

Abstract
Background and Aims Betula L. (birch) is a genus of approx. 60 species, subspecies or varieties with a wide distribution in the northern hemisphere, of ecological and economic importance. A new classification of Betula has recently been proposed based on morphological characters. This classification differs somewhat from previously published molecular phylogenies, which may be due to factors such as convergent evolution, hybridization, incomplete taxon sampling or misidentification of samples. While chromosome counts have been made for many species, few have had their genome size measured. The aim of this study is to produce a new phylogenetic and genome size analysis of the genus.
Methods Internal transcribed spacer (ITS) regions of nuclear ribosomal DNA were sequenced for 76 Betula samples verified by taxonomic experts, representing approx. 60 taxa, of which approx. 24 taxa have not been included in previous phylogenetic analyses. A further 49 samples from other collections were also sequenced, and 108 ITS sequences were downloaded from GenBank. Phylogenetic trees were built for these sequences. The genome sizes of 103 accessions representing nearly all described species were estimated using flow cytometry.
Key Results As expected for a gene tree of a genus where hybridization and allopolyploidy occur, the ITS tree shows clustering, but not resolved monophyly, for the morphological subgenera recently proposed. Most sections show some clustering, but species of the dwarf section Apterocaryon are unusually scattered. Betula corylifolia (subgenus Nipponobetula) unexpectedly clusters with species of subgenus Aspera. Unexpected placements are also found for B. maximowicziana, B. bomiensis, B. nigra and B. grossa. Biogeographical disjunctions were found within Betula between Europe and North America, and also disjunctions between North-east and South-west Asia. The 2C-values for Betula ranged from 0·88 to 5·33 pg, and polyploids are scattered widely throughout the ITS phylogeny. Species with large genomes tend to have narrow ranges.
Conclusions Betula grossa may have formed via allopolyploidization between parents in subgenus Betula and subgenus Aspera. Betula bomiensis may also be a wide allopolyploid. Betula corylifolia may be a parental species of allopolyploids in the subsection Chinenses. Placements of B. maximowicziana, B. michauxii and B. nigra need further investigation. This analysis, in line with previous studies, suggests that section Apterocaryon is not monophyletic and thus dwarfism has evolved repeatedly in different lineages of Betula. Polyploidization has occurred many times independently in the evolution of Betula.
Keywords: Betula, convergent evolution, genome size, hybridization, ITS, phylogeny, polyploidy
INTRODUCTION
Phylogenetic trees based on individual genes (gene trees) provide useful data for systematics even though the evolutionary history of a particular gene is not necessarily the same as the history of other parts of the genome, or the species (Nichols, 2001). When gene trees contradict classifications based on morphological characters, two broad categories of factors can underlie this discordance. First, a gene tree may be discordant with the species tree due to the effects of hybridization, gene duplication, polyploidy and incomplete lineage sorting (Tate and Simpson, 2003; Koonin, 2005; Degnan and Rosenberg, 2009). Secondly, morphological similarities may give a misleading phylogenetic signal due to convergence (Day et al., 2014). In addition, specimens may be occasionally misidentified (Wiens, 2004), and insufficient sampling can be a problem when interpreting phylogenetic relationships (Pick et al., 2010). Phylogenetic analysis of Betula L. (Betulaceae) is likely to be subject to these problems as Betula species are reported to hybridize frequently, include a number of polyploids and encompass several species that are similar morphologically (Ashburner and McAllister, 2013).
Betula, a genus of trees and shrubs, occupies a broad latitudinal range in the northern hemisphere, from the sub-tropics to the arctic, populating various habitats, including bogs, highlands, tundra and forests. Species of this genus occur in natural landscapes and play important roles in horticulture and forestry (Ashburner and McAllister, 2013). Although several Betula species have wide ranges, some have narrow ranges and are evaluated as endangered in the IUCN Red List (Ashburner and McAllister, 2013; Shaw et al., 2014). The estimated species number within the genus ranges from 30 to 120 (Furlow, 1990; Koropachinskii, 2013), and new species have been described recently (Zeng et al., 2008; McAllister and Rushforth, 2011; Zeng et al., 2014).
The taxonomy of this genus is difficult and controversial, and several classifications have been proposed (Regel, 1865; Winkler, 1904; De Jong, 1993; Skvortsov, 2002). Regel (1865) divided it into subgenus Alnaster and subgenus Eubetula, with the former having the single section Acuminatae and the latter consisting of six sections (Albae, Costatae, Dahuricae, Fruticosae, Lentae and Nanae). Winkler (1904) lowered the two subgenera proposed by Regel (1865) to two sections and merged section Dahuricae and section Fruticosae of Regel (1865) into subsection Albae, and placed section Lentae into subsection Costatae. De Jong (1993) divided the genus into five subgenera: Betula, Betulaster, Betulenta, Chamaebetula and Neurobetula. Based on previous publications and specimens collected from northern Asia, Skvortsov (2002) proposed a classification of four subgenera and eight sections, namely Asperae (sections Asperae, Chinenses and Lentae), Betula (sections Acuminatae, Apterocaryon, Betula, Costatae and Dahuricae), Nipponobetula and Sinobetula. More recently, in a monograph of Betula (Ashburner and McAllister, 2013), a classification into four subgenera and eight sections was proposed. These subgenera are: Acuminata (section Acuminatae), Aspera (sections Asperae and Lentae), Betula (sections Apterocaryon, Betula, Costatae and Dahuricae) and Nipponobetula (section Nipponobetula), with section Asperae being further divided into two subsections: subsection Asperae and subsection Chinenses. This classification largely agrees with the one proposed by Skvortsov (2002), but places section Acuminatae (subgenus Betula) of Skvortsov (2002) as subgenus Acuminata and treats sections Asperae, Chinenses and Lentae of Skvortsov (2002) as subsections Asperae, Chinenses and section Lentae, respectively. Subgenus Sinobetula is not included in this recent classification since the sole species included was proposed based only on a single specimen (Skvortsov, 2002), which is considered to belong to subsection Asperae (Ashburner and McAllister, 2013).
Several molecular phylogenies have been published for the family Betulaceae (Bousquet et al., 1992; Chen et al., 1999; Forest et al., 2005; Grimm and Renner, 2013) and for its constituent genera: Alnus (Navarro et al., 2003), Corylus (Erdogan and Mehlenbacher, 2000; Forest and Bruneau, 2000; Whitcher and Wen, 2001), Carpinus (Yoo and Wen, 2002) and Betula (see references above). It is generally agreed that genus Betula is sister to Alnus, and the remaining four genera (Carpinus, Corylus, Ostryopsis and Ostrya) form another group (Bousquet et al., 1992; Chen et al., 1999). Within Betula, current understanding of phylogenetic relationships is based primarily on five studies with only a sub-set of currently identified species sampled in each study (Järvinen et al., 2004; Li et al., 2005; Nagamitsu et al., 2006; Li et al., 2007; Schenk et al., 2008). To our knowledge, approx. 24 taxa were not included in any previous phylogenetic studies, some because they have been recently discovered or are of limited distribution, including B. ashburneri, B. bomiensis, B. hainanensis and B. murrayana. Some species placements in these phylogenies remain debated, such as the placement of B. schmidtii (Järvinen et al., 2004; Li et al., 2005), the grouping of B. costata and B. alleghaniensis, and the placement of B. glandulosa within section Asperae (Li et al., 2005).
Previous comparisons of morphological and molecular classifications in Betula reveal that they are partially inconsistent and contradictory (Li et al., 2005; Schenk et al., 2008). One potential cause of this, hybridization, is known to occur frequently between Betula species (Dehond and Campbell, 1987; Dehond and Campbell, 1989; Nagamitsu et al., 2006; Karlsdottir et al., 2009; Wang et al., 2014a) and has been shown to occur across sections and even subgenera within Betula (Johnsson, 1945; Dancik and Barnes, 1972; Czernicka et al., 2014; Thomson et al., 2015), potentially causing discordance in phylogenetic relationships.
The recent monograph of Betula (Ashburner and McAllister, 2013) includes determinations of the ploidy level of Betula species based on chromosome counts, with levels ranging from diploid to dodecaploid and counted chromosome numbers from 2n = 28 to 2n = 168. Ploidy level is an important factor in distinguishing some of the morphologically similar species in the genus, such as diploid B. pendula (2n = 2x = 28) and tetraploid B. pubescens (2n = 4x = 56); and diploid B. ashburneri (2n = 2x = 28) and tetraploid B. utilis (2n = 4x = 56). Although the ploidy level has been estimated for nearly all species of Betula, there are only five counts of genome size in the Plant DNA C-values Database (Bennett and Leitch, 2010), representing two diploid species, two tetraploid species and one triploid hybrid. Three of these five counts are from Anamthawat-Jónsson et al. (2010) where the genome size of 12 plants was measured. The genome size of another three species has been reported recently elsewhere (Bai et al., 2012). Of these genome size measurements of which we are aware for Betula, species considered to be diploid appear to have very different genome sizes: the 2C-values of diploid species B. populifolia, B. nana and B. nigra were estimated to be 0·40, 0·91 and 2·90 pg, respectively (Bennett and Leitch, 2010; Bai et al., 2012). Hence, there is a need for complete genome size information for the genus carried out under standard conditions with reliable identification of the specimens used.
Here, we constructed a genus-level phylogeny based on the nuclear ribosomal internal transcribed spacer (ITS) region for the genus Betula using only samples that have been verified by the authors of the recent monograph of the genus, Ashburner and McAllister, except in the case of four species where samples were obtained from three researchers highly familiar with them. We used the ITS region because its high level of polymorphism can help to distinguish species for phylogenetic analyses (Álvarez and Wendel, 2003) although it may suffer from complicating factors such as pseudogenes and biparental signals in recent hybrids (Razafimandimbison et al., 2004). We also conducted broader analyses with samples from living collections or GenBank that have not been previously verified by the monographers. We also measured the genome size of each taxon using flow cytometry.
MATERIALS AND METHODS
Taxon sampling
In order to ensure a complete correspondence between the species names of Ashburner and McAllister (2013) and the taxa included in this study, we obtained species from living collections at the Stone Lane Gardens in Devon (SL hereafter) and University of Liverpool Botanic Gardens at Ness (N hereafter) since these have been collected and curated by Ashburner and McAllister. In addition, we obtained four species (B. alnoides, B. delavayi, B. glandulosa and B. hainanensis) from Jie Zeng (Institute of Tropical Forestry, Chinese Academy of Forestry), Paul Grogan (Queen’s University, Canada) and Zhikun Wu (Kunming Institute of Botany, Chinese Academy of Sciences) who have studied them over many years. We built our main phylogenetic tree using these, which we designate for the purposes of this study the ‘verified’ sample set. We then also built a phylogenetic tree including additional samples obtained from the Royal Botanic Gardens Kew, the Royal Botanic Garden Edinburgh, the Helsinki Botanic Garden, field collections (Supplementary Data Table S1) and GenBank sequences from previous published phylogenetic analyses.
DNA extraction, amplification and sequencing
Genomic DNA was isolated from silica-dried cambial tissue (green vascular tissue located beneath the outer bark of woody stems) or leaves following a modified 2× CTAB (cetyltrimethylammonium bromide) protocol (Wang et al., 2013). The isolated DNA was assessed with 1·0 % agarose gels and measured with a Qubit 2.0 Fluorometer (Invitrogen, Life Technologies) using broad-range assay reagents. The quantified DNA was then diluted to a final concentration of 10–20 ng μL–1 for subsequent use. The nuclear ribosomal internal transcribed spacer (nrITS) region (ITS1, 5·8S and ITS2) was amplified using primers ITS4 (White et al., 1990) and ITSLeu (Baum et al., 1998). The volume of the reaction mix was 20 μL containing: 0·4 μL of AmpliTaq polymerase, 2·0 μL of 10× NH4 buffer (Bioline), 1·6 μL of 50 mm MgCl2 (Bioline), 0·5 μL of 100 mm dNTP, 0·8 μL of each primer (10 mm), 12·9 μL of ddH2O and 1 μL of diluted DNA (10–20 ng). The PCR was carried out using a touchdown program, consisting of an initial denaturation at 95 °C for 3 min, followed by 32 cycles of 1 min at 94 °C, 50 s at 56–52 °C, 1·5 min at 72 °C, and was ended with an extension step of 10 min at 72 °C. The PCR products were purified by binding a 0·8 vol. of Ampure beads (Beckman Coulter Inc.). The purified PCR products were diluted to approx. 20 ng μL–1 in ddH2O prior to sending them to Eurofins (Ebersberg, Germany) for sequencing.
Phylogenetic analyses
ITS tree based on the ‘verified’ sample set.
Seventy-six ‘verified’ accessions representing approx. 60 Betula species and various subspecies, varieties and natural hybrids were Sanger sequenced. Their ITS sequences were checked for recombination in the RPD4 program (Martin et al., 2015) using seven automated detection methods: Bootscanning (Salminen et al., 1995); Chimaera (Posada and Crandall, 2001); GENECONV (Padidam et al., 1999); MaxChi (Smith, 1992); RDP (Martin et al., 2005); SiScan (Gibbs et al., 2000); and 3SEQ (Boni et al., 2007). No signals of recombination were detected using these methods. We downloaded ITS sequences of nine species from other genera of Betulaceae from GenBank, for use as an outgroup. In total, 85 sequences were aligned using BioEdit v 7.0.9.0 (Hall, 1999) with default parameters and the alignment edited manually where necessary. A maximum likelihood (ML) analysis was conducted in PhyML v.3.0 with the default settings (Guindon and Gascuel, 2003) and with the best-fit substitution model GTR + G selected in jModelTest 2.0 (Guindon and Gascuel, 2003; Darriba et al., 2012) using the Akaike information criterion (AIC). A Bayesian inference (BI) analysis was also conducted using the program MrBayes v.3.2 (Ronquist et al., 2012). Two independent runs were performed. For each run, ten million generations were completed with four chains (three heated, one cold). Trees were sampled every 1000 generations, and the first 25 % of runs were discarded as burn-in. Convergence was assessed by determining that the average standard deviation of split frequencies reached a value of <0·01. A majority-rule consensus of the remaining trees from the two runs was produced and used as the BI tree with posterior probabilities (PPs).
ITS tree based on all samples.
In addition to the ‘verified’ sample set, another 49 accessions were Sanger sequenced (Supplementary Data Table S1) and 99 ITS sequences of Betula species were retrieved from GenBank. A total of 233 sequences were aligned and analysed with ML and BI as described above. The consensus trees generated using the above methods were visualized in FigTree v.1.3.1 (http://tree.bio.ed.ac.uk/software/figtree) and edited in Adobe Illustrator CS4 (Adobe Systems).
ITS tree based on diploid samples.
We also conducted phylogenetic analyses exclusively on ‘verified’ species that our C-value measurements (see below) showed to be diploid. Thirty-three Betula accessions were included. An ML analysis was conducted using the same parameters as described above.
Genome size analysis
We measured the genome size of nearly all samples collected from SL and N to correlate them with ploidy levels obtained from chromosome counts (Ashburner and McAllister, 2013). Fresh leaves or cambial tissue were co-chopped with internal standards: Oryza sativa ‘IR36’ (Bennett and Smith, 1991), Solanum lycopersicum L. ‘Stupiké polní rané’ (Doležel et al., 1998), Petroselinum crispum (Mill.) Nyman ex A.W.Hill ‘Champion Moss Curled’ (Obermayer et al., 2002) and Pisum sativum L. ‘Minerva Maple’ (Bennett and Smith, 1991) in 1 mL of Extraction Buffer (Cystain PI absolute P, Partec GmbH, Germany) and then filtered into a tube containing 2·0 mL of Staining Solution (Cystain PI absolute P, Partec GmbH) with 12 μL of propidium iodide (PI). Samples were incubated at room temperature in the dark for approx. 30 min. Three to five replicates were analysed per sample; for each replicate, >5000 nuclei were measured using a Partec CyFlow Space flow cytometer (Partec GmbH) fitted with a 100 mW green solid-state laser (Cobolt Samba; Cobolt, Sweden). Four taxa were analysed with less than three replicates (Supplementary Data Table 1). The resulting histograms were analysed with the Flow-Max software (v.2.4, Partec GmbH).
The ranges of the species for which we measured genome size were divided into four loose categories: narrow (species occurring in a single or a few localities and tending to be endangered), medium (species occurring commonly in multiple areas), widespread (species occupying several parts of a continent) and very widespread (species spread extensively within a continent or across continents) (Supplementary Data Table S2) based on distribution information in the recent monograph of Betula (Ashburner and McAllister, 2013). For species in which multiple individuals were measured, the mean genome size was used for subsequent analysis. Using the average ploidy level and the mean 2C-value of each range category, statistically significant differences between categories were tested using analysis of variance (ANOVA). Tukey HSD post-hoc tests were performed at P < 0·05 when results of ANOVA indicated significance (α ≤ 0·05). All analyses and plots were performed in R 3.1.0 (R Develoment Core Team, 2012) and the package ‘ggplot2' (Wickham, 2009).
To investigate further the evolution of genome size in Betula, we calculated the monoploid genome size, 1Cx (found by dividing the 2C-value by the ploidy level of the species) (Greilhuber et al., 2005), for each of the 71 verified accessions plus each accession of B. pubescens and B. tianshanica from RBGE. These 1Cx-values were grouped according to the ITS clade membership of the species; for each group, 1Cx-values were plotted against ploidy level. We also compared the homogeneity of variance for 1Cx-values among diploid (2x), tetraploid (4x), hexaploid (6x), and octoploid and above (8x–12x) accessions, with R package ‘lawstat’ using the modified robust Brown–Forsythe Levene-type test with 1000 bootstraps (Hui et al., 2008).
RESULTS
The phylogeny of ‘verified’ Betula accessions based on ITS sequences
The aligned ITS data matrix for the ‘verified’ sample set contains 85 ITS sequences and 618 characters, of which 157 characters are variable and 111 informative. There is broad agreement between our ML (Fig. 1) and Bayesian (Supplementary Data Fig. S1) analyses; below we discuss our results based on the ML analyses as these give greater resolution. To facilitate discussion, we have labelled six main clades. Clades I, II and III consist of species of section Lentae (subgenus Aspera). Betula alleghaniensis is sister to B. murrayana whereas B. insignis is sister to B. insignis ssp. fansipanensis, forming clade I and II, respectively. Clade III consists of B. lenta, B. megrelica and B. medwediewii. Clade IV includes species of section Asperae and B. corylifolia, the single species of subgenus Nipponobetula, which appears to be sister to Aspera subsection Chinenses. Clade V contains all species of the subgenus Acuminata together with a sub-clade of B. bomiensis (subsection Asperae) and B. nigra (section Dahuricae), the latter being on a long branch. Clade VI contains all but one of the species in subgenus Betula plus B. grossa (subgenus Aspera, section Lentae) and B. maximowicziana (subgenus Acuminata). The only species of subgenus Betula not found in Clade VI is B. michauxii, which forms a polytomy with clades IV, V and VI. Within Clade VI, the various sections of subgenus Betula do not form unique sub-clades, though B. costata, B. utilis and B. ashburneri from section Costatae cluster together, and B. pubescens, B. pendula and their subspecies/varieties cluster together (Fig. 1). Phylogenetic relationships within the above clades are not fully resolved.
Fig. 1.

Phylogenetic tree from the maximum likelihood analysis of ‘verified’ Betula L. specimens using ITS sequences. Species were classified according to Ashburner and McAllister (2013). Values above branches are bootstrap percentages of ≥50 %. The bars on the right-hand side indicate genome sizes, with colours corresponding to the taxonomy. Bars with a black outline indicate a tentative genome size of the individual.
The phylogeny of all available Betula ITS sequences
The aligned ITS data matrix for all accessions contains 233 ITS sequences and 622 characters, of which 188 characters are variable and 132 informative. The phylogeny of all samples (Fig. 2) reveals a similar overall topology to that of the phylogeny based only on the ‘verified’ sample set. However, 24 (16 %) of the 148 unverified samples have unexpected phylogenetic positions. Of these 24, half were downloaded from GenBank and half were sequenced from samples collected from botanic gardens. Putative B. lenta (GenBank accession FJ011775.1) and B. costata (GenBank accession AY352337.1) appear within Clade II, whereas verified accessions for these species are in Clade III and Clade VI, respectively (Fig. 2). One putative accession of B. glandulosa (GenBank accession AY761110.1) appeared within Clade IV, a clade of species mainly of subsection Chinenses, whereas another three unverified B. glandulosa accessions (GenBank accession FJ011774.1, RBG Kew DNA bank ID: 19950 and Helsinki Botanic Garden accession 1986-0630) are placed in Clade VI. One accession of B. insignis (GenBank accession KP092744) and of B. delavayi (RBG Kew accession 1993-3034) are unexpectedly placed within Clade V, whereas the ‘verified’ samples for these species are in Clade I and Clade IV, respectively. An accession of putative B. dahurica (GenBank accession FJ011773) and one of putative B. skvortsovii are clustered with B. utilis in Clade VI and one accession of B. chinensis (GenBank accession AY761105.1) is clustered with seven accessions of B. dahurica in Clade VI (Fig. 2). All the remaining 12 non-verified accessions found unexpectedly in Clade VI cluster with B. pubescens, B. pendula and their subspecies/varieties (Fig. 2).
Fig. 2.

Phylogenetic tree from the maximum likelihood analysis of all Betula L. samples using ITS sequences. Species were classified according to Ashburner and McAllister (2013). Values above branches are bootstrap percentages of ≥50 %. Names marked in red, blue and black represent potentially misidentified accessions, potentially correctly identified accessions and ‘verified’ accessions, respectively. Included in parentheses are the original labels of potentially misidentified species and their sources; the suggested correct identification of these species is placed before the parentheses.
The phylogeny of diploid Betula accessions
Betula diploids reveal similar phylogenetic positions to when polyploids were included, with a few exceptions: B. corylifolia is in a polytomy with subsection Asperae; B. lenta and B. lenta f. uber are sister to species of subgenera Betula and Acuminata whereas B. costata clusters with subgenus Acuminata (Supplementary Data Fig. S2).
Genome sizes
We found the 2C genome sizes of Betula species to range from 0·88 pg in B. nigra to 5·33 pg in B. insignis ssp. fansipanensis, thus the 1C-value ranges from 0·44 pg (430 Mbp) to 2·67 pg (2611 Mbp). We found Chinese B. alnoides to have a 2C genome size of 1·95 pg, indicating that it is tetraploid rather than diploid (Fig. 1; Supplementary Data Table S1). The fact that B. alnoides is tetraploid has been confirmed by chromosome counting and microsatellite genotyping (Hugh McAllister and Jie Zeng, pers. comm.). We found a genome size of 0·91 pg for B. hainanensis, indicating for the first time that this recently discovered species is diploid. If all other ploidy levels given in Ashburner and McAllister (2013) are correct, the monoploid genome size of Betula (1Cx-value) ranges from 371 Mbp for B. murrayana to 616 Mbp for B. dahurica (Fig. 3). The monoploid genome size is similar among all diploids except for B. potaninii. Variance in monoploid genome size is greater among polyploid accessions. There is a significant difference in the variance of 1Cx-values among the groups of 2x, 4x, 6x and 8x–12x accessions [Fig. 4, P < 0·05; treated pairwise, all groups are significantly non-homogenous in their variances except 4x and 6x (P = 0·15) and 6x and 8x–12x (P = 0·38)]. The proportion of polyploid species of this genus is approx. 0·60, if only species, subspecies/varieties and different cytotypes are included and species having synonyms are treated as one.
Fig. 3.

The monoploid genome size (1Cx-value) of Betula species and cytotypes measured from ‘verified’ samples. Ploidy levels were taken from Ashburner and McAllister (2013). Species are grouped according to the clades shown in Fig. 1.
Fig. 4.
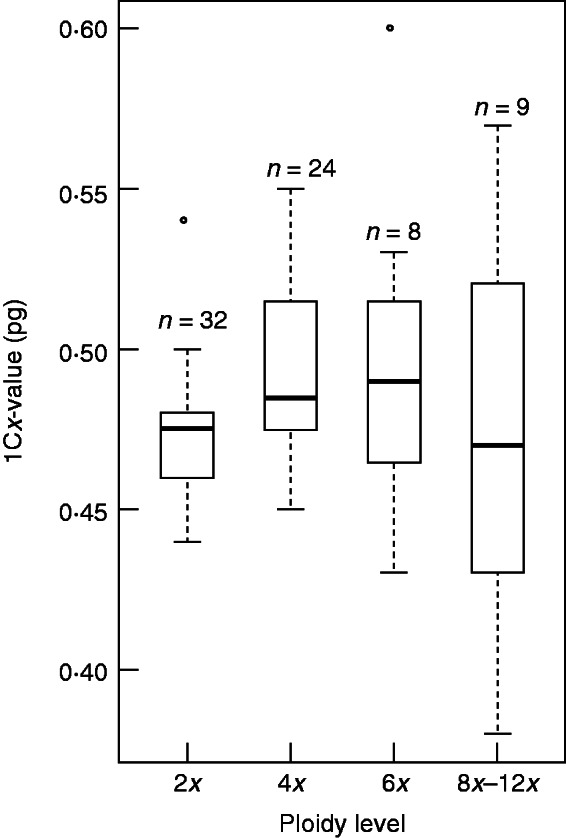
Boxplots showing Betula monoploid (1Cx) genome size of differing ploidy level groups: 2x, 4x, 6x and 8x and above. The number of individuals in each group is shown above the boxplot.
There is a significant difference in the average ploidy level between species with narrow ranges and species with medium, widespread and very widespread ranges (Fig. 5A, P < 0·05), with species with narrow ranges tending to have higher ploidy levels. There is no significant difference in the average ploidy level for species with medium, widespread and very widespread ranges (Fig. 5A, P > 0·05). Similar results also hold true for 2C-values (Fig. 5B).
Fig. 5.
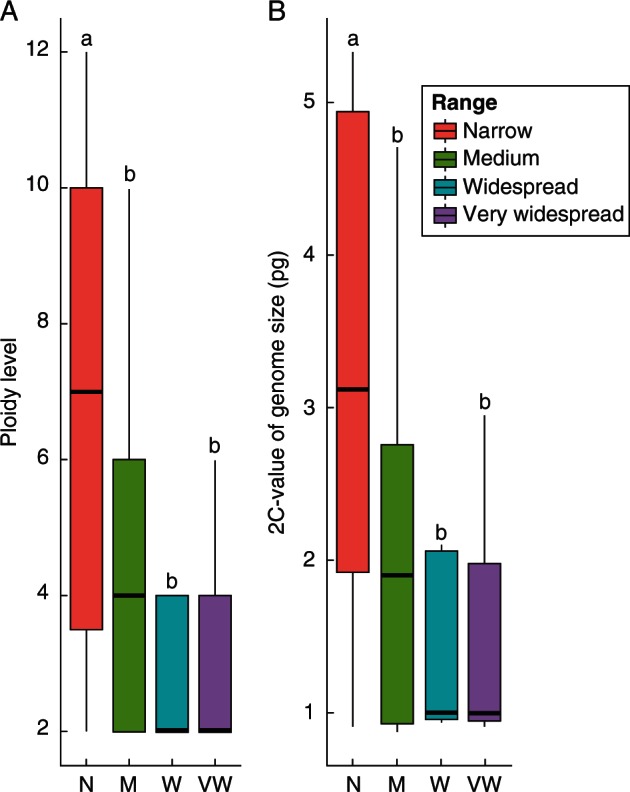
The average ploidy level (A) and the 2C value of genome size (B) among species of different ranges followed by the number of species in parentheses: narrow (12), medium (21), widespread (5) and very widespread (7), respectively. Letters a and b indicate differences at the significance level of P ≤ 0·05. There is no significant difference in the average ploidy level or the 2C-value of genome size if different categories share the same letter.
DISCUSSION
Phylogenetics and taxonomy
Subgenus Aspera.
Ashburner and McAllister (2013) divided subgenus Aspera into two sections: section Lentae (from Regel 1865) and section Asperae. Our ITS data support this division, as the majority of species in these two sections fall into distinct ITS clades, though section Lentae is further subdivided into three unresolved clades. The amplified fragment length polymorphism (AFLP) data of Schenk et al. (2008) also agree with the division of sections Lentae and Asperae. Ashburner and McAllister (2013) further divided section Asperae into subsections Chinenses and Asperae, which are synonymous with section Chinenses and section Asperae of Skvortsov (2002), respectively. Our ITS data broadly support this division. Our ITS data do not support Winkler’s (1904) combination of sections Lentae and Costatae of Regel (1865) into subsection Costatae, nor do the data support subgenus Neurobetula of De Jong (1993), which consists of species from section Asperae, section Costatae and section Dahuricae of Ashburner and McAllister (2013). In addition, our ITS data do not support subgenus Betulenta of De Jong (1993) including species such as B. lenta, B. lenta f. uber and B. globispica as B. globispica is placed in a distinct clade (Fig. 1).
The tetraploid species B. bomiensis, which Ashburner and McAllister (2013) place within section Asperae, is clustered by ITS into a group of species of subgenus Acuminata, but as sister to B. nigra which Ashburner and McAllister (2013) place in section Dahuricae. As Ashburner and McAllister (2013) note, B. bomiensis is morphologically similar to B. potaninii (section Asperae), suggesting that this diploid species may be a parent of B. bomiensis. Our genome size data support this hypothesis, in that the monoploid genome size (1Cx) is unusually large for both species (0·54 pg for B. potaninii and 0·55 pg for B. bomiensis) (Fig. 3; Supplementary Data Table S2). The hypothesis that B. bomiensis was formed via hybridization between B. potaninii and a species of subgenus Acuminata merits further research with additional genetic loci.
Decaploid species B. medwediewii and dodecaploid species B. megrelica form a well-supported clade with diploid species B. lenta and B. lenta f. uber (Figs 1 and 2). This suggests that B. lenta or its ancestral lineage may have been a parent of these two polyploid species. The morphology of the three species also supports this hypothesis (Hugh McAllister, unpubl. res). The study of Li et al. (2005) found a similar result that B. lenta and B. lenta f. uber formed a clade with B. medwediewii. It has previously been suggested (Barnes and Dancik, 1985) that the octoploid species B. murrayana is a recent allopolyploid derivative from B. × purpusii, an inter-subgenus hybrid between B. alleghaniensis (8x) and B. pumila (4x). We find it to form a clade with B. alleghaniensis in the ITS tree, supporting this species as one of its parents (Ashburner and McAllister, 2013).
Interestingly, B. delavayi, a hexaploid species, clustered with the diploid species B. calcicola and B. potaninii, indicating that one of these species or their common ancestor could be a parental species of B. delavayi. Interestingly, both B. potaninii (1Cx = 0·54 pg) and B. delavayi (1Cx = 0·53 pg) have an unusually large monoploid genome size, which could be evidence favouring B. potaninii as its parental species rather than B. calcicola (1Cx = 0·46 pg). Further research is needed to confirm whether other species may also be potential progenitors of B. delavayi.
Ashburner and McAllister (2013) place the hexaploid species B. grossa in section Lentae due to clear morphological similarities, but is not clustered with species of that section by ITS sequences (Fig. 1). This is consistent with AFLP data of Schenk et al. (2008) and the ITS sequences of Nagamitsu et al. (2006). In our case, both B. grossa accessions are from different botanic gardens but each shows the same result (Fig. 2), making misidentification less likely. The unexpected placement of B. grossa into a clade of species of subgenus Betula may indicate that one of the progenitors of this polyploid belongs to subgenus Betula. It is perhaps an allopolyploid formed from hybridization with a species of section Lentae to which it has morphological similarity, causing McAllister and Ashburner (2013) to place it in that section. The ITS sequences from B. grossa may be homogenized from one parent (Nagamitsu et al., 2006). This hypothesis for the parentage of B. grossa deserves further investigation with a larger number of genetic loci.
Subgenus Nipponobetula.
Subgenus Nipponobetula, which comprises the single species B. corylifolia, with distinctive morphology, forms a moderately supported clade (IV) with species of subgenus Aspera in this study, with which it shares some morphological features (Ashburner and McAllister, 2013). Our data do not support the placement of B. corylifolia in section Costatae as in Regel (1865), or subsection Costatae as in Winkler (1904), or subgenus Betulenta as in De Jong (1993). The placement of B. corylifolia with subgenus Aspera was also indicated in two previous phylogenetic studies (Li et al., 2005; Nagamitsu et al., 2006). However, we note that B. corylifolia is found in an ITS clade within Aspera that is composed of the polyploid species B. chinensis (hexaploid and octoploid), B. fargesii and B. globispica, and this clade of four species is sister to a clade containing the diploid Aspera species, of subsection Asperae. We cannot therefore exclude the possibility that B. corylifolia is a parental species of allopolyploids B. chinensis (hexaploid and octoploid), B. fargesii and B. globispica, through hybridization with a species from section Asperae, and may appear nested in the subgenus Aspera as a result. Indeed, in phylogenetic analyses that include only diploid species, B. corylifolia is not nested within subgenus Aspera, but in a polytomy with that clade.
Subgenus Acuminata.
The subgenus Acuminata does not form a distinct clade in our ITS phylogenies. Four of its species appear in a clade with B. nigra, an outlier from subgenus Betula, and B. bomiensis, an outlier from subgenus Aspera. Of these four species, B. alnoides and B. cylindrostachya are tetraploid and B. hainanensis and B. luminifera are diploid species, suggesting that one or both of the two diploids or their common ancestor could be parental species of the tetraploids. A fifth species of Acuminata, B. maximowicziana, appears in the subgenus Betula. A close relationship of B. maximowicziana with species of section Costate (subgenus Betula) is also supported by AFLP markers (Schenk et al., 2008) (though other species of subgenus Acuminata were not included in the AFLP study of Schenk et al., 2008). In contrast, the low-copy nuclear gene NIA supports the grouping of B. maximowicziana with B. alnoides, another species of subgenus Acuminata (Li et al., 2007), making the phylogenetic position of this species questionable. Two lines of evidence in addition to our ITS results may suggest that B. maximowicziana is closely related to species of subgenus Betula. First, a crossing experiment apparently showed that fertile hybrids can form between B. maximowicziana and B. pendula ssp. mandshurica (Johnsson, 1945), indicating that no post-zygotic barriers exist; however, this result has not been convincingly reproduced and we thus cannot exclude the possibility that pollen contamination could have occurred. Secondly, the autumn fruiting and much thicker male catkins of B. maximowicziana are distinct from other species of subgenus Acuminata (Ashburner and McAllister, 2013). Although the overall appearance and detailed characteristics of B. maximowicziana suggest a close relationship with other species of subgenus Acuminata, it does stand apart from them in several features, suggesting an ancient genetic contribution from another evolutionary line within the genus. If the subgenus Acuminata is not monophyletic, the racemose pistillate inflorescence which characterizes it is possibly due to convergent evolution.
Subgenus Betula.
The majority of the species of the subgenus Betula form a single clade, but the four sections of this subgenus have complex relationships in the ITS tree. Section Costatae shows a close relationship with section Betula, and section Apterocaryon species are intermixed with section Betula (Figs 1 and 2). Species of section Betula may have diverged from a lineage of section Costatae recently as the reproductive barrier between the two sections is incomplete: hybrids have been created and reported to be fertile, such as B. pubescens × B. ermanii, B. pubescens × B. albosinensis and B. pendula × B. ermanii (Johnsson, 1945). The status of section Apterocaryon, containing B. michauxii and B. apoiensis, B. nana, B. ovalifolia, B. fruticosa, B. pumila, B. humilis and B. glandulosa, defined by dwarf character, is not supported by the ITS tree, which indicates that the dwarf birches are heterogeneous (Figs 1 and 2). This study, together with several other studies (Li et al., 2005, 2007; Schenk et al., 2008), suggests that dwarfism is a convergent trait, perhaps due to adaptation to cold temperature as evidenced by the existence of bud scales (De Jong, 1993). Betula nana shows a closer relationship with B. pubescens/B. pendula than with B. humilis (Fig. 1). A similar result has been indicated by ADH (Järvinen et al., 2004) and NIA (Li et al., 2007). In addition, the more similar flavonoid profiles of the buds of B. nana and B. pubescens compared with those between B. nana and B. humilis (Wollenweber, 1975) suggest a closer relationship of the former pair than the latter. Surprisingly, B. michauxii, a species morphologically almost identical to B. nana, is not placed within subgenus Betula (Fig. 1), which is consistent with the NIA phylogeny (Li et al., 2007). Further research is needed to decipher the phylogenetic position of B. michauxii.
The taxonomies of the widespread species B. pendula and its tetraploid relative B. pubescens have been particularly controversial in the past, with several subspecies or varieties of both being described and sometimes classified as independent species. Our analysis (Figs 1 and 2) supports the taxonomic treatment of these two species suggested by Ashburner and McAllister (2013), where taxa within the two species are not given species status. Betula pubescens is a tetraploid species; its close relationship with B. pendula indicates the possible involvement of B. pendula in its formation, as has previously been suggested (Howland et al., 1995). The morphological diversity found within these species is probably due to their wide distribution ranges, with morphological variation shaped by overall climatic factors, similar to the variation found within B. papyrifera in North America (Pyakurel and Wang, 2013). Another factor may be hybridization and gene flow between Betula species in different areas of their distributions.
Within section Costatae, B. costata forms a well-supported clade with other species of section Costatae such as B. utilis based on ITS data (Fig. 1). This supports the inclusion of B. costata and B. utilis in section Costatae (Skvortsov, 2002; Ashburner and McAllister, 2013). Within Clade V, the tetraploid species B. alnoides and B. cylindrostachya form an unresolved cluster with the two diploid species, B. luminifera and B. hainanensis, indicating their common ancestry (Fig. 1).
Betula nigra is placed outside the subgenus Betula in all of our ITS phylogenies, both with and without unverified samples, and with and without polyploids in the analyses (Figs 1 and 2; Supplementary Data Fig. S2). In contrast, a phylogenetic study based on NIA suggests that it is more closely related to species of subgenus Betula than B. alnoides (Li et al., 2007), and morphologically B. nigra is most similar to B. dahurica (subgenus Betula). The phylogenetic position of B. nigra needs further research based on multiple loci.
Genome size and ploidy evolution
Different ploidy levels are present in all subgenera and sections of Betula except subgenus Nipponobetula, indicating several independent occurrences of polyploidy in the evolution of the genus (Järvinen et al., 2004). Only subgenus Aspera contains ploidy levels above octoploid (Fig. 3; Supplementary Data Table S1).
The narrow ranges of these species of subgenus Aspera with high ploidy level (e.g. B. insignis, B. megrelica, B. globispica and B. fargesii) may indicate that they are of recent origin or have low invasiveness perhaps due to a low growth rate, which has been associated with larger genome size (Lavergne et al., 2010; Fridley and Craddock, 2015), or their lack of, or very narrow, seed wings (Ashburner and McAllister, 2013). The narrow distributions of these relatively large genomes may also be influenced by available nutrients, such as nitrogen or phosphorus which may select against plants with large genome sizes (Knight et al., 2005; Leitch and Leitch, 2012), and low temperature, which may influence the rate of cell division (Grime and Mowforth, 1982). On the other hand, these high ploidy level birches occur in areas known to harbour many relictual species, and their small populations may be relicts from larger distributions in the past. In contrast, the most diversified, widespread and ‘successful’ species are members of subgenus Betula with low ploidy levels (such as B. pendula, B. nana and B. glandulosa). Hybridization and adaptive introgression occur frequently within subgenus Betula (Thórsson et al., 2010), which may play an important role in colonization of new habitats.
Our genome size results agree with published genome sizes for Icelandic birches, B. nana and B. pubescens, which suggest that no significant genome downsizing has occurred in tetraploid B. pubescens (Anamthawat-Jónsson et al., 2010). However, our results for the 2C-value of B. populifolia are over twice as large as those measured by Feulgen microdensitometry (Olszewska and Osiecka, 1984). This is unlikely to be simply due to the difference in methodology, as flow cytometry and Feulgen microdensitometry were shown to give congruent measurements for Icelandic birches (Anamthawat-Jónsson et al., 2010). Specimen misidentification is also unlikely to be the cause of the differences, as all of the Betula species that we measured have a 2C-value of more than twice the measure of the 2C-value of B. populifolia (Olszewska and Osiecka, 1984); perhaps chemical interference (Greilhuber, 2008) is the explanation for their unusual result. We also found the previously reported 2C-value of B. nigra at 2·90 pg (Bai et al., 2012) to be large compared with the 2C-value of 0·88 pg for B. nigra here, and the specimen measured by Bai et al. (2012) has now been identified as B. alleghaniensis through checking the voucher specimen (DOB0420) (Professor Waller, pers. comm.), which is congruent with the 2C-value of 2·97 pg of B. alleghaniensis found here (Supplementary Data Table S1).
We found the monoploid genome size (1Cx-value) for most species of Betula to be between 0·42 pg and 0·57 pg. Four outlier species, two with lower 1Cx-values and two with higher 1Cx-values, all have higher ploidy levels: octoploid B. murrayana (1Cx = 0·38 pg), octoploid B. chinensis (1Cx = 0·39 pg), hexaploid B. dahurica (1Cx = 0·60 pg) and octoploid B. dahurica (1Cx = 0·57 pg). The chromosome counts of these accessions need to be double-checked, but, assuming they are correct, we found a general pattern that the variance of 1Cx genome sizes is greater in the species of Betula with higher ploidy levels than it is in the diploid species. This suggests that upsizing or downsizing of the sizes of the genomes is occurring in the polyploid birches, perhaps through loss of genome fragments (Buggs et al., 2009, 2012), or proliferation of transposable elements (Bennetzen et al., 2005).
Biogeography
The phylogeography of several species of Betula has been extensively studied. In general, widespread species, such as B. pubescens/B. pendula (Maliouchenko et al., 2007) in Europe and B. papyrifera/B. alleghaniensis in North America (Thomson et al., 2015) show little population subdivision even at large scale, perhaps due to rapid population growth and high levels of gene flow, due to dispersal of pollen and seeds over long distances. In contrast, species likely to have lower dispersal ability, such as B. nana (Wang et al., 2014a), B. humilis (Jadwiszczak et al., 2012) and B. maximowicziana (Tsuda and Ide, 2005; Tsuda et al., 2015), reveal a more subdivided genetic population structure. In addition, geographic barriers in the past and present may play an important role in causing genetic discontinuity (Eidesen et al., 2013).
To our knowledge, biogeographical disjunctions among Betula species have only been mentioned in Li et al. (2005), based on a smaller sample size. Species of Clade III have disjunct distributions (Ashburner and McAllister 2013), with B. medwediewii and B. megrelica in Georgia and Turkey, and B. lenta in North America. We speculate that their common ancestor may have been continuously distributed over the northern hemisphere. Subsequent climate change may have eliminated it in intervening regions, causing geographical disjunctions. In addition, this genus contains three groups with disjunct distributions between North-east Asia and South-west Asia: a common disjunction in groups of related species (Ran et al., 2006). Within subsection Asperae, B. schmidtii and B. chichibuensis occur in North-east Asia whereas B. calcicola, B. potaninii and B. delavayi occur only in South-west China. In the clade comprising subsection Chinenses, B. globispica occurs in North-east Asia, whereas B. fargesii occurs in South-west and central China. In the clade comprising B. costata, B. utilis and B. ashburneri (section Costatae), the first species occurs in North-east Asia whereas the latter two are in South-west and central China.
Unexpected phylogenetic positions of unverified accessions
Unexpected phylogenetic signals for a subset of taxa in our phylogeny of all samples led us to re-appraise their identification. The B. fruticosa and B. nana ssp. exilis (synonym B. glandulosa) samples from Helsinki Botanic Garden were determined to be a subspecies of B. pendula and B. pumila, respectively, based on ITS and morphology (examined by H.A.M.). The putative B. skvortsovii sample was determined to be B. ashburneri based on ITS, morphology (examined by H.A.M.) and genome size of 1·00 pg (2C-value). The nesting of two accessions of B. glandulosa into a clade including B. pumila, whereas the verified B. glandulosa was placed into a distinct clade, was probably caused by the misidentification of B. pumila as B. glandulosa due to their morphological similarity (Fig. 2). Similarly, B. pendula is sometimes misidentified as B. pubescens, and vice versa, as there is a continuum of leaf variations between the two (Wang et al., 2014b).
In addition, of the 12 sequences downloaded from GenBank, we think that at least five were possibly misidentified: B. costata (AY352337.1), B. insignis (KP092744.1), B. glandulosa (AY761110.1), B. dahurica (FI011773) and B. chinensis (AY761105.1). The fact that B. dahurica (FI011773) was collected from the Himalaya region is a strong signal of its misidentification because B. dahurica is distributed in North-east Asia. This species is more likely to be B. utilis as B. utilis is common in the Himalaya region, and this fits with the ITS data. There are 12 accessions clustered with a clade of B. pubescens/B. pendula, showing unexpected phylogenetic signals (Fig. 2). Besides the one labelled as B. fruticosa that is a clear misidentification, the remaining unexpected placements may be caused by hybridization or gene flow between B. pubescens/B. pendula, as these species (such as B. nana, B. glandulosa, B. humilis, B. occidentalis, B. turkstanica and B. papyrifera) can hybridize naturally or in cultivation with B. pubescens/B. pendula (Barnes et al., 1974; Sulkinoja, 1990; Truong et al., 2007; Jadwiszczak et al., 2012; Ashburner and McAllister, 2013).
Concluding remarks
Phylogenentic analyses of the genus Betula based on ITS sequences provide broad agreement with Ashburner and McAllister’s (2013) taxonomical treatment of this genus. This study gives us some new information about the possible origins of some polyploids in the genus, such as B. alnoides, B. chinensis, B. delavayi, B. medwediewii and B. megrelica, but the origins of B. bomiensis and B. grossa remain ambiguous. The phylogenetic positions of B. michauxii, B. maximowicziana and B. nigra remain questionable. The phylogenetic relationships of the genus Betula needs to be further addressed using multiple loci and next-generation sequencing methods such as restriction site-associated DNA markers, which have been successfully applied to Betula species in a pilot study (Wang et al., 2013).
SUPPLEMENTARY DATA
Supplementary data are available online at www.aob.oxfordjournals.org and consist of the following. Table S1: detailed information of the taxa used for ITS sequencing and taxa used for genome size estimation. Table S2: detailed information of the taxa used for comparing the average ploidy level and the mean 2C value of genome size of different ranges. Figure S1: Bayesian analysis of verified Betula species using ITS sequences. Figure S2: phylogenetic tree from the maximum likelihood analysis of Betula diploids using ITS.
ACKNOWLEDGEMENTS
We are grateful to Jie Zeng, Martyn Rix, Paul Grogan, Peter Brownless and Zhikun Wu for providing several Betula species, to Maité Guignard for helpful discussion on genome size analysis, and to Laura Kelly for commenting on the manuscript. This work was funded by Natural Environment Research Council Fellowship NE/G01504X/1 to R.J.A.B. and by Queen Mary University of London. N.W. was funded by the Chinese Scholarship Council (CSC).
LITERATURE CITED
- Álvarez I, Wendel JF. 2003. Ribosomal ITS sequences and plant phylogenetic inference. Molecular Phylogenetics and Evolution 29: 417–434. [DOI] [PubMed] [Google Scholar]
- Anamthawat-Jónsson K, Thórsson Æ, Temsch EM, Greilhuber J. 2010. Icelandic birch polyploids – the case of perfect fit in genome size. Journal of Botany 2010: 347254. [Google Scholar]
- Ashburner K, McAllister HA. 2013. The genus Betula: a taxonomic revision of birches. London: Kew Publishing. [Google Scholar]
- Bai CK, Alverson WS, Follansbee A, Waller DM. 2012. New reports of nuclear DNA content for 407 vascular plant taxa from the United States. Annals of Botany 110: 1623–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes BV, Dancik BP. 1985. Characteristics and origin of a new birch species, Betula murrayana, from southeastern Michigan. Canadian Journal of Botany 63: 223–226. [Google Scholar]
- Barnes BV, Bruce PD, Sharik TL. 1974. Natural hybridization of yellow birch and white birch. Forest Science 20: 215–221. [Google Scholar]
- Baum DA, Small RL, Wendel JF. 1998. Biogeography and floral evolution of Baobabs (Adansonia, Bombacaceae) as inferred from multiple data sets. Systematic Biology 47: 181–207. [DOI] [PubMed] [Google Scholar]
- Bennett MD, Leitch IJ. 2010. Plant DNA C-values Database (release 5.0, December 2010). http://data.kew.org/cvalues/. [Google Scholar]
- Bennett MD, Smith JB. 1991. Nuclear DNA amounts in angiosperms. Philosophical Transactions of the Royal Society B: Biological Sciences 334: 309–345. [DOI] [PubMed] [Google Scholar]
- Bennetzen JL, Ma JX, Devos KM. 2005. Mechanisms of recent genome size variation in flowering plants. Annals of Botany 95: 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni MF, Posada D, Feldman MW. 2007. An exact non-parametric method for inferring mosaic structure in sequence triplets. Genetics 176: 1035–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet J, Strauss SH, Li P. 1992. Complete congruence between morphological and rbcL-based molecular phylogenies in birches and related Species (Betulaceae). Molecular Biology and Evolution 9: 1076–1088. [DOI] [PubMed] [Google Scholar]
- Buggs RJA, Doust AN, Tate JA, et al. 2009. Gene loss and silencing in Tragopogon miscellus (Asteraceae): comparison of natural and synthetic allotetraploids. Heredity 103: 73–81. [DOI] [PubMed] [Google Scholar]
- Buggs R, Chamala S, Wu W, et al. 2012. Rapid, repeated, and clustered loss of duplicate genes in allopolyploid plant populations of independent origin. Current Biology 22: 248–252. [DOI] [PubMed] [Google Scholar]
- Chen ZD, Manchester SR, Sun HY. 1999. Phylogeny and evolution of the Betulaceae as inferred from DNA sequences, morphology, and paleobotany. American Journal of Botany 86: 1168–1181. [PubMed] [Google Scholar]
- Czernicka M, Pławiak J, Muras P. 2014. Genetic diversity of F1 and F2 interspecific hybrids between dwarf birch (Betula nana L.) and Himalayan birch (B. utilis var. jacquemontii (Spach) Winkl. ‘Doorenbos’) using RAPD-PCR markers and ploidy analysis. Acta Biochimica Polonica 61: 195–199. [PubMed] [Google Scholar]
- Dancik BP, Barnes BV. 1972. Natural variation and hybridization of yellow birch and bog birch in southeastern Michigan. Silvae Genetica 21: 1–9. [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods 9: 772–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day PD, Berger M, Hill L, et al. 2014. Evolutionary relationships in the medicinally important genus Fritillaria L. (Liliaceae). Molecular Phylogenetics and Evolution 80: 11–19. [DOI] [PubMed] [Google Scholar]
- Degnan JH, Rosenberg NA. 2009. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends in Ecology and Evolution 24: 332–340. [DOI] [PubMed] [Google Scholar]
- Dehond PE, Campbell CS. 1987. Natural hybridization between Betula cordifolia and B . populifolia (Betulaceae) in Maine. American Journal of Botany 74: 731–731. [Google Scholar]
- Dehond PE, Campbell CS. 1989. Multivariate analyses of hybridization between Betula cordifolia and B. populifolia (Betulaceae). Canadian Journal of Botany 67: 2252–2260. [Google Scholar]
- De Jong PC. 1993. An introduction to Betula: its morphology, evolution, classification and distribution, with a survey of recent work. In: D Hunt, ed. Proceedings of the IDS Betula symposium, 2–4 October 1992 International Dendrology Society; Richmond, UK. [Google Scholar]
- Doležel J, Greilhuber J, Lucretti S, et al. 1998. Plant genome size estimation by flow cytometry: inter-laboratory comparison. Annals of Botany 82: 17–26. [Google Scholar]
- Eidesen PB, Ehrich D, Bakkestuen V, et al. 2013. Genetic roadmap of the Arctic: plant dispersal highways, traffic barriers and capitals of diversity. New Phytologist 200: 898–910. [DOI] [PubMed] [Google Scholar]
- Erdogan V, Mehlenbacher SA. 2000. Phylogenetic relationships of Corylus species (Betulaceae) based on nuclear ribosomal DNA ITS region and chloroplast matK gene sequences. Systematic Botany 25: 727–737. [Google Scholar]
- Forest F, Bruneau A. 2000. Phylogenetic analysis, organization, and molecular evolution of the nontranscribed spacer of 5S ribosomal RNA genes in Corylus (Betulaceae). International Journal of Plant Sciences 161: 793–806. [Google Scholar]
- Forest F, Savolainen V, Chase MW, et al. 2005. Teasing apart molecular- versus fossil-based error estimates when dating phylogenetic trees: a case study in the birch family (Betulaceae). Systematic Botany 30: 118–133. [Google Scholar]
- Fridley JD, Craddock A. 2015. Contrasting growth phenology of native and invasive forest shrubs mediated by genome size. New Phytologist 207: 659–668. [DOI] [PubMed] [Google Scholar]
- Furlow J. 1990. The genera of Betulaceae in the south eastern United States. Journal of Arnold Arboretum 71: 1–67. [Google Scholar]
- Gibbs MJ, Armstrong JS, Gibbs AJ. 2000. Sister-Scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16: 573–582. [DOI] [PubMed] [Google Scholar]
- Greilhuber J. 2008. Cytochemistry and C-values: the less-well-known world for nuclear DNA amounts. Annals of Botany 101: 791–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greilhuber J, Dolezel J, Lysak MA, Bennett MD. 2005. The origin, evolution and proposed stabilization of the terms ‘Genome Size’ and ‘C-Value’ to describe nuclear DNA contents. Annals of Botany 95: 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grime JP, Mowforth MA. 1982. Variation in genome size – an ecological interpretation. Nature 299: 151–153. [Google Scholar]
- Grimm GW, Renner SS. 2013. Harvesting Betulaceae sequences from GenBank to generate a new chronogram for the family. Botanical Journal of the Linnean Society 172: 465–477. [Google Scholar]
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology 52: 696–704. [DOI] [PubMed] [Google Scholar]
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41: 95–98. [Google Scholar]
- Howland DE, Oliver RR, Davy AJ. 1995. Morphological and molecular variation in natural populations of Betula. New Phytologist 130: 117–124. [Google Scholar]
- Hui W, Gel YR, Gastwirth JL. 2008. lawstat: an R package for law, public policy and biostatistics. Journal of Statistical Software 28: 1–26. [Google Scholar]
- Järvinen P, Palmé AE, Morales LO, et al. 2004. Phylogenetic relationships of Betula species (Betulaceae) based on nuclear ADH and chloroplast matK sequences. American Journal of Botany 91: 1834–1845. [DOI] [PubMed] [Google Scholar]
- Jadwiszczak K, Banaszek A, Jabłońska E, Sozinov O. 2012. Chloroplast DNA variation of Betula humilis Schrk. in Poland and Belarus. Tree Genetics and Genomes 8: 1017–1030. [Google Scholar]
- Johnsson H. 1945. Interspecific hybridization within the genus Betula. Hereditas 31: 163–176. [DOI] [PubMed] [Google Scholar]
- Karlsdottir L, Hallsdottir M, Thórsson Æ, Anamthawat-Jónsson K. 2009. Evidence of hybridisation between Betula pubescens and B. nana in Iceland during the early Holocene. Review of Palaeobotany and Palynology 156: 350–357. [Google Scholar]
- Knight CA, Molinari NA, Petrov DA. 2005. The large genome constraint hypothesis: evolution, ecology, and phenotype. Annals of Botany 95: 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. 2005. Orthologs, paralogs, and evolutionary genomics. Annual Review of Genetics 39: 309–338. [DOI] [PubMed] [Google Scholar]
- Koropachinskii IY. 2013. Natural hybridization and taxonomy of birches in North Asia. Contemporary Problems in Ecology 6: 350–369. [Google Scholar]
- Lavergne S, Muenke NJ, Molofsky J. 2010. Genome size reduction can trigger rapid phenotypic evolution in invasive plants. Annals of Botany 105: 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch AR, Leitch IJ. 2012. Ecological and genetic factors linked to contrasting genome dynamics in seed plants. New Phytologist 194: 629–646. [DOI] [PubMed] [Google Scholar]
- Li JH, Shoup S, Chen ZD. 2005. Phylogenetics of Betula (Betulaceae) inferred from sequences of nuclear ribosomal DNA. Rhodora 107: 69–86. [Google Scholar]
- Li JH, Shoup S, Chen ZD. 2007. Phylogenetic relationships of diploid species of Betula (Betulaceae) inferred from DNA sequences of nuclear nitrate reductase. Systematic Botany 32: 357–365. [Google Scholar]
- Maliouchenko O, Palmé AE, Buonamici A, Vendramin GG, Lascoux M. 2007. Comparative phylogeography and population structure of European Betula species, with particular focus on B . pendula and B. pubescens. Journal of Biogeography 34: 1601–1610. [Google Scholar]
- Martin DP, Williamson C, Posada D. 2005. RDP2: recombination detection and analysis from sequence alignments. Bioinformatics 21: 260–262. [DOI] [PubMed] [Google Scholar]
- Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. 2015. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evolution 1: vev003. doi:010.1093/ve/vev1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister HA, Rushforth K. 2011. Betula ashburneri. Curtis’s Botanical Magazine 28: 111–118. [Google Scholar]
- Nagamitsu T, Kawahara T, Kanazashi A. 2006. Endemic dwarf birch Betula apoiensis (Betulaceae) is a hybrid that originated from Betula ermanii and Betula ovalifolia. Plant Species Biology 21: 19–29. [Google Scholar]
- Navarro E, Bousquet J, Moiroud A, Munive A, Piou D, Normand P. 2003. Molecular phylogeny of Alnus (Betulaceae), inferred from nuclear ribosomal DNA ITS sequences. Plant and Soil 254: 207–217. [Google Scholar]
- Nichols R. 2001. Gene trees and species trees are not the same. Trends in Ecology and Evolution 16: 358–364. [DOI] [PubMed] [Google Scholar]
- Obermayer R, Leitch IJ, Hanson L, Bennett MD. 2002. Nuclear DNA C-values in 30 species double the familial representation in Pteridophytes. Annals of Botany 90: 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewska MJ, Osiecka R. 1984. The relationship between 2C DNA content, life cycle type, systematic position, and the level of DNA endoreplication in nuclei of parenchyma cells during growth and differentiation of roots in some monocotyledonous species. Biochemie und Physiologie der Pflanzen 177: 319–336. [Google Scholar]
- Padidam M, Sawyer S, Fauquet CM. 1999. Possible emergence of new geminiviruses by frequent recombination. Virology 265: 218–225. [DOI] [PubMed] [Google Scholar]
- Pick KS, Philippe H, Schreiber F, et al. 2010. Improved phylogenomic taxon sampling noticeably affects nonbilaterian relationships. Molecular Biology and Evolution 27: 1983–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D, Crandall KA. 2001. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proceedings of the National Academy of Sciences, USA 98: 13757–13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyakurel A, Wang JR. 2013. Leaf morphological variation among paper birch (Betula papyrifera Marsh.) genotypes across Canada. Open Journal of Ecology 3: 284–295. [Google Scholar]
- Ran JH, Wei XX, Wang XQ. 2006. Molecular phylogeny and biogeography of Picea (Pinaceae): implications for phylogeographical studies using cytoplasmic haplotypes. Molecular Phylogenetics and Evolution 41: 405–419. [DOI] [PubMed] [Google Scholar]
- Razafimandimbison SG, Kellogg EA, Bremer B. 2004. Recent origin and phylogenetic utility of divergent ITS putative pseudogenes: a case study from Naucleeae (Rubiaceae). Systematic Biology 53: 177–192. [DOI] [PubMed] [Google Scholar]
- R Develoment Core Team. 2012. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Regel E. 1865. Bemerkungen über die Gattungen Betula und Alnus nebst Beschreibung einiger neuer Arten. Bulletin of Society of Naturalist (Moscou) 38: 388–434. [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, et al. 2012. MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen MO, Carr JK, Burke DS, Mccutchan FE. 1995. Identification of breakpoints in intergenotypic recombinants of HIV type-1 by Bootscanning. Aids Research and Human Retroviruses 11: 1423–1425. [DOI] [PubMed] [Google Scholar]
- Schenk MF, Thienpont CN, Koopman WJM, Gilissen LJWJ, Smulders MJM. 2008. Phylogenetic relationships in Betula (Betulaceae) based on AFLP markers. Tree Genetics and Genomes 4: 911–924. [Google Scholar]
- Shaw K, Stritch L, Rivers M, Roy S, Wilson B, Govaerts R. 2014. The red list of Betulaceae. BGCI; Richmond. UK. [Google Scholar]
- Skvortsov AK. 2002. A new system of the genus Betula L. – the birch. Bulletin of Moscow Society of Naturalist 107: 73–76. [Google Scholar]
- Smith JM. 1992. Analyzing the mosaic structure of genes. Journal of Molecular Evolution 34: 126–129. [DOI] [PubMed] [Google Scholar]
- Sulkinoja M. 1990. Hybridization, introgression and taxonomy of the mountain birch in SW Greenland compared with related results from Iceland and Finnish Lapland. Meddelelser om Grönland Bioscience 33: 21–29. [Google Scholar]
- Tate JA, Simpson BB. 2003. Paraphyly of Tarasa (Malvaceae) and diverse origins of the polyploid species. Systematic Botany 28: 723–737. [Google Scholar]
- Thórsson Æ, Pálsson S, Lascoux M, Anamthawat-Jónsson K. 2010. Introgression and phylogeography of Betula nana (diploid), B. pubescens (tetraploid) and their triploid hybrids in Iceland inferred from cpDNA haplotype variation. Journal of Biogeography 37: 2098–2110. [Google Scholar]
- Thomson AM, Dick CW, Dayanandan S. 2015. A similar phylogeographical structure among sympatric North American birches (Betula) is better explained by introgression than by shared biogeographical history. Journal of Biogeography 42: 339–350. [Google Scholar]
- Truong C, Palmé AE, Felber F. 2007. Recent invasion of the mountain birch Betula pubescens ssp. tortuosa above the treeline due to climate change: genetic and ecological study in northern Sweden. Journal of Evolutionary Biology 20: 369–380. [DOI] [PubMed] [Google Scholar]
- Tsuda Y, Ide Y. 2005. Wide-range analysis of genetic structure of Betula maximowicziana, a long-lived pioneer tree species and noble hardwood in the cool temperate zone of Japan. Molecular Ecology 14: 3929–3941. [DOI] [PubMed] [Google Scholar]
- Tsuda Y, Nakao K, Ide Y, Tsumura Y. 2015. The population demography of Betula maximowicziana, a cool-temperate tree species in Japan, in relation to the last glacial period: its admixture-like genetic structure is the result of simple population splitting not admixing. Molecular Ecology 24: 1403–1418. [DOI] [PubMed] [Google Scholar]
- Wang N, Thomson M, Bodles WJA, et al. 2013. Genome sequence of dwarf birch (Betula nana) and cross-species RAD markers. Molecular Ecology 22: 3098–3111. [DOI] [PubMed] [Google Scholar]
- Wang N, Borrell JS, Bodles WJA, Kuttapitiya A, Nichols RA, Buggs RJA. 2014a. Molecular footprints of the Holocene retreat of dwarf birch in Britain. Molecular Ecology 23: 2771–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Borrell JS, Buggs RJA. 2014b. Is the Atkinson discriminant function a reliable method for distinguishing between Betula pendula and B . pubescens? New Journal of Botany 4: 90–94. [Google Scholar]
- Whitcher IN, Wen J. 2001. Phylogeny and biogeography of Corylus (Betulaceae): inferences from ITS sequences. Systematic Botany 26: 283–298. [Google Scholar]
- White TJ, Bruns T, Lee S, Taylor T. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: MA Innis, DH Gelfland, JJ Sninsky, TJ White, eds. PCR protocols: a guide to methods and applications. New York: Academic Press, 315–322. [Google Scholar]
- Wickham H. 2009. ggplot2: elegant graphics for data analysis , 3rd printing 2010 edn. New York: Springer. [Google Scholar]
- Wiens JJ. 2004. The role of morphological data in phylogeny reconstruction. Systematic Biology 53: 653–661. [DOI] [PubMed] [Google Scholar]
- Winkler H. 1904. Betulaceae. Das Pflanzenreich 19: 1–149. [Google Scholar]
- Wollenweber E. 1975. Flavonidmuster in Knospenexkret der Betulaceen. Biochemical Systematics and Ecology 3: 47–52. [Google Scholar]
- Yoo K, Wen J. 2002. Phylogeny and biogeography of Carpinus and subfamily Coryloideae (Betulaceae). International Journal of Plant Sciences 163: 641–650. [Google Scholar]
- Zeng J, Li JH, Chen ZD. 2008. A new species of Betula section Betulaster (Betulaceae) from China. Botanical Journal of the Linnean Society 156: 523–528. [Google Scholar]
- Zeng J, Ren BQ, Zhu JY, Chen ZD. 2014. Betula hainanensis (Betulaster, Betulaceae), a new species from Hainan Island, China. Annales Botanici Fennici 51: 399–402. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.