

Abstract
Background
Tumor cells use aerobic glycolysis to rapidly generate ATP and growth substrate which expenses a large amount of glucose. However, how tumor cells take in enough glucose from the tumor microenvironment of insufficient blood supply remains poorly understood. The cellular FLICE-like inhibitory protein (FLIP), a cell apoptosis inhibiting molecule, is highly expressed in hepatocellular carcinoma (HCC) and is implicated in HCC development.
Methods
The effects of FLIPL (the long form of FLIP) on aerobic glycolysis and glucose uptake were assessed in HCC cells and xenograft tumors. The correlations between FLIPL expression and sodium/glucose cotransporter 1 (SGLT1) expression in clinical HCC tissues were analyzed. The consequences of FLIPL-induced regulation of SGLT1 at the transcription and translation levels and the interaction between FLIPL and SGLT1 were examined. FLIPL-mediated tolerance upon glucose limitation and its mechanism were detected.
Results
We report a novel role for FLIPL in promoting the aerobic glycolysis of HCC cells. FLIPL overexpression induced a significant increase in cell aerobic glycolysis indexes including glucose uptake, glucose consumption, and lactate production. FLIPL co-localized and interacted with SGLT1, a major active glucose transporter in HCC cells. FLIPL increased the stability of SGLT1 protein by inhibiting its ubiquitination and degradation. The expression level of FLIPL was positively correlated with the expression level of SGLT1 in 79 HCC tissues from surgical operation. Furthermore, FLIPL increased cell tolerance ability and decreased cell apoptosis to low glucose by regulating SGLT1.
Conclusions
Our results indicate that FLIPL plays an essential role in HCC aerobic glycolysis and that SGLT1 is required for FLIPL-modulated tumor proliferation under low glucose conditions. Targeting the actions of FLIPL in cell glucose-dependent aerobic glycolysis may provide an attractive strategy for therapeutic intervention in HCC.
Electronic supplementary material
The online version of this article (doi:10.1186/s13046-016-0358-3) contains supplementary material, which is available to authorized users.
Keywords: Hepatocellular carcinoma, Cellular FLICE-like inhibitory protein, Sodium-glucose cotransporter 1, Aerobic glycolysis, Glucose uptake
Background
Hepatocellular carcinoma (HCC) is a highly aggressive malignant tumor worldwide. Due to the resistance of HCC to conventional therapies, such as radiotherapy and chemotherapy, hepatic resection remains the most effective method for treating HCC [1, 2]. However, many patients have already lost the opportunity for hepatic resection by the time that HCC is diagnosed. Therefore, continued efforts are needed to find new targets for HCC treatment.
Tumor cells convert most glucose into lactate regardless of whether oxygen supply is sufficient. This aerobic glycolytic metabolism is known as the Warburg effect [3]. Tumor cells utilize aerobic glycolysis to rapidly generate ATP and other substrates to meet the need of tumor growth [4]. In view of the fact that aerobic glycolysis is much less efficient than oxidative phosphorylation for generating ATP [5], tumor cells need to increase glucose absorption to meet the high requirement for glucose metabolism [6]. Multiple oncogenes and tumor suppressor genes participate in the glucose uptake and metabolism processes, including PI3K, AKT, AMPK, p53, c-Myc, and HIF-1α [7, 8]. These regulators increase glucose uptake by enhancing the transcription and membrane translocation of glucose transporters in tumor cells. There are two classes of glucose transporters in humans, the facilitated transporters (GLUTs) and the active transporters (SGLTs). Previous studies have revealed that ectopic expression of glucose transporters in many tumors is beneficial to cell proliferation and survival under low glucose condition [9]. It has been reported that there is abnormal GLUT1 expression in HCC [10]. Despite glucose transporters is significant in tumor aerobic glycolysis, the key molecules and precise mechanisms controlling the glucose transporters are still largely unknown.
Cellular FLICE-like inhibitory protein (FLIP) is a potential oncogene in HCC, which highly homologous to caspase-8 containing two death effector domains [11]. The human FLIP gene is located on chromosome 2q33-q34. Three splice variants of the FLIP protein have been identified, a 55 kDa long form FLIP (FLIPL), a 27 kDa short form FLIP (FLIPS) and a 25 kDa FLIP Raji (FLIPR) [12]. These isoforms perform different cellular functions [12], with FLIPL being the most well-studied isoform. Elevated FLIPL is observed in many cancers, including HCC, malignant melanoma, breast cancer, prostate cancer, bladder cancer, and non-Hodgkin lymphoma [13]. FLIPL has been shown to be a multifunctional protein involved in death receptor-mediated apoptosis, the NF-κB pathway [14, 15], necrosis [16], autophagy [17], inflammation [18], the ubiquitin-proteasome system [19], and endoplasmic reticulum morphology [20]. Recently, FLIPL was shown to be up-regulated following the disruption of glycolysis with 2-deoxy-D-glucose (2-DG) [21]. However, whether and how FLIPL participates in glucose metabolism in cancer cells is unclear.
In the current study, we first demonstrated the function of FLIPL in facilitating HCC cells’ aerobic glycolysis by modulating SGLT1-mediated glucose uptake. We found SGLT1 is required for FLIPL induced cell aerobic glycolysis and survival to low glucose conditions. We also extended our findings to clinical HCC patients. In 79 HCC cases, FLIPL expression level was positively correlated with SGLT1 expression level. Therefore, our results suggest that FLIPL is a potential therapeutic target for HCC [22].
Methods
Cells and reagents
HepG2, MHCC97-H, Huh-7, SMMC-7721, BEL-7704 HCC, and HEK293 cell lines were obtained from the Department of Biochemistry and Molecular Biology (The Fourth Military Medical University) and cultured in a humidified incubator under 5 % CO2 in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, Carlsbad, CA, USA) supplemented with 10 % fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37 °C. The proteasome inhibitor, MG132, was from Sigma-Aldrich (Shanghai, China). Mouse anti-GLUT1, rabbit anti-SGLT1 and mouse anti-FLIP antibodies were obtained from Abcam (New Territories, HK). Rabbit anti-FLIP and mouse anti-HA-Tag antibodies were from Cell Signaling Technology (Danvers, MA). 2-NBDG (2-[N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy -D-glucose) was purchased from Cayman Chemical Company (Michigan, US).
Cell transfections
FLIPL expression plasmid (Flag-FLIPL), FLIPS expression plasmid (Flag-FLIPS), SGLT1 expression plasmid (Myc-SGLT1), HA-ubiquitin expression plasmid, shRNA targeting FLIPL, and shRNA targeting SGLT1 were constructed by Jikai biotechnology (Shanghai, China). Cells (1 × 106) were seeded into six-well plates and transfected using Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, USA) according to the manufacturer’s instructions. The target sequences of shRNA of FLIPL and SGLT1 are 5’ CAGAATAGACCTGAAGACA3’ and 5’ CTTCCGCATCCAGGT CAAT3’, respectively.
Real-time PCR
RNA was reverse transcribed with the PrimeScript™ 1st Strand cDNA Synthesis Kit (Takara, Dalian, China) according to the manufacturer's instructions. Real-time PCR was performed using SYBR Premix Ex Taq™ II (Takara, Dalian, China). The relative gene expression was calculated using the 2−ΔΔCt method, in which Ct represented the threshold cycle. β-actin RNA was used as the reference. The primers were used as following. FLIPL: Forward 5’GGCTCCCCCTGCATCAC3’, Reverse 5’ TTTGGCTT CCTGCTAGATAAGG3’; SGLT1: Forward 5’ATGGACAGTAGCACCTTGAGCC3’; Reverse 5’TAGCCCCAGAGAAGATGTCTGC3’; GLUT1: Forward 5’ AAGTCTCC TTTACCCACATCC 3’; Reverse 5’ GAGTGTCCGTGTCTTCTTGAGT 3’.
Immunoblot analysis
Cells were harvested and lysed with RIPA buffer (0.15 M NaCl, 1 % NP40, 0.01 M deoxycholate, 0.1 % SDS, 0.05 M Tris-HCl pH 8.0, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 mg/ml aprotinin, 10 mg/ml pepstatin and 10 mg/ml leupeptin). Protein concentrations were determined using the BCA Protein Assay kit (Beyotime, China). The total protein was electrophoresed on 8 or 12 % SDS-PAGE gels and transferred to polyvinylidene fluoride membranes. Membranes were blocked with 5 % nonfat dry milk or BSA in TBST buffer (20 mM Tris-HCl pH 7.4, 8 g/L NaCl and 0.1 % Tween 20) for 1 h at room temperature, and then incubated with the indicated primary antibody in TBST buffer containing 2 % BSA at 4 °C overnight. The membrane was then probed with the appropriate horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h, and the proteins were detected with a chemiluminescence imaging analysis system.
Glucose uptake assay in vitro
Cells were plated at 1×106/well in 6-well plates and cultured for 24 h. After the specified treatments, cells were refreshed with serum-starved (0.1 % FBS) and glucose-free DMEM. 16 h later, cells were grown in the presence of 50 mM 2-NBDG for 30 or 60 min, respectively, and glucose uptake was quantified using FACS analysis.
Tumorigenesis and glucose uptake assay in vivo
5×106 of HepG2 cells were injected into the hind legs of athymic male nude mice (8 weeks, seven mice per group). The tumor size was measured weekly with a caliper, and the tumor volume was determined with the standard formula: 0.5×length×width2. The tumor growth curve was then derived from these data. 18F-FDG was injected into the mice via a tail vein and PET/CT images were collected on a nanoScan PET/CT system (Mediso, Budapest, Hungary).
Lactate production assay
1×105 cells were plated in 6-well plates and cultured overnight. Culture medium was removed from cells and the lactate concentration was determined using the Lactate Kit (Nangjing Jiancheng Bioengineering, Nanjing, China) according to manufacturer’s protocol. Cells were then harvested, stained with trypan blue, and the number of viable cells was counted using a hemocytometer. The rate of lactate production was determined (lactate production rate=lactate concentration/cells/time) and normalized to the rate detected in the control group.
Glycolysis assay
Extracellular acidification rate (ECAR) was conducted using a Seahorse XF24 Analyzer. 20,000 cells were seeded into Seahorse 24-well microplates and cultured at 37 °C with 5 % CO2 for 24 h. Culture medium was replaced with 700 μl of assay medium, composed of DMEM without FBS and sodium bicarbonate, and cells were then incubated at 37 °C without CO2 for 1 h. To measure the glycolytic capacity of the cells, additions were injected to the wells at final concentrations of 11 mM glucose, 1 μM oligomycin, and 20 mM 2-DG.
Immunofluorescence assay
Cells were fixed in a freshly prepared 4 % paraformaldehyde, rinsed with 0.1 % PBS and permeabilized with Triton X-100. Cells were then incubated with horse serum in PBS to block nonspecific binding. After being washed with PBS, cells were incubated overnight at 4 °C with mouse anti-FLIP antibody (1:200), rabbit anti-SGLT1 antibody (1:200). Subsequently, fluorescein isothiocyanate-conjugated anti-mouse antibody (diluted 1:400; Sigma-Aldrich) or cyanine 3-conjugated anti-rabbit antibody (diluted 1:400; Sigma-Aldrich) were incubated with the cells at room temperature for 2 h. The isotype mouse and rabbit IgG were used as negative controls. Fluorescence detection was performed using a laser confocal microscope (Nikon, US) after cell nucleus counterstaining with 4’, 6-diamidino-2-phenylindole.
Immunoprecipitation
Cells were incubated with 1 ml of lysis buffer containing (50 mM HEPES, pH 7.4; 150 mM NaCl; 0.1 % Triton X-100; 1.5 mM MgCl2; 1 mM EDTA; and 1 mM phenylmethylsulfonyl fluoride), as well as protease inhibitors for 30 min at 4 °C. The insoluble fraction was eliminated through centrifugation at 12,000 rpm for 30 min at 4 °C. The lysates were incubated with the antibody after centrifugation and protein A/G-conjugated sepharose (Santa Cruz Bio Technology, USA) at 4 °C overnight. 20 μl of protein was saved to quantify the total amount of protein loaded. Beads were washed four times with lysis buffer. Proteins were eluted in SDS-PAGE sample buffer and separated by SDS-PAGE for immunoblot analysis.
Cell proliferation assay
Cells were seeded into 96-well cell culture plates. Each well contained 1×103 cells in 100 μl culture media. After the specified treatments, the medium in each well was replaced with 100 μl fresh medium with 10 % CCK8 (Dojindo, Japan), and the cells incubated at 37 °C for additional 2 h. The absorbance was measured at 450 nm wavelength.
Cell apoptosis assay
Cells were cultured in MEM containing low glucose (0.75 mM) or high glucose (25 mM) for 48 h after cell transfection. For Annexin staining, cells were trypsinized and resuspended in 1 ml Annexin-binding buffer (BD Biosciences, US) to which was added 5 μl Annexin V-FITC (BD Biosciences, US). After incubation in the dark at room temperature for 15 min, 50 μl PI (Sigma, US) was added to discriminate dead cells and the samples were analyzed on a FACS Caliber flow cytometer.
Tissue samples and study cohort and immunohistochemistry (IHC)
All patients from whom we obtained the 79 pairs of HCC and adjacent normal liver tissue specimens provided their full consent to participate in the study at the Xijing Hospital of the Fourth Military Medical University (Xi’an, China). IHC was performed using the avidin-biotin-peroxidase complex method. All sections were deparaffinized in xylenes and dehydrated through a gradient concentration of alcohol before endogenous peroxidase activity was blocked using 0.5 % H2O2 in methanol for 10 min. After nonspecific binding was blocked, the slides were incubated with FLIPL antibody (1:200) or SGLT1 antibody (1:200) in phosphate-buffered saline (PBS) at 4 °C overnight in a humidified container. Biotinylated goat anti-rabbit immunoglobulin G (IgG) (1:400; Sigma-Aldrich) was incubated with the sections for 1 h at room temperature and detected using a streptavidin-peroxidase complex. The brown color indicative of peroxidase activity was developed by incubation with 0.1 % 3, 3-diaminobenzidine in PBS with 0.05 % H2O2 for 5 min at room temperature. An immunoreactivity score system was provided on the basis of the proportion and intensity of positively stained cancer cells. The extensional standard used was as follows: (1) the number of cells with positive staining (0–5 %: 0; 6–25 %: 1; 26–50 %: 2; 51–75 %: 3; and>75 %: 4) and (2) the staining intensity (colorless: 0; pallide-flavens: 1; yellow: 2; brown: 3). We multiplied the numbers scored (1) and (2) from the extensional standard, and the staining grade was stratified as absent (0 score), weak (1–4 score), moderate (5–8 score) or strong (9–12 score). Two pathologists blinded to the clinicopathologic information and outcome of the patients scored the multiple tissue arrays. Tumors with moderate or strong immunostaining intensity were classified as staining positive (+), whereas tumors with absent were classified as staining negative (−).
Statistical analysis
Data are reported as the mean ± S.D. or S.E.M. analyses were performed using GraphPad Prism software. The significance of differences was determined by one-way analysis of variance followed by Scheffe’s post hoc test, independent samples t-test or Student’s t-test. Associations between protein expression and categorical variables were analyzed using the χ2 test or Fisher’s exact test as appropriate. P < 0.05 was considered statistically significant.
Results
FLIPL facilitates aerobic glycolysis by promoting glucose uptake
To investigate whether FLIP could affect aerobic glycolysis in HCC, FLIPL and FLIPS, the two main isoforms of FLIP, were overexpressed in HepG2 cells (Fig. 1a, b). Overexpressed FLIPL significantly increased aerobic glycolytic capacity in response to glucose stimulation compared with the control group, but overexpressed FLIPS had no effect on glycolysis (Fig. 1c, d). We also found that FLIPL, but not FLIPS, induced lactate production (Fig. 1e), which was consistent with the ECAR results. These findings suggest that FLIPL is involved in aerobic glycolysis in HCC.
Fig. 1.
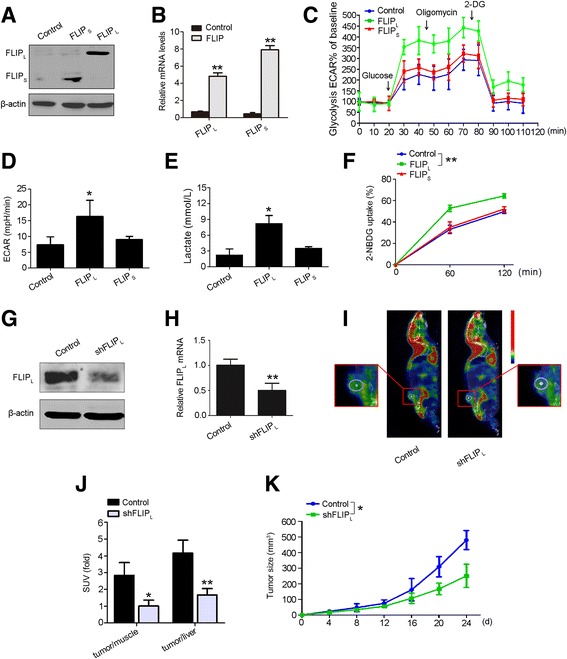
FLIPL promotes aerobic glycolysis by up-regulating glucose uptake. a and b HepG2 cells were transfected with FLIPL or FLIPs overexpression vectors. Expression was detected 48 h after transfection in protein and mRNA levels. Data are the mean ± S.E.M. from three independent experiments. **p < 0.01 versus the control group. Cell ECAR in c and d was measured by the glycolysis stress test in HepG2 cells with FLIPL or FLIPS overexpression. Data represent the mean ± S.E.M. from three independent experiments, each performed in quadruplicate. *p < 0.05 versus the control group. e Lactate production was measured in HepG2 cells with FLIPL or FLIPS overexpression. Data represent the mean ± S.E.M. from three independent experiments, each performed in quadruplicate. *p < 0.05 versus the control group. f Glucose uptake was measured in HepG2 cells with FLIPL or FLIPS overexpression. Cells were grown in the presence of the fluorescent analog 2-NBDG for various time points, and glucose uptake was quantified using FACS analysis. Data represent the mean ± S.E.M. Similar results were obtained from three independent experiments. **p < 0.01 versus the control group. g and h HepG2 cells were transfected with shFLIPL vector. Expression was detected 48 h after transfection in protein and mRNA levels. Data are the mean ± S.E.M. from three independent experiments. **p < 0.01 versus the control group. i Representative PET/CT images in nude mice bearing liver tumors with control or shFLIPL plasmid treatment. Yellow dotted lines indicate area of the liver tumors. j Glucose uptake in HCC was normalized with muscle or liver tissues in each mouse (n=8). Data are presented as the mean ± S.D. *p < 0.05, **p < 0.01 versus control. k Tumor development in nude mice bearing with FLIPL-silent HepG2 cells (n=8). Data are presented as the mean ± S.D. *p < 0.05 versus control
Glucose uptake is a key step in aerobic glycolysis, which is critical for cancer cell survival. Most cancer cells generate high levels of ATP through increasing the glucose uptake [23]. Therefore, we examined the effect of FLIP on glucose uptake. Ectopic expression of FLIPL significantly induced glucose uptake in HepG2 cells, but glucose uptake was not altered in cells overexpressing FLIPS (Fig. 1f). Moreover, to evaluate the influence of FLIPL on glucose uptake and HCC progression in vivo, nude mice bearing FLIPL down-regulated HepG2 cells (Fig. 1g, h) were imaged by 18F-FDG PET/CT scanning. The scan images demonstrated that 18F-FDG accumulation was notably decreased with the deletion of FLIPL (Fig. 1i, j). We also analyzed the effect of FLIPL on HCC development by continuously measuring the size of tumors in the xenograft mouse model. As shown in Fig. 1k, the growth of transplanted tumors with FLIPL silenced was significantly slower, compared with the control group. These results indicated that FLIPL promotes tumor cell survival and aerobic glycolysis by increasing glucose uptake both in vivo and in vitro.
FLIPL expression positively correlated with SGLT1 expression in hepatic carcinoma
Glucose is transported into cells by two families of transporters, the facilitative-type glucose transporters and the active-type glucose transporters [24]. GLUT1 and SGLT1, two well-studied glucose transporters in the respective families, are highly expressed in several cancers and participate in cellular glucose uptake. To determinate the mechanisms by which FLIPL promotes the aerobic glycolysis and survival of HCC cells, we measured the GLUT1 and SGLT1 mRNA and protein levels after shRNA-mediated depletion of FLIPL expression in HepG2 cells. Although the mRNA level of SGLT1 did not change, its protein expression was notably decreased following FLIPL loss. However, neither mRNA nor protein levels of GLUT1 altered with the down-regulation of FLIPL (Fig. 2a, b). These findings suggested that SGLT1, but not GLUT1, is involved in FLIPL-mediated HCC glucose uptake.
Fig. 2.
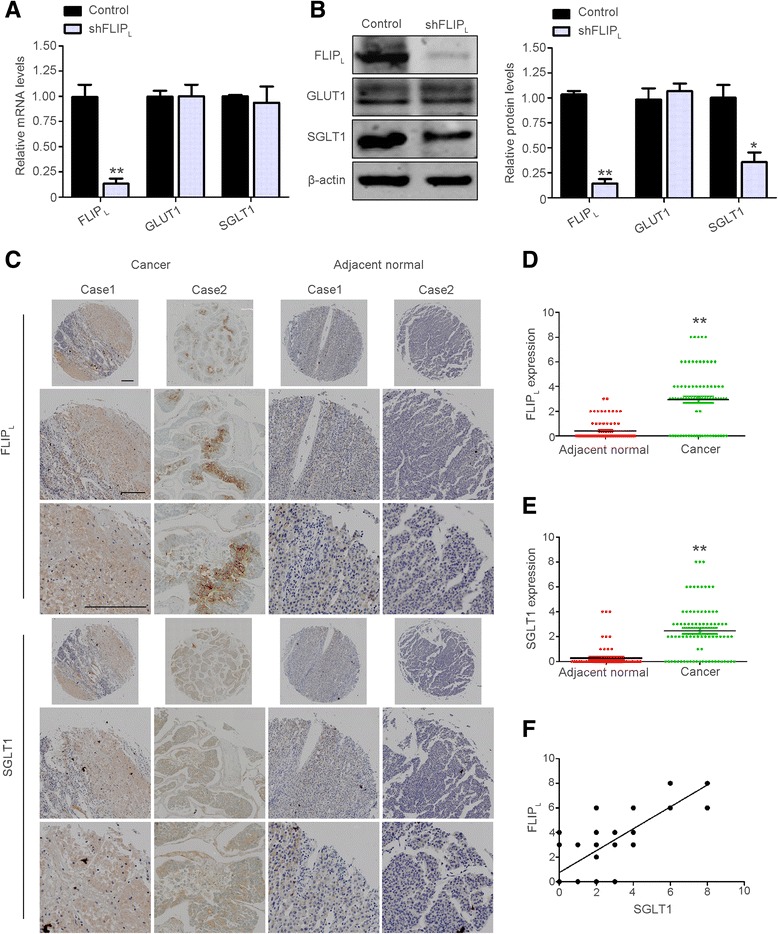
FLIPL expression positively correlated with SGLT1 expression in HCC. GLUT1 and SGLT1 expression were determined by real-time PCR (a) and immunoblot analysis (b) in HepG2 cells with FLIPL knockdown treatment. Left panel: Immunoblotting. Right panel: Densitometric quantification of the data in b, normalized to β-tubulin. Data are the mean ± S.E.M. from three independent experiments. *p < 0.05, **p < 0.01 versus the control group. c IHC staining of FLIPL and SGLT1 in adjacent normal and liver cancer samples from patients at different stages. Case 1 is from a stage II patient, and Case 2 is from a stage I patient. Bar = 200 μm. (d) and (e) Quantification of the expression levels of FLIPL and SGLT1 in human HCC samples. **p < 0.01 versus adjacent normal tissues. f Correlation between FLIPL and SGLT1 expression with linear regression and Pearson’s correlation significance (p < 0.01)
Dysregulated expression of SGLT1 has been identified in various human cancers. However, the expression status of SGLT1 in HCC has not been investigated. To assess SGLT1 protein expression levels and its potential association with FLIPL in HCC, we compared 79 pairs of HCC and adjacent normal tissue samples using immunohistochemistry (IHC). The results showed that SGLT1 and FLIPL were highly expressed in liver cancer tissue compared with adjacent normal liver tissue (Fig. 2c). In normal versus liver cancer tissue, FLIPL levels were 0.41 versus 2.97 (Fig. 2d), and SGLT1 were 0.28 versus 2.47 (Fig. 2e). Moreover, in these 79 pairs of HCC and adjacent normal tissue samples, SGLT1 and FLIPL expression were correlated with tumor stage (Tables 1 and 2) in liver cancer tissues, but the correlation between the expression and sex or age was not statistically significant in both liver cancer tissues and their adjacent normal specimens (Tables 1 and 2, Additional file 1: Table S1 and Table S2).
Table 1.
Statistical results of FLIPL expression in 79 liver cancer specimens
| N | - | +to+++ | P-value | |
|---|---|---|---|---|
| Total | 79 | 24 | 55 | |
| Sex | 0.605* | |||
| Male | 66 | 20 | 46 | |
| Female | 13 | 4 | 9 | |
| Age at diagnosis, years | 0.437** | |||
| <60 | 61 | 18 | 35 | |
| ≥60 | 18 | 6 | 20 | |
| TNM stage | 0.015** | |||
| I | 22 | 12 | 10 | |
| II | 40 | 11 | 29 | |
| III | 17 | 1 | 16 |
Abbreviation: TNM tumor node metastasis. *P-value when expression levels were compared using Fisher’ exact test; **P-value when expression levels were compared using the Kruskal-Wallis test
Table 2.
Statistical results of SGLT1 expression 79 liver cancer specimens
| N | - | +to+++ | P-value | |
|---|---|---|---|---|
| Total | 79 | 23 | 56 | |
| Sex | 0.490* | |||
| Male | 66 | 21 | 45 | |
| Female | 13 | 2 | 9 | |
| Age at diagnosis, years | 0.769* | |||
| <60 | 61 | 17 | 44 | |
| ≥60 | 18 | 6 | 12 | |
| TNM stage | 0.046** | |||
| I | 22 | 9 | 13 | |
| II | 40 | 13 | 27 | |
| III | 17 | 1 | 16 |
Abbreviation: TNM tumor node metastasis. *P-value when expression levels were compared using Fisher’ exact test; **P-value when expression levels were compared using the Kruskal-Wallis test
Because knocking down FLIPL expression led to the suppression of SGLT1 protein levels, we speculated whether there is a correlation between the protein levels of FLIPL and SGLT1. We analyzed the correlation between FLIPL and SGLT1 expression using spearman analysis. The expression of FLIPL was demonstrated to positively correlate with SGLT1 expression (Fig. 2f), suggesting that FLIPL is involved in the post-transcriptional regulation of SGLT1.
FLIPL interacts with SGLT1
Given that FLIPL could regulate the protein level of SGLT1, but not the SGLT1 transcript, we investigated whether the FLIPL protein interacts with the SGLT1 protein. To address this question, we examined the subcellular distribution of FLIPL and SGLT1 in HepG2 cells using immunofluorescence. Both FLIPL and SGLT1 were predominantly localized in the cytoplasm and there was a strong co-localization of the two proteins (Fig. 3a). The co-localization of FLIPL and SGLT1 prompted us to further analyze whether FLIPL could bind with SGLT1. Co-immunoprecipitation was performed using lysates prepared from HepG2 cells, and showed that endogenous FLIPL interacted with endogenous SGLT1, and vice versa (Fig. 3b).
Fig. 3.
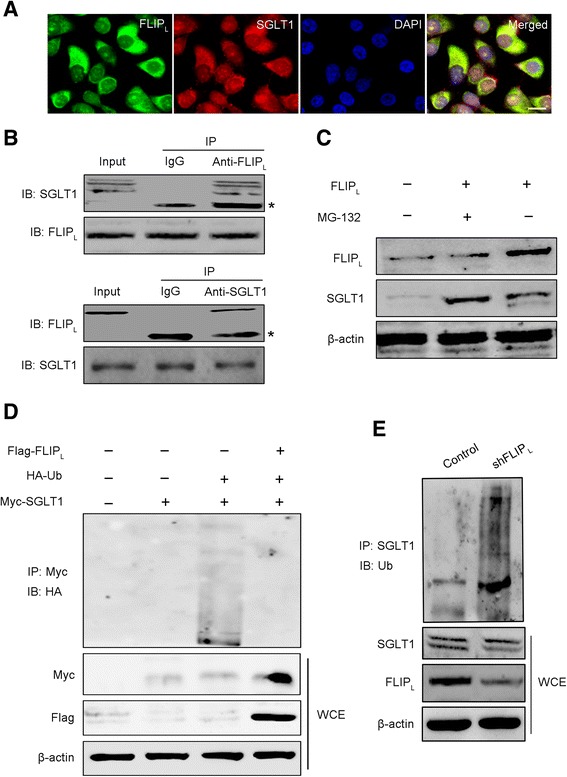
FLIPL inhibits the ubiquitination and degradation of SGLT1 by interacting with SGLT1. a The localization of FLIPL and SGLT1 were determined in HepG2 cells by laser confocal scanning microscopy. Green fluorescence indicated FLIPL expression, red fluorescence indicated SGLT1 expression, and blue fluorescence indicated nuclear staining with DAPI. Bar = 20 μm. b HepG2 cells were harvested for co-immunoprecipitation assays followed by immunoblot (IB) analysis. Immunoprecipitation (IP) assays were performed with whole-cell lysates (WCE) pretreated with A/G–conjugated Sepharose beads. WCE were probed for input. The antibodies used for IP and IB are as indicated. c SGLT1 expression was determined in HepG2 cells with FLIPL overexpression for 48 h and/or 5 μM MG132 for another 4 h. d HEK293 cells were transfected with hemagglutinin (HA)-ubiquitin plasmid, Flag-FLIPL plasmid, and Myc-SGLT1 plasmid for 48 h. Subsequently, the cell lysates were collected and analyzed by IP and IB. e HepG2 cells were transfected with HA-ubiquitin and shFLIPL plasmids for 48 h, and cell lysates were collected and analyzed by IP and IB
FLIPL inhibits the ubiquitination and degradation of SGLT1
FLIPL has been shown to up-regulate the levels of some proteins by inhibiting the ubiquitin-proteasome degradation pathway [19]. We hypothesized that FLIPL stabilizes SGLT1 protein by regulating its degradation. First we determined the protein degradation pathway of SGLT1. As shown in Fig. 3c, FLIPL increased the SGLT1 protein level and MG132, a proteasome inhibitor, induced further accumulation of SGLT1 protein, indicating that the degradation of SGLT1 protein occurs via a proteasome associated pathway. In addition, we examined the effect of FLIPL on SGLT1 ubiquitination in HEK293 cells and HepG2 cells. In HEK293 cells, exogenous FLIPL inhibited the ubiquitination of exogenous SGLT1 and increased the total levels of SGLT1 protein (Fig. 3d). Endogenous SGLT1 ubiquitination was also examined in HepG2 cells and, consistent with the results with the exogenous proteins in HEK293 cells, we found that down-regulation of the expression of FLIPL led to increased SGLT1 ubiquitination and accelerated SGLT1 degradation (Fig. 3e). Collectively, these results suggested that FLIPL interacts with SGLT1 in the cytoplasm and increases the stability of SGLT1 protein by inhibiting its ubiquitination and proteasome-mediated degradation. This promotes the membrane translocation and function of SGLT1.
FLIPL promotes the tolerance of HCC cells to low glucose
High level of glycolysis could not only rapidly provide ATP for cancer cell proliferation but also allow cancer cells to adapt low glucose condition [9]. We investigated whether and how FLIPL regulates the tolerance for low glucose of HCC cells. We first assessed the ability of several HCC cell lines to tolerate low glucose. The apoptosis levels of HepG2, MHCC97-H, Huh-7, SMMC-7721 and BEL-7704 were detected under high glucose condition (25 mM) and low glucose condition (0.75 mM). Of these cell lines, HepG2, MHCC97-H, and Huh-7 cells exhibited higher tolerance of the low glucose condition (Fig. 4a). We next examined the FLIPL and SGLT1 expressions in these cell lines. FLIPL and SGLT1 protein levels were higher in two of the cell lines tolerant to low glucose, HepG2 and Huh-7 cells (Fig. 4b), so we used the HepG2 cells to perform the following experiments.
Fig. 4.
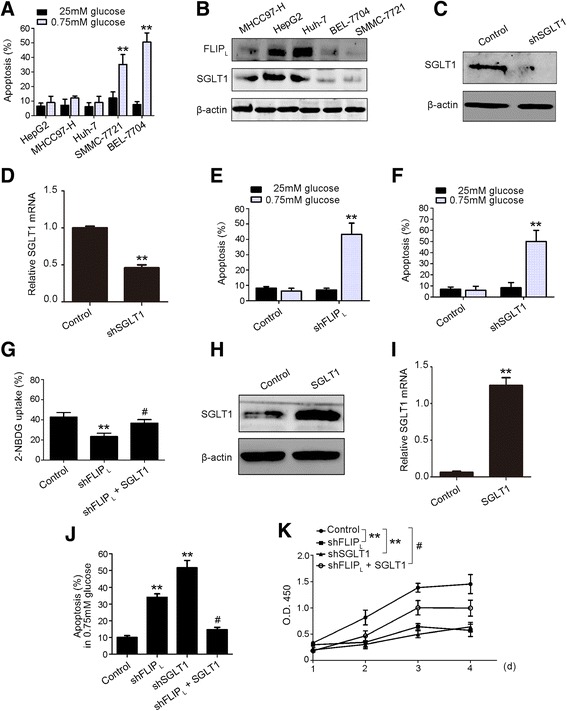
FLIPL promotes low glucose tolerance in HCC cells. a FACS analysis was performed to determine the apoptosis of HepG2, MHCC97-H, Huh-7, SMMC-7721 and BEL-7704 cells under low glucose (0.75 mM) or normal glucose (25 mM) treatment. b Immunoblot was used to detect the FLIPL and SGLT1 expression in MHCC97-H, HepG2, Huh-7, BEL-7704, and SMMC-7721 cell lines. c, d HepG2 cells were transfected with shSGLT1 vector. Expression was detected 48 h after transfection at protein and mRNA levels. **p < 0.01 versus control. e The apoptosis was measured in HepG2 cell with FLIPL knockdown treatment. Cells were cultured in low glucose (0.75 mM) or normal glucose (25 mM) and the levels of apoptosis were determined by FACS analysis. f Apoptosis was measured in HepG2 cell with SGLT1 knockdown. Cells were cultured in low glucose (0.75 mM) or normal glucose (25 mM), apoptosis was determined by FACS analysis. g Glucose uptake was measured in HepG2 cell with FLIPL knockdown or FLIPL knockdown plus SGLT1 overexpression treatment. h, i FLIPL-deficient HepG2 cells were transfected with the SGLT1 overexpression vector. Expression was detected 48 h after transfection at protein and mRNA levels. **p < 0.01 versus control. j Apoptosis was measured in HepG2 cell with FLIPL knockdown, SGLT1 knockdown, and FLIPL knockdown plus SGLT1 overexpression treatment. k Cell proliferation was measured by CCK8 assay in HepG2 cells with FLIPL knockdown, SGLT1 knockdown, and FLIPL knockdown plus SGLT1 overexpression treatment. (a, e, and f) Data represent the mean ± S.E.M. from three independent experiments, each performed in quadruplicate. **p < 0.01 versus 25 mM glucose group. (g, i, j, and k) Data represent the mean ± S.E.M. from three independent experiments, each performed in quadruplicate. *p < 0.05, **p < 0.01 versus control group; # p < 0.05 versus shFLIPL group
To determine whether FLIPL and SGLT1 affect the susceptibility of HCC cells to low glucose conditions, FLIPL and SGLT1 were separately down-regulated in HepG2 cells (Figs. 1g, h, and 4c, d). Examination of cell apoptosis, as shown in Fig. 4e, f, showed that apoptosis rates were significantly increased in FLIPL-silenced or SGLT1-silenced HepG2 cells. In addition, we found that the loss of FLIPL inhibited glucose uptake in HepG2 cells (Fig. 4g), which is consistent with the results of FLIPL overexpression in Fig. 1f. We next rescued the SGLT1 expression in FLIPL-deficient HepG2 cells (Fig. 4h, i), and the reduced glucose uptake was substantially recovered (Fig. 4g). The increased apoptosis rate was also attenuated with the restoration of SGLT1 expression in FLIPL-deficient HepG2 cells (Fig. 4j). In addition, we examined the survival of HepG2 cells in a low glucose environment. FLIPL-deficient HepG2 cells exhibited a decreased cell proliferation rate, and rescued SGLT1 expression significantly recovered cell survival under low glucose treatment (Fig. 4k). These results demonstrated that FLIPL promotes glucose uptake and the survival of HCC cells under low glucose conditions through regulation of SGLT1.
Discussion
In the present study, we determined a previous unknown role for FLIPL in HCC cells aerobic glycolysis. We have made three novel observations here. First, we demonstrated that FLIPL interacted with SGLT1 to inhibit the ubiquitination of SGLT1, thereby promoting glucose uptake and glycolysis in HCC cells. Second, we found that FLIPL was highly expressed in HCC tissue compared with adjacent normal tissue and that FLIPL expression positively correlated with SGLT1 expression. Third, FLIPL increased tumor cell tolerance to glucose limitation through the regulation of SGLT1 protein stability.
Glucose, as the main energy source of cells, is vital to tumor cell proliferation. Aerobic glycolysis is a hallmark in various tumors [25, 26]. Glucose uptake is the first step in glycolysis. HCC enhanced glucose uptake to meet the metabolic requirement of cells [27]. A previous study has shown that a glucose transporter can be a novel and attractive therapeutic target for HCC [28]. In the current study, we found that SGLT1 was highly expressed in HCC. Our findings support the notion that SGLT1 can be a promising intervention target for HCC.
Multiple molecules are involved in the regulation of tumor glucose metabolism. Investigating the factors that control cancer cell aerobic glycolysis could not only advance the knowledge of the regulation of glucose metabolism in cancer cells but also provide new approaches for cancer therapy. Many factors have been shown to regulate or affect aerobic glycolysis, oxidative phosphorylation, pentose phosphate pathway, and glutamine metabolism. The well-studied PI3K/AKT pathway [29] stimulates glycolysis by increasing glucose transporter expression and membrane translocation and by phosphorylating the key enzymes of glycolysis [30]. Hypoxia-inducible factor-1a (HIF-1a) increases the glycolytic capacity by promoting the transcription of genes encoding glucose transporters and key enzyme of glycolysis [31]. FLIPL is aberrantly expressed in HCC, and plays an important role in tumor progression [11]. There is evidence showing that FLIPL up-regulates AKT [32] and HIF-1 [19] expression. Considering that AKT/HIF-1 regulate glucose transporter expression, we cannot rule out that FLIPL might regulate glycolysis through the HIF-1 or AKT signaling pathways. Further studies are needed to explore this possibility.
As tumor cells proliferate, they consume large amounts of nutrients, particularly glucose. However, the glucose concentration of cancer tissues is frequently 3-6-fold lower than normal tissue [33]. Therefore, tumor cells must have the capacity to adapt to low levels of nutrients [8]. Aerobic glycolysis is less efficient than oxidative phosphorylation for generating ATP. However, this less efficient ATP generation is beneficial to cell proliferation [34]. Different tumor cells exhibit diverse responses to glucose limitation, and the glucose utilization and mitochondrial function determine the sensitivity to glucose limitation. Increased glucose uptake promotes glycolysis, thereby increasing the capacity to utilize glucose. GLUTs transport glucose into cells depending on the glucose concentration gradient across the cell membrane. In contrast, SGLTs do not rely on the glucose concentration [35]. In prostate cancer, SGLT1 enhanced the survival of tumor cells in low glucose conditions by increasing the intracellular glucose level [36]. We found that SGLT1 also promotes the survival of HCC cells exposed to low glucose levels. Our findings indicate that SGLT1 plays an important role in the adaptation of HCC cells to glucose limitation.
The pathology of HCC is complicated due to the involvement of multiple and distinct signaling pathways in the initiation and development of HCC. This study revealed that FLIPL not only promotes cell survival by modulating cell apoptosis, autophagy, and necrosis [37] but also affects glucose metabolism through the regulation of SGLT1 in HCC. Our findings suggest that FLIPL could be a potential intervention target for HCC therapy in the future.
Conclusions
In the current study, we provided evidence strongly suggesting that FLIPL, an apoptosis suppressor, is positively correlated with SGLT1 expression in HCC. This correlation is associated with FLIPL-induced glucose uptake and glucose-dependent aerobic glycolysis. We have further demonstrated that FLIPL plays a significant role in the tolerance ability of HCC to low glucose, during which FLIPL interacts with and protects SGLT1 from proteasomal degradation. Although the contribution of FLIPL to tumor onset and progression is complicated, our findings first identified that FLIPL is involved in HCC energy metabolism and is a promising therapeutic target for future intervention for HCC.
Acknowledgements
This work was supported by the grants from National Natural Science Foundation of China (81172287, 81372859, and 81471110), China Postdoctoral Science Foundation funded project (2015 T81094 and 2014 M562599), Youth Science and Technology New Star Project of Shaanxi Province (2015KJXX-49), and Science and Technology Project of Liaoning Province (2015020412).
Additional files
Table S1. Statistical results for FLIPL expression in the adjacent normal specimens of 79 HCC. Table S2. Statistical results of SGLT1 expression in the adjacent normal specimens of 79 HCC. (DOCX 17 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors participated in the conception and design of this study. SXL, JDY, and CC performed most of the experiments. JCS and LY analyzed the data. HLT, TY, and AC provided formalin-fixed, paraffin-embedded, archived patient materials and conducted pathologic reviews and clinical data evaluations. HDZ reviewed the data. XLD and YL initiated the project and wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Yan Li, Phone: +86-29-84774516-8024, FAX: +86-29-84774513, Email: liyann@fmmu.edu.cn.
Xilin Du, Phone: +86-29-84777732, FAX: +86-29-84777731, Email: duxl0705@fmmu.edu.cn, Email: Dxlin0715@163.com.
References
- 1.Roayaie S, Jibara G, Tabrizian P, Park JW, Yang J, Yan L, Schwartz M, Han G, Izzo F, Chen M, et al. The role of hepatic resection in the treatment of hepatocellular cancer. Hepatology. 2015;62:440–51. doi: 10.1002/hep.27745. [DOI] [PubMed] [Google Scholar]
- 2.Hayashi H, Beppu T, Okabe H, Kuroki H, Nakagawa S, Imai K, Nitta H, Chikamoto A, Ishiko T, Baba H. Functional assessment versus conventional volumetric assessment in the prediction of operative outcomes after major hepatectomy. Surgery. 2015;157:20–6. doi: 10.1016/j.surg.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 3.Warburg O, Wind F, Negelein E. The metabolism of tumors in the Body. J Gen Physiol. 1927;8:519–30. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 2008;13:472–82. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008;49(Suppl 2):24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- 7.Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 9.Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–12. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amann T, Kirovski G, Bosserhoff AK, Hellerbrand C. Analysis of a promoter polymorphism of the GLUT1 gene in patients with hepatocellular carcinoma. Mol Membr Biol. 2011;28:182–6. doi: 10.3109/09687688.2011.554447. [DOI] [PubMed] [Google Scholar]
- 11.Du X, Bao G, He X, Zhao H, Yu F, Qiao Q, Lu J, Ma Q. Expression and biological significance of c-FLIP in human hepatocellular carcinomas. J Exp Clin Cancer Res. 2009;28:24. doi: 10.1186/1756-9966-28-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozturk S, Schleich K, Lavrik IN. Cellular FLICE-like inhibitory proteins (c-FLIPs): fine-tuners of life and death decisions. Exp Cell Res. 2012;318:1324–31. doi: 10.1016/j.yexcr.2012.01.019. [DOI] [PubMed] [Google Scholar]
- 13.Shirley S, Micheau O. Targeting c-FLIP in cancer. Cancer Lett. 2013;332:141–50. doi: 10.1016/j.canlet.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Hu WH, Johnson H, Shu HB. Activation of NF-kappaB by FADD, Casper, and caspase-8. J Biol Chem. 2000;275:10838–44. doi: 10.1074/jbc.275.15.10838. [DOI] [PubMed] [Google Scholar]
- 15.Kataoka T, Budd RC, Holler N, Thome M, Martinon F, Irmler M, Burns K, Hahne M, Kennedy N, Kovacsovics M, et al. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr Biol. 2000;10:640–8. doi: 10.1016/S0960-9822(00)00512-1. [DOI] [PubMed] [Google Scholar]
- 16.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–5. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 17.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11:1355–62. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu YH, Kuo WC, Wu YJ, Yang KT, Chen ST, Jiang ST, Gordy C, He YW, Lai MZ. Participation of c-FLIP in NLRP3 and AIM2 inflammasome activation. Cell Death Differ. 2014;21:451–61. doi: 10.1038/cdd.2013.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishioka T, Katayama R, Kikuchi R, Nishimoto M, Takada S, Takada R, Matsuzawa S, Reed JC, Tsuruo T, Naito M. Impairment of the ubiquitin-proteasome system by cellular FLIP. Genes Cells. 2007;12:735–44. doi: 10.1111/j.1365-2443.2007.01087.x. [DOI] [PubMed] [Google Scholar]
- 20.Marini ES, Giampietri C, Petrungaro S, Conti S, Filippini A, Scorrano L, Ziparo E. The endogenous caspase-8 inhibitor c-FLIPL regulates ER morphology and crosstalk with mitochondria. Cell Death Differ. 2015;22:1131–43. doi: 10.1038/cdd.2014.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Huang F, Wang J, Luo H, Wang Z. 2-DG-Regulated RIP and c-FLIP Effect on Liver Cancer Cell Apoptosis Induced by TRAIL. Med Sci Monit. 2015;21:3442–8. doi: 10.12659/MSM.895034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4 doi: 10.1038/cddis.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferreira LM. Cancer metabolism: the Warburg effect today. Exp Mol Pathol. 2010;89:372–80. doi: 10.1016/j.yexmp.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. 2012;24:650–4. doi: 10.1097/CCO.0b013e328356da72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Massari F, Ciccarese C, Santoni M, Brunelli M, Piva F, Modena A, Bimbatti D, Fantinel E, Santini D, Cheng L, et al. Metabolic alterations in renal cell carcinoma. Cancer Treat Rev. 2015. [DOI] [PubMed]
- 26.Shuch B, Linehan WM, Srinivasan R. Aerobic glycolysis: a novel target in kidney cancer. Expert Rev Anticancer Ther. 2013;13:711–9. doi: 10.1586/era.13.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parikh U, Marcus C, Sarangi R, Taghipour M, Subramaniam RM. FDG PET/CT in pancreatic and hepatobiliary carcinomas: value to patient management and patient outcomes. PET Clin. 2015;10:327–43. doi: 10.1016/j.cpet.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–46. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 29.Paplomata E, O'Regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol. 2014;6:154–66. doi: 10.1177/1758834014530023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–9. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 31.Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–6. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quintavalle C, Incoronato M, Puca L, Acunzo M, Zanca C, Romano G, Garofalo M, Iaboni M, Croce CM, Condorelli G. c-FLIPL enhances anti-apoptotic Akt functions by modulation of Gsk3beta activity. Cell Death Differ. 2010;17:1908–16. doi: 10.1038/cdd.2010.65. [DOI] [PubMed] [Google Scholar]
- 33.Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, Kinoshita T, Saito N, Ochiai A, Tomita M, et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009;69:4918–25. doi: 10.1158/0008-5472.CAN-08-4806. [DOI] [PubMed] [Google Scholar]
- 34.Lu J, Tan M, Cai Q. The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015;356:156–64. doi: 10.1016/j.canlet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91:733–94. doi: 10.1152/physrev.00055.2009. [DOI] [PubMed] [Google Scholar]
- 36.Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ, Hung MC. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13:385–93. doi: 10.1016/j.ccr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He MX, He YW. A role for c-FLIP(L) in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes. Cell Death Differ. 2013;20:188–97. doi: 10.1038/cdd.2012.148. [DOI] [PMC free article] [PubMed] [Google Scholar]