

Abstract
Background
Neuroinflammation is a key phenomenon in the pathogenesis of many neurodegenerative diseases. Understanding the mechanisms by which brain inflammation is engaged and delineating the key players in the immune response and their contribution to brain pathology is of great importance for the identification of novel therapeutic targets for these devastating diseases. Gaucher disease, the most common lysosomal storage disease, is caused by mutations in the GBA1 gene and is a significant risk factor for Parkinson’s disease; in some forms of Gaucher disease, neuroinflammation is observed.
Methods
An unbiased gene profile analysis was performed on a severely affected brain area of a neurological form of a Gaucher disease mouse at a pre-symptomatic stage; the mouse used for this study, the Gbaflox/flox; nestin-Cre mouse, was engineered such that GBA1 deficiency is restricted to cells of neuronal lineage, i.e., neurons and macroglia.
Results
The 10 most up-regulated genes in the ventral posteromedial/posterolateral region of the thalamus were inflammatory genes, with the gene expression signature significantly enriched in interferon signaling genes. Interferon β levels were elevated in neurons, and interferon-stimulated genes were elevated mainly in microglia. Interferon signaling pathways were elevated to a small extent in the brain of another lysosomal storage disease mouse model, Krabbe disease, but not in Niemann-Pick C or Sandhoff mouse brain. Ablation of the type I interferon receptor attenuated neuroinflammation but had no effect on GD mouse viability.
Conclusions
Our results imply that the type I interferon response is involved in the development of nGD pathology, and possibly in other lysosomal storage diseases in which simple glycosphingolipids accumulate, and support the notion that interferon signaling pathways play a vital role in the sterile inflammation that often occurs during chronic neurodegenerative diseases in which neuroinflammation is present.
Electronic supplementary material
The online version of this article (doi:10.1186/s12974-016-0570-2) contains supplementary material, which is available to authorized users.
Background
Type I interferons (IFNs; Ifnα1-13 and Ifnβ), a large family of structurally related cytokines, are key components of the innate immune response and are the fundamental cellular defense mechanism against viral infection [1]. IFNs are currently used therapeutically for a number of viral diseases, for numerous malignancies, and for a number of chronic inflammatory disorders such as the demyelinating disease and multiple sclerosis [2]. However, even though activation of the type 1 IFN response has been intensely studied as part of the host response to viral and bacterial infection, induction of this response has also been observed in the absence of infection [3]. In the central nervous system (CNS), IFN activation occurs in amyotrophic lateral sclerosis (ALS) [4], in Alzheimer’s disease (AD) [5], during aging [6], in Aicardi-Goutières syndrome (AGS) [7, 8], and upon axonal transection [9]. Activation of the IFN response under sterile conditions has led to a paradigm shift in our understanding of the role of these cytokines and their role in inflammation inasmuch as the type I interferonopathies are now believed to comprise a heterogeneous group of genetically determined diseases characterized by inappropriate activation of the type I IFN response [10].
A wide range of neurodegenerative conditions are characterized by brain inflammation, including Alzheimer’s and Parkinson’s diseases (PD) [11], and the less common lysosomal storage diseases (LSDs) [12–14], which are normally caused by the defective activity of a lysosomal hydrolase. One such LSD is Gaucher disease (GD), caused by mutations in the GBA1 gene, which encodes the lysosomal enzyme, acid-β-glucosidase (glucocerebrosidase, GCase) [15], resulting in the accumulation of the sphingolipid glucosylceramide (GlcCer) and its deacylated form, glucosylsphingosine (GlcSph) [16, 17]. GD is the most common LSD and ~10 % of GD patients worldwide present with neurological symptoms. However, the distinction between neurological and non-neurological forms of the disease has become blurred over the recent years based on the finding that heterozygous mutations in GBA1 are a major risk factor for PD [18]. Thus, understanding the pathological pathways activated upon alteration of GCase activity in the brain might have significant ramifications for understanding more common neurodegenerative diseases such as PD.
The neuronopathic forms of GD, types 2 and 3, are characterized by astrogliosis, neuronophagia (i.e., brain inflammation), and neuronal loss [19, 20]. We now demonstrate elevation of IFNβ levels in neurons and activation of the type 1 IFN response in mouse models of neuronopathic Gaucher disease (nGD), but to a much lower extent, or absent completely, in other LSD mouse models. This discovery was made during an unbiased gene profile analysis of a severely affected brain area of an nGD mouse at a pre-symptomatic stage. The mouse used for this study, the Gbaflox/flox; nestin-Cre mouse [21], was engineered such that GBA1 deficiency is restricted to cells of neuronal lineage, i.e., neurons and macroglia. Robust induction of type I IFN-stimulated genes (ISGs), including pathogen recognition receptors (PRRs) and antiviral genes was observed; the lack of activation of this pathway in other LSDs suggests that GlcCer and/or GlcSph specifically activate the antiviral response. Moreover, our data suggest that a key player in the pathway of programmed necrosis, the protein mixed lineage kinase domain-like (MLKL), is a novel ISG which is induced down-stream to the IFN receptor in cells of myeloid lineage. Together, our data demonstrate that the IFN response can be activated in neuroinflammation under sterile conditions and that this pathway might be involved in nGD pathogenesis, although whether this pathways is detrimental or beneficial remains to be established.
Results
Induction of the type I interferon response
To determine changes in gene expression in the brain of the Gbaflox/flox; nestin-Cre mouse, herein referred to as the Gba−/− mouse, we performed a microarray analysis on the ventral posteromedial/posterolateral (VPM/VPL) region of the thalamus, the area which is most affected in the Gba−/− mouse, at 14 days of age, an age at which the first signs of neuroinflammation and neuronal loss are observed but prior to overt signs of disease [22]. Approximately 10 % of the genes detected by the array were differentially expressed (absolute fold-change ≥1.5, p value ≤0.05), with 907 genes up-regulated in Gba−/− versus Gba+/− mice and 481 genes down-regulated (for a complete list of the differentially-expressed transcripts, see Additional file 1: Table S1). Changes in expression of Gfap, Lgals3, Kcnk4, Sptssb, and Ryr3 were confirmed by quantitative polymerase chain reaction (qPCR) using the same RNA samples used for the microarray analysis, and their fold-changes were ~100, ~230, ~0.35, ~0.35, and ~0.40, respectively. Up-regulated genes were next subjected to gene ontology (GO) analysis [23] (Fig. 1a). Among the up-regulated genes, innate immune response pathways, and surprisingly, type I IFN-related pathways, were highly enriched (Fig. 1a). The robust induction of the immune response, and in particular of the type I IFN response, was confirmed by the fact that the 10 most up-regulated genes were all genes that are induced during neuroinflammation, with most induced by type I IFN (Table 1).
Fig. 1.
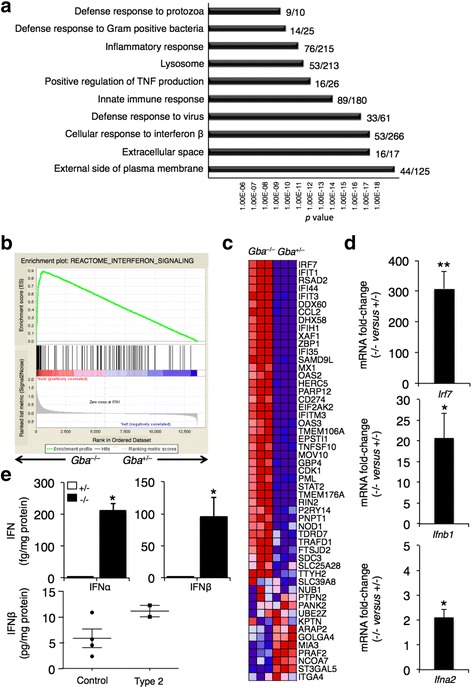
Activation of the type I IFN response in nGD. a The 10 most significantly enriched GO terms for up-regulated genes illustrating enrichment of type I IFN-related pathways. The plot shows the enrichment p values. The number of differentially expressed genes out of the total number of genes included in the term detected by the array are indicated. n = 3, 14-day-old mice. b, c Gene set enrichment analysis (GSEA) of gene expression in Gba −/−compared to Gba +/– mice. b GSEA enrichment plot. False discovery rate (FDR) q value; normalized enrichment scores (NES). c Heat map displaying the relative expression level of proteins in the interferon signaling set, sorted by their fold change. Depicted are the top 51 genes. d qPCR analysis of Irf7 mRNA in the VPM/VPL obtained from 16-day-old mice and of Ifnb1 and Ifna2 mRNA in cortical tissues obtained from 21-day-old mice. **p < 0.001, *p < 0.005. Values are shown as fold change in mRNA levels (Gba −/− versus control) and are means ± s.e.m, n = 3–4. e ELISA analysis of IFNα and IFNβ protein levels in cerebral brain tissue obtained from 21-day-old control and Gba −/− mice. IFNα and IFNβ were not detected in control samples. *p < 0.005. Values are means ± s.e.m, n = 3 (upper panel). IFNβ protein levels in cerebellum of two human type 2 GD patients (n = 2) compared to age-matched control brains (n = 4). p value = 0.06 (lower panel)
Table 1.
Top ten up-regulated genes
| Gene symbol | Gene name | Fold change (−/− vs. +/−) | p value | Referencea |
|---|---|---|---|---|
| Cxcl10* (IP-10) | Chemokine (C-X-C motif) ligand 10 | 86.3 | <0.001 | [68] |
| Usp18* | Ubiquitin specific peptidase 18 | 79.2 | <0.005 | [30] |
| Ifit1* | Interferon-induced protein with tetratricopeptide repeats 1 | 75.2 | <0.001 | [69] |
| Irf7* | Interferon regulatory factor 7 | 71.7 | <0.005 | [30] |
| Cxcl13* | Chemokine (C-X-C motif) ligand 13 | 67.2 | <0.05 | [70] |
| Gpnmb | Glycoprotein (Transmembrane) Nmb | 63.0 | <0.005 | |
| Lcn2 | Lipcalin 2 | 60.1 | <0.005 | |
| Oasl2* | 2′-5′ oligoadenylate synthetase-like 2 | 56.0 | <0.005 | [30] |
| Tgm1 | Transglutaminase 1, K polypeptide | 46.5 | <0.005 | |
| Ifit3* | Interferon-induced protein with tetratricopeptide repeats 3 | 42.1 | <0.001 | [69] |
Type I IFN-induced genes are indicated by asterisks
aReferences refer to studies demonstrating that these genes are induced by type I IFN
Gene set enrichment analysis (GSEA) confirmed that the gene expression signature of the Gba−/− mouse was significantly enriched in IFN signaling genes, with an approximate false discovery rate of zero and a normalized enrichment score of 2.27 (Fig. 1b, c). Innate immunity is the first line of defense against infection and cell damage and is triggered by the recognition of pathogen/danger-associated molecular patterns (PAMPs or DAMPs) by PRRs. Genes encoding various PRRs were elevated in the Gba−/− brain, including Toll-like receptors (Tlr), which encode transmembrane PRRs, other membrane-bound PRRs such as the C-type lectin receptors, Clec7a (Dectin-1) and Clec5a (MDL-1) [24], and the scavenger receptors Cd36 and Msr1 (SR-A1) [25] (Additional file 1: Table S1). Genes encoding cytosolic nucleic-acid sensing PRRs [26] were also up-regulated (Additional file 1: Table S1). PRR stimulation leads to IFN transcription and secretion [27] by interferon regulatory factor (IRF) transcription factors [28], of which Irf7, Irf8, Irf9, Irf1, and Irf5 were up-regulated. Irf7, which is among the most highly-elevated genes (Table 1), is the master regulator of type I IFN signaling, and its expression is further up-regulated by type I IFN [29]. qPCR analysis confirmed that Irf7 was highly induced in the VPM/VPL of Gba−/− mice (Fig. 1d). Both Ifnb1 and Ifna2 mRNA levels were also elevated (~20 and ~2-fold, respectively) in the cerebral cortex of Gba−/− mice (Fig. 1d), another highly affected brain area in nGD mice [22]. Moreover, IFNα and IFNβ were detected by enzyme-linked immunosorbent assay (ELISA) in cerebral tissue from Gba−/− mice, but not from control mice (Fig. 1e). Crucially, levels of IFNβ were also elevated in the two available cerebella of human patients who succumbed to type 2 GD (Fig. 1e).
IFNα and IFNβ bind the Ifnα/β receptor (IFNAR) to induces gene expression via the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway, resulting in expression of a large spectrum of ISGs [27]. Phosphorylated Stat2 was up-regulated in an affected Gba−/− brain area (Fig. 2a), and its levels were increased in both MAC2-positive (i.e. activated microglia/macrophages) and MAC2-negative cells (Fig. 2a). P-Stat2 rarely co-localized with the neuronal nuclear marker NeuN, but neuronal cell bodies were frequently encircled by P-Stat2 staining, likely indicating P-Stat2 elevation in phagocytic cells (Fig. 2a). Finally, P-Stat2 staining was also detected in the nuclei of cells that were not labeled with the neuronal marker, NeuN (Fig. 2a).
Fig. 2.
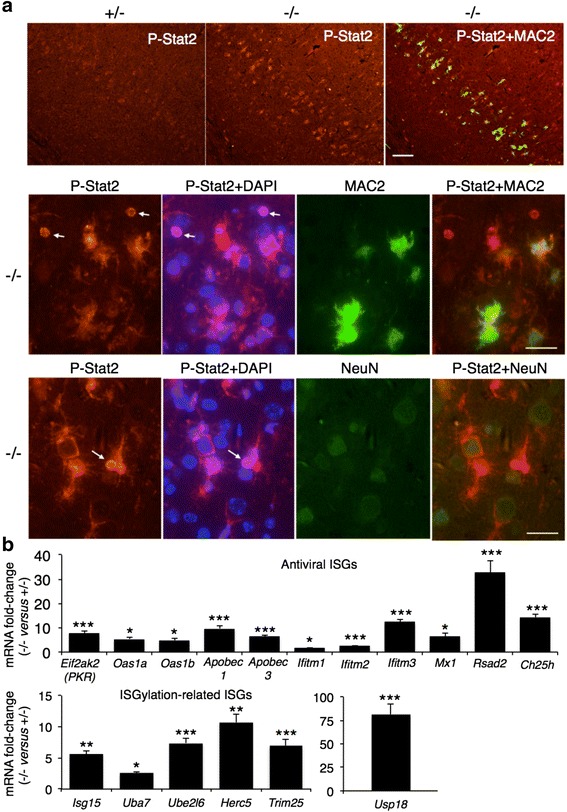
Induction of the JAK/STAT pathway in nGD. a P-Stat2 is increased in the nGD brain. Double immunofluorescence of cortical layer V in 16-day-old mice using anti-P-Stat2 (red) and anti-MAC2 (green, upper and middle panels) or anti-NeuN (green, lower panel) antibodies and the nuclear marker, DAPI (blue). Arrows indicate nuclear staining of P-Stat2. Scale bar, 25 μm. Results are representative of three experiments. b Microarray analysis of ISG expression in the nGD brain. Data is presented as fold change of mRNA levels (Gba −/− versus Gba +/–). Values are means ± s.e.m, n = 3. *p < 0.05, **p < 0.05, ***p < 0.005. Janus kinase (JAK); signal transducer and activator of transcription (STAT)
A number of ISGs down-stream to JAK-STAT [30] were up-regulated, indicating activation of IFNAR and the antiviral response (Fig. 2b). Isg15, which mediates IFN-regulated ubiquitination of cellular and viral targets termed ISGylation [1], was up-regulated (Fig. 2b), as were genes encoding proteins which sequentially catalyze the conjugation of Isg15 to proteins [1] (Fig. 2b). ISGylation is reversible, and Usp18, a negative regulator of type I IFN signaling which catalyzes the hydrolysis of Isg15 [30], was one of the most highly up-regulated genes (Table 1 and Fig. 2b), consistent with a robust activation of the type I IFN response.
The double-stranded RNA-dependent protein kinase (PKR), encoding by the ISG [1] Eif2ak2, is a major antiviral effector due to its role in blocking protein synthesis by phosphorylating the α subunit of eukaryotic translation initiation factor 2α (eIF2α) [31]. PKR protein levels were highly elevated in the cerebral cortex of Gba−/− mice (Fig. 3a), confirming the microarray results (Fig. 2b). PKR levels were also elevated in tissues from the two available cerebella obtained from human patients who succumbed to type 2 GD (Fig. 3b). Interestingly, PKR has recently been shown to play a role in programmed necrosis mediated by IFNs [32]. IFN induces association of PKR with receptor-interacting protein 1 (RIP1; RIPK1) to form assembly of the necrosome complex which contains RIP1, RIP3, and MLKL, and to initiate programmed necrosis and/or activation of the inflammasome; recently, we demonstrated that RIP3 is involved in nGD pathology [33]. PKR was selectively up-regulated in layer V of the cortex (Fig. 3c), another brain area which displays significant pathology in the Gba−/− mouse, and MAC2-positive cells expressed high levels of PKR (Fig. 3c), similar to that observed for P-Stat2 (Fig. 2a) and for RIP3 [33]. PKR also co-localized with the astrocyte marker, GFAP (Fig. 3c).
Fig. 3.
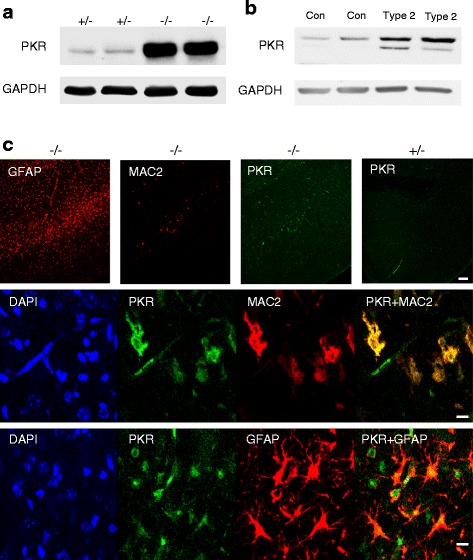
Elevation of PKR (Eif2ak2) in Gba −/− mice. a Western blot of homogenates (150 μg of protein) from the cortex of 21-day-old Gba −/− mice (n = 5). GAPDH was used as loading control. b Western blot of homogenates (50 μg of protein) from the cerebellum of two human patients who succumbed to type 2 GD compared to an age-matched control brain. c PKR levels in microglia/macrophages and in astrocytes in the Gba −/− brain. Immunofluorescence of cortical layer V in 16-day-old Gba −/− using anti-PKR (green), anti-MAC2 (red), or anti-GFAP (red) antibodies. Upper panel PKR staining is located in pathological areas as shown by staining of both MAC2 and GFAP. Scale bar, 100 μm. Middle and lower panels Double immunofluorescence of 16-day-old Gba −/− mice using either anti-PKR and anti-MAC2 (middle panel) or anti-PKR and anti-GFAP (lower panel) antibodies. PKR is in green, MAC2 and GFAP are in red, and areas of overlap are indicated on the right. Scale bar, 10 μm. Results are representative of three experiments
Ablation of the type I IFN receptor attenuates neuroinflammation but has no effect on the viability of nGD mice
To determine the role of the IFN-I receptor (IFNAR) in GD, IFN-I receptor-deficient (Ifnar1−/−) mice [34] were injected with conduritol B epoxide (CBE), an irreversible GCase inhibitor, which can be used to induce GD in mice with different severities depending on the dose used [35, 36]. Unlike Rip3−/− mice, which develop GD upon CBE injection much more slowly than their littermate controls [33], no differences in the lifespan or in the weight of Ifnar1−/− mice were observed upon CBE injection (Fig. 4a, b). However, the extent of elevation of genes down-stream to IFNAR, such as Irf7, Ifnb1, and Eif2ak2, was significantly lower in Ifnar1−/− mice injected with CBE compared to Ifnar1+/− mice. mRNA levels of other inflammatory markers that are elevated in nGD mice [12, 37] (i.e., Ccl2 (MCP-1), Ccl5 (RANTES), and glycoprotein non-metastatic B (GPNMB)) were significantly reduced in Ifnar1−/− mice upon CBE injection (Fig. 4c), although no differences were observed in levels of Ccl3 (MIP1α) and the astrocyte marker, Gfap (Fig. 4c). Levels of activated microglia/macrophages were higher upon CBE injection in both Ifnar1−/− and Ifnar1+/− mice (Fig. 4d), although blocking the type I IFN response caused a small but significant decrease in levels of activated microglia/macrophages (Fig. 4d), confirming a role for IFN in activation of the innate immune response in nGD.
Fig. 4.
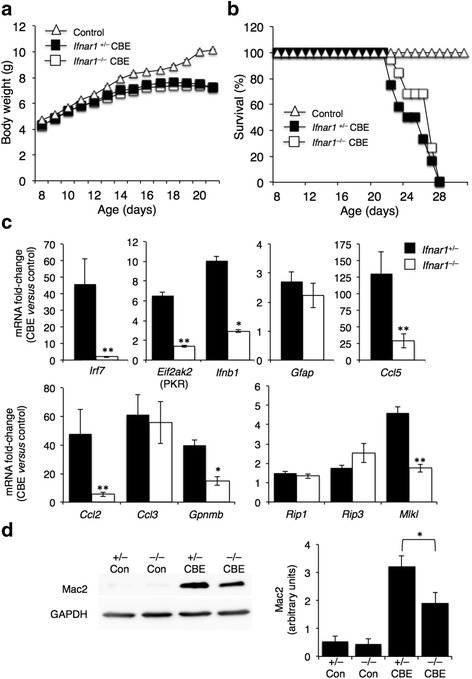
Effect of IFNAR on nGD progression and on down-stream signaling pathways. a Body weight of Ifnar +/− (n = 12) and Ifnar −/− (n = 19) mice treated with CBE (37.5 mg/kg per day) from 8 days of age or untreated mice (control, n = 9). Results are means ± s.e.m. b Kaplan-Meyer survival curves for Ifnar +/− (n = 12) and Ifnar −/− (n = 19) mice. c qPCR analysis of various genes in cortical homogenates from ~30-day-old Ifnar +/− (n = 2) and Ifnar −/− (n = 3) mice treated with CBE (37.5 mg/kg per day) from 8 days of age. Results are expressed as fold change of CBE-treated versus control and are means ± s.e.m. CT values were normalized to levels of HPRT. *p < 0.005, **p < 0.001. d Left, Western blots of homogenates (80 μg of protein) from cortex of ~30-day-old Ifnar +/− (+/−) and Ifnar −/− (−/−) (control) mice treated with or without CBE (+/− CBE, −/− CBE) from 8 days of age. Blots were probed with an anti-MAC2 antibody. Results are representative of three experiments for control mice and five for CBE treated mice. GAPDH was used as loading control. Right Densitometer analysis of the blots. *p < 0.05
mRNA levels of both Rip1 and Rip3 were up-regulated to a similar extent in both Ifnar1−/− and Ifnar1+/− mice upon CBE injection (Fig. 4c). Interestingly, mRNA levels of Mlkl, a crucial protein involved in necroptosis induction [38] and required for activity of the NLRP3 inflammasome, was significantly lower in Ifnar1−/− mice injected with CBE (Fig. 4c), suggesting that Mlkl is an ISG.
IFNβ production in neurons that accumulate GlcCer leads to ISG expression in neighboring microglia
To determine in which cell types IFNβ was elevated in nGD mice, Ifnb1tm1Lky/J mice, which co-express Ifnb1 and EYFP from the Ifnb1 locus, were injected with CBE. IFNβ immunoreactivity was significantly elevated in layer V of the cerebral cortex of CBE-treated mice (Fig. 5a), mainly in neurons and occasionally in microglia, but no elevation was detected in astrocytes. To determine the contribution of microglia to ISG expression down-stream to IFNAR, nGD was induced by CBE injection in Ifnarflox/flox; CX3CR1-Cre mice [39, 40], which lack the IFN receptor in mononuclear phagocytic cells (i.e. microglia in the brain). While mRNA levels of Gpnmb, Rip1, and Rip3 were up-regulated to a similar extent as in control (Ifnarflox/flox) mice upon CBE treatment, levels of Ccl2, Ccl5, Irf7, Eif2ak2 (PKR), Usp18, Oas1b, and Mlkl were significantly lower in the Ifnarflox/flox; CX3CR1-Cre mice (Fig. 5b), indicating that their transcription is down-stream to IFNAR in microglia in nGD brain.
Fig. 5.
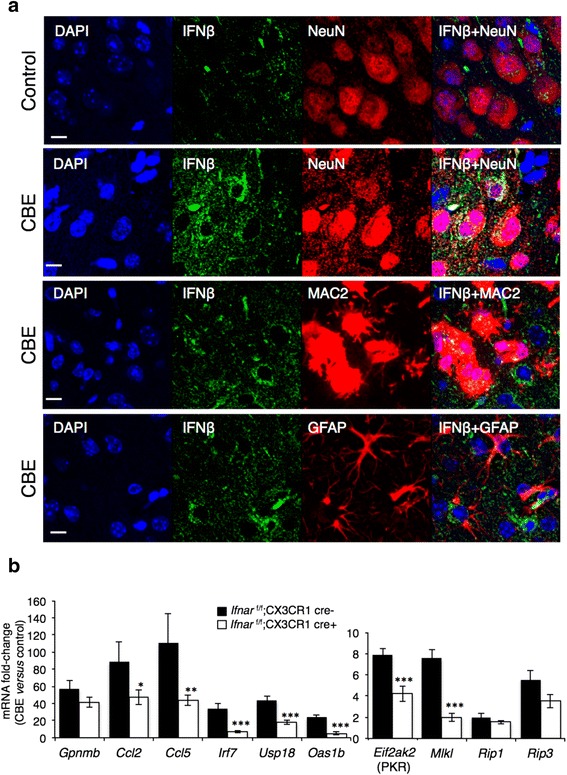
Induction of the IFN response in neurons and downstream signaling in microglia. a Elevation of IFNβ in neurons in the cerebral cortex of 18-day-old CBE-treated mice (100 mg/kg, from 8 days of age) compared to untreated mice (control). Double immunofluorescence using either anti-GFP (IFNβ, green) and anti-NeuN (red), anti-GFP and anti-MAC2 (red), or anti-GFP and anti-GFAP (red) antibodies and the nuclear marker, DAPI (blue). Scale bar, 10 μm. Results are representative of two experiments. b qPCR analysis of various genes in cortical homogenates from 18-day-old Ifnar flox/flox; CX3CR1-Cre + (n = 4) and Ifnar flox/flox; CX3CR1-Cre- (n = 4) mice treated with CBE (100 mg/kg per day) from 8 days of age for 10 days. Results are expressed as fold change of CBE-treated versus control (n = 3–4) mice and are means ± s.e.m. CT values were normalized to levels of HPRT. *p < 0.05, **p < 0.01, ***p < 0.001
Distinct inflammatory profiles in brains from different LSD mouse models
Brain inflammation and microglial activation are shared features of many LSDs [13, 14, 41–43]. Thus, we examined whether IFNs and antiviral ISGs are up-regulated in other LSDs which present significant brain inflammation. Expression levels of the astrocyte marker, Gfap, and the myeloid-lineage cell marker, Adgre1 (F4/80), as well as the inflammatory chemokines Ccl3, Ccl5, Gpnmb, Il-1β, and Tnfα were up-regulated in cerebral hemispheres obtained from the most severe stage of mouse models of nGD [21], Krabbe’s disease [44], Niemann-Pick type C1 disease (NPC1) [5] and Sandhoff disease [45], indicative of neuroinflammation (Table 2). However, levels of Ifnb1 were only significantly up-regulated in cerebral hemispheres from nGD mice, and to a small extent in Krabbe mice; no significant elevation was detected in either NPC1 or in Sandhoff disease (Table 2), suggesting that the antiviral response is elevated specifically in diseases in which simple glycosphingolipids are elevated. Genes related to the RIPK pathway were likewise only elevated in GD and Krabbe’s disease (Table 2).
Table 2.
qPCR analysis of genes in cerebral hemispheres obtained from the end-stage of various LSDs
| Gene | nGD | Krabbe | NPC1 | Sandhoff | ||||
|---|---|---|---|---|---|---|---|---|
| Fold change (−/− vs. +/−) | p value | Fold change (−/− vs. +/−) | p value | Fold change (−/− vs. +/−) | p value | Fold change (−/− vs. +/−) | p value | |
| Inflammation | ||||||||
| Gfap | 6.8 ± 1.6 | <0.005 | 2.6 ± 0.8 | <0.05 | 2.1 ± 0.6 | 0.059 | 2 ± 0.5 | <0.05 |
| Adgre1 (F4/80) | 6.6 ± 0.4 | <0.001 | 3.9 ± 0.7 | <0.005 | 3.6 ± 1.1 | <0.05 | 2.7 ± 0.4 | <0.05 |
| Gpnmb | 78.4 ± 15.6 | <0.001 | 77.2 ± 45.4 | <0.05 | 12.6 ± 4.9 | <0.05 | 2.6 ± 0.4 | <0.005 |
| Tnf a | 11.2 ± 2.5 | <0.005 | 9.3 ± 5.12 | <0.05 | 1.7 ± 0.3 | <0.05 | 1.6 ± 0.2 | <0.05 |
| Il-1β | 7.5 ± 4.6 | <0.005 | 4.4 ± 1.0 | <0.005 | 5.8 ± 1.4 | <0.05 | 2.8 ± 0.1 | <0.005 |
| Ccl2 | 129.2 ± 46.3 | <0.005 | 25.4 ± 16.1 | <0.05 | 3.9 ± 1.2 | <0.05 | 3.5 ± 1.5 | <0.05 |
| Ccl3 | 371.3 ± 131.7 | <0.05 | 18.0 ± 9.3 | <0.001 | 11.8 ± 3.5 | <0.05 | 6.7 ± 1.3 | <0.001 |
| Ccl5 | 240 ± 19.3 | <0.001 | 11.3 ± 2.2 | <0.001 | 4.7 ± 1.2 | <0.05 | 2 ± 0.3 | <0.001 |
| Il-10 | 1.8 ± 0.4 | <0.05 | 2.0 ± 0.6 | ns | 0.9 ± 0.2 | ns | 1.1 ± 0.2 | ns |
| Il-6 | 22.1 ± 7.0 | <0.005 | 7.4 ± 3.1 | <0.05 | 1.7 ± 0.8 | ns | 1.1 ± 0.3 | ns |
| Anti-viral response | ||||||||
| Ifnα | 1.7 ± 0.8a | ns | 2.0 ± 1.3 | ns | 0.9 ± 0.5 | ns | 1.5 ± 0.7 | ns |
| Ifnβ | 3.9 ± 1.7 | <0.05 | 2.1 ± 1.7 | ns | 1 ± 0.5 | ns | 1 ± 0.4 | ns |
| Irf7 | 130.9 ± 18.4 | <0.001 | 7.3 ± 2.9 | <0.05 | 4.6 ± 0.8 | <0.001 | 1.6 ± 0.2 | <0.05 |
| Usp18 | 104.4 ± 13.3 | <0.001 | 11.2 ± 2.4 | <0.001 | 7.1 ± 0.5 | <0.001 | 1.6 ± 0.2 | <0.05 |
| Oas1b | 22.6 ± 5.6 | <0.001 | 5. 7 ± 2.1 | <0.05 | 2.6 ± 1.2 | ns | 1.1 ± 0.4 | ns |
| Eif2ak2 | 11.7 ± 1.1 | <0.001 | 3.5 ± 1 | <0.05 | 1.9 ± 0.3 | <0.05 | 1.4 ± 0.1 | <0.05 |
| RIPK pathway | ||||||||
| Rip1 | 2.3 ± 0.4 | <0.005 | 2 ± 0.36 | <0.05 | 1.4 ± 0.2 | <0.05 | 0.9 ± 0.04 | ns |
| Rip3 | 9.1 ± 0.9 | <0.001 | 2.3 ± 0.27 | <0.05 | 1.5 ± 0.2 | <0.05 | 0.7 ± 0.1 | ns |
| Mlkl | 9.4 ± 1.8 | <0.001 | 4.74 ± 1.46 | <0.05 | 1.8 ± 0.2 | 0.00898 | 1.2 ± 0.3 | ns |
Results are expressed as fold change of −/− versus control mice and are means ± s.e.m. CT values were normalized to levels of HPRT. Genes that were elevated >2-fold with a p value of <0.05 are in bold. nGD mice (Gba flox/flox; nestin-Cre mice, 3 weeks of age, n = 4); Krabbe’s disease (Galc −/− mice, 5 weeks of age, n = 3); Niemann-Pick type C1 disease mice (NPC1−/−), 10 weeks of age, n = 3–4; Sandhoff disease (HexB −/− mice) 17 weeks of age, n = 2–3
aIt should be noted that there is a significant difference in the extent of elevation of mRNA levels depending on whether a pathological area of the brain was used compared to half cerebral hemispheres. For instance, Ifnα was elevated 2.1-fold in layer V of the cortex, but 1.7-fold in a cerebral hemisphere. However, even when using whole cerebral hemispheres, it is apparent that the extent of the inflammatory response is much higher in nGD compared to the other LSDs
Discussion
Type I IFNs are mainly recognized as part of the host response to viral and bacterial infection. Much less is known about the role of the type I IFN response under conditions of sterile inflammation, particularly in the CNS [3–6]. Activation of this response in the absence of infection can either be detrimental, as in AD, ALS, and AGS, or beneficial, as in multiple sclerosis. Interestingly, depletion of IFNβ causes spontaneous neurodegeneration resembling sporadic Lewy body and PD [46]. In the current study, we demonstrate massive induction of the type I IFN response in nGD brain at a pre-symptomatic stage, suggesting a key role for the antiviral response in the initial stages of brain pathology.
The mechanism by which the immune response is triggered in nGD is not known. Importantly, in the Gbaflox/flox; nestin-Cre mouse, microglia do not accumulate GlcCer [21] and their activation cannot therefore be due to intrinsic GlcCer accumulation, but must rather be a response to extrinsic signals from other cells that do accumulate GlcCer, such as neurons and/or astrocytes. PRRs, ISGs, and other components of innate immunity are expressed in astrocytes, microglia, and neurons [47, 48], and our data is consistent with the idea that the initial activation of the immune response is up-stream to IFNAR in microglia. A remaining challenge is to determine if the immune response is triggered by PRRs which directly sense increased GlcCer levels or by PRRs that sense known PAMPs/DAMPs such as dsRNA, cytosolic DNA, glycoproteins, and nucleic acids, which for unknown reasons may be altered in nGD (Fig. 6). Constitutive up-regulation of type I IFN production was found in a group of monogenic diseases characterized by neurological and dermatological features, known as type I interferonopathies, which result from accumulation of endogenous nucleic acid products, which are subsequently sensed as non-self by the innate immune machinery [8]. Cross-talk between changes in lipid composition and TLR signaling was recently demonstrated, with GD fibroblasts displaying a hyper-inflammatory response to TLR-induced IL-6 release [49]. This, together with our data, suggests a scenario by which GlcCer is recognized as a DAMP upon its accumulation in nGD leading to IFN induction and secretion. Subsequently, IFN activates surrounding microglia via binding to IFNAR, resulting in the phosphorylation of STAT and induction of ISGs [27]. Neurons from distinct brain regions have unique innate immune signatures which determine their susceptibility to viral infection [50], and we speculate that different neuronal populations have different immunological properties which might explain their selective vulnerability to GlcCer accumulation in nGD [16] and to other lipids and/or substrates in other LSDs.
Fig. 6.
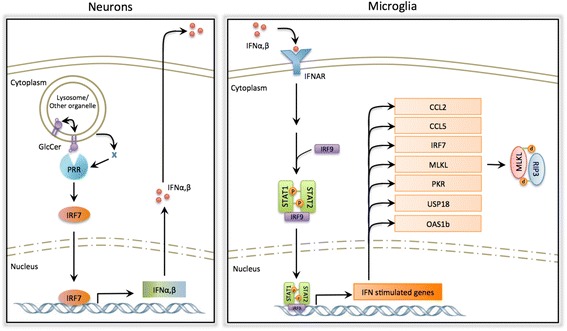
Proposed mechanism by which type I IFN is activated in nGD. Left Induction of the IFN response in neurons upon GlcCer accumulation. Elevated levels of GlcCer are sensed directly or indirectly by a PRR resulting in activation of the antiviral response and production of IFNα and IFNβ. IFN is secreted from neurons and engages IFNAR in neighboring cells, i.e., microglia. Right Upon engagement, IFNAR activates phosphorylation, dimerization, and nuclear translocation of STAT proteins, which subsequently induce the expression of ISGs, among them MLKL, which forms the necrosome complex together with phosphorylated RIP3
Brain inflammation is a universal feature of many neurodegenerative diseases, including LSDs with brain involvement. The inflammatory response can take many guises, being initiated in response to different stimuli, can be chronic or acute, beneficial or detrimental, all of which can be mediated by different inflammatory factors. Each LSD has its own pattern of accumulation of storage material and its own pattern of neuroinflammation [42]. It is notable that of the four LSDs that we studied, nGD gave the most severe inflammatory response, although this might, at least in part, be due to the rapid onset of disease symptoms in the Gbaflox/flox; nestin-Cre mouse compared to the other mouse models examined. The Gbaflox/flox; nestin-Cre mouse displayed by far the most robust type 1 IFN response, as exemplified by highly elevated levels of Irf7 and Usp18. Ours is of course not the first study to examine patterns of gene activation in the brains of LSD mice or patients, but lack of consistency in the age of the brain material used, and in the age of animals, renders quantitative comparison difficult. Nevertheless, some information can be gleaned from such a comparison. For instance, in a gene microarray analysis using spinal cord tissue from 4-month-old Sandhoff disease mice [51], 58 genes were up-regulated, of which 32 are common to nGD, although only one of these is involved in IFN signaling (Fig. 7 and Table 3). A microarray analysis on Npc1−/− mouse brain at different stages of disease severity [52] revealed up-regulation of 112 genes, of which 88 were also up-regulated in nGD, and 9 of these are in the IFN signaling pathway. IFNβ secretion and increased STAT levels were demonstrated in cultured human NPC fibroblasts [53]. The similarity of the lipids that accumulate in nGD (GlcCer/GlcSph), Krabbe (galactosylsphingosine), and NPC (in which small but significant amounts of GlcCer accumulate in addition to the primary storage material, cholesterol) suggest that more complex GSLs such as ganglioside GM2, which accumulate in Sandhoff disease, cannot trigger the IFN response, whereas the more simple GSLs do trigger this response, although to a different extent.
Fig. 7.
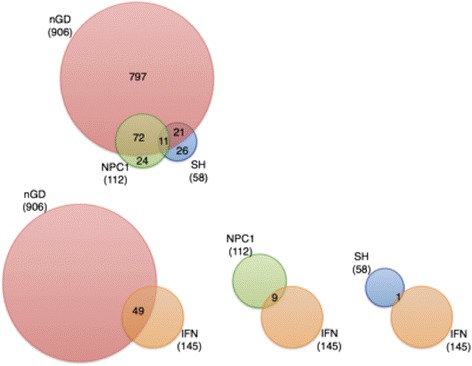
Gene changes in LSDs. Upper panel Venn diagram of differentially-expressed genes in spinal cord of Sandhoff disease, brain of NPC1 disease, and the VPM/VPL of nGD. Lower panel Venn diagram of differentially expressed genes related to the type I IFN pathway to up-regulated genes in other diseases. The enrichment IFN signaling lists were obtained from GSEA (REACTOME_INTERFERON_SIGNALING). Overlapping regions were drawn to scale and the number of shared and unique genes listed. Overlap was visualized using BioVenn
Table 3.
Comparison of gene arrays for NPC1, Sandhoff, and nGD
| Sandhoff [51] | NPC [52] | nGD | |
|---|---|---|---|
| Tissue | Spinal cord | Brain | VPM/VPL |
| Age/disease severity | 4 months old (late-symptomatic stage) | 20 to 80 days old (asymptomatic-, early-, and late-symptomatic stages) | 14 days old (asymptomatic stage) |
| Analysis cut-off | 2-fold | 1.5-fold | 1.5-fold |
| Number of up-regulated genes | 58 | 112 | 907 |
| Number (and percent) of up-regulated genes common to nGD | 32 (55.2 %) | 88 (78.5 %) | |
| Number (and percent) of IFN-related genes among the up-regulated genes | 1 (1.7 %) | 9 (8 %) | 49 (5.4 %) |
| Percentage of IFN-related gene out of total of 145 IFN-related genes | 0.70 % | 6.20 % | 33.80 % |
We recently demonstrated a critical role for RIP3 in nGD pathology [33], although we have so far not been able to determine the precise mechanistic role of RIP3. Of some interest is the observation that viral and bacterial pathogens induce programmed necrosis via a RIP3-dependent cell death mechanism [54–57], which can involve type I IFN [58]. Unlike RIP3, ablating the type 1 IFN response, using the Ifnar1-deficient mouse, did not alleviate disease symptoms in nGD mice, and moreover, mRNA levels of Rip1 and Rip3 were essentially unaltered. In contrast, expression of Mlkl, the key down-stream player to RIP1-RIP3 in programmed necrosis, was significantly lower in Ifnar1−/− mice, indicating that Mlkl is a novel ISG. We would like to verify this by examining the phosphorylation state of RIP3 and MLKL, but unfortunately, no commercial antibodies against the phosphorylated forms of mouse RIP3 and mouse MLKL are currently available [59].
At this stage, we do not know if activation of type I IFN signaling in nGD brain is beneficial or detrimental. The lack of benefit upon induction of nGD in Ifnar1−/− mice is somewhat surprising and might be due to activation of a compensatory inflammatory mechanism upon interfering with IFN signaling pathways; alternatively, endogenous activation of proteins that block interferon signaling, such as Usp18 (Fig. 4), might alleviate any beneficial effect of IFN. Usp18 is arguably the ISG with the most important role in establishing and maintaining long-term desensitization to type I IFN signaling [30]. This desensitized state allows cells to recover from IFN signaling, whereas dysregulation of IFN production and signaling manifests in autoimmune disorders such as systemic lupus erythematosus and Sjögren’s syndrome [60]. Finally, we cannot exclude the possibility that mice treated with CBE succumb to systemic disease, rather than a disease of the CNS, and therefore, amelioration of neurological disease might not be sufficient to alleviate disease symptoms in the case of the Ifnar1−/− mouse.
Methods
Mice
Gbaflox/flox; nestin-Cre mice were used as a genetic model of nGD [21]. Gbaflox/flox mice were crossed with Gbaflox/wt; nestin-Cre mice to generate Gbaflox/flox; nestin-Cre mice (referred to as Gba−/− mice) and Gbaflox/wt; nestin-Cre mice (referred to as Gba+/− mice), which served as healthy controls since they do not show any overt pathology [22]. IFN-I receptor-deficient (Ifnar1−/−) mice [34] were backcrossed with C57BL/6 mice to generate Ifnar1+/− mice. Ifnar1+/− mice were crossed with Ifnar1−/− mice to generate Ifnar1+/− and Ifnar1−/− littermates. Ifnar1+/− and Ifnar1−/− mice were injected daily i.p. with 37.5 mg/kg body weight CBE, an irreversible GCase inhibitor [36], from 8 days of age. Ifnb1tm1Lky/J mice were obtained from Jackson and were injected daily i.p. with 100 mg/kg body weight CBE from 8 days of age. Ifnarflox/flox mice [61] were crossed with CX3CR1-Cre mice [40] to generate Ifnarflox/wt; CX3CR1-Cre mice. Ifnarflox/flox; CX3CR1-Cre mice were crossed with Ifnarflox/flox mice to generate Ifnarflox/flox; CX3CR1-Cre and Ifnarflox/flox littermates. Ifnarflox/flox; CX3CR1-Cre and Ifnarflox/flox mice were injected daily i.p. with 100 mg/kg body weight CBE from 8 days of age. Mice deficient in the β subunit of β-hexosaminidase A and B were used as mouse model of Sandhoff disease [45]. Brains from a mouse model of Krabbe’s disease [44], defective in β-galactocerebrosidase (the Twitcher mouse), were provided by Dr. Timothy M. Cox (University of Cambridge). Brains from Niemann-Pick disease type C1 mice, defective in the Npc1 gene [62], were provided by Dr. Nick Platt (Oxford University).
Human brain tissue
A control human brain from an infant who died at birth was provided by the University of Miami Brain and Tissue Bank for Developmental Disorders. The control brain was frozen within 6 and 26 h of death. Brains from Gaucher patients were obtained postmortem with informed consent between 7 and 22 h after death [63]. After removal, brains were frozen on dry ice.
Gene expression profiling microarray analysis
Mice were sacrificed and their brains removed and placed on a Young Mouse Brain Slicer Matrix (BSMYS001-1, Zivic instruments, Pittsburgh, PA, USA). Single-edge razor blades were inserted into the matrix to generate 1-mm coronal sections. Sections containing the VPM/VPL were snap-frozen on dry ice and a 17-g (1 mm diameter) blunt needle attached to a syringe was used to remove the VPM/VPL; the cerebral cortex was separated using a spatula. Each sample was placed in a microcentrifuge tube, snap-frozen in liquid N2, and stored at −80 °C.
Total RNA was isolated using the RNeasy Mini Kit (QIAGEN GmbH, Hilden, Germany) according to manufacturer’s instructions, which included DNase treatment and addition of β-mercaptoethanol. A microarray experiment was performed on 100-ng RNA per sample purified from the VPM/VPL region of Gba+/– and Gba−/− mice at 14 days (n = 3) of age. Purified RNA was reverse-transcribed and amplified using an Ambion WT expression kit and labeled with an Affymetrix GeneChip® WT Terminal Labeling kit. Labeled cDNA was hybridized to Affymetrix Mouse Gene 1.0 microarrays according to manufacturer’s instructions. Microarrays were scanned using a GeneChip® scanner 30007G and statistical analysis performed using Partek® Genomics Suite software (Partek Inc., St. Louis, MI, USA). For each age group, CEL files (containing raw expression measurements) were imported to Partek GS. The data was processed and normalized using the RMA (Robust Multichip Average) algorithm [64] with GC correction, and a three-way ANOVA model was used to identify differentially expressed genes; fold-changes were calculated. Analysis of Gba gene probe sets verified that the mRNA expression of Gba flanked exons [21] was reduced in Gba−/− samples. Gene lists were created by filtering the genes based on an absolute linear fold change ≥1.5, p ≤ 0.05, and signal above background in at least one microarray (log2 intensity ≥6) (Additional file 1: Table S1). The gene lists were analyzed for enriched pathways using GSEA (www.broadinstitute.org/gsea), Ontologizer (Elim algorithm) [23], DAVID [65] against the KEGG pathways database, and IPA (Ingenuity® Systems, www.ingenuity.com). The enrichment of the lists was tested against the expressed genes (above background signal) when implementing the Ontologizer tool. Microarray data were deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE46866). The BioVenn tool (http://www.cmbi.ru.nl/cdd/biovenn/) [66] was used to identify common and exclusively expressed genes between groups.
qPCR
cDNA synthesis was performed using the Reverse-iT First-Strand Synthesis Kit (Thermo Fisher Scientific, Surrey, UK) using random decamers. cDNA products were stored at −20 °C. qPCR was performed using PerfeCTa SYBR Green FastMix (Quanta BioSciences, Gaithersburg, MD, USA) and an ABI Prism 7300 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The primer concentration was 13 nM in a reaction volume of 20 μl and cDNA equivalent to 2–20 ng of total RNA, or 300 ng for Ifnb1 and Ifna2. Each reaction was performed in triplicate. The thermal cycling parameters were as follows: step 1, 95 °C for 10 min; step 2, 95 °C for 15 s, 60 °C for 30 s, and 68 °C for 30 s. Step 2 was repeated for 40 cycles and was followed by a dissociation step. Fold change in mRNA levels was calculated using the comparative cycle threshold method using TATA box binding protein (TBP) or hypoxanthine phosphoribosyltransferase 1 (HPRT) for normalization. p values were calculated using a two-tailed, two-independent sample Student’s t test. Primers for Ifnb1, Ifna2, Il-1β, and Il-10 were from QIAGEN. Primers sequences are in Table 4.
Table 4.
Primers used for qPCR
| Gene name | Primer sequence |
|---|---|
| Tbp | F: 5′-TGCTGTTGGTGATTGTTGGT-3′ |
| R: 5′-CTGGCTTGTGTGGGAAAGAT-3′ | |
| Hprt | F: 5′-TGCTCGAGATGTCATGAAGG-3′ |
| R: 5′-AATCCAGCAGGTCAGCAAAG-3′ | |
| Lgals3 (Mac2) | F: 5′-CACTGACGGTGCCCTATGAC-3′ |
| R: 5′-AACAATCCTGTTTGCGTTGGG-3′ | |
| Kcnk4 | F: 5′-GCAGGCTCAGAAGAAAATGG-3′ |
| R: 5′-TGGTCCCTCAGAAACTGGTC-3′ | |
| Sptssb | F: 5′-TCATTCTAAGGCAGGAGACGA-3′ |
| R: 5′-AAGCTGGGAAAAGTCTGCCT-3′ | |
| Ryr3 | F: 5'-GGTGGGCTATTACTGCCTGA-3' |
| R: 5'-CATCCTCAGATGGCTGTTCA -3' | |
| Gfap | F: 5′-TAGTCCAACCCGTTCCTCCA-3′ |
| R: 5′-CCAGTTGTCGACTAGGACCG-3′ | |
| Irf7 | F: 5′-CAATGGCTGAAGTGAGGGGG-3′ |
| R: 5′-GACCGAAATGCTTCCAGGGT-3′ | |
| Eif2ak2 (PKR) | F: 5′-GATGGAAAATCCCGAACAAGGAG-3′ |
| R: 5′-AGGCCCAAAGCAAAGATGTCCAC-3′ | |
| Ccl5 | F: 5′-TGCCTACCTCTCCCTCGCGC-3′ |
| R: 5′-GGCACACACTTGGCGGTTCCT-3′ | |
| Ccl2 | F: 5′-TCACCTGCTGCTACTCATTCACCA-3′ |
| R: 5′-AGCACAGACCTCTCTCTTGAGCTT-3′ | |
| Ccl3 | F: 5′-TTTTGAAACCAGCAGCCTTT-3′ |
| R: 5′-CTCAAGCCCCTGCTCTACAC-3′ | |
| Gpnmb | F: 5′-AGCACAACCAATTACGTGGC-3′ |
| R: 5′-CTTCCCAGGAGTCCTTCCA-3′ | |
| Rip1 | F: 5′-AGTCGAGACTGAAGGACACAGCACT-3′ |
| R: 5′-TCCAGCAGGTCACTGGATGCCAT-3′ | |
| Rip3 | F: 5′-CTTGAACCCTCCGCTCCTGC-3′ |
| R: 5′-CGGCTCACCAGAGGAACCGCAT-3′ | |
| Mlkl | F: 5′-TCACAGATCTCCAGTTACCATC-3′ |
| R: 5′-ACGCAAGATGTTGGGAGAATCG-3′ | |
| Tnfα | F: 5′-CTTGTGGCAGGGGCCACCAC-3′ |
| R: 5′-CCATGCCGTTGGCCAGGAGG-3′ | |
| Usp18 | F: 5′-CAGGAGTCCCTGATTTGCGT-3′ |
| R: 5′-GGGCTGGACGAAACATCTCA-3′ | |
| Oas1b | F: 5′-GCGTAGGCCTGTTATGCTCT-3′ |
| R: 5′-CCACTGGGCTCACAGGAAAA-3′ |
ELISA
Frozen cerebral brain tissues were homogenized in 0.1 M phosphate buffer (PBS) supplemented with a protease inhibitor cocktail (1:200, Sigma). Samples were sonicated and centrifuged at 14,000gav for 10 min at 4 °C, and the supernatant was collected. Protein was quantified using the BCA protein assay reagent (Pierce Chemical Co., Rockford, Illinois, USA). Homogenates containing ~3–4 mg of protein were applied to a VeriKine™ mouse Interferon Alpha ELISA kit (PBL Interferon Source, Piscataway, NJ, USA), ~2 mg of protein were applied to Verikine-HS™ mouse Interferon Beta Elisa kit (PBL Interferon Source), and 1 mg was applied to a VeriKine™ Human Interferon Beta ELISA kit (PBL Interferon Source, Piscataway, NJ, USA) according to manufacturer’s guidelines.
Immunohistochemistry
Brains were removed and immersion fixed in 2.5 % paraformaldehyde in 0.1 M PBS, pH 7.4 for 2 days, embedded in paraffin blocks, and 4-μm coronal sections prepared on Superfrost + slides. Sections were deparaffinized and rehydrated prior to further use. Antigen retrieval was performed using 10 mM citric acid (pH 6.0) for 10 min at ~100 °C. Sections were blocked using 20 % normal horse serum (NHS) (Vector, Burlingame, CA, USA) and 0.3 % Triton X-100. Sections were incubated with either rabbit antiphospho-Y690-Stat2 (1:50, Abcam, Cambridge, MA), mouse anti-NeuN (1:50, Chemicon, Temecula, CA, USA), rat anti-MAC2 (1:250, Cedarlane, Ontario, Canada), mouse anti-PKR (1:100, Santa Cruz), rabbit anti-GFAP (1:100, Dako), or with a goat anti-GFP antibody (Biotin, 1:100, Abcam, Cambridge, MA). Antibody mixtures were diluted in 2 % normal horse serum (NHS) containing 0.3 % Triton X-100. Sections were incubated with the first antibodies overnight at room temperature. For P-Stat2 detection, sections were incubated with a biotinylated donkey antirabbit secondary antibody (1:200, Jackson, West Grove, PA, USA) and for PKR with a biotinylated donkey antimouse secondary antibody (1:200, Jackson, West Grove, PA, USA) for 2 h followed by Cy3-conjugated streptavidin (1:200, Jackson) for 1 h. NeuN-stained samples were incubated with a Cy2-conjugated donkey antimouse antibody (1:200, Jackson ImmunoResearch), MAC2 with a Cy2-conjugated donkey antirat antibody (1:200, Jackson ImmunoResearch), and GFAP with a Cy2-conjugated donkey antirabbit antibody (1:200, Jackson ImmunoResearch) for 1 h. Counterstaining was performed with DAPI (Molecular Probes, Eugene, OR, USA) (1:1000, 4 min), sections were rinsed with PBS, mounted with Flouromount-G (SouthernBiotech), and coverslipped.
Protein extraction and Western blotting
Homogenates were prepared as described [67]. Blots were incubated with the following antibodies: mouse anti-PKR (1:1,000, Santa Cruz), rat anti-MAC2 (1:1,000, Cell Signaling), and a mouse anti-GAPDH (1:10,000, Chemicon), followed by incubation with a horseradish peroxidase-conjugated secondary antibody (antimouse, Jackson ImmunoResearch). Bound antibodies were detected using the SuperSignal West Pico chemiluminescent substrate (Thermo Scientific).
Conclusions
The type I IFN response appears to be a significant player in the development of nGD pathology and possibly in other LSDs in which simple GSLs accumulate. Our data support the developing notion that the IFN signaling pathway (Fig. 6) plays a vital role during the sterile inflammation that often occurs during chronic neurodegenerative diseases in which neuroinflammation is present.
Ethics approval
Mice were maintained under specific pathogen-free conditions and handled according to protocols approved by the Weizmann Institute Animal Care Committee according to international guidelines.
Consent for publication
Not applicable.
Availability of data and materials
All material used in this manuscript will be made available to researchers subject to confidentiality.
Acknowledgements
We thank Dr. Stefan Karlsson for providing the Gba flox/flox; nestin-Cre nGD mice, Dr. Shirley Horn-Saban and Anna Weismann for conducting the microarray experiment, Vladmir Kiss (Weizmann Institute of Science) for help with fluorescence microscopy, Dr. Timothy M. Cox (University of Cambridge) for providing Krabbe mouse tissues, Dr. Nick Platt (Oxford University) for providing NPC mouse tissues, Dr. Steffen Jung for providing CX3CR1-Cre mice, and Dr. Ulrich Kalinke (Hannover Medical School) for providing Ifnar flox/flox mice. We thank Iris Zelnik for help with Fig. 6. A.H. Futerman is the Joseph Meyerhoff Professor of Biochemistry at the Weizmann Institute of Science.
Funding
This work was generously supported by the Children’s Gaucher Research Fund and by the Minerva Foundation.
Abbreviations
- GD
Gaucher disease
- GCase
acid-β-glucosidase
- GlcCer
glucosylceramide
- Ifn
interferon
- Irf
interferon regulatory factor
- ISG
interferon-stimulated gene
- LSD
lysosomal storage disease
- nGD
neuronopathic Gaucher disease
- qPCR
quantitative polymerase chain reaction
- STAT
signal transducers and activators of transcription
- VPM/VPL
ventral posteromedial/posterolateral
Additional file
List of the differentially-expressed transcripts. (XLSX 330 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EBV and TFB planned and performed most of the experiments and helped with writing the manuscript. NSF injected mice and performed PCR and Western blot analysis. DL helped with gene array data analysis. PS and KSL performed initial studies injecting Ifnar1 −/− mice with CBE, and KSL helped with experimental design and with writing the manuscript. AHF obtained funding for the study, led the research team, and wrote the manuscript. All authors read and approved the final manuscript.
References
- 1.Sadler AJ, Williams BRG. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–68. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975–90. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–35. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang R, Yang B, Zhang D. Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia. 2011;59:946–58. doi: 10.1002/glia.21167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor JM, Minter MR, Newman AG, Zhang M, Adlard PA, Crack PJ. Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer’s disease. Neurobiol Aging. 2014;35:1012–23. doi: 10.1016/j.neurobiolaging.2013.10.089. [DOI] [PubMed] [Google Scholar]
- 6.Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T, et al. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014;346:89–93. doi: 10.1126/science.1252945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lebon P, Badoual J, Ponsot G, Goutieres F, Hemeury-Cukier F, Aicardi J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J Neurol Sci. 1988;84:201–8. doi: 10.1016/0022-510X(88)90125-6. [DOI] [PubMed] [Google Scholar]
- 8.Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429–40. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- 9.Khorooshi R, Owens T. Injury-induced type I IFN signaling regulates inflammatory responses in the central nervous system. J Immunol. 2010;185:1258–64. doi: 10.4049/jimmunol.0901753. [DOI] [PubMed] [Google Scholar]
- 10.Lee-Kirsch MA, Wolf C, Kretschmer S, Roers A. Type I interferonopathies—an expanding disease spectrum of immunodysregulation. Semin Immunopathol. 2015;37:349–57. doi: 10.1007/s00281-015-0500-x. [DOI] [PubMed] [Google Scholar]
- 11.Amor S, Peferoen LAN, Vogel DYS, Breur M, van der Valk P, Baker D, van Noort JM. Inflammation in neurodegenerative diseases—an update. Immunology. 2014;142:151–66. doi: 10.1111/imm.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vitner EB, Farfel-Becker T, Eilam R, Biton I, Futerman AH. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain. 2012;135:1724–35. doi: 10.1093/brain/aws095. [DOI] [PubMed] [Google Scholar]
- 13.Smith D, Wallom K-L, Williams IM, Jeyakumar M, Platt FM. Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1. Neurobiol Dis. 2009;36:242–51. doi: 10.1016/j.nbd.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 14.Jeyakumar M, Thomas R, Elliot-Smith E, Smith DA, van der Spoel AC, d’Azzo A, Perry VH, Butters TD, Dwek RA, Platt FM. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain. 2003;126:974–87. doi: 10.1093/brain/awg089. [DOI] [PubMed] [Google Scholar]
- 15.Beutler E, Grabowski GA. Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Diseases. New York: McGraw-Hill; 2001. p. 3635. [Google Scholar]
- 16.Farfel-Becker T, Vitner EB, Kelly SL, Bame JR, Duan J, Shinder V, Merrill AH, Dobrenis K, Futerman AH. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum Mol Genet. 2014;23:843–54. doi: 10.1093/hmg/ddt468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nilsson O, Svennerholm L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J Neurochem. 1982;39:709–18. doi: 10.1111/j.1471-4159.1982.tb07950.x. [DOI] [PubMed] [Google Scholar]
- 18.Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–61. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conradi NG, Sourander P, Nilsson O, Svennerholm L, Erikson A. Neuropathology of the Norrbottnian type of Gaucher disease. Morphological and biochemical studies. Acta Neuropathol. 1984;65:99–109. doi: 10.1007/BF00690463. [DOI] [PubMed] [Google Scholar]
- 20.Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, Morrison A, Lwin A, Colegial C, Allman JM, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. 2004;82:192–207. doi: 10.1016/j.ymgme.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 21.Enquist IB, Bianco Lo C, Ooka A, Nilsson E, Mansson JE, Ehinger M, Richter J, Brady RO, Kirik D, Karlsson S. Murine models of acute neuronopathic Gaucher disease. Proc Natl Acad Sci U S A. 2007;104:17483–8. doi: 10.1073/pnas.0708086104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farfel-Becker T, Vitner EB, Pressey SNR, Eilam R, Cooper JD, Futerman AH. Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Hum Mol Genet. 2011;20:1375–86. doi: 10.1093/hmg/ddr019. [DOI] [PubMed] [Google Scholar]
- 23.Bauer S, Grossmann S, Vingron M, Robinson PN. Ontologizer 2.0—a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics. 2008;24:1650–1. doi: 10.1093/bioinformatics/btn250. [DOI] [PubMed] [Google Scholar]
- 24.Osorio F, Reis e Sousa C. Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity. 2011;34:651–64. doi: 10.1016/j.immuni.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol. 2013;13:621–34. doi: 10.1038/nri3515. [DOI] [PubMed] [Google Scholar]
- 26.Broz P, Monack DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol. 2013;13:551–65. doi: 10.1038/nri3479. [DOI] [PubMed] [Google Scholar]
- 27.Liu S-Y, Sanchez DJ, Cheng G. New developments in the induction and antiviral effectors of type I interferon. Curr Opin Immunol. 2011;23:57–64. doi: 10.1016/j.coi.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paun A, Pitha PM. The IRF family, revisited. Biochimie. 2007;89:744–53. doi: 10.1016/j.biochi.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun. 2011;12:399–414. doi: 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–45. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dalet A, Gatti E, Pierre P. Integration of PKR-dependent translation inhibition with innate immunity is required for a coordinated anti-viral response. FEBS Lett. 2015;589:1539–45. doi: 10.1016/j.febslet.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 32.Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, Rall GF, Degterev A, Balachandran S. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013;110:E3109–18. doi: 10.1073/pnas.1301218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vitner EB, Salomon R, Farfel-Becker T, Meshcheriakova A, Ali M, Klein AD, Platt FM, Cox TM, Futerman AH. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat Med. 2014;20:204–8. doi: 10.1038/nm.3449. [DOI] [PubMed] [Google Scholar]
- 34.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–21. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 35.Farfel-Becker T, Vitner EB, Futerman AH. Animal models for Gaucher disease research. Dis Model Mech. 2011;4:746–52. doi: 10.1242/dmm.008185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanfer JN, Legler G, Sullivan J, Raghavan SS, Mumford RA. The Gaucher mouse. Biochem Biophys Res Commun. 1975;67:85–90. doi: 10.1016/0006-291X(75)90286-7. [DOI] [PubMed] [Google Scholar]
- 37.Zigdon H, Savidor A, Levin Y, Meshcheriakova A, Schiffmann R, Futerman AH. Identification of a biomarker in cerebrospinal fluid for neuronopathic forms of Gaucher disease. PLoS One. 2015;10:e0120194. doi: 10.1371/journal.pone.0120194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 39.Detje CN, Meyer T, Schmidt H, Kreuz D, Rose JK, Bechmann I, Prinz M, Kalinke U. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J Immunol. 2009;182:2297–304. doi: 10.4049/jimmunol.0800596. [DOI] [PubMed] [Google Scholar]
- 40.Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vitner EB, Futerman AH, Platt N. Innate immune responses in the brain of sphingolipid lysosomal storage diseases. Biol Chem. 2015;396:659–67. doi: 10.1515/hsz-2014-0301. [DOI] [PubMed] [Google Scholar]
- 42.Vitner EB, Platt FM, Futerman AH. Common and uncommon pathogenic cascades in lysosomal storage diseases. J Biol Chem. 2010;285:20423–7. doi: 10.1074/jbc.R110.134452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeyakumar M, Smith DA, Williams IM, Borja MC, Neville DCA, Butters TD, Dwek RA, Platt FM. NSAIDs increase survival in the Sandhoff disease mouse: synergy with N-butyldeoxynojirimycin. Ann Neurol. 2004;56:642–9. doi: 10.1002/ana.20242. [DOI] [PubMed] [Google Scholar]
- 44.Suzuki K, Taniike M. Murine model of genetic demyelinating disease: the twitcher mouse. Microsc Res Tech. 1995;32:204–14. doi: 10.1002/jemt.1070320304. [DOI] [PubMed] [Google Scholar]
- 45.Sango K, Yamanaka S, Hoffmann A, Okuda Y, Grinberg A, Westphal H, McDonald MP, Crawley JN, Sandhoff K, Suzuki K, et al. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat Genet. 1995;11:170–6. doi: 10.1038/ng1095-170. [DOI] [PubMed] [Google Scholar]
- 46.Ejlerskov P, Hultberg JG, Wang J, Carlsson R, Ambjorn M, Kuss M, Liu Y, Porcu G, Kolkova K, Friis Rundsten C, et al. Lack of neuronal IFN-beta-IFNAR causes Lewy Body and Parkinson’s Disease-like dementia. Cell. 2015;163:324–39. doi: 10.1016/j.cell.2015.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–45. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 48.Okun E, Griffioen KJ, Lathia JD, Tang S-C, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Rev. 2009;59:278–92. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koberlin MS, Snijder B, Heinz LX, Baumann CL, Fauster A, Vladimer GI, Gavin A-C, Superti-Furga G. A conserved circular network of coregulated lipids modulates innate immune responses. Cell. 2015;162:170–83. doi: 10.1016/j.cell.2015.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho H, Proll SC, Szretter KJ, Katze MG, Gale MJ, Diamond MS. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat Med. 2013;19:458–64. doi: 10.1038/nm.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci U S A. 2000;97:10954–9. doi: 10.1073/pnas.97.20.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alam MS, Getz M, Safeukui I, Yi S, Tamez P, Shin J, Velazquez P, Haldar K. Genomic expression analyses reveal lysosomal, innate immunity proteins, as disease correlates in murine models of a lysosomal storage disorder. PLoS One. 2012;7:e48273. doi: 10.1371/journal.pone.0048273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki M, Sugimoto Y, Ohsaki Y, Ueno M, Kato S, Kitamura Y, Hosokawa H, Davies JP, Ioannou YA, Vanier MT, et al. Endosomal accumulation of Toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal transducers and activators of transcription in Niemann-Pick disease type C (NPC) fibroblasts: a potential basis for glial cell activation in the NPC brain. J Neurosci. 2007;27:1879–91. doi: 10.1523/JNEUROSCI.5282-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cho Y, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK-M. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sridharan H, Upton JW. Programmed necrosis in microbial pathogenesis. Trends Microbiol. 2014;22:199–207. doi: 10.1016/j.tim.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 56.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–7. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, Mocarski ES, Pouliot K, Chan FK-M, Kelliher MA, et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci U S A. 2014;111:7391–6. doi: 10.1073/pnas.1403477111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–62. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jouan-Lanhouet S, Riquet F, Duprez L, Vanden Berghe T, Takahashi N, Vandenabeele P. Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol. 2014;35:2–13. doi: 10.1016/j.semcdb.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 60.Ronnblom L. The type I interferon system in the etiopathogenesis of autoimmune diseases. Ups J Med Sci. 2011;116:227–37. doi: 10.3109/03009734.2011.624649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kamphuis E, Junt T, Waibler Z, Forster R, Kalinke U. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood. 2006;108:3253–61. doi: 10.1182/blood-2006-06-027599. [DOI] [PubMed] [Google Scholar]
- 62.Pentchev PG, Boothe AD, Kruth HS, Weintroub H, Stivers J, Brady RO. A genetic storage disorder in BALB/C mice with a metabolic block in esterification of exogenous cholesterol. J Biol Chem. 1984;259:5784–91. [PubMed] [Google Scholar]
- 63.Pelled D, Trajkovic-Bodennec S, Lloyd-Evans E, Sidransky E, Schiffmann R, Futerman AH. Enhanced calcium release in the acute neuronopathic form of Gaucher disease. Neurobiol Dis. 2005;18:83–8. doi: 10.1016/j.nbd.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 64.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 65.Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–75. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hulsen T, de Vlieg J, Alkema W. BioVenn—a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics. 2008;9:488. doi: 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vitner EB, Dekel H, Zigdon H, Shachar T, Farfel-Becker T, Eilam R, Karlsson S, Futerman AH. Altered expression and distribution of cathepsins in neuronopathic forms of Gaucher disease and in other sphingolipidoses. Hum Mol Genet. 2010;19:3583–90. doi: 10.1093/hmg/ddq273. [DOI] [PubMed] [Google Scholar]
- 68.Petry H, Cashion L, Szymanski P, Ast O, Orme A, Gross C, Bauzon M, Brooks A, Schaefer C, Gibson H, et al. Mx1 and IP-10: biomarkers to measure IFN-beta activity in mice following gene-based delivery. J Interferon Cytokine Res. 2006;26:699–705. doi: 10.1089/jir.2006.26.699. [DOI] [PubMed] [Google Scholar]
- 69.Diamond MS, Farzan M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol. 2013;13:46–57. doi: 10.1038/nri3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cohen KW, Dugast A-S, Alter G, McElrath MJ, Stamatatos L. HIV-1 single-stranded RNA induces CXCL13 secretion in human monocytes via TLR7 activation and plasmacytoid dendritic cell-derived type I IFN. J Immunol. 2015;194:2769–75. doi: 10.4049/jimmunol.1400952. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All material used in this manuscript will be made available to researchers subject to confidentiality.