligomeric protein consisting of a catalytic α-subunit, a β-subunit required for translocation of the protein complex to the plasma membrane, and a small regulatory protein belonging to the FXYD family (for reviews, see Refs. 4 and 5). The α-subunit is made up of a membrane part containing transmembrane helices 1–10 (M1–M10)2 and a cytoplasmic “head” formed from three subdomains: actuator (A), nucleotide binding (N), and phosphorylation (P) domains (Fig. 1B). During the Na+,K+-pump cycle (reaction scheme in Fig. 1A), ATP binds to the N- and P-domains, and the γ-phosphate is transferred to a conserved aspartic acid residue of the P-domain. This phosphorylation is triggered by Na+ binding from the cytoplasmic side of the membrane, whereas dephosphorylation with release of inorganic phosphate is triggered by K+ binding from the extracellular side. The Na+ and K+ binding residues are found in the transmembrane helices M4, M5, M6, and M8 (6–8). Na+,K+-ATPase alternates between so-called E1 and E2 states (1, 2) (see reaction scheme in Fig. 1A). E1 states bind Na+ at three sites denoted I, II, and III, whereas E2 states prefer K+, binding at two sites, I and II, that overlap considerably with Na+ sites I and II. The Na+ selectivity of site III is thought to arise in part from interactions of the two C-terminal tyrosines, stabilizing the position of the M5 helix, such that Na+, but not the larger K+ ion, can fit into site III, which is the first of the three Na+ sites to be occupied when Na+ binds from the intracellular side (6, 8, 9).
FIGURE 1.

Reaction cycle and structure of Na+,K+-ATPase. A, simplified scheme of the Na+,K+-ATPase reaction cycle (reactions 1–6). E1 and E2 denote the two major conformational states of the enzyme. P represents phosphorylation of the conserved aspartate in the catalytic site of the P-domain. Occluded ions are shown in parentheses, and boxed ATP indicates low affinity binding of ATP. B, overall structure of the Na+-bound E1 form of Na+,K+-ATPase with the L8-9 loop containing the potential PKA site highlighted in yellow. Details of this region (framed by a broken line) are shown in C. The cytoplasmic A-, P-, and N-domains of the α-subunit as well as the β- and γ-subunits are indicated. C, close-up showing the relations of the potential PKA site at Ser-938 in L8-9 (yellow), the C terminus (light blue), and the ion-binding transmembrane segments M4, M5, M6, and M8. The view is from the cytoplasmic side of the membrane. The residues studied by mutagenesis are depicted as sticks (Ser-938, Asp-997, Glu-998, Arg-1000, Lys-1001, and Arg-1005). In addition, Thr-1016 and the C-terminal tyrosine (Tyr-1018) are shown as sticks as are the interaction partners of the C terminus, Arg-935 in L8-9 and Lys-768 in M5. The three bound Na+ ions are depicted as purple spheres labeled I, II, and III according to site nomenclature. B and C were prepared from the Protein Data Bank structure with code 3WGV (8) using PyMOL.
In addition to the autophosphorylation of the P-domain conserved aspartate, which is central to the reaction cycle of Na+,K+-ATPase, the Na+,K+-ATPase activity depends on regulatory phosphorylation by protein kinases (for a review, see Ref. 10). In cardiac myocytes, “kinase-mediated” phosphorylation of the FXYD1 protein stimulates Na+,K+-ATPase (11). Furthermore, phosphorylation of the Na+,K+-ATPase α-subunit mediated by protein kinases A (PKA) and C (PKC) may occur. Several lines of evidence point to Ser-938 (rat α1 numbering), a conserved PKA consensus site, as a possible target for PKA-mediated phosphorylation reducing the Na+,K+-ATPase activity (12–15). The serine present at the homologous sequence position in the H+,K+-ATPase also seems to be a target for regulatory phosphorylation (16). Ser-938 is located within the cytoplasmic loop between transmembrane segments M8 and M9 (L8-9) at the interphase between the membrane and the cytoplasm (Fig. 1C) in the vicinity of the C terminus. Molecular dynamics computational studies based on the K+-bound E2 structure have suggested that the introduction of the bulky and negatively charged phosphate group at Ser-938 will affect the structure of the C terminus and promote hydration of a cavity in the transmembrane region that connects the cytoplasm and the ion-binding sites (17), which might lead to defective interaction with Na+. This idea was examined by electrophysiological measurements of the apparent affinity for Na+ in a mutant with glutamate replacement of Ser-938, which due to the negative charge and considerable size of the glutamate commonly is considered to mimic phosphorylation. However, no significant effect of the phosphomimetic glutamate substitution was revealed in the electrophysiological study (17). Therefore the mechanism underlying the putative effect of Ser-938 phosphorylation on Na+,K+-ATPase function has remained elusive. A limitation of the electrophysiological study is that only the affinity for Na+ binding from the extracellular side was examined, thus leaving the effect of the phosphomimetic mutation on the affinity for activating Na+ binding from the intracellular side uncharacterized. In fact, it is the interaction with intracellular Na+ and extracellular K+, and not extracellular Na+, that fine-tunes the Na+,K+-ATPase activity in vivo.
The present study was undertaken to provide a deeper understanding of the function and mechanism of the potential Ser-938 PKA site on the basis of the recently solved structure of Na+,K+-ATPase in the E1 form with bound Na+ (8), which is the most relevant structure in relation to possible effects on Na+ affinity. Fig. 1C shows the location in this structure of Ser-938 and surrounding charged residues that might be affected by the introduction of the negatively charged phosphate group. Closest to the side chain hydroxyl group of Ser-938 are the Glu-998 carboxylate group and a guanidino group nitrogen of Arg-1005 of transmembrane segment M10. A bit further away are Asp-997, Arg-1000, and Lys-1001. We have studied the functional consequences (including Na+ affinities on both sides of the membrane) of alanine scanning mutagenesis of these residues as well as other mutations to Arg-1005 and Ser-938, including the phosphomimetic S938D and S938E mutations. Furthermore, we have combined S938E with mutations to Glu-998 or Arg-1005 to investigate the importance of electrostatic interaction between the phosphate group on the phosphorylated Ser-938 (Ser(P)-938) and the glutamate and arginine, respectively. Our results demonstrate that the phosphomimetic mutation reduces the affinity for cytoplasmic Na+ significantly as do mutations to some of the charged residues in the vicinity. The positive charge of Arg-1005 is shown to be essential for accommodation of the negative charge of S938E, indicating a critical role of the arginine-phosphate electrostatic interaction in Ser(P)-938. This arginine is fully conserved among all Na+,K+-ATPases and H+,K+-ATPases. Hence, the electrostatic interaction may well be part of a general regulatory mechanism that also pertains to the closely related H+,K+-ATPases.
Experimental Procedures
Mutagenesis and Expression
The QuikChange site-directed mutagenesis procedure was used to introduce the desired base substitutions into the cDNA encoding the ouabain-resistant rat α1 isoform of Na+,K+-ATPase. The resultant cDNA constructs were transfected into COS-1 cells by the calcium phosphate precipitation method (18). Because the endogenous Na+,K+-ATPase of the COS-1 cells is ouabain-sensitive, stable cell lines expressing the recombinant ouabain-resistant wild type or mutants could be isolated by ouabain selection (19–21). To confirm the stable integration of the cDNA carrying the intended single or double mutations, the genomic DNA was isolated, and the integrated cDNA was amplified by PCR using primers that span exon-exon boundaries as described previously (22). In a few cases where the expressed exogenous mutant Na+,K+-ATPase did not support cell growth in the presence of ouabain, transient co-expression with siRNA to knock down the endogenous COS-1 Na+,K+-ATPase was carried out as described previously (23, 24).
Plasma Membrane Vesicles
Plasma membrane vesicles, harboring the recombinant wild type or mutant Na+,K+-ATPase, were isolated by differential centrifugation (20). Prior to functional analysis, the vesicles were permeabilized by treatment with sodium deoxycholate or alamethicin to make both sides of the membrane accessible to substrates and inhibitors.
ATPase Activity Assays
ATPase activity measurements were carried out at 37 °C for 15 min in 30 mm histidine (pH 7.4), 3 mm MgCl2, 1 mm EGTA, and various concentrations of NaCl, KCl, ATP, and ouabain according to a modification (20–22) of the method originally described by Baginski and co-workers (25). For determination of the maximal catalytic turnover rate, the respective NaCl, KCl, and ATP concentrations were 130, 20, and 3 mm, and the specific activity was related to the active site concentration obtained by stoichiometric phosphorylation from [γ-32P]ATP at 0 °C in the presence of 150 mm NaCl and oligomycin (see below) (26). In studies of K+ dependence of Na+,K+-ATPase activity, 40 mm NaCl, 3 mm ATP, and 0–30 mm KCl were included in the reaction buffer, whereas 130 mm NaCl, 20 mm KCl, and 0.01–3 mm ATP were used in studies of ATP dependence. In measurements of so-called “Na+-ATPase” activity, KCl was omitted from the reaction mixture, which included 3 mm ATP and 0–1000 mm NaCl. All measurements of ATPase activity were carried out in the presence of 10 μm ouabain, which specifically inhibits the endogenous ouabain-sensitive COS-1 Na+,K+-ATPase. Background ATPase measurements were performed in the presence of 10 mm ouabain, which inhibits all Na+,K+-ATPase activity, and subtracted from that measured at 10 μm ouabain.
Phosphorylation and Dephosphorylation Assays
The autophosphorylation of the P-type ATPase signature aspartate in the P-domain and dephosphorylation of wild type and mutants were studied at 0 °C using [γ-32P]ATP. To examine the Na+ dependence of phosphorylation, the enzyme was incubated for 15 s in 20 mm Tris (pH 7.5), 3 mm MgCl2, 1 mm EGTA, 2 μm [γ-32P]ATP, 20 μg/ml oligomycin (to block dephosphorylation), and various concentrations of NaCl as described previously (22, 24, 26). In this assay, the ionic strength was kept constant at 150 mm by addition of various concentrations of N-methyl-d-glucamine.
The distribution of the phosphoenzyme between the ADP-sensitive E1P and ADP-insensitive/K+-sensitive E2P was examined following phosphorylation for 15 s in 20 mm Tris (pH 7.5), 150 mm NaCl, 3 mm MgCl2, 1 mm EGTA, and 2 μm [γ-32P]ATP. The time course of dephosphorylation was followed by adding 2.5 mm ADP and 1 mm unlabeled ATP (9, 22). Under these conditions, phosphoenzyme present as the E1P form will react rapidly with ADP in the backward direction (Fig. 1A, reaction 2), whereas that present as the E2P form will dephosphorylate slowly in the forward direction by hydrolysis (Fig. 1A, reaction 5), thus giving rise to biphasic curves for which the amplitudes of the fast and slow components represent E1P and E2P, respectively.
In all phosphorylation and dephosphorylation assays, 10 μm ouabain was included to inhibit the endogenous COS-1 Na+,K+-ATPase. The background level of phosphorylation was determined by phosphorylation in the presence of 50 mm KCl and absence of NaCl.
Phosphorylation of Na+,K+-ATPase by PKA
Plasma membrane vesicles containing stably expressed wild type or mutant were preincubated for 30 min at 30 °C in the presence of 20 mm Tris-HCl (pH 7.5), 1 mm EGTA, and 10 mm MgCl2 followed by addition of 100 ng of PKA (bovine PKA catalytic subunit from Sigma-Aldrich), 2% (v/v) Triton X-100, and 1 mm dithiothreitol (DTT). The phosphorylation reaction was started by addition of 50 μm [γ-32P]ATP and allowed to proceed for 15 min at 30 °C in a total volume of 15 μl. The reaction was stopped by addition of 5 μl of 4× sample buffer (200 mm Tris-HCl (pH 6.8), 8% SDS, 32% glycerol, 400 mm DTT, and bromphenol blue), and the reaction products were separated by gel electrophoresis using 4–15% polyacrylamide gels (Bio-Rad). Purified pig kidney Na+,K+-ATPase (27) was used as positive control to indicate the migration position of PKA-phosphorylated Na+,K+-ATPase. In this case, the Triton X-100 concentration present during the phosphorylation reaction was 0.2%.
Data Analysis
Data were processed and analyzed using the SigmaPlot program (SPSS, Inc.) for non-linear regression using the complete set of normalized data as described in detail previously (21). Ligand concentration dependences were fitted by use of the appropriate form of the Hill equation. For Na+ dependence of phosphorylation, the following equation was used.
EP is the phosphorylation level, EPmax is the maximum level of phosphorylation, K0.5 is the Na+ concentration giving half-maximum activation (“apparent affinity”), and n is the Hill coefficient (ranging between 1.0 and 2.0 in the present study). For ATP dependence of ATPase activity, the following equation was used.
V is the ATPase activity, Vmax is the extrapolated activity corresponding to infinite ATP concentration, K0.5 is the ATP concentration giving half-maximum activation (apparent affinity), and n is the Hill coefficient (ranging between 0.9 and 1.2 in the present study). For K+ dependence of ATPase activity, a two-component Hill equation was used.
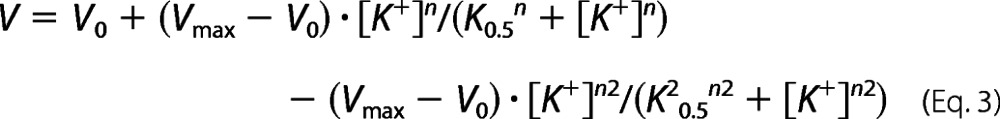 |
where the second term representing inhibition was omitted for wild type and mutants not showing inhibition. V is the ATPase activity, V0 is the activity in the absence of K+ (“Na+-ATPase activity”), Vmax is the extrapolated activity corresponding to infinite K+ concentration in the absence of inhibition, K0.5 is the K+ concentration giving half-maximum activation (apparent affinity corresponding to the rising phase), K20.5 is the K+ concentration giving half-maximum inhibition (apparent affinity for inhibition), n and n2 are the corresponding Hill coefficients. Time courses of dephosphorylation were fitted using a double exponential decay function.
EP is the total amount of phosphoenzyme, E1P and E2P are the ADP-sensitive and -insensitive fractions, and k1 and k2 are the respective decay constants. The results of the non-linear regression analysis (K0.5 values and E2P fraction) ±S.E. from the regression are reported in Table 1.
TABLE 1.
Functional analysis of mutants
-Fold changes relative to WT are shown in red parentheses. The number of independent experiments n/total number of data points and mean ± S.E. are indicated. ++, pronounced inhibition: +, partial inhibition; and −, no inhibition of ATPase activity by high K+ or Na+ concentrations.

Results
Ser-938 Is Important for Binding of Intracellular Na+ but Not Extracellular Na+
Ser-938 of the potential PKA site was replaced by either aspartate or glutamate to mimic phosphorylation by PKA. To study the importance of side chain size and charge, the serine was furthermore replaced by alanine and arginine. All four mutants S938A/D/E/R could be stably expressed in COS-1 cells at levels similar to that of the wild type enzyme under ouabain selection pressure, showing that the α- and β-subunits are correctly assembled and transported to the plasma membrane where they are capable of transporting Na+ and K+ across the membrane at rates compatible with cell viability. The maximal catalytic turnover rate, calculated as the ratio between the specific Na+,K+-ATPase activity measured under optimal conditions and the active site concentration obtained by phosphorylation (26), was wild type-like for mutant S938A and slightly increased (1.2- and 1.3-fold) for S938D, S938E, and S938R (Table 1).
To obtain information about the Na+ binding properties of the sites on the E1 form that in the intact cell face the intracellular side (Fig. 1A, reaction 1), advantage was taken of the dependence of activation of phosphorylation from ATP (Fig. 1A, reaction 2) on the binding of all three Na+ ions at these sites. The measurements of the Na+ dependence of phosphorylation were carried out in the presence of oligomycin to prevent dephosphorylation and in the absence of K+ to exclude competition from K+. Whereas mutant S938A displayed a slight (1.7-fold) decrease of the apparent affinity for Na+ relative to wild type, the effects of the substitutions S938D, S938E, and S938R were more pronounced, resulting in 2.3-, 3.1-, and 3.8-fold reductions, respectively, of the apparent affinity for Na+ (Fig. 2A and Table 1). In principle, such a reduction of apparent Na+ affinity can be due to either a reduction of the intrinsic affinity of the E1 form for intracellular Na+ or a shift of the E1-E2 conformational equilibrium away from the Na+-binding E1 form (Fig. 1A, reaction 6). To distinguish between these two possibilities, we determined the ATP concentration dependence of Na+,K+-ATPase activity, which can be used as a measure of the distribution of the enzyme between the E1 and E2 conformations because E1 binds ATP with high affinity, whereas E2 only binds ATP with low affinity and without being phosphorylated (1, 28). As seen in Fig. 2B, the mutations S938D, S938E, and S938R caused a left shift of the ATP titration curves, which is indicative of an increased apparent ATP affinity relative to wild type and hence a shift of the E1-E2 equilibrium in favor of E1 (Fig. 2B and Table 1). Mutant S938A, in contrast, displayed a wild type-like apparent affinity for ATP (Fig. 2B and Table 1). These latter findings allow us to conclude that the reduced apparent Na+ affinities observed for mutants S938D, S938E, and S938R are caused by mutation-induced disturbance of the binding of Na+ to the E1 form rather than being caused indirectly by a shift of the conformational equilibrium in favor of E2.
FIGURE 2.

The phosphomimetic S938D/E mutations interfere with the binding of Na+ at the intracellularly facing E1 sites. A, Na+ dependence of phosphorylation from [γ-32P]ATP. Each line shows the best fit of the Hill equation, and the extracted K0.5 values are listed in Table 1. B, ATP dependence of Na+,K+-ATPase activity. Each line shows the best fit of the Hill equation, and the extracted K0.5 values are listed in Table 1. C, Na+ dependence of Na+-ATPase activity. Inhibition of Na+-ATPase activity by high Na+ concentrations is indicated semiquantitatively in Table 1. D, K+ dependence of Na+,K+-ATPase activity. Each line shows the best fit of the Hill equation, and the extracted K0.5 values are listed in Table 1. E, distribution of the phosphoenzyme between E1P and E2P at 150 mm NaCl. Each line shows the best fit of a biexponential decay function. The initial amounts of E2P, which correspond to the amplitude of the slow phase, are listed in Table 1. A–E, experimental conditions and equations used for data fitting are described under “Experimental Procedures.” Statistical information is given in Table 1. Symbol and error bars (seen only when larger than the size of the symbols) represent mean ± S.E. Dotted lines reproduce the wild type for direct comparison in the same panel.
Experiments with sided membrane vesicles have shown that besides binding to the intracellularly facing Na+ sites, activating phosphorylation from ATP, Na+ also binds to extracellularly facing activating and inhibitory sites (29–31). Having observed that the phosphomimetic mutations S938E and S938D as well as mutation S938R disturb the interaction with Na+ at the intracellularly facing sites of the E1 form, we next investigated whether these mutations also interfere with the binding of Na+ to the E2P form of the enzyme at the sites that in the intact cell face the extracellular side. To this end, we determined the Na+ concentration dependence of the ATPase activity in the absence of K+. When K+ is omitted from the reaction medium, Na+ is able to bind to E2P, activating dephosphorylation, although much less efficiently than K+ (Fig. 1A, reactions 4 and 5) (1). Consequently, the catalytic turnover rate measured in the mere presence of Na+ (Na+-ATPase activity) is only a small fraction of that measured with K+ present (less than 5% for the wild type). For the wild type enzyme, Na+ concentrations in the 50–400 mm range activate dephosphorylation, whereas high Na+ concentrations are inhibitory (Fig. 2C) due to the binding of Na+ to the E2P form driving the E1P → E2P transition backward (Fig. 1A, reaction 3) (30–32). Similar results with both an activating and an inhibitory phase were obtained for the S938A/D/E/R mutants (Fig. 2C and Table 1). For the S938A/D/E mutants, the apparent affinity for Na+ inhibition was similar to that of the wild type or slightly higher. Thus, the extracellularly facing Na+ sites on E2P appear to be intact in all S938 mutants, indicating that this serine is not essential to binding of extracellular Na+ and that the phosphomimetic mutations S938D and S938E only interfere with Na+ binding from the intracellular side of the membrane and not with Na+ binding from the extracellular side. Mutant S938R displayed a slight right shift of the inhibitory part of the curve relative to wild type, indicating a minor reduction of the affinity for binding of extracellular Na+.
Ser-938 Is Less Important for Binding of K+ than for Binding of Na+
By binding at sites on E2P that in the intact cell face the extracellular side, K+ triggers dephosphorylation, thereby stimulating the ATPase activity (Fig. 1A, reactions 4 and 5). Compared with the significant effects on the binding of Na+ from the intracellular side, only relatively small changes in the apparent K+ affinities for activation were detected (Fig. 2D and Table 1): the S938D, S938E, and S938R mutants exhibited 1.6–1.8-fold reductions of the K+ affinity relative to wild type, whereas S938A displayed wild type-like K+ affinity. Thus, the substitutions S938D, S938E, and S938R seem primarily to affect the interaction with Na+ binding from the intracellular side.
There are in principle two possible explanations of the above described slight reductions of the apparent K+ affinity observed for the Ser-938 mutants: 1) reduced intrinsic affinity of E2P for K+ or 2) a conformational shift away from the K+-binding E2 or E2P form in favor of E1 and E1P, respectively. As described above, ATP titrations of Na+,K+-ATPase activity (Fig. 2B) disclosed a shift of the E2-E1 equilibrium in favor of E1. This shift can account for the slightly reduced K+ affinities of mutants S938D, S938E, and S938R; hence, there is no need to assume any reduction of the intrinsic K+ affinity of E2P. We furthermore investigated the distribution of the phosphoenzyme between the intermediates E1P and E2P by taking advantage of the ability of E1P to dephosphorylate upon addition of ADP (1, 9, 33). None of the Ser-938 mutants differed significantly from the wild type with respect to the fraction of the phosphoenzyme that was ADP-insensitive (compare 52–59 with 62% for the wild type; Fig. 2E and Table 1), meaning that the slightly reduced K+ affinities of S938D, S938E, and S938R cannot be attributed to a displacement of the E1P-E2P equilibrium toward E1P.
Charged Residues Asp-997, Glu-998, Arg-1000, and Lys-1001 in the Vicinity of Ser-938 Are Crucial for Na+ Binding
As shown in Fig. 1C, transmembrane segment M10 contains two negatively charged residues, Asp-997 and Glu-998, and three positively charged residues, Arg-1000, Lys-1001, and Arg-1005, relatively close to Ser-938. These residues might be electrostatically repulsed and attracted, respectively, when a negatively charged phosphate group is attached at Ser-938. To investigate the function of Asp-997, Glu-998, Arg-1000, Lys-1001, and Arg-1005, alanine scanning mutagenesis of these residues was performed. In addition, Asp-997 and Glu-998 were simultaneously replaced by alanine. The results obtained with R1005A encouraged us to study also the mutants R1005K/M/S. All the above mentioned single mutations were compatible with cell viability in COS-1 cells in the presence of ouabain, whereas the double mutant D997A/E998A did not sustain cell growth. Functional analysis of the maximal turnover rates revealed wild type-like catalytic rates for D997A, E998A, R1000A, K1001A, and R1005A/K/M/S (0.8–1.3-fold changes relative to wild type) (Table 1).
Investigation of the Na+ dependences of activation of phosphorylation from ATP disclosed marked effects of mutations D997A, E998A, R1000A, and K1001A on the binding of Na+, amounting to 4.4-, 8.8-, 15-, and 7.6-fold reductions of the Na+ affinity, respectively, compared with wild type (Fig. 3A and Table 1). These effects are not caused by a conformational shift away from the Na+-binding E1 form as the apparent affinity for ATP was in fact increased, indicating that the E1-E2 equilibrium is shifted in favor of E1 (Fig. 3B and Table 1). The effect is rather specific for Na+ binding, as D997A, E998A, R1000A, and K1001A all displayed a close to wild type-like apparent affinity for K+ activation of Na+,K+-ATPase activity (1.1–1.5-fold reductions of the K+ affinity relative to wild type; Fig. 3C and Table 1). As regards Arg-1005, none of the investigated mutations replacing Arg-1005 with alanine, lysine, methionine, or serine affected the Na+ or K+ binding properties substantially (Figs. 3 and 4A and Table 1).
FIGURE 3.

Several charged residues in M10 near Ser-938, but not Arg-1005, are crucial to binding of Na+ at the intracellularly facing E1 sites. A, Na+ dependence of phosphorylation from [γ-32P]ATP. Each line shows the best fit of the Hill equation, and the extracted K0.5 values are listed in Table 1. B, ATP dependence of Na+,K+-ATPase activity. Each line shows the best fit of the Hill equation, and the extracted K0.5 values are listed in Table 1. C, K+ dependence of Na+,K+-ATPase activity. Each line shows the best fit of the Hill equation or, for the mutants showing inhibition at high K+ concentrations, a two-component Hill equation with the inhibition represented by a negative term. K0.5 values for the rising parts of the curves are listed in Table 1. A–C, experimental conditions and equations used for data fitting are described under “Experimental Procedures.” Statistical information is given in Table 1. Symbol and error bars (seen only when larger than the size of the symbols) represent mean ± S.E. Dotted lines reproduce the wild type for direct comparison in the same panel.
FIGURE 4.

Several charged residues in M10 near Ser-938, but not Arg-1005, are important for binding of Na+ from the extracellular side of the membrane. A, Na+ dependence of Na+-ATPase activity. Inhibition of Na+-ATPase activity by high Na+ concentrations is indicated semiquantitatively in Table 1. B, distribution of the phosphoenzyme between E1P and E2P at 150 mm NaCl. Each line shows the best fit of a biexponential decay function. The initial amounts of E2P, which correspond to the amplitude of the slow phase, are listed in Table 1. A and B, experimental conditions and equations used for data fitting are described under “Experimental Procedures.” Statistical information is given in Table 1. Symbol and error bars (seen only when larger than the size of the symbols) represent mean ± S.E. Dotted lines reproduce the wild type for direct comparison in the same panel.
Unlike the wild type, the mutants D997A, E998A, and R1000A were inhibited at high K+ concentrations; this was most distinct for D997A and R1000A (Fig. 3C). This inhibition may be explained by the low intrinsic Na+ affinity of these mutants (see Table 1), allowing K+ to bind in competition with Na+ at a site(s) on the E1 form, thus leading to displacement of the E1-E2 equilibrium toward E2 (34). However, the inhibition is not strictly correlated with the reduction of Na+ affinity (e.g. the D997A mutant shows more K+ inhibition than E998A, whereas the order is reversed for Na+ affinity). Therefore, the Na+ selectivity of site III might be affected in these mutants (see Discussion).
Mutations D997A, E998A, and R1000A also had an impact on binding of extracellular Na+ as seen from the Na+-ATPase measurements in Fig. 4A. In contrast with the wild type enzyme, which is completely inhibited at a Na+ concentration of 1 m, these mutants were not inhibited at all (R1000A) or only partly inhibited (D997A and E998A) at high Na+ concentrations (Fig. 4A and Table 1), indicating that the extracellularly facing Na+ sites on E2P are less efficient in binding Na+ and reversing the E1P → E2P transition and thereby impeding ATPase activity (Fig. 1A, reaction 3). Conversely, K1001A and the Arg-1005 mutants displayed wild type-like behavior with respect to binding of extracellular Na+. Based on these findings, it may be concluded that Asp-997, Glu-998, and Arg-1000, the latter in particular, are important for the binding of Na+ from both sides of the membrane, whereas Lys-1001 only is important for Na+ binding from the intracellular side, and Arg-1005 is not, per se, crucial for cation interaction.
These mutations had limited influence on the distribution of the phosphoenzyme intermediates between E1P and E2P; the largest deviation was seen for R1000A (83% E2P versus 62% in wild type). K1001A and the various Arg-1005 mutants showed slightly lower accumulation of E2P than the wild type (33–52%), thus indicating slowing of the E1P to E2P conversion (Fig. 4B and Table 1). The increased steady-state level of E2P seen for R1000A is likely the consequence of the inability of this mutant to react with Na+ at the extracellularly facing sites, which would have driven the E1P-E2P equilibrium in the direction of E1P.
The Phosphomimetic Mutation S938E Mediates Its Effect Through Interaction with Arg-1005
To examine whether the phosphorylated side chain of Ser-938 senses the potential interaction partners Arg-1005 and Glu-998, we made several double mutants where the phosphomimetic mutation S938E was combined with either of the single mutations R1005A/K/M/S and E998A (see Table 1 for an overview). Of the four mutants combining S938E with substitutions of Arg-1005, only S938E/R1005K was capable of sustaining cell viability in the presence of ouabain (Table 1). Hence, mutants S938E/R1005A, S938E/R1005M, and S938E/R1005S are unable to transport Na+ and K+, or the transport rate is below a critical level (5–10% of wild type activity seems to be required to sustain cell viability based on previous experience of our laboratory). Expression of these mutants was then carried out by transient co-transfection with siRNA knocking down the endogenous Na+,K+-ATPase in the COS-1 cells, a technique allowing analysis of the partial reaction steps in the enzyme cycle despite severe reduction of transport activity that we previously used for other transport-deficient mutants (23, 24). Hence, the ability to form a phosphorylated intermediate from ATP at the conserved aspartate in the catalytic site was examined (Fig. 1A, reaction 2). As seen in Fig. 5A, none of the transiently expressed mutants S938E/R1005A, S938E/R1005M, and S938E/R1005S could be phosphorylated in the presence of oligomycin and a Na+ concentration of 100 or 300 mm with the latter being more than 20-fold higher than required for saturation of the wild type enzyme. This finding is remarkable in the context of the more or less wild type-like behavior of the mutants R1005A, R1005M, and R1005S with single substitutions.
FIGURE 5.

The phosphomimetic mutation S938E mediates its effect through interaction with Arg-1005. A, representative phosphorimaging autoradiograph of transiently expressed double mutants. The double mutants S938E/R1005A, S938E/R1005M, and S938E/R1005S, which could not sustain cell viability in the presence of ouabain, were transiently expressed in the presence of siRNA-targeted knockdown of the endogenous COS-1 Na+,K+-ATPase. The autoradiograph shows 32P incorporation from [γ-32P]ATP at the catalytic site aspartate in the P-domain following separation by SDS-PAGE of enzyme phosphorylated in the presence of the Na+ concentrations indicated in mm. It can be seen that all the double mutants tested were phosphorylation-inactive. B, Na+ dependence of phosphorylation from [γ-32P]ATP. Each line shows the best fit of the Hill equation, and the extracted K0.5 values are listed in Table 1. C, ATP dependence of Na+,K+-ATPase activity. Each line shows the best fit of the Hill equation, and the extracted K0.5 values are listed in Table 1. D, K+ dependence of Na+,K+-ATPase activity. Each line shows the best fit of a two-component Hill equation with the inhibition represented by a negative term. K0.5 values for the rising parts of the curves are listed in Table 1. E, Na+ dependence of Na+-ATPase activity in the absence of K+. Inhibition of Na+-ATPase activity by high Na+ concentrations is indicated semiquantitatively in Table 1. B–E, experimental conditions and equations used for data fitting are described under “Experimental Procedures.” Statistical information is given in Table 1. Symbol and error bars (seen only when larger than the size of the symbols) represent mean ± S.E. Dotted lines reproduce the wild type for direct comparison in the same panel.
The viable double mutant S938E/R1005K exhibited a maximal catalytic turnover rate only slightly decreased compared with that of the wild type (7313 versus 8474 min−1; Table 1), whereas a substantial disturbance of Na+ binding at the sites on E1 was found, amounting to as much as a 10-fold reduction of the apparent affinity for Na+ activation of phosphorylation (Fig. 5B and Table 1). This effect is particularly noteworthy because the single mutation S938E reduced the Na+ affinity only 3-fold, and R1005K was without effect, suggesting that the presence of the lysine reinforced the effect of the phosphomimetic S938E mutation. Again the effects on apparent Na+ affinity are not caused by a conformational shift away from the Na+-binding E1 form as the apparent affinity for ATP was increased (Fig. 5C and Table 1). These findings support the hypothesis that electrostatic interaction between the introduced phosphate on Ser-938 and the positive side chain of Arg-1005 is central to the PKA regulatory effect on the binding of intracellular Na+. The K+ dependence of the Na+,K+-ATPase activity of S938E/R1005K showed only a minor reduction of K+ affinity for activation on par with the K+ affinity observed for the S938E single mutation (Fig. 5D and Table 1). Furthermore, the extracellularly facing low affinity sites on E2P were still capable of binding Na+, leading to inhibition of Na+-ATPase activity; however, the apparent affinity for the inhibitory Na+ appears slightly reduced relative to wild type, and the maximal Na+-ATPase activity was clearly reduced in S938E/R1005K (Fig. 5E), indicating a reduced efficacy of Na+ activation of dephosphorylation. Thus, the combination of S938E with R1005K also leads to an altered Na+ interaction at the extracellularly facing sites, although neither of the single mutations had such an effect.
Information about the influence of the electrostatic repulsion between S938E and Glu-998 was obtained by studying the double mutation S938E/E998A, which turned out to further compromise Na+ affinity relative to the single mutations S938E and E998A (14-fold in the double mutant versus 3- and 9-fold in the respective mutants with single substitutions) (Fig. 5B and Table 1). It is, however, important to note that the E998A mutation does not diminish the effect of the S938E substitution on the Na+ affinity. This result indicates that electrostatic interaction between S938E and Glu-998 is of minor importance for the effect of the S938E mutation on Na+ affinity. The S938E/E998A mutant displayed a wild type-like turnover rate and apparent affinity for K+ activation (Fig. 5D). A distinct inhibition phase was observed at high K+ concentrations, which is in accordance with the marked reduction of Na+ affinity because, as explained above, inhibition by K+ results from K+ binding in competition with Na+ (Fig. 5D and Table 1).
In addition, we constructed the swap mutant S938R/R1005S, which also displayed a larger reduction of Na+ affinity (6-fold; Fig. 5B and Table 1) than the corresponding mutants with single substitutions S938R and R1005S (4- and 2-fold, respectively). Hence, the swap of the serine and the arginine did not recover the Na+ binding properties of the Na+,K+-ATPase, indicating that interaction of the unphosphorylated serine with the arginine is not a major determinant of the normal Na+ affinity in the absence of phosphorylation.
Direct Measurement of PKA-mediated Phosphorylation of Mutants
To examine whether Arg-1005 is required for PKA-mediated phosphorylation of Ser-938, PKA phosphorylation experiments were performed with selected mutants (Fig. 6). Conditions were chosen under which almost no phosphorylation was observed in the absence of PKA (Fig. 6, A, lanes 1 and 4, and B, lanes 1, 3, and 5). As a positive control, highly purified renal Na+,K+-ATPase was subjected to PKA-mediated phosphorylation, giving rise to a single phosphorylated band (Fig. 6B, lanes 8 and 9). PKA-mediated phosphorylation of the microsomes from COS-1 cells expressing the wild type Na+,K+-ATPase showed a distinct band migrating at the same position as the band corresponding to the purified Na+,K+-ATPase in addition to a number of other bands reflecting the fact that the expressed Na+,K+-ATPase constitutes only a few percent of the total microsomal protein (Fig. 6A, lane 2). The microsomes from cells expressing the phosphorylation-deficient S938A mutant showed a weaker phosphorylated Na+,K+-ATPase band corresponding to the background of endogenous COS-1 cell Na+,K+-ATPase (Fig. 6, A, lane 3, and B, lane 2). For all the Arg-1005 mutants examined (R1005A, R1005K, and R1005M; Fig. 6B, lanes 4, 6, and 7, respectively), a distinct phosphorylated Na+,K+-ATPase band stronger than the background corresponding to S938A could be seen. Hence, the presence of Arg-1005 is not obligatory for the PKA-mediated phosphorylation even though the arginine appears to be mechanistically involved in the signal transduction.
FIGURE 6.

PKA-mediated phosphorylation of Na+,K+-ATPase mutants. Representative phosphorimaging autoradiographs showing 32P incorporation following incubation with [γ-32P]ATP with PKA or without PKA catalytic subunit and separation by SDS-PAGE of COS-1 cell plasma membrane proteins containing expressed Na+,K+-ATPase mutants or wild type (WT) or purified Na+,K+-ATPase from pig kidney. See “Experimental Procedures” for further details. Two SDS-PAGE gels, A and B, are shown. A, lane 1, WT − PKA; lane 2, WT + PKA; lane 3, S938A + PKA; lane 4, S938A − PKA. B, lane 1, S938A − PKA; lane 2, S938A +PKA; lane 3, R1005A − PKA; lane 4, R1005A + PKA; lane 5, R1005K − PKA; lane 6, R1005K + PKA; lane 7, R1005M + PKA; lane 8, purified Na+,K+-ATPase (15 μg) +PKA; lane 9, purified Na+,K+-ATPase (20 μg) + PKA. The arrow indicates the migration position corresponding to the purified Na+,K+-ATPase. For both gel A and gel B, 35 μg of total plasma membrane protein from COS-1 cells expressing mutant or wild type were loaded in each lane. Plasma membrane preparations containing approximately equal amounts (±5%) of expressed exogenous mutant or wild type rat Na+,K+-ATPase per mg of total protein were used based on determination of the active site concentration.
Discussion
Although it is well established that PKA regulates Na+,K+-ATPase activity in vivo, the underlying molecular mechanism has remained obscure. The current study provides new information about this mechanism. Our data show that the phosphomimetic mutations S938D and S938E reduce the apparent affinity of the E1 form for Na+ 2–3-fold relative to wild type (Fig. 2A and Table 1). In the intact cell, the Na+ sites on E1 face the intracellular side. The sites on E2P that bind extracellular Na+ and K+ were unaffected by these mutations (Fig. 2, C and D, and Table 1). In agreement with our findings, previous electrophysiological investigations of S938E did not disclose any defect in the binding of extracellular Na+, which was attributed to the smaller size and charge of the glutamate substituent compared with a phosphate group having a charge of −2 at physiological pH (17). However, the latter study did not investigate the binding of Na+ from the intracellular side to the E1 form probably due to the difficulty in controlling the intracellular Na+ concentration in whole cells. The presently observed reduction of Na+ affinity of the intracellularly facing E1 sites caused by the phosphomimetic mutations S938D and S938E is within the range expected for physiological regulation of the Na+,K+-ATPase (15). Because the Na+,K+-ATPase works under suboptimal conditions in vivo where the intracellularly facing Na+ sites are not saturated with respect to Na+, the moderate 2–3-fold reduction of Na+ affinity observed here is expected to have a significant effect on the Na+,K+-ATPase turnover rate in intact cells. Thus, altogether, the present results suggest that PKA phosphorylation of Na+,K+-ATPase, mimicked by the glutamate substituent, exerts its role in cells through modification of the intracellular Na+ affinity. According to a recent study from our group, this modification will have consequences for the intracellular concentration of Na+, which we have shown correlates with the apparent Na+ affinity (24). Our findings are in agreement with a previous study showing that the Na+ clearance rate decreases upon modulation of the Na+,K+-ATPase α3 isoform by PKA (35).
In the vicinity of Ser-938 in L8-9 are five charged residues, Asp-997, Glu-998, Arg-1000, Lys-1001, and Arg-1005, of M10 (Fig. 1C). Our results demonstrate that alanine substitution of Asp-997, Glu-998, and Arg-1000 affects the binding of Na+ from both sides of the membrane, whereas alanine substitution of Lys-1001 only affects the binding of Na+ from the intracellular side. In contrast, Arg-1005 is not required for proper Na+ or K+ binding (Figs. 3 and 4 and Table 1). The double sidedness of the effects seen for Asp-997, Glu-998, and Arg-1000 indicates that these residues are involved in stabilizing the Na+-occluded state, which is reached from both sides of the membrane. The most defective Na+ binding is seen for the R1000A mutant, which displays a 15-fold reduced apparent Na+ affinity of the E1 form (Fig. 3A) as well as a complete disruption of Na+ binding from the extracellular side (Fig. 4A). These observations suggest a key role for the cytoplasmic half of M10 in the regulation of Na+ binding. M10 is physically connected to the C terminus whose interaction with M5 through Lys-768 (see Fig. 1C) is known to stabilize the so-called Na+ site III, which is specific for Na+ and is believed to be the first Na+-binding site occupied during the sequential binding of three Na+ ions to Na+,K+-ATPase (8). Because replacement of each of the residues Asp-997, Glu-998, Arg-1000, and Lys-1001 only affects the interaction with Na+ and not the interaction with K+, the observed mutational effects may be ascribed to modification of the Na+-specific site III (Fig. 3). In the Na+,K+-ATPase structure with bound Na+, the side chain of Asp-997 seems to form salt bridges with the side chains of both Arg-1000 and Lys-1001 (Figs. 1C and 7). Arg-1000 is hydrogen-bonded to Thr-1016 near the C terminus (Fig. 1C), and Lys-1001 is bonded to backbone carbonyl oxygens of M8 (Thr-934 and Ile-931; Fig. 7). Furthermore, Glu-998 is hydrogen-bonded to the backbone amide of Val-939 in L8-9, and the C terminus is tied up to Arg-935 in L8-9 (6, 9). Thus, Asp-997, Glu-998, Arg-1000, and Lys-1001 of M10 are part of an elaborate bonding network that involves M10, L8-9, the C terminus, and the M5 and M8 helices providing liganding residues (Tyr-773, Thr-776, and Ser-777 in M5, Gln-925, and Asp-928 in M8) at Na+ site III (8, 34, 36). This extended network likely allows changes in the configuration of the M10 residues to be transmitted to Na+ site III.
FIGURE 7.

Structural relations of L8-9 containing the potential PKA site Ser-938. Shown is the relevant part of the structure of the Na+-bound E1 form of Na+,K+-ATPase (Protein Data Bank code 3WGV) (8) viewed from the side along the membrane surface. Depicted is the extensive bonding network that connects L8-9 (yellow) harboring the potential PKA site at Ser-938 with M10 (gray), the C terminus (light blue), and the Na+ binding segment M8 (gray). The residues studied by mutagenesis are depicted as sticks (Ser-938, Asp-997, Glu-998, Arg-1000, Lys-1001, and Arg-1005). Stick representation of backbone atoms (blue nitrogen and red oxygen atoms) is shown for the entire L8-9. In addition, Gln-925 and Asp-928 known to contribute to the binding of the Na+ ion at site III are shown as sticks as are Ile-931 and Thr-934 that interact with Lys-1001 in M10. The Na+ ion bound at site III is depicted as a large purple sphere. The figure was prepared using PyMOL.
The inhibition at high K+ concentrations, which is most distinct for the alanine substitutions of Arg-1000 and its interaction partner Asp-997 (Fig. 3C), may also arise from disturbance of the above mentioned bonding network. Several mutations affecting the C terminus have been found to exhibit this pattern of K+ inhibition (6, 9, 24, 34, 36), and the marked K+ inhibition of D997A and R1000A may be seen as a consequence of interference by the mutations with the bond between Arg-1000 and Thr-1016 next to the C-terminal tyrosines (Fig. 1C). Hence, analysis of the crystal structure has indicated that K+ binding at Na+ site III and resulting inhibition, i.e. loss of the Na+ selectivity of site III, will occur if the cytoplasmic half of M5 is inclined toward M10 by 10° as a consequence of disturbance of the stabilizing interaction of the C terminus with M5 (8).
In this context, it is worth mentioning that a mutation replacing the arginine corresponding to Arg-1000 with glutamine in the Na+,K+-ATPase α2 isoform has been found in patients with familial hemiplegic migraine (37). Asp-997 has also been implicated in neurological disease as substitutions of this residue have been identified both in α2 in association with familial hemiplegic migraine and in the α3 isoform of patients with alternating hemiplegia of childhood (38, 39).
The side chain hydroxyl of Ser-938 seems to form hydrogen bonds to the two backbone amides of Phe-940 and Gln-941 that may serve to stabilize a rigid loop structure of L8-9 (Figs. 1C and 7). The loss of these hydrogen bonds may be the reason for the 2-fold reduction of Na+ affinity caused by the S938A mutation. The introduction of the phosphomimetic S938E mutation led to a more pronounced, 3-fold reduction of Na+ affinity. In the Na+-bound structure, the side chains of Glu-998 and Arg-1005 are 4.12 and 4.91 Å away, respectively, from the Ser-938 hydroxyl group. The glutamate substituent or in vivo phosphorylation of the serine might disrupt the interactions with the L8-9 backbone, which would provide opportunities for electrostatic attraction to the arginine side chain or repulsion by the glutamate Glu-998. To test these ideas, we generated double mutants of S938E combined with substitutions of either Arg-1005 or Glu-998 to probe the interactions. None of the three double mutants S938E/R1005A, S938E/R1005M, and S938E/R1005S could support cell growth (Table 1) despite the fact that the mutants R1005A, R1005M, and R1005S with single substitutions behaved more or less wild type-like (Figs. 3 and 4 and Table 1). Even when expressed transiently, S938E/R1005A, S938E/R1005M, and S938E/R1005S failed to phosphorylate in a Na+-dependent reaction at the conserved P-domain aspartate, explaining their inability to support cell growth (Fig. 5A). In contrast, mutant S938E/R1005K was able to transport Na+ and K+ across the membrane at rates sufficiently high to support cell growth, and the maximal turnover rate of this mutant was almost wild type-like (Table 1), thus underscoring the importance of the positive charge of the side chain at position 1005, which apparently interacts with the phosphomimetic negatively charged glutamate. Arginine side chains are well known and common interaction partners of phosphate groups as analysis of phosphate interactions in several proteins has shown that phosphate groups preferentially interact with one or more arginine residues (40–42). Due to the planar structure of the guanidino group of the side chain, which has the ability to form several hydrogen bonds, the phosphate-arginine interaction is extraordinarily strong. Besides arginines, phosphate groups can also interact with lysines although generally less strongly. It is therefore of note that even though R1005K exhibited wild type-like Na+ affinity the Na+ affinity was even more reduced in S938E/R1005K (10-fold) than in single mutant S938E (3-fold) where arginine is present at position 1005 (Figs. 2, 3, and 5 and Table 1). Hence, replacement of Arg-1005 with lysine reinforced the effect of the phosphomimetic S938E mutation on Na+ affinity. The lysine side chain is more flexible than that of arginine, which may increase the probability of “catching” its electrostatic interaction partner. Furthermore, due to the shorter length of the bridging lysine side chain compared with that of arginine, the interaction of the phosphomimetic glutamate of S938E with the lysine at position 1005, near the edge of M10, may cause larger structural perturbation of the elaborate bonding network that connects L8-9 to M10, the C terminus, and the Na+ binding region than the interaction with the longer arginine side chain.
The importance of Arg-1005 in the signal transduction resulting from the PKA-mediated phosphorylation of Ser-938 raises the question whether Arg-1005 is obligatory as interaction partner with the phosphate group for the PKA-mediated phosphorylation to occur? Our results showed that the PKA-mediated phosphorylation is possible in the Arg-1005 mutants (Fig. 6), which is reasonable because the latter residue is located in transmembrane helix M10 outside the PKA consensus sequence in the loop between transmembrane helices M8 and M9. In the wild type Na+,K+-ATPase, the phosphorylation of Ser-938 conceivably occurs without requirement for the arginine in the binding of the phosphate, but upon phosphorylation of the serine the phosphate group attracts the arginine, thereby propagating structural perturbation via the surrounding binding network to Na+ site III.
Although the structural closeness of Glu-998 to Ser-938 might suggest a role for electrostatic repulsion between a negative charge at position 938 and Glu-998 in the mechanism generating reduced Na+ affinity, such an effect was not confirmed experimentally. Hence, the substitution of Glu-998 with alanine did not attenuate the effect of S938E on Na+ affinity (Figs. 2, 3, and 5 and Table 1). The marked effect of the E998A single substitution seems to result from disruption of a hydrogen bond between one of the side chain oxygen atoms of Glu-998 and the backbone amide of Val-939 (Fig. 7), thus again pointing to the importance of bonds that stabilize a rigid L8-9 loop structure.
The swap mutant S938R/R1005S was constructed to test whether S938R and R1005S interact in the absence of phosphorylation of the serine. Because the swap mutant displayed a larger reduction of Na+ affinity than each of the single mutants S938R and R1005S, interaction of the unphosphorylated serine with the arginine is most likely not a major determinant of the normal Na+ affinity in the absence of phosphorylation.
In conclusion, the biochemical data presented in this study, particularly the effects of combining the phosphomimetic S938E mutation with Arg-1005 mutations, support a scenario where a phosphate group introduced at Ser-938 by PKA-mediated phosphorylation would reduce the affinity of Na+ site III in the internally facing configuration by way of a mechanism involving electrostatic interaction with the Arg-1005 side chain and destabilization of the rigid loop structure of L8-9, thereby conveying changes to M8 with its Na+-liganding residues. The role of the positive charge at position 1005 is emphasized by the strengthening of the effect of the phosphomimetic mutation on Na+ affinity seen upon substitution of Arg-1005 with lysine. The reduced Na+ affinity at the internally facing sites resulting from phosphorylation of Ser-938 is expected to increase the intracellular Na+ concentration.
Author Contributions
A. P. E., H. N. N., and B. V. designed the research. A. P. E., H. N. N., R. H., and M. S. T.-J. performed the experiments. A. P. E., H. N. N., R. H., M. S. T.-J., and B. V. analyzed the data and interpreted the results. A. P. E. and B. V. drafted the manuscript, which was finalized and approved by all authors.
Acknowledgments
We thank Janne Petersen, Kirsten Lykke Pedersen, Randi Scheel, and Tina Anna-Margrethe Kahr for expert technical assistance.
This work was supported in part by grants from the Danish Medical Research Council, L'Oréal Denmark, the Novo Nordisk Foundation, the Riisfort Foundation, and the Lundbeck Foundation. The authors declare that they have no conflict of interest with the contents of this article.
- M1–M10
- transmembrane helices 1–10
- A-domain
- actuator domain
- L8-9
- cytoplasmic loop between transmembrane segments M8 and M9
- N-domain
- nucleotide binding domain
- P-domain
- phosphorylation domain.
References
- 1. Post R. L., Hegyvary C., and Kume S. (1972) Activation by adenosine triphosphate in the phosphorylation kinetics of sodium and potassium ion transport adenosine triphosphatase. J. Biol. Chem. 247, 6530–6540 [PubMed] [Google Scholar]
- 2. Albers R. W. (1967) Biochemical aspects of active transport. Annu. Rev. Biochem. 36, 727–756 [DOI] [PubMed] [Google Scholar]
- 3. Glynn I. M. (1993) Annual review prize lecture. ‘All hands to the sodium pump.’ J. Physiol. 462, 1–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaplan J. H. (2002) Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 71, 511–535 [DOI] [PubMed] [Google Scholar]
- 5. Garty H., and Karlish S. J. (2006) Role of FXYD proteins in ion transport. Annu. Rev. Physiol. 68, 431–459 [DOI] [PubMed] [Google Scholar]
- 6. Morth J. P., Pedersen B. P., Toustrup-Jensen M. S., Sørensen T. L., Petersen J., Andersen J. P., Vilsen B., and Nissen P. (2007) Crystal structure of the sodium-potassium pump. Nature 450, 1043–1049 [DOI] [PubMed] [Google Scholar]
- 7. Shinoda T., Ogawa H., Cornelius F., and Toyoshima C. (2009) Crystal structure of the sodium-potassium pump at 2.4 Å resolution. Nature 459, 446–450 [DOI] [PubMed] [Google Scholar]
- 8. Kanai R., Ogawa H., Vilsen B., Cornelius F., and Toyoshima C. (2013) Crystal structure of a Na+-bound Na+,K+-ATPase preceding the E1P state. Nature 502, 201–206 [DOI] [PubMed] [Google Scholar]
- 9. Toustrup-Jensen M. S., Holm R., Einholm A. P., Schack V. R., Morth J. P., Nissen P., Andersen J. P., and Vilsen B. (2009) The C terminus of Na+,K+-ATPase controls Na+ affinity on both sides of the membrane through Arg935. J. Biol. Chem. 284, 18715–18725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Therien A. G., and Blostein R. (2000) Mechanisms of sodium pump regulation. Am. J. Physiol. Cell Physiol. 279, C541–C566 [DOI] [PubMed] [Google Scholar]
- 11. Despa S., Bossuyt J., Han F., Ginsburg K. S., Jia L. G., Kutchai H., Tucker A. L., and Bers D. M. (2005) Phospholemman-phosphorylation mediates the β-adrenergic effects on Na/K pump function in cardiac myocytes. Circ. Res. 97, 252–259 [DOI] [PubMed] [Google Scholar]
- 12. Poulsen H., Morth P., Egebjerg J., and Nissen P. (2010) Phosphorylation of the Na+,K+-ATPase and the H+,K+-ATPase. FEBS Lett. 584, 2589–2595 [DOI] [PubMed] [Google Scholar]
- 13. Sweadner K. J., and Feschenko M. S. (2001) Predicted location and limited accessibility of protein kinase A phosphorylation site on Na-K-ATPase. Am. J. Physiol. Cell Physiol. 280, C1017–C1026 [DOI] [PubMed] [Google Scholar]
- 14. Fisone G., Cheng S. X., Nairn A. C., Czernik A. J., Hemmings H. C. Jr., Höög J. O., Bertorello A. M., Kaiser R., Bergman T., Jörnvall H., Aperia A., and Greengard P. (1994) Identification of the phosphorylation site for cAMP-dependent protein kinase on Na+,K+-ATPase and effects of site-directed mutagenesis. J. Biol. Chem. 269, 9368–9373 [PubMed] [Google Scholar]
- 15. Cheng X. J., Fisone G., Aizman O., Aizman R., Levenson R., Greengard P., and Aperia A. (1997) PKA-mediated phosphorylation and inhibition of Na+-K+-ATPase in response to β-adrenergic hormone. Am. J. Physiol. Cell Physiol. 273, C893–C901 [DOI] [PubMed] [Google Scholar]
- 16. Codina J., Liu J., Bleyer A. J., Penn R. B., and DuBose T. D. Jr. (2006) Phosphorylation of S955 at the protein kinase A consensus promotes maturation of the α subunit of the colonic H+,K+-ATPase. J. Am. Soc. Nephrol. 17, 1833–1840 [DOI] [PubMed] [Google Scholar]
- 17. Poulsen H., Nissen P., Mouritsen O. G., and Khandelia H. (2012) Protein kinase A (PKA) phosphorylation of Na+/K+-ATPase opens intracellular C-terminal water pathway leading to third Na+-binding site in molecular dynamics simulations. J. Biol. Chem. 287, 15959–15965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen C., and Okayama H. (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7, 2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jewell E. A., and Lingrel J. B. (1991) Comparison of the substrate dependence properties of the rat Na,K-ATPase α1, α2, and α3 isoforms expressed in HeLa cells. J. Biol. Chem. 266, 16925–16930 [PubMed] [Google Scholar]
- 20. Vilsen B. (1992) Functional consequences of alterations to Pro328 and Leu332 located in the 4th transmembrane segment of the α-subunit of the rat kidney Na+,K+-ATPase. FEBS Lett. 314, 301–307 [DOI] [PubMed] [Google Scholar]
- 21. Toustrup-Jensen M., Hauge M., and Vilsen B. (2001) Mutational effects on conformational changes of the dephospho- and phospho-forms of the Na+,K+-ATPase. Biochemistry 40, 5521–5532 [DOI] [PubMed] [Google Scholar]
- 22. Einholm A. P., Andersen J. P., and Vilsen B. (2007) Importance of Leu99 in transmembrane segment M1 of the Na+,K+-ATPase in the binding and occlusion of K+. J. Biol. Chem. 282, 23854–23866 [DOI] [PubMed] [Google Scholar]
- 23. Beuschlein F., Boulkroun S., Osswald A., Wieland T., Nielsen H. N., Lichtenauer U. D., Penton D., Schack V. R., Amar L., Fischer E., Walther A., Tauber P., Schwarzmayr T., Diener S., Graf E., et al. (2013) Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat. Genet. 45, 440–444 [DOI] [PubMed] [Google Scholar]
- 24. Toustrup-Jensen M. S., Einholm A. P., Schack V. R., Nielsen H. N., Holm R., Sobrido M. J., Andersen J. P., Clausen T., and Vilsen B. (2014) Relationship between intracellular Na+ concentration and reduced Na+ affinity in Na+,K+-ATPase mutants causing neurological disease. J. Biol. Chem. 289, 3186–3197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baginski E. S., Foà P. P., and Zak B. (1967) Microdetermination of inorganic phosphate, phospholipids, and total phosphate in biologic materials. Clin. Chem. 13, 326–332 [PubMed] [Google Scholar]
- 26. Vilsen B. (1997) Leucine 332 at the boundary between the fourth transmembrane segment and the cytoplasmic domain of Na+,K+-ATPase plays a pivotal role in the ion translocating conformational changes. Biochemistry 36, 13312–13324 [DOI] [PubMed] [Google Scholar]
- 27. Jorgensen P. L. (1974) Purification and characterization of (Na+ + K+)-ATPase. III. Purification from the outer medulla of mammalian kidney after selective removal of membrane components by SDS. Biochim. Biophys. Acta 356, 36–52 [DOI] [PubMed] [Google Scholar]
- 28. Karlish S. J., and Yates D. W. (1978) Tryptophan fluorescence of (Na+ + K+)-ATPase as a tool for study of the enzyme mechanism. Biochim. Biophys. Acta 527, 115–130 [DOI] [PubMed] [Google Scholar]
- 29. Garrahan P. J., and Glynn I. M. (1967) The stoicheiometry of the sodium pump. J. Physiol. 192, 217–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaplan J. H., and Hollis R. J. (1980) External Na dependence of ouabain-sensitive ATP:ADP exchange initiated by photolysis of intracellular caged-ATP in human red cell ghosts. Nature 288, 587–589 [DOI] [PubMed] [Google Scholar]
- 31. Yoda S., and Yoda A. (1986) ADP- and K+-sensitive phosphorylated intermediate of Na,K-ATPase. J. Biol. Chem. 261, 1147–1152 [PubMed] [Google Scholar]
- 32. Taniguchi K., and Post R. L. (1975) Synthesis of adenosine triphosphate and exchange between inorganic phosphate and adenosine triphosphate in sodium and potassium ion transport adenosine triphosphatase. J. Biol. Chem. 250, 3010–3018 [PubMed] [Google Scholar]
- 33. Post R. L., Kume S., Tobin T., Orcutt B., and Sen A. K. (1969) Flexibility of an active centre in sodium-plus-potassium adenosine triphosphatase. J. Gen. Physiol. 54, 306–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Einholm A. P., Toustrup-Jensen M. S., Holm R., Andersen J. P., and Vilsen B. (2010) The rapid-onset dystonia Parkinsonism mutation D923N of the Na+,K+-ATPase α-3 isoform disrupts Na+ interaction at the third Na+ site. J. Biol. Chem. 285, 26245–26254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Azarias G., Kruusmägi M., Connor S., Akkuratov E. E., Liu X. L., Lyons D., Brismar H., Broberger C., and Aperia A. (2013) A specific and essential role for Na,K-ATPase α3 in neurons co-expressing α1 and α3. J. Biol. Chem. 288, 2734–2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holm R., Einholm A. P., Andersen J. P., and Vilsen B. (2015) Rescue of Na+ affinity in aspartate 928 mutants of Na+,K+-ATPase by secondary mutation of glutamate 314. J. Biol. Chem. 290, 9801–9811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jen J. C., Klein A., Boltshauser E., Cartwright M. S., Roach E. S., Mamsa H., and Baloh R. W. (2007) Prolonged hemiplegic episodes in children due to mutations in ATP1A2. J. Neurol. Neurosurg. Psychiatry 78, 523–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heinzen E. L., Arzimanoglou A., Brashear A., Clapcote S. J., Gurrieri F., Goldstein D. B., Jóhannesson S. H., Mikati M. A., Neville B., Nicole S., Ozelius L. J., Poulsen H., Schyns T., Sweadner K. J., van den Maagdenberg A., et al. (2014) Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol. 13, 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fernandez D. M., Hand C. K., Sweeney B. J., and Parfrey N. A. (2008) A novel ATP1A2 gene mutation in an Irish familial hemiplegic migraine kindred. Headache 48, 101–108 [DOI] [PubMed] [Google Scholar]
- 40. Johnson L. N., and Lewis R. J. (2001) Structural basis for control by phosphorylation. Chem. Rev. 101, 2209–2242 [DOI] [PubMed] [Google Scholar]
- 41. Frame S., Cohen P., and Biondi R. M. (2001) A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 7, 1321–1327 [DOI] [PubMed] [Google Scholar]
- 42. Woods A. S., and Ferré S. (2005) Amazing stability of the arginine-phosphate electrostatic interaction. J. Proteome Res. 4, 1397–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]