

Abstract
Although several animal models have been developed to study human pulmonary fibrosis, lack of a perfect model has raised the need for various animal models of pulmonary fibrosis. In this study, we evaluated the pulmonary effect of polyhexamethyleneguanidine phosphate instillation into the lungs of mice to determine the potential of these mice as a murine model of pulmonary fibrosis. Intratracheal instillation of polyhexamethyleneguanidine phosphate induced severe lung inflammation manifested by the infiltration of mononuclear cells and neutrophils and increased production of IL-6, TNF-α, CCL2 and CXCL1. The lung inflammation gradually increased until 28 days after polyhexamethyleneguanidine phosphate exposure, and increases of collagen deposition and TGF-β production, which are indicators of pulmonary fibrosis, were seen. Our study showed that intratracheal instillation of polyhexamethyleneguanidine phosphate induces pulmonary inflammation and fibrosis in mice.
Keywords: lung inflammation, mice, polyhexamethyleneguanidine phosphate, pulmonary fibrosis
Introduction
Idiopathic pulmonary fibrosis (IPF), the most common type of interstitial lung disease (ILD) with unknown etiology, affects 132,000–200,000 people in the USA1. Approximately 50,000 new cases are diagnosed and as many as 40,000 Americans die from IPF each year1. Pulmonary fibrosis (PF) is the formation of excessive fibrous connective tissue in the lung2. A wide range of causes, including viral infections and exposure to radiotherapy, chemotherapeutic drugs and aerosolized environmental toxins, may induce PF as a secondary effect3,4,5. Although the relative importance of inflammation in the progression of pulmonary fibrosis has been debated, many forms of PF are believed to be induced by inflammatory responses6. In the early stage after tissue damage, epithelial cells release inflammatory mediators that allow recruitment of inflammatory cells such as neutrophils and macrophages to the site of injury. The recruited inflammatory cells produce a variety of cytokines and chemokines that trigger fibroblast proliferation and recruitment. Once fibroblasts become activated, they transform into α-smooth muscle actin–expressing myofibroblasts that secrete extracellular matrix (ECM) components leading to fibrosis when the wound is severe, the tissue-damaging irritant persists, or the repair process becomes deregulated2.
Although anti-inflammatory medicine has been suggested as a potential therapy for PF based on evidence showing involvement of inflammation in PF, it is often ineffective7. Moreover, the limited knowledge of PF pathogenesis leads to lack of effective treatment for PF8. To understand the pathogenesis of PF and develop a successful therapy, an animal model that can produce the characteristic features of human PF is necessary. To date, several animal models have been developed for PF, and these models have been used to identify cells and mediators involved in the process of PF. However, these animal models cannot represent human PF appropriately, and the need for various animal models of PF has been raised9. Recent case reports showed that lung injury and fibrosis can occur as a result of inhaling a humidifier disinfectant, polyhexamethyleneguanidine phosphate (PHMG-P)10,11,12,13. PHMG-P, a member of the polymeric guanidine family, is widely used as an antimicrobial additive in paper and plastics and as a disinfectant for sanitation in food processing plants14. It has broad-spectrum activity against Gram-positive and Gram-negative bacteria, fungi, yeasts and human immunodeficiency virus by disrupting the cell membrane15,16,17,18. Although PHMG-P is known as a disinfectant that is noncorrosive and nontoxic to humans and animals16, 17, 19, a recent report showed that more than 12,500 patients were admitted to hospital owing to drinking illegal cheap “vodka” that was mixed with PHMG, and 9.4% of the patients died20.
In the present study, we evaluated the pulmonary effect of PHMG-P instillation into the mouse lung to determine potential of these mice as a murine model of PF.
Materials and Methods
Mice
Six-week-old female wild-type C57BL/6 mice were purchased from Koatech (Pyeongtaek, Republic of Korea). All animal experiments were approved and followed the regulations of the Institutional Animal Care and Use Committee (IACUC) of Konyang University (Daejeon, Republic of Korea).
PHMG-P inhalation
Intratracheal instillation of PHMG-P (1.5 mg/kg, SK Chemicals, Seoul, Republic of Korea) into mice was performed as described previously21. Mice were euthanized with excessive anesthesia 7, 14 or 28 days after instillation, and lung tissues were collected for further analysis (Fig. 1A).
Fig. 1.
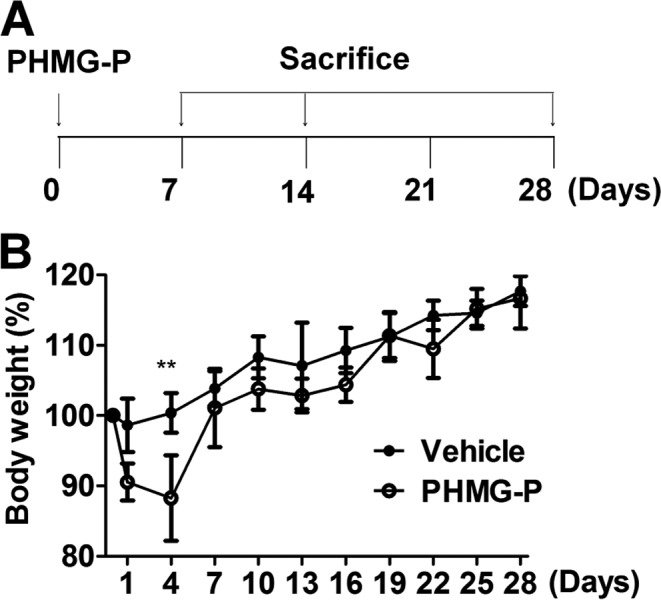
Body weight changes caused by PHMG-P instillation in mice. A schematic diagram of the experimental design described in Materials and Methods (A). Mice were intratracheally instilled with PHMG-P (1.5 mg/kg) or PBS, and body weight changes were monitored daily (B). N = 5 mice per group. *P<0.05; **P<0.01.
Histopathology and Masson’s trichrome stain
The left lung tissues from each mouse were fixed with 10% neutral-buffered formalin for 24 hours and embedded in paraffin. Tissue sections (2 μm thick) were prepared and stained with hematoxylin and eosin (HE) or Masson’s trichrome and then examined under a light microscope.
Measurement of cytokines and chemokines
The right lung tissues from each mouse were homogenized, and the supernatant was obtained after centrifugation. The concentrations of IL-6, TNF-α, CCL2, CXCL1, and TGF-β in the supernatant were determined using commercial ELISA kits (IL-6, TNF-α, CCL2 and CXCL1, R&D System, Minneapolis, MN, USA; TGF-β, eBioscience Inc., San Diego, CA, USA) according to the manufacturers’ manuals.
Statistical analysis
The differences in mean values among different groups were assessed, and all data are expressed as the mean ± SD. All statistical analyses were performed by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test or by the unpaired Student’s test using the GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA, USA). Values of P<0.05 were considered statistically significant.
Results
Clinical signs and body weight changes caused by PHMG-P exposure in mice
To determine the optimal dosages for the experiment, mice were exposed to various dosages (0, 1.5, 3 and 6 mg/kg) of PHMG-P, and body weight loss and survival rate were monitored up to 30 days after PHMG-P exposure. All mice (N ≥ 5 per group) except for mice exposed to 6 mg/kg of PHMG-P (survival rate of about 40%) survived to the end of the 30-day period (data not shown). However, exposure to 3 mg/kg of PHMG-P induced nearly 30% body weight loss in mice (data not shown). According to the recommendation of our IACUC, we exposed mice to 1.5 mg/kg of PHMG-P in the subsequent. Mild physical signs of ill health such as body weight loss, decreased movement and ruffled fur were seen in PHMG-P-exposed mice until 4 days after exposure. These clinical signs began to disappear 4 days after exposure (data not shown). The loss of body weight prominent 1 day after exposure and continued until 4 days after exposure, and then most mice gradually regained their body weight until it was in a range comparable to that of vehicle-exposed mice (vehicle = phosphate buffered saline [PBS]) at 7 days after exposure (Fig. 1B).
Histopathology of lung tissue from PHMG-P-exposed mice
Histopathologic examination of lung tissue was performed at 7, 14 and 28 days after PHMG-P exposure. PHMG-P exposure induced the infiltration of polymorphonuclear (PMN) cells and macrophages into the alveolar sac and interstitium of the peribronchiolar area and activation of pneumocytes from 7 days after PHMG-P exposure with 100% incidence (Fig. 2B). The severity of lung lesion gradually increased until 28 days after PHMG-P exposure (Fig. 2C-E). However, neither the infiltration of inflammatory cells nor the activation of pneumocytes was seen in lung tissue from vehicle-treated mice (Fig. 2A).
Fig. 2.

Histopathology of lung caused by PHMG-P instillation in mice. At 7, 14 and 28 days after PHMG-P exposure, lung tissues were processed and stained with hematoxylin and eosin for histopathologic examination. Representative images of each group are shown at a magnification of 400×. Small quadrangles within each image are at low magnification (40×). (A) Vehicle, (B) 7 days, (C) 14 days and (D) 28 days after PHMG-P exposure. (E) The severity of the inflammation in the lung is presented as the mean ± SD. (*P<0.05; ***P<0.001).
Production of cytokines and chemokines in lung tissue caused by PMHG-P exposure
The levels of IL-6, TNF-α, CCL1 and CCL2 in the supernatants of lung homogenates were measured 7, 14 and 28 days after PHMG-P exposure. All cytokines and chemokines measured gradually increased until 28 days after PHMG-P exposure (Fig. 3). Compared with vehicle exposure, PHMG-P exposure significantly increased the levels of TNF-α and CCL2 at 7 and 14 days after treatment respectively (Fig. 3A-D), and the levels of CXCL1 and IL-6 were significantly higher at 28 days after PHMG-P exposure (Fig. 3B-C).
Fig. 3.

Production of cytokines and chemokines caused by PHMG-P instillation in the mouse lung. Lung homogenates from vehicle-exposed mice or PHMG-P-treated mice at 7, 14 and 28 days after exposure were collected, and the concentrations of IL-6, TNF-α, CCL2 and CXCL1 in supernatants were determined by ELISA. Vehicle-treated (N = 3), PHMG-P-treated groups (N = 5) mice. (*P<0.05; **P<0.01)
Pulmonary fibrosis caused by PHMG-P inhalation
We evaluated the level of collagen deposition in the lung, a hall marker of lung fibrosis, by Masson’s trichrome staining. The level of Masson’s trichrome positivity was increased from 14 days after PHMG-P exposure and became stronger at 28 days after PHMG-P exposure (Fig. 4A-E). Only 2 of 5 mice showed slight Masson’s trichrome positivity at 7 days after PHMG-P exposure (Fig. 4B, E). Moreover, the level of TGF-β, which is known to play an important role in lung fibrosis22, in lung homogenates was gradually increased after exposure to PHMG-P (Fig. 5).
Fig. 4.

The degree of collagen deposition in the lung after PHMG-P instillation. At 7, 14 and 28 days after PHMG-P exposure, lung tissues were collected. The degree of collagen deposition was assessed by Masson’s trichrome staining. Representative images of each group are shown at a magnification of 400×. (A) Vehicle and (B) 7 days, (C) 14 days and (D) 28 days after PHMG-P exposure. (E) The severity of fibrosis in the lung is presented as the mean ± SD. (**P<0.01; ***P<0.001)
Fig. 5.
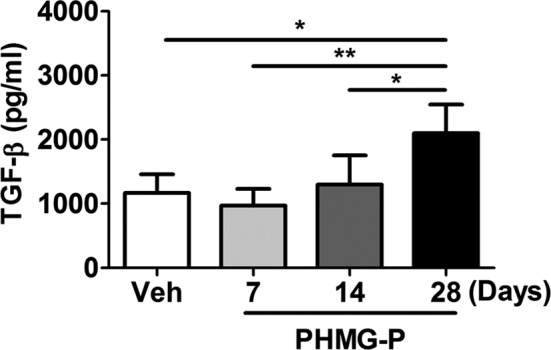
Level of TGF-β in the lung after PHMG-P instillation. At 7, 14 and 28 days after PHMG-P exposure, lung tissues were collected. The level of TGF-β were assessed by ELISA. (A) Vehicle and (B) 7 days, (C) 14 days and (D) 28 days after PHMG-P exposure.
Discussion
Recently, the toxic effects of PHMG-P, including pulmonary complications, were reported in epidemiological studies23. However, the mechanism of toxicity caused by PHMG-P was not fully elucidated, although one study using human cells and zebrafish embryos reported that PHMG-P showed acute cardiovascular toxicity by inducing severe inflammation, atherogenesis and aging, with embryo toxicity24. In our mouse study, we observed severe lung inflammation manifested by the infiltration of mononuclear cells and neutrophils and the elevated production of IL-6, TNF-α, CCL2 and CXCL1 after intratracheal inhalation of PHMG-P. The lung inflammation gradually increased up to 28 days after PHMG-P treatment, and increased collagen deposition and TGF-β production were seen. We did not clarify the elaborate mechanism of PHMG-P-induced lung inflammation in this study. However, the production of reactive oxygen species caused by PHMG-P treatment demonstrated in previous studies with zebrafish embryos11, 24 could be key, even in the case of lung inflammation caused by PHMG-P inhalation resulting in pulmonary fibrosis.
Inflammatory cytokines and chemokines have been considered to play an important role in both the initiation and progression of some forms of pulmonary fibrosis23. Clinical samples from patients with pulmonary fibrosis displayed elevated levels of TNF-α, and mice genetically modified to overexpress TNF-α in the lung developed progressive pulmonary fibrosis25,26,27. IL-1β also contributes to the progression of pulmonary fibrosis by crosstalking with TNF-α or inducing neutrophil chemoattractants16, 28. Moreover, T helper cell-associated cytokines also play important role in pulmonary fibrosis29,30,31. Whereas a Th1-associated cytokine, IFN-γ, has been known to inhibit fibrosis, Th2- and Th17-associated cytokines, IL-4, IL-5 and IL13 (Th2) and IL-17A (Th17), respectively, are linked to pulmonary fibrosis30, 31. Both IL-4 and IL-13 have a profibrotic function by activating myofibroblasts32, 33, and IL-5 promotes pulmonary fibrosis by recruiting eosinophils that produce profibrotic mediators34, 35. An experiment using mice with bleomycin-induced fibrosis showed that IL-17A and IL-23, an IL-17A inducing cytokine, are important for the development of pulmonary fibrosis30. In addition, chemokines and their signaling receptors, including CCL2, CXCL12, CCL12 and CXCR2, promote pulmonary fibrosis by recruiting mononuclear cells, neutrophils or collagen-secreting fibrocytes to the lung36,37,38. The increased production of IL-6, TNF-α, CCL2 and CXCL1 after PHMG-P treatment in our study might function as an initiator of pulmonary fibrosis.
Intratracheal inhalation of PHMG-P induced the production of TGF-β, which has been known to play a central role in the pathogenesis of pulmonary fibrosis by promoting the activation, proliferation and differentiation of collagen-producing myofibroblasts39, 40. TGF-β stimulates production of collagens and other extracellular matrix proteins in mesenchymal cells through a classic or nonclassic pathway41. In the classic TGF-β pathway, binding of TGF-β to the TGF-β type II receptor (TGF-βRII) recruits a TGF-β type I receptor (TGF-βRI), activating its phosphorylation. Activated TGF-βRI phosphorylates Smad2 and Smad3, which then form a complex with Smad4, resulting in their transport to the nucleus. The Smad complex binds to specific DNA-binding sites in the promoter regions of target genes such as the 1 and 2 chains of type I collagen or extracellular matrix regulatory proteins and modulates their transcriptional activity39, 42. In the non-classic TGF-β pathway, TGF-βRI phosphorylates cellular Abelson nonreceptor kinase (c-Abl), resulting in activation of Smad1, early growth response gene (EgR), or protein kinase C-δ (PKC-δ), all of which contribute to the fibrotic response43. Although collagen deposition was significantly increased from 14 days after PHMG-P exposure, the level of TGF-β was comparable with the vehicle-exposed group 14 days after exposure and significantly increased 28 days after exposure. This could be explained by the difference in sensitivity between the two methods of measurement used.
Several agents, including bleomycin, asbestos, silica, FITC and radiation, have been known to induce pulmonary fibrosis in mouse models36. Among them, bleomycin is the best characterized one in studying murine pulmonary fibrosis44. Intratracheal treatment of bleomycin results in direct damage of alveolar epithelial cells followed by alveolar infiltration of neutrophils19. DNA strand breakage and oxidant injury45 are thought to be a main mechanism of bleomycin-induced alveolar cell damage. Subsequently, fibroblast proliferation and synthesis of extracellular matrix appear46. During development of pulmonary fibrosis caused by bleomycin, CCL2 recruits inflammatory cells, including monocytes, lymphocytes and fibrocytes, and TGF-β stimulates the production of ECM47, 48. The progression of pulmonary fibrosis in our PHMG-P model was very similar to that in the bleomycin model.
In the current study, we characterized PHMG-P-induced lung inflammation and pulmonary fibrosis in mice, and our observations should provide basic data concerning the toxicity of PHMG-P. As we did not clarify the mechanism of PHMG-P-induced lung inflammation in detail, further study is necessary to clarify it in detail.
Acknowledgments
This study was supported by the Korea Environmental Industry and Technology Institute (grant number 2012001370006).
Footnotes
Disclosure of Potential Conflicts of Interest: The authors declare that they have no financial or commercial conflicts of interest.
References
- 1.Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, and Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 176: 277–284. 2007. [DOI] [PubMed] [Google Scholar]
- 2.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 208: 1339–1350. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Denham JW, and Hauer-Jensen M. The radiotherapeutic injury—a complex ‘wound’. Radiother Oncol. 63: 129–145. 2002. [DOI] [PubMed] [Google Scholar]
- 4.Fubini B, and Hubbard A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic Biol Med. 34: 1507–1516. 2003. [DOI] [PubMed] [Google Scholar]
- 5.Kelly BG, Lok SS, Hasleton PS, Egan JJ, and Stewart JP. A rearranged form of Epstein-Barr virus DNA is associated with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 166: 510–513. 2002. [DOI] [PubMed] [Google Scholar]
- 6.Crystal RG, Bitterman PB, Mossman B, Schwarz MI, Sheppard D, Almasy L, Chapman HA, Friedman SL, King TE, Jr , Leinwand LA, Liotta L, Martin GR, Schwartz DA, Schultz GS, Wagner CR, and Musson RA. Future research directions in idiopathic pulmonary fibrosis: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med. 166: 236–246. 2002. [DOI] [PubMed] [Google Scholar]
- 7.Chmiel JF, and Konstan MW. Anti-inflammatory medications for cystic fibrosis lung disease: selecting the most appropriate agent. Treat Respir Med. 4: 255–273. 2005. [DOI] [PubMed] [Google Scholar]
- 8.Korfei M, Schmitt S, Ruppert C, Henneke I, Markart P, Loeh B, Mahavadi P, Wygrecka M, Klepetko W, Fink L, Bonniaud P, Preissner KT, Lochnit G, Schaefer L, Seeger W, and Guenther A. Comparative proteomic analysis of lung tissue from patients with idiopathic pulmonary fibrosis (IPF) and lung transplant donor lungs. J Proteome Res. 10: 2185–2205. 2011. [DOI] [PubMed] [Google Scholar]
- 9.Moore BB, and Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 294: L152–L160. 2008. [DOI] [PubMed] [Google Scholar]
- 10.Hong SB, Kim HJ, Huh JW, Do KH, Jang SJ, Song JS, Choi SJ, Heo Y, Kim YB, Lim CM, Chae EJ, Lee H, Jung M, Lee K, Lee MS, and Koh Y. Korean Unknown Severe Respiratory Failure Collaborative Korean Study Group of Respiratory Failure A cluster of lung injury associated with home humidifier use: clinical, radiological and pathological description of a new syndrome. Thorax. 69: 694–702. 2014. [DOI] [PubMed] [Google Scholar]
- 11.Kim HJ, Lee MS, Hong SB, Huh JW, Do KH, Jang SJ, Lim CM, Chae EJ, Lee H, Jung M, Park YJ, Park JH, Kwon GY, Gwack J, Youn SK, Kwon JW, Yang BG, Jun BY, Kim Y, Cheong HK, Chun BC, Kim H, Lee K, and Koh Y. A cluster of lung injury cases associated with home humidifier use: an epidemiological investigation. Thorax. 69: 703–708. 2014. [DOI] [PubMed] [Google Scholar]
- 12.Lee JH, Kim YH, and Kwon JH. Fatal misuse of humidifier disinfectants in Korea: importance of screening risk assessment and implications for management of chemicals in consumer products. Environ Sci Technol. 46: 2498–2500. 2012. [DOI] [PubMed] [Google Scholar]
- 13.Song JA, Park HJ, Yang MJ, Jung KJ, Yang HS, Song CW, and Lee K. Polyhexamethyleneguanidine phosphate induces severe lung inflammation, fibrosis, and thymic atrophy. Food Chem Toxicol. 69: 267–275. 2014. [DOI] [PubMed] [Google Scholar]
- 14.Rosin M, Welk A, Bernhardt O, Ruhnau M, Pitten FA, Kocher T, and Kramer A. Effect of a polyhexamethylene biguanide mouthrinse on bacterial counts and plaque. J Clin Periodontol. 28: 1121–1126. 2001. [DOI] [PubMed] [Google Scholar]
- 15.Broxton P, Woodcock PM, and Gilbert P. A study of the antibacterial activity of some polyhexamethylene biguanides towards Escherichia coli ATCC 8739. J Appl Bacteriol. 54: 345–353. 1983. [DOI] [PubMed] [Google Scholar]
- 16.Krebs FC, Miller SR, Ferguson ML, Labib M, Rando RF, and Wigdahl B. Polybiguanides, particularly polyethylene hexamethylene biguanide, have activity against human immunodeficiency virus type 1. Biomed Pharmacother. 59: 438–445. 2005. [DOI] [PubMed] [Google Scholar]
- 17.Müller G, and Kramer A. Effect of selected wound antiseptics on adult articular cartilage (bovine sesamoid bone) in the presence of Escherichia coli and Staphylococcus aureus. J Orthop Res. 23: 127–133. 2005. [DOI] [PubMed] [Google Scholar]
- 18.Zhou ZX, Wei DF, Guan Y, Zheng AN, and Zhong JJ. Damage of Escherichia coli membrane by bactericidal agent polyhexamethylene guanidine hydrochloride: micrographic evidences. J Appl Microbiol. 108: 898–907. 2010. [DOI] [PubMed] [Google Scholar]
- 19.Li Q, Park PW, Wilson CL, and Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 111: 635–646. 2002. [DOI] [PubMed] [Google Scholar]
- 20.Ostapenko YN, Brusin KM, Zobnin YV, Shchupak AY, Vishnevetskiy MK, Sentsov VG, Novikova OV, Alekseenko SA, Lebed’ko OA, and Puchkov YB. Acute cholestatic liver injury caused by polyhexamethyleneguanidine hydrochloride admixed to ethyl alcohol. Clin Toxicol (Phila). 49: 471–477. 2011. [DOI] [PubMed] [Google Scholar]
- 21.Kim SN, Lee J, Yang HS, Cho JW, Kwon S, Kim YB, Her JD, Cho KH, Song CW, and Lee K. Dose-response Effects of Bleomycin on Inflammation and Pulmonary Fibrosis in Mice. Toxicol Rev. 26: 217–222. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolb M, Margetts PJ, Anthony DC, Pitossi F, and Gauldie J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. 107: 1529–1536. 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bringardner BD, Baran CP, Eubank TD, and Marsh CB. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid Redox Signal. 10: 287–301. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim JY, Kim HH, and Cho KH. Acute cardiovascular toxicity of sterilizers, PHMG, and PGH: severe inflammation in human cells and heart failure in zebrafish. Cardiovasc Toxicol. 13: 148–160. 2013. [DOI] [PubMed] [Google Scholar]
- 25.Hasegawa N, Oka Y, Nakayama M, Berry GJ, Bursten S, Rice G, and Raffin TA. The effects of post-treatment with lisofylline, a phosphatidic acid generation inhibitor, on sepsis-induced acute lung injury in pigs. Am J Respir Crit Care Med. 155: 928–936. 1997. [DOI] [PubMed] [Google Scholar]
- 26.Miyazaki Y, Araki K, Vesin C, Garcia I, Kapanci Y, Whitsett JA, Piguet PF, and Vassalli P. Expression of a tumor necrosis factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J Clin Invest. 96: 250–259. 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piguet PF, Ribaux C, Karpuz V, Grau GE, and Kapanci Y. Expression and localization of tumor necrosis factor-alpha and its mRNA in idiopathic pulmonary fibrosis. Am J Pathol. 143: 651–655. 1993. [PMC free article] [PubMed] [Google Scholar]
- 28.Kolb M, Margetts PJ, Sime PJ, and Gauldie J. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am J Physiol Lung Cell Mol Physiol. 280: L1327–L1334. 2001. [DOI] [PubMed] [Google Scholar]
- 29.Simonian PL, Roark CL, Wehrmann F, Lanham AK, Diaz del Valle F, Born WK, O’Brien RL, and Fontenot AP. Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. J Immunol. 182: 657–665. 2009. [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, and Wynn TA. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med. 207: 535–552. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 4: 583–594. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rankin JA, Picarella DE, Geba GP, Temann UA, Prasad B, DiCosmo B, Tarallo A, Stripp B, Whitsett J, and Flavell RA. Phenotypic and physiologic characterization of transgenic mice expressing interleukin 4 in the lung: lymphocytic and eosinophilic inflammation without airway hyperreactivity. Proc Natl Acad Sci USA. 93: 7821–7825. 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, and Elias JA. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 103: 779–788. 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho JY, Miller M, Baek KJ, Han JW, Nayar J, Lee SY, McElwain K, McElwain S, Friedman S, and Broide DH. Inhibition of airway remodeling in IL-5-deficient mice. J Clin Invest. 113: 551–560. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reiman RM, Thompson RW, Feng CG, Hari D, Knight R, Cheever AW, Rosenberg HF, and Wynn TA. Interleukin-5 (IL-5) augments the progression of liver fibrosis by regulating IL-13 activity. Infect Immun. 74: 1471–1479. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore BB, Kolodsick JE, Thannickal VJ, Cooke K, Moore TA, Hogaboam C, Wilke CA, and Toews GB. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 166: 675–684. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, and Strieter RM. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 114: 438–446. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith RE, Strieter RM, Phan SH, Lukacs NW, Huffnagle GB, Wilke CA, Burdick MD, Lincoln P, Evanoff H, and Kunkel SL. Production and function of murine macrophage inflammatory protein-1 alpha in bleomycin-induced lung injury. J Immunol. 153: 4704–4712. 1994. [PubMed] [Google Scholar]
- 39.Border WA, and Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 331: 1286–1292. 1994. [DOI] [PubMed] [Google Scholar]
- 40.Datta A, Scotton CJ, and Chambers RC. Novel therapeutic approaches for pulmonary fibrosis. Br J Pharmacol. 163: 141–172. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verrecchia F, and Mauviel A. TGF-beta and TNF-alpha: antagonistic cytokines controlling type I collagen gene expression. Cell Signal. 16: 873–880. 2004. [DOI] [PubMed] [Google Scholar]
- 42.Nathan SD. Therapeutic management of idiopathic pulmonary fibrosis: an evidence-based approach. Clin Chest Med. 27(Suppl 1): S27–S35, vi. 2006. [DOI] [PubMed] [Google Scholar]
- 43.Pannu J, Nakerakanti S, Smith E, ten Dijke P, and Trojanowska M. Transforming growth factor-beta receptor type I-dependent fibrogenic gene program is mediated via activation of Smad1 and ERK1/2 pathways. J Biol Chem. 282: 10405–10413. 2007. [DOI] [PubMed] [Google Scholar]
- 44.Ishikawa H, Takeda K, Okamoto A, Matsuo S, and Isobe K. Induction of autoimmunity in a bleomycin-induced murine model of experimental systemic sclerosis: an important role for CD4+ T cells. J Invest Dermatol. 129: 1688–1695. 2009. [DOI] [PubMed] [Google Scholar]
- 45.Limoli CL, Kaplan MI, Phillips JW, Adair GM, and Morgan WF. Differential induction of chromosomal instability by DNA strand-breaking agents. Cancer Res. 57: 4048–4056. 1997. [PubMed] [Google Scholar]
- 46.Manni ML, Czajka CA, Oury TD, and Gilbert TW. Extracellular matrix powder protects against bleomycin-induced pulmonary fibrosis. Tissue Eng Part A. 17: 2795–2804. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thannickal VJ, Toews GB, White ES, Lynch JP, 3rd , and Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. 55: 395–417. 2004. [DOI] [PubMed] [Google Scholar]
- 48.Vannella KM, Luckhardt TR, Wilke CA, van Dyk LF, Toews GB, and Moore BB. Latent herpesvirus infection augments experimental pulmonary fibrosis. Am J Respir Crit Care Med. 181: 465–477. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]