

Abstract
Strain M27-SA2 was isolated from the deep-sea salt-saturated anoxic lake Medee, which represents one of the most hostile extreme environments on our planet. On the basis of physiological studies and phylogenetic positioning this extremely halophilic euryarchaeon belongs to a novel genus ‘Halanaeroarchaeum’ within the family Halobacteriaceae. All members of this genus cultivated so far are strict anaerobes using acetate as the sole carbon and energy source and elemental sulfur as electron acceptor. Here we report the complete genome sequence of the strain M27-SA2 which is composed of a 2,129,244-bp chromosome and a 124,256-bp plasmid. This is the second complete genome sequence within the genus Halanaeroarchaeum. We demonstrate that genome of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 harbors complete metabolic pathways for acetate and sulfur catabolism and for de novo biosynthesis of 19 amino acids. The genomic analysis also reveals that ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 harbors two prophage loci and one CRISPR locus, highly similar to that of Kulunda Steppe (Altai, Russia) isolate ‘H. sulfurireducens’ HSR2T. The discovery of sulfur-respiring acetate-utilizing haloarchaeon in deep-sea hypersaline anoxic lakes has certain significance for understanding the biogeochemical functioning of these harsh ecosystems, which are incompatible with life for common organisms. Moreover, isolations of Halanaeroarchaeum members from geographically distant salt-saturated sites of different origin suggest a high degree of evolutionary success in their adaptation to this type of extreme biotopes around the world.
Keywords: Sulfur reduction, Strictly anaerobic, Extremely halophilic archaea, Hypersaline lake, Anoxic habitats
Introduction
‘Halanaeroarchaeum sulfurireducens’ M27-SA2 was isolated from the deep-sea hypersaline anoxic lake Medee (Ionian Sea, Eastern Mediterranean, water depth 3105 m). Together with other five strains, previously isolated from shallow and terrestrial athalassic hypersaline sites of Russia and Spain [1], this haloarchaeon possesses maximum of 91–93 % 16S rDNA sequence similarity to the nearest cultured members of Halobacteriaceae. All Halanaeroarchaeum isolates represent a novel type of strictly anaerobic haloarchaea that grow best in NaCl brines close to saturation and use acetate as sole electron donor and carbon source with elemental sulfur as the only electron acceptor. Little is known about anaerobic sulfur metabolism at saturated salt conditions [2]. There is some evidence suggesting that bacterial sulfate reduction is possible under salt-saturated conditions [3], but sulfur respiration under such conditions has so far been very poorly investigated, except for the ‘Halanaeroarchaeum’ strain HSR2T and two extremely haloalkaliphilic bacteria of the order Halanaerobiales, Halarsenatibacter silvermanii and Natroniella sulfidigena [1, 4, 5]. Following the fact, that we were able to isolate these haloarchaea from various geographically and physico-chemically distinct hypersaline sites [1], the sulfidogenic anaerobic oxidation of acetate is likely a common feature in anoxic salt-saturated habitats, overlooked so far.
In this paper we describe the genome properties of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 providing details on carbon and sulfur metabolism, on clustered regularly interspaced short palindromic repeats (CRISPR) and on presence of prophage loci and genomic islands.
Organism information
Classification and features
‘Halanaeroarchaeum sulfurireducens’ M27-SA2 has typical haloarchaeal pleomorphic cell morphology, ranging from flattened rods to coccoid or irregular forms (Fig. 1). The pleomorphism of M27-SA2 strain increased with the cultivation time, as is often observed for members of the family Halobacteriaceae. The 16S rRNA gene of M27-SA2 exhibited 99.58 % sequence similarity with H. sulfurireducens strain HSR2T and 97-98 % sequence similarity with clones of uncultured haloarchaea obtained from hypersaline anoxic soils, brines and sediments around the world [1] (Fig. 2).
Fig. 1.

Morphology of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 cells grown on acetate (a) and pyruvate (b) as electron donors and elemental sulphur as electron acceptor. The scale bars represent 5 μm
Fig. 2.
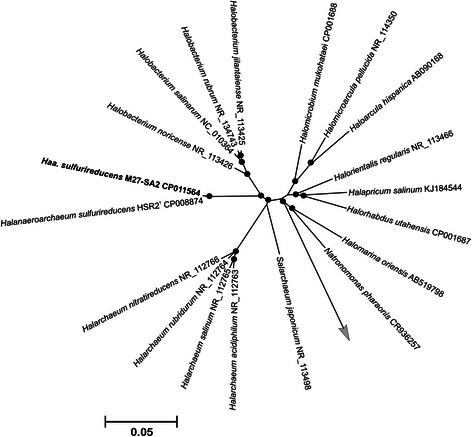
Phylogenetic tree of 16S rRNA gene sequences showing the position of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2. Tree was inferred from a 16S rRNA gene sequence alignment with PAUP*4.b10 [59] using a LogDet/paralinear distance method. Support for nodes in the tree corresponds to bootstrap values for 1000 pseudo-replicates. Only bootstrap values greater than 75 % are displayed as solid circles. The tree has been arbitrarily rooted on sequence of Natronomonas pharaonis (D87971) and Halomarina oriensis (AB519798). The 16S rRNA gene sequence of Methanohalophilus halophilus (FN870068) was used as the outgroup. The scale bars represent a 5 % nucleotide sequence divergence
Together with other Halanaeroarchaeum isolates, M27-SA2 represents the only type of obligate and strictly anaerobic haloarchaea. Most of the known cultivated extremely halophilic euryarchaeota are aerobic heterotrophs except for a few examples of facultatively anaerobic species capable of growth by fermentation [6], denitrification [7], fumarate, DMSO and TMAO reduction [8, 9]. Strain M27-SA2 was isolated from the brine (320 g l−1 of total salt content) of deep-sea Lake Medee (Eastern Mediterranean) collected in September 2012 at depth of 3,010 m. The collected Medee brine was transferred into the serum vials (120 ml) prefilled with the artificial brine to attain 230 g l−1 of final salinity. The artificial brine has the following composition: NaCl 200 g l−1; KH2PO4 0.33 g l−1; yeast extract 50 mg l−1; Na2S 0.5 g l−1; acetate 15 mmol l−1; S° 2.5 g l−1, 10 ml l−1 trace elements solution (DSMZ medium 320); and 10 ml l−1 vitamin solution (DSMZ medium 141); pH values were adjusted to 6.7 corresponding to in situ values of the brine. Similar to all known ‘Halanaeroarchaeum’ isolates, strain M27-SA2 grew between pH 6.7 and 8.0 (with the optimum at pH 7.2–7.5), 3.0 and 5.0 M of NaCl with the optimum growth observed at total salinity of 250 g l−1. Notwithstanding the isolation from the environment with permanent temperature of 15 °C [10], strain M27-SA2 has the optimal temperature of growth at 40 °C (Table 1). The isolate has a very limited metabolic profile restricted to acetate and pyruvate as the only available sources of carbon and energy and elemental sulfur as a electron acceptor [1]. Nevertheless, yeast extract should be added to the medium in concentrations of at least 10 mg l−1, as supplemental source of some amino acids, vitamins and cofactors which M27-SA2 likely cannot synthetize.
Table 1.
Classification and general features of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2T [48]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Archaea | TAS [49] | |
| Phylum Euryarchaeota | TAS [50] | ||
| Class Halobacteria | TAS [51, 52] | ||
| Order Halobacteriales | TAS [53–55] | ||
| Family Halobacteriaceae | TAS [56, 57] | ||
| Genus Halanaeroarchaeum | TAS [1] | ||
| Species ‘Halanaeroarchaeum sulfurireducens’ | TAS [1] | ||
| Type strain M27-SA2T (CP011564, CP011565) | TAS [1] | ||
| Cell shape | Pleomorphic | TAS [1] | |
| Motility | Non-motile | TAS [1] | |
| Sporulation | Non-sporulating | NAS | |
| Temperature range | 15–50 °C | TAS [1, 10] | |
| Optimum temperature | 40 °C | TAS [1] | |
| pH range; Optimum | 6.7–8.0; 7.2–7.5 | TAS [1] | |
| Carbon source | Acetate, pyruvate | TAS [1] | |
| MIGS-6 | Habitat | Hypersaline anoxic lake sediments (brine) | TAS [1, 10] |
| MIGS-6.3 | Salinity | 3.0–5.0 M NaCl | TAS [1] |
| MIGS-22 | Oxygen requirement | Strictly anaerobic | TAS [1] |
| MIGS-15 | Biotic relationship | Free-living | TAS [1] |
| MIGS-14 | Pathogenicity | Non-pathogen | NAS |
| MIGS-4 | Geographic location | Lake Medee, Ionian Sea, Eastern Mediterranean | TAS [1, 10] |
| MIGS-5 | Sample collection | 24 September 2012 | TAS [1, 10] |
| MIGS-4.1 | Latitude | 34°26.250N | TAS [1, 10] |
| MIGS-4.2 | Longitude | 22°19.783E | TAS [1, 10] |
| MIGS-4.3 | Depth | 3105 m | TAS [1, 10] |
aEvidence codes – IDA Inferred from Direct Assay (first time in publication), TAS Traceable Author Statement (i.e., a direct report exists in the literature), NAS Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [58]. If the evidence is IDA, then the property was directly observed for a live isolate by one of the authors or an expert mentioned in the acknowledgements
Genome sequencing information
Genome project history
‘Halanaeroarchaeum sulfurireducens’ strain M27-SA2 was selected for sequencing on the basis of its phylogenetic positions, its particular feature as a novel strictly anaerobic haloarchaeon from the deep-sea anoxic salt-saturated lake and the interest of studying this unique mechanism of anaerobic respiration, recently discovered for the first time among entire Archaea domain [1]. The respective genome project is deposited on the NCBI BioProject PRJNA284332 and the complete genome sequence in GenBank CP011564 and CP011565 (chromosome and plasmid) is available since 30 of September 2015. The main project information is summarized in Table 2.
Table 2.
Project information for ‘Halanaeroarchaeum sulfurireducens’ M27-SA2
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Illumina standard library, Miseq Reagent kit v2. |
| MIGS-29 | Sequencing platforms | Illumina MiSeq System |
| MIGS-31.2 | Fold coverage | 634x chromosome, 691x plasmid |
| MIGS-30 | Assemblers | Velvet 1.2.10, Geneious 7.1 |
| MIGS-32 | Gene calling method | Geneious 7.1, Glimmer 3.02, tRNAScan-SE |
| Locus Tag | HLASA | |
| GenBank ID | CP011564 (chromosome) | |
| CP011565 (plasmid) | ||
| GenBank date of release | 30/09/2015 | |
| BIOPROJECT | PRJNA284332 | |
| MIGS-13 | Source material identifier | Isolated from the deep-sea hypersaline lake Medee, Ionian Sea, Eastern Mediterranean, water depth 3105 m. Salinity: 230 g/l; pH 6.8. Coordinates 34°26.250N, 22°19.783E. |
| Project relevance | Extremophile hypersaline environments |
Growth conditions and genomic DNA preparation
Strain M27-SA2 was routinely grown anaerobically to early stationary phase in 120-ml flasks using a protocol described elsewhere [1]. Genomic DNA was isolated from the cell paste according to extraction method from Urakawa et al. [11]. DNA quality and quantity were determined with a Nanodrop spectrometer (Thermo Scientific, Wilmington, USA).
Genome sequencing and assembly
The M27-SA2 genome was sequenced with MiSeq System technology of Illumina Inc. (San Diego, CA, USA) using paired-end 250-bp reads. The library was prepared from 1 μg of genomic DNA with NEBNext Ultra DNA library preparation kit (NewEngland Biolabs, Ipswich, USA) according to manufacturer’s instructions with insert size range of 250–750 bp and maximum of insert size distribution of 470 bp. Sequencing run resulted in 6,480,650 paired-end reads with an average read length of 250 bp, yielding 1.62 Gbp. These reads were assembled using both Velvet 1.2.10 [12] and Geneious 7.1 software. Gaps between contigs were closed with a conventional PCR-based gap closure approach and supported by manual refining with Geneious 7.1 embedded tools, resulting in a fully closed circular chromosome of 2,129,244 bp, and a circular plasmid of 124,256 bp. Together, all matching sequences provided 634× coverage for chromosome and 691× for plasmid.
Genome annotation
Protein-coding genes were predicted by Glimmer 3.02 [13]; rRNA genes by RNAmmer 1.2 Server online tool [14]; tRNA-coding sequences by tRNAscan-SE 1.21 online tool [15]; while operon prediction was performed by the FgenesB online tool [16]. Some of structural and functional annotations were performed as it was described by Toshchakov et al. [17]. For each predicted gene similarity search was performed by Geneious 7.1 BLAST embedded tool against public amino acid sequence databases (nr, SwissProt), conserved domains families databases (Pfam, COG). Finally, annotations were manually curated using the Artemis 16.0 program [18] and refined for each gene with NCBI blastx against nr database (only for control) [19].
Genome properties
The genome of strain M27-SA2 comprises two circular replicons: a 2,129,244-bp chromosome and a 124,256-bp plasmid (Fig. 3 and Table 3). The chromosome has a 63.19 % GC content. Of the 2,200 predicted genes (88.36 % of coding density), 2,151 were protein coding genes (84.3 % started with an ATG codon, 12.9 % with a GTG, and 2.7 % with a TTG), and 49 RNAs genes (a single rRNA operon and 46 tRNAs, see Table 4). The majority of the protein-coding genes (60.72 %) were assigned with a putative function, while remaining sequences were annotated as hypothetical proteins. An assignment of genes by COGs functional categories is presented in Table 5. The plasmid has 55.39 % GC content and contains 119 protein-coding genes. Only 24 of them (20.16 %) were assigned to COGs (Table 6 and Table 7) while the remaining genes were annotated as hypothetical proteins.
Fig. 3.
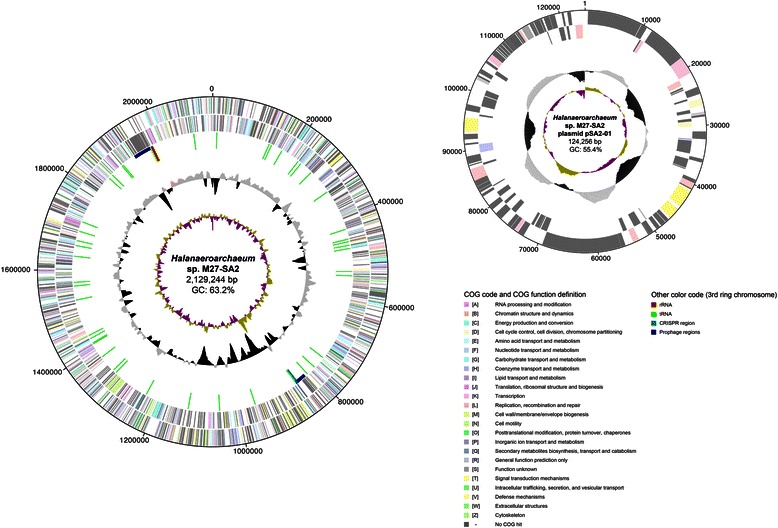
Graphical circular map of the genome of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2. From outside to center: genes on forward strand (COG color-coded), genes on reverse strand (COG color-coded), RNA genes and other (only for chromosome: tRNAs green, rRNAs red, CRISPR azure, Prophages dark blue), GC content, GC skew [18, 60]
Table 3.
Genome composition for ‘Halanaeroarchaeum sulfurireducens’ M27-SA2
| Label | Size (Mb) | Topology | INSDC identifier | RefSeq ID |
|---|---|---|---|---|
| Chromosome | 2.129 | circular | CP011564.1 | NZ_CP011564.1 |
| Plasmid | 0.124 | circular | CP011565.1 | NZ_CP011565.1 |
Table 4.
Chromosome statistics for ‘Halanaeroarchaeum sulfurireducens’ M27-SA2
| Attribute | Value | % of total |
|---|---|---|
| Chromosome size (bp) | 2,129,244 | |
| DNA coding (bp) | 1,860,079 | 87.36 % |
| DNA G + C (bp) | 1,345,472 | 63.19 % |
| Total genes | 2,200 | |
| Protein-coding genes | 2,151 | 97.77 % |
| tRNA genes | 46 | 2.09 % |
| rRNA genes (5S-16S-23S) | 3 | 0.14 % |
| Genes assigned to COGs | 1,306 | 60.72 % |
| CRISPR repeats | 1 | |
| Average length (bp) | 861 | |
| Max length (bp) | 5,619 | |
| ATG initiation codon proteins | 1,814 | 84.33 % |
| GTG initiation codon proteins | 278 | 12.93 % |
| TTG initiation codon proteins | 59 | 2.74 % |
Table 5.
Number of genes associated with the general COG functional categories for chromosome
| Code | Value | % age | COG category |
|---|---|---|---|
| J | 129 | 5.99 % | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.05 % | RNA processing and modification |
| K | 79 | 3.67 % | Transcription |
| L | 91 | 4.23 % | Replication, recombination and repair |
| B | 3 | 0.14 % | Chromatin structure and dynamics |
| D | 8 | 0.37 % | Cell cycle control, cell division, chromosome partitioning |
| V | 10 | 0.46 % | Defense mechanisms |
| T | 46 | 2.14 % | Signal transduction mechanisms |
| M | 46 | 2.14 % | Cell wall/membrane/envelope biogenesis |
| N | 28 | 1.30 % | Cell motility |
| U | 9 | 0.42 % | Intracellular trafficking, secretion, and vesicular transport |
| O | 58 | 2.70 % | Posttranslational modification, protein turnover, chaperones |
| C | 93 | 4.32 % | Energy production and conversion |
| G | 27 | 1.26 % | Carbohydrate transport and metabolism |
| E | 133 | 6.18 % | Amino acid transport and metabolism |
| F | 52 | 2.42 % | Nucleotide transport and metabolism |
| H | 80 | 3.72 % | Coenzyme transport and metabolism |
| I | 29 | 1.35 % | Lipid transport and metabolism |
| P | 57 | 2.65 % | Inorganic ion transport and metabolism |
| Q | 7 | 0.33 % | Secondary metabolites biosynthesis, transport and catabolism |
| R | 177 | 8.23 % | General function prediction only |
| S | 143 | 6.65 % | Function unknown |
| - | 845 | 39.28 % | Not in COGs |
Table 6.
Plasmid statistics for ‘Halanaeroarchaeum sulfurireducens’ M27-SA2
| Attribute | Value | % of total |
|---|---|---|
| Plasmid size (bp) | 124,256 | |
| DNA coding (bp) | 103,887 | 83.61 % |
| DNA G + C (bp) | 68,831 | 55.39 % |
| Total genes | 119 | |
| Protein-coding genes | 119 | |
| Genes assigned to COGs | 24 | 20.16 % |
| Average length (bp) | 873 | |
| Max length (bp) | 4,326 | |
| ATG initiation codon proteins | 78 | 65.55 % |
| GTG initiation codon proteins | 28 | 23.53 % |
| TTG initiation codon proteins | 13 | 10.92 % |
Table 7.
Number of genes associated with the general COG functional categories for plasmid
| Code | Value | % age | COG category |
|---|---|---|---|
| K | 4 | 3.36 % | Transcription |
| L | 7 | 5.88 % | Replication, recombination and repair |
| D | 2 | 1.68 % | Cell cycle control, cell division, chromosome partitioning |
| T | 4 | 3.36 % | Signal transduction mechanisms |
| H | 2 | 1.68 % | Coenzyme transport and metabolism |
| P | 1 | 0.84 % | Inorganic ion transport and metabolism |
| R | 2 | 1.68 % | General function prediction only |
| S | 2 | 1.68 % | Function unknown |
| - | 95 | 79.84 % | Not in COGs |
Insights from the genome sequence
Genome comparisons: M27-SA2 vs HSR2 T
As a demonstration of their extreme similarity, the genomes of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 and ‘Halanaeroarchaeum sulfurireducens’ HSR2T, were compared with three different methods: the Artemis Comparison Tool program [20], the LAST web service [21], and the Multiple Genome Alignment system software (Mauve) [22]. Additionally, Double ACT web service was used to generate the required ACT comparison file. The results of these tools are shown in Fig. 4. It follows that the chromosome of M27-SA2 has high average nucleotide identities, over 97 %, to the corresponding replicon of HSR2T, whereas the plasmids of both isolates possess even higher average nucleotide identities values, over 99 %. The only difference with respect to the HSR2T chromosome (the gap visible for all methods used) was due to the presence of an extra phage-like region (prophage 2, see Phage-like elements below). Similarly to what was found in HSR2T, the genome analysis of M27-SA2 identified two blocks of genes responsible for the oxidation of acetate to CO2 with elemental sulfur as the electron acceptor. The acetate oxidation pathway occurred by means of an ATP-dependent acetyl-CoA synthase and TCA cycle, while sulfur dissimilation could be accomplished by four different operons, coding for molybdopterin oxidoreductases (HLASA_0051-0056; HLASA_0525-0529; HLASA_0688-0694; HLASA_1275-1271). Notwithstanding, these two strains were isolated apparently from very different and geographically very distant habitats, e.g. top 10 cm-layer sediments of Kulunda Steppe (Central Russia) terrestrial hypersaline lakes (HSR2T), and from hypersaline brine at 3105 m depth of Lake Medee (Eastern Mediterranean) M27-SA2, we failed to find any genetic determinants reflecting such significant difference in environmental settings of these two habitats.
Fig. 4.
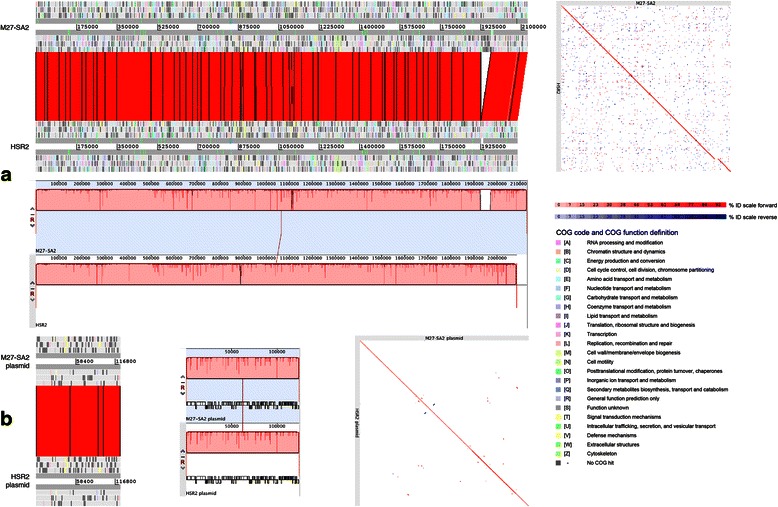
Comparison of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 vs. ‘Halanaeroarchaeum sulfurireducens’ HSR2T: chromosomes in (a) and plasmids in (b) ACT [20] comparisons at left (98 % ID, 40 bp minimum bitscore cutoff), LAST [21] comparisons at right (default parameters), and Mauve [22] alignments in the middle (default parameters). Genes COG color-coded, comparisons forward ID scale in red tones and reverse ID scale in blue tones
Amino acid biosynthesis pathways
The addition of yeast extract in amounts 10–20 mg l−1 is necessary for growth of strain M27-SA2, which is likely indicating that some amino acids are not synthesized or their biosynthesis could be arduous and they should be imported from the environment. We reconstructed the amino acid biosynthetic pathways of M27-SA2 using both the SEED subsystem [23] and KEGG orthology [24] assignments (Fig. 5). Similarly to what found on strain HSR2T (genomes were nearly identical) this analysis indicated that the genome of strain M27-SA2 harbors all the genes required for complete synthesis of at least 19 amino acids. Seven of the eight genes involved in conversion of aspartate to lysine via tetrahydrodipicolinate, which should involve succinylated intermediates, were found. The gene dapC encoding N-succinyldiaminopimelate-aminotransferase was not identified in M27-SA2 genome. The pathway for the biosynthesis of isoleucine, valine, and leucine from pyruvate seems to be fully present and all genes were detected in the analyzed genome. Interestingly, a branched-chain amino acid transport system related to the permease protein Liv (HLASA_0776-0780), was detected in the M27-SA2 genome, suggesting that non-secreted amino acids could be imported via various transporters.
Fig. 5.
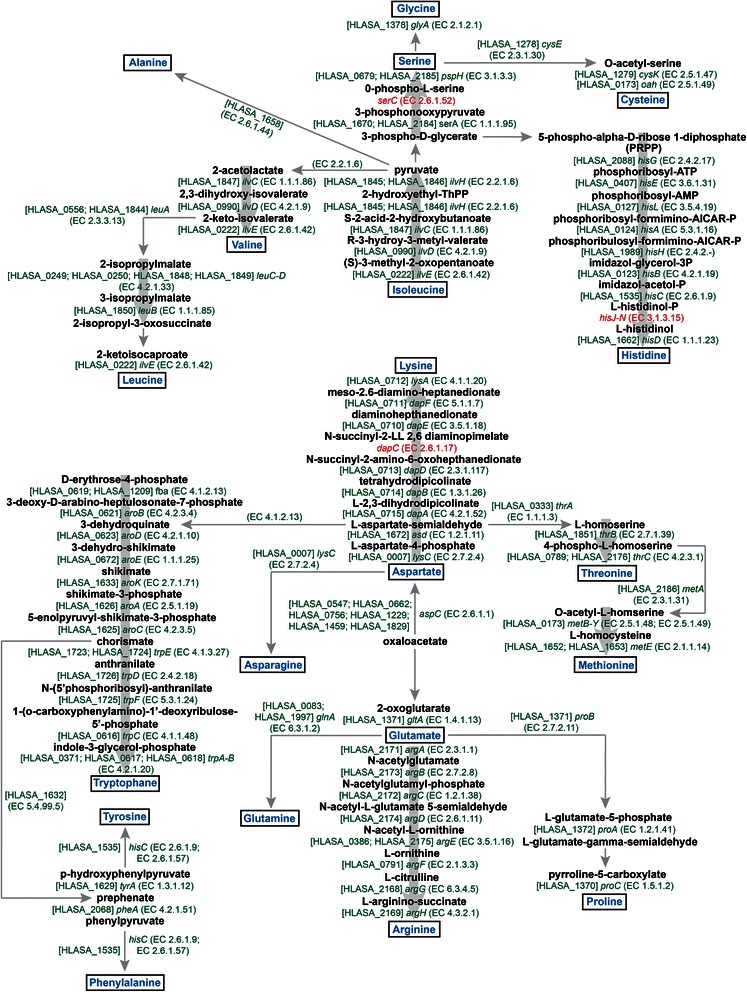
Overview of amino acid biosynthesis pathways in the genome of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2. The green colour indicates the presence of a homolog coding an enzyme that may catalyse this reaction. Red colour indicates the absence of the corresponding gene in M27-SA2 genome. EC numbers are shown in parentheses, while M27-SA2 gene locus_tags are in brackets
CRISPR analysis
Pilercr v1.02 [25] with default parameters was used to identify Clustered Regularly Interspaced Short Palindromic Repeats array in M27-SA2 genome. The CRISPRfinder tool was used for CRISPR search as a control [26]. Cas genes were identified with the NCBI Blastn online tool [19]. Spacer sequences detected in CRISPR array were analyzed in order to find similarities with plasmids, phages or haloarchaeal chromosomes. Spacer sequences were blasted against nt, env_nt and wgs databases using NCBI BLAST+ blastn tool installed into web-based Galaxy platform. Additionally, spacer sequences were blasted against a local database made of several hypersaline metagenomes, including those from the anoxic hypersaline lakes Kryos (M. Yakimov, unpublished results) and Thetis [27], the hypersaline Australian lake Tyrrell [28], and solar salterns of Santa Pola [29] and South Bay Salt [30]. Spacers with ≤7 SNPs (80 % match or 30/37 nucleotides) were considered as positive hits. Obtained matches of at least 100 bp-long were compared to the nr and nt NCBI databases using NCBI Blast + blastx and blastn, respectively.
Most sequenced so far archaeal genomes contain at least one CRISPR-Cas system [26]. DNA fragment of 13.1 kbp that included CRISPR array and associated cas genes was detected in the M27-SA2 genome. The CRISPR array found in M27-SA2 was practically identical to that found in ‘Halanaeroarchaeum sulfurireducens’ HSR2T, contained the same 30-bp direct repeat sequence (5′- GTTCCAGACGGACCCTTGAGGGGTTGAAGC -3′), and carried 57 spacers instead of 55 detected in HSR2T, with an average length of 37 nucleotides (individual spacer length ranged from 35 bp to 43 bp). Similarly, eight cas vgenes were detected in vicinity of the CRISPR array: cas6, cas8b/csh1, cas7/csh2, cas5, cas3, cas4, cas1, cas2 (Fig. 6). All cas genes had high level of similarity to cas genes of Halorhabdus tiamateaandHaloarcula argentinensis (with e-value ranged from 1e−37 to 0.0), and thus were highly conserved between closely relative haloarchaeal genera. According to the current classification, this system was affiliated to I-B subtype or CASS7 [31].
Fig. 6.
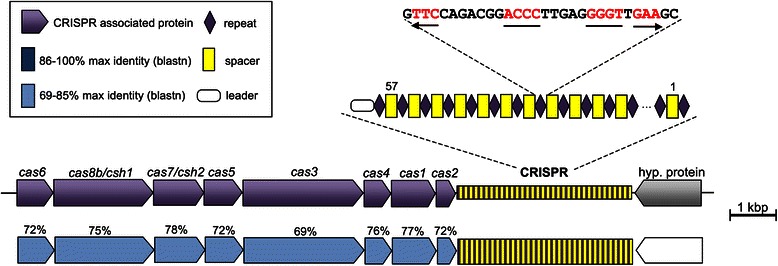
Structure of CRISPR system identified in ‘Halanaeroarchaeum sulfurireducens’ M27-SA2, with description of associated protein and repeat region found
BLASTn analysis of repeat sequence of M27-SA2 revealed several matches to haloarchaeal genomes with identical or similar (up to 3 mismatches) sequences of repeats: Natronomonas pharaonisDSM 2160 plasmid PL131, Haloarcula marismortuiATCC 43049 plasmid pNG300, Haloarcula hispanica N601 plasmid pHH126 and Halorhabdus tiamatea SARL4B.
Spacers extracted from ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 were identical to that found in HSR2T, although two spacers (#36 and #37) were found only in M27-SA2.
When we compared 57 spacers extracted from ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 to nt, env_nt and wgs Genbank databases, no matches were obtained. We blasted the spacers against metagenomic sequences of samples obtained from aforementioned hypersaline lake environments. This analysis identified six spacers that matched the metagenomic sequences (protospacers) obtained from the salt-saturated lakes Kryos and Tyrrel (Table 8). Spacer #37 matched to DNA fragment from lake Kryos metagenome that contained CRISPR array with repeat type of H. marismortuiATCC 43049 plasmid pNG400. These results suggest the occurrence of bacteria or/and viruses transfer between hypersaline biotopes. The spacer #7 matched to metagenomic sequences from viral fraction of lake Tyrrell. When those reads were compared to nt database, hits to viral contigs eHP-1, eHP-4, eHP-15 and eHP-19 from Santa-Pola solar saltern [29], and to the sequence of uncultured virus clone from Tunisian solar salterns [32] were found. Four other spacers (#24, #34, #52 and #54) matched to sequences from viral fraction of metagenome from lake Tyrrell. No homology in the GenBank was found. These results demonstrate that ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 CRISPR spacers, likewise HSR2T CRISPR spacers, target mobile genetic elements that have been identified in distant solar and deep-sea hypersaline lakes, suggesting that this haloarchaeon could have been adapted to yet unexplored haloviruses.
Table 8.
CRISPR spacers analysis in the chromosome of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2
| Spacer # | Match to metagenomic library | # of mismatches | 5′ PAM sequence | Match to GenBank nt database |
|---|---|---|---|---|
| 7 | Lake Tyrrell (SRR402046) | 4 | TTT | gb|JQ807236.1|, environmental Halophage eHP-15 |
| 24 | Lake Tyrrell (SRR402045) | 3 | CTC/TGC | no |
| 34 | Lake Tyrrell (SRR402046) | 2 | TTC/TTT | no |
| 37 | Lake Kryos (unpublished) | 0 | gb|AY596293.1|, Haloarcula marismortui ATCC 43049 plasmid pNG400, CRISPR array | |
| 52 | Lake Tyrrell (SRR402046) | 5 | GTG | no |
| 54 | Lake Tyrrell (SRR402045) | 5 | TTC | no |
The presence of a short protospacer adjacent motif located upstream of the protospacer is required for immunity of type I CRISPR-Cas system [33]. The PAM sequence varies in different CRISPR subsystems. It has been shown that archaeal I-B CRISPR-Cas system has several different PAM sequences upstream of the protospacer: TTC, ACT, TAA, TAT, TAG and CAC for Haloferaxvolcani [34], TTC for Haloquadratum walsbyi [34], TTT, TTC, TTG, and CCC for H. hispanica [35]. Our analysis detected several variants of trinucleotide sequence upstream of different protospacers (Table 8). Interestingly, most of them had TTC/TTT sequence, which are highly conserved among archaeal I-B subsystems reported so far [36]. This fact suggests that CRISPR-Cas system is likely active in ‘Halanaeroarchaeum sulfurireducens’ M27-SA2.
Phage-like elements
It has been estimated that 60–70 % of prokaryotic genomes deposited to GenBank contain prophage sequences [37]. We analyzed the genome of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 in terms of presence of prophages. Apparently, there were two fragments of 28.5 kbp (prophage 1) and 49.5 kbp (prophage 2) that contained clusters of genes of viral origin. Manual annotation of prophage 1 gave best matches to putative ORFs of haloarchaeal pleomorphic phages HRPV1, HRPV2, HRPV3, HRPV6, HHPV1 and HHPV2 of Haloferax lucentense, pHK2 plasmid, and prophages in the genomes of Halomicrobium mukohataei and Haloferax volcanii. The genome of prophage 1, presents in both M27-SA2 and HSR2T strains, was located near the tRNA gene, a common site for prophage insertions [38], and contained a putative XerC/D integrase/recombinase gene on the opposite to tRNA gene flank. Presence of an integrase and a tRNA insertion site could be interpreted as indicator of an active prophage. The alignment of the genome of prophage 1 and its closest relatives shows that prophage 1 contains ORFs homologous to the whole set of core genes in the genomes of lytic pleoviruses HRPV-1, HRPV-2, HRPV-3 and HHPV-1 (Fig. 7 and Tables 9 and 10). However, we have found a single CRISPR array and eight associated cas genes of I-B subtype inside the genome of prophage 1. The insert is ~14 kbp long and occupies half of the prophage 1 genome (~28.5 kbp). Earlier, the entire CRISPR-Cas system including cas genes of I-F type and a CRISPR array was found in the genome of myovirus ICP1 of Vibriocholera serogroup O1 [39]. Most of the spacers from ICP1 CRISPR array targeted PICI-like element from the genome of V. cholera, an excised circular DNA fragment, which becomes induced and interferes with phage reproduction during infection. Therefore, bacteriophages can acquire CRISPR-Cas systems from the host genome or from the environment through natural transformation of the host cell and use it to abolish anti-phage cellular mechanisms. Two opposite suggestions could be made based on the presence of the CRISPR-Cas system in the prophage 1. On one hand, the insert of DNA fragment containing CRISPR-Cas system equal to the length of the “viral” fragment of the prophage 1 as well as presence of a number of small ORFs of host origin interspaced by long non-coding regions with numerous stop codons on the right wings of its genome (see Fig. 7) would compromise release of viral particles. On the other hand, the pleomorphic nature of the prophage 1 could allow of formation viable particles with extended genomes, as viral packaging and release are driven by a budding vesicle from the plasma membrane, and the size of the genome therefore dictates the size of the vesicle [40]. According to this logic, CRISPR-Cas system of prophage 1 would not be active during lysogenic stage of infection as the expression of most of the lytic genes of a prophage are usually shut off [41], but could become active during lytic stage of infection and be used to overcome bacterial defense. Additional experiments can be designed and performed to distinguish between these two scenarios.
Fig. 7.
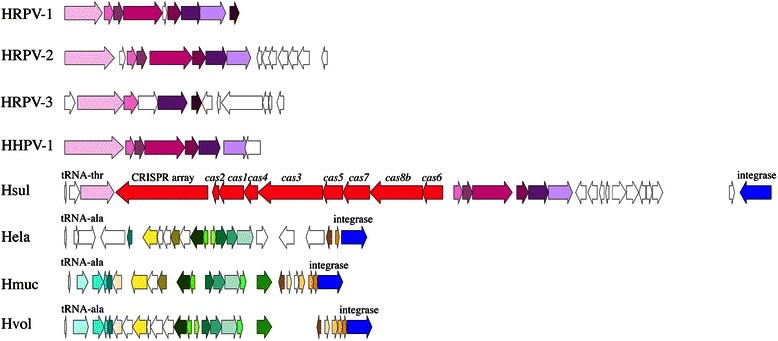
Comparative genome map with genome alignment results. Relative lytic pleomorphic viruses HRPV1, HRPV2, HRPV3, HHPV1 have seven core genes (colored in shades of magenta) that are shared between them and a prophage 1 from ‘Halanaeroarchaeum sulfurireducens’ M27-SA2. Relative prophages of Haloferax Hela, Hmuc, Hvol (coloured in shades of green) have tRNA and XerC/D integrase/recombinase flanking their genomes. The same flanks has the genome of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 prophage Hsul. CRISPR array and cas genes are colored in red. XerC/D integrase/recombinase is colored in blue. The same colors of the genes in genomes represent homologous genes. Short designations used: HRPV1, Halorubrum pleomorphic virus 1; HRPV2, Halorubrum pleomorphic virus 2; HRPV3, Halorubrum pleomorphic virus 3; HHPV1, Haloarcula pleomorphic virus 1; Hsul, ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 prophage 1; Hela; Haloferax elongans contig AOLK01000011 [38]; Hmuc, Haloferax mucosum contig AOLN01000011 [42]; Hvol, Haloferax volcanii chrom1 [42]
Table 9.
NCBI blastp results for prophage 1 region of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2
| locus_tag | Length (bp) | NCBI blastp best hit | e-value | gb accession |
|---|---|---|---|---|
| HLASA_0843 | 1239 | integrase [Halorhabdus utahensis] | 6.00e-176 | WP_015789480.1 |
| HLASA_0844 | 213 | hypothetical protein [Halomicrobium katesii] | 5.00e-05 | WP_018258875.1 |
| HLASA_0845 | 417 | DNA-binding protein [Halostagnicola larsenii XH-48] | 1.00e-79 | AHG02386.1 |
| HLASA_0846 | 231 | hypothetical protein [Salinarchaeum sp. Harcht-Bsk1] | 5.00e-37 | WP_020446487.1 |
| HLASA_0847 | 267 | hypothetical protein [uncultured archaeon A07HR60] | 6.00e-07 | WP_023506388.1 |
| HLASA_0848 | 489 | hypothetical protein [Halarchaeum acidiphilum] | 4.00e-94 | WP_020221057.1 |
| HLASA_0849 | 552 | hypothetical protein [Halarchaeum acidiphilum] | 9.00e-119 | WP_044957257.1 |
| HLASA_0850 | 141 | MULTISPECIES: hypothetical protein [Haloarcula] | 2.00e-21 | WP_004594507.1 |
| HLASA_0851 | 189 | hypothetical protein [Haloquadratum walsbyi] | 4.00e-33 | WP_021056874.1 |
| HLASA_0852 | 381 | hypothetical protein [Halorubrum sp. T3] | 2.00e-82 | WP_026046123.1 |
| HLASA_0853 | 423 | hypothetical protein [Halorubrum saccharovorum] | 3.00e-44 | WP_004048594.1 |
| HLASA_0854 | 954 | hypothetical protein [Halomicrobium mukohataei] | 6.00e-27 | WP_015761810.1 |
| HLASA_0855 | 798 | hypothetical protein HRPV-1_gp7 [Halorubrum pleomorphic virus 1] | 2.00e-26 | YP_002791892.1 |
| HLASA_0856 | 474 | unknown [Haloarcula hispanica pleomorphic virus 1] | 6.00e-19 | YP_003411999.1 |
| HLASA_0857 | 1581 | MULTISPECIES: hypothetical protein [Haloferax] | 8.00e-52 | WP_008576942.1 |
| HLASA_0858 | 399 | gp3 [Haloarcula hispanica pleomorphic virus 2] | 6.00e-47 | YP_009008689.1 |
| HLASA_0859 | 351 | hypothetical protein [Haloarcula argentinensis] | 5.00e-38 | WP_005538312.1 |
| HLASA_0860 | 813 | CRISPR-associated protein Cas6 [Halorhabdus tiamatea] | 7.00e-151 | WP_008524857.1 |
| HLASA_0861 | 2112 | CRISPR-associated protein Csh1 [Halorhabdus tiamatea] | 0 | WP_008524855.1 |
| HLASA_0862 | 1068 | CRISPR-associated protein, Csh2 family [Natronorubrum sulfidifaciens] | 0 | WP_008164963.1 |
| HLASA_0863 | 801 | CRISPR-associated protein Cas5 [Halorhabdus tiamatea] | 2.00e-124 | WP_008524852.1 |
| HLASA_0864 | 2589 | CRISPR-associated helicase, Cas3 [Halorhabdus tiamatea] | 0 | WP_020936219.1 |
| HLASA_0865 | 555 | CRISPR-associated protein Cas4 [Halorhabdus utahensis] | 4.00e-99 | WP_015789200.1 |
| HLASA_0866 | 993 | CRISPR-associated protein Cas1 [Halorhabdus utahensis] | 0 | WP_015789199.1 |
| HLASA_0867 | 264 | CRISPR-associated endonuclease Cas2 [Halorhabdus utahensis] | 1.00e-37 | WP_015789198.1 |
| repeat_region | 3798 | CRISPR repeat region | ||
| HLASA_0868 | 1338 | replication-related protein [Natrinema versiforme] | 5.00e-145 | WP_006432249.1 |
| HLASA_0869 | 411 | PREDICTED: multidrug and toxin extrusion protein 1 [Cavia porcellus] | 0.1 | XP_003465357.1 |
Table 10.
NCBI blastp results for prophage 2 region of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2
| locus_tag | Length (bp) | NCBI blastp best hit | e-value | gb accession |
|---|---|---|---|---|
| HLASA_2001 | 1347 | integrase [Natronobacterium gregoryi] | 3.00e-178 | WP_005577816.1 |
| HLASA_2002 | 186 | no | ||
| HLASA_2003 | 1125 | hypothetical protein [Haloferax sp. ATB1] | 5.00e-11 | WP_042662540.1 |
| HLASA_2004 | 996 | zinc finger SWIM domain protein [Haloarcula vallismortis] | 5.00e-14 | WP_004517574.1 |
| HLASA_2005 | 219 | hypothetical protein [Halosimplex carlsbadense] | 4.00e-15 | WP_006885565.1 |
| HLASA_2006 | 1071 | ORC / cell division control protein 6 [Haloarcula amylolytica] | 2.00e-98 | WP_008307569.1 |
| HLASA_2007 | 660 | hypothetical protein [Haloarcula amylolytica] | 9.00e-59 | WP_008312971.1 |
| HLASA_2008 | 978 | hypothetical protein [Haloarcula amylolytica] | 3.00e-144 | WP_008312969.1 |
| HLASA_2009 | 639 | PHP domain-containing protein [Haloarcula amylolytica] | 3.00e-106 | WP_008312967.1 |
| HLASA_2010 | 912 | decaprenyl-phosphate phosphoribosyltransferase [Haloarcula amylolytica] | 0 | WP_008312966.1 |
| HLASA_2011 | 423 | hypothetical protein [Haloarcula vallismortis] | 2.00e-29 | WP_004518340.1 |
| HLASA_2012 | 1092 | NAD-dependent epimerase/dehydratase [Haloarcula amylolytica] | 0 | WP_008312962.1 |
| HLASA_2013 | 1299 | hypothetical protein [Anaerolinea thermophila] | 2.00e-35 | WP_013559525.1 |
| HLASA_2014 | 810 | concanavalin A-like lectin/glucanases family protein [Halorubrum sp. AJ67] | 2.00e-18 | CDK38289.1 |
| HLASA_2015 | 597 | hypothetical protein OSG_eHP34_00135 [Environmental halophage eHP-34] | 1.00e-28 | AFH22760.1 |
| HLASA_2016 | 576 | hypothetical protein HGTV1_28 [halovirus HGTV-1] | 2.00e-11 | YP_008059236.1 |
| HLASA_2017 | 912 | hypothetical protein PhiCh1p32 [Natrialba phage PhiCh1] | 2.00e-29 | NP_665949.1 |
| HLASA_2018 | 1263 | baseplate J protein [haloarchaeon 3A1_DGR] | 2.00e-178 | WP_039401004.1 |
| HLASA_2019 | 363 | hypothetical protein [haloarchaeon 3A1_DGR] | 7.00e-15 | WP_021074727.1 |
| HLASA_2020 | 681 | hypothetical protein [Natrialba magadii] | 3.00e-34 | WP_004268274.1 |
| HLASA_2021 | 879 | hypothetical protein [haloarchaeon 3A1_DGR] | 4.00e-97 | WP_021074730.1 |
| HLASA_2022 | 345 | hypothetical protein [haloarchaeon 3A1_DGR] | 3.00e-43 | WP_039401001.1 |
| HLASA_2023 | 564 | hypothetical protein [haloarchaeon 3A1_DGR] | 1.00e-35 | WP_021075289.1 |
| HLASA_2024 | 3123 | prophage pi3 protein 14 [Halalkalicoccus jeotgali] | 1.00e-55 | WP_008414607.1 |
| HLASA_2025 | 477 | hypothetical protein [haloarchaeon 3A1_DGR] | 1.00e-13 | WP_021074542.1 |
| HLASA_2026 | 1296 | hypothetical protein [haloarchaeon 3A1_DGR] | 2.00e-155 | WP_039400994.1 |
| HLASA_2027 | 588 | hypothetical protein [Natrialba magadii] | 1.00e-37 | WP_004268261.1 |
| HLASA_2028 | 447 | hypothetical protein [haloarchaeon 3A1_DGR] | 1.00e-45 | WP_039400992.1 |
| HLASA_2029 | 285 | hypothetical protein [haloarchaeon 3A1_DGR] | 6.00e-29 | WP_021074547.1 |
| HLASA_2030 | 387 | hypothetical protein EL22_16975 [Halostagnicola sp. A56] | 2.00e-05 | KDE59819.1 |
| HLASA_2031 | 366 | hypothetical protein HHTV1_22 [halovirus HHTV-1] | 2.00e-05 | YP_008058712.1 |
| HLASA_2032 | 1140 | major capsid protein go21 [halovirus HHTV-1] | 3.00e-91 | YP_008058711.1 |
| HLASA_2033 | 453 | acyl dehydratase [Haloferax mediterranei] | 0.032 | WP_014732690.1 |
| HLASA_2034 | 1221 | PREDICTED: myosin-9-like [Nelumbo nucifera Gaertn.] | 2.00e-04 | XP_010274858.1 |
| HLASA_2035 | 294 | no | ||
| HLASA_2036 | 216 | no | ||
| HLASA_2037 | 417 | no | ||
| HLASA_2038 | 339 | hypothetical protein [Natronobacterium gregoryi] | 5.00e-09 | WP_005577927.1 |
| HLASA_2039 | 669 | hypothetical protein [Haloterrigena thermotolerans] | 9.00e-23 | WP_006649646.1 |
| HLASA_2040 | 381 | hypothetical protein [Halovivax ruber] | 2.00e-08 | WP_015300135.1 |
| HLASA_2041 | 1605 | uncharacterized protein BN903_58 [Halorubrum sp. AJ67] | 1.00e-48 | CDK39659.1 |
| HLASA_2042 | 1710 | hypothetical protein HALG_00007 [Halorubrum phage CGphi46] | 1.00e-131 | YP_008126542.1 |
| HLASA_2043 | 444 | hypothetical protein HCTV2_15 [halovirus HCTV-2] | 4.00e-29 | YP_008058377.1 |
| HLASA_2044 | 1320 | DNA methylase [Halostagnicola sp. A56] | 4.00e-120 | KDE56926.1 |
| HLASA_2045 | 219 | hypothetical protein OSG_eHP14_00030 [Environmental halophage eHP-14] | 4.00e-08 | AFH21986.1 |
| HLASA_2046 | 171 | hypothetical protein [Haloarcula argentinensis] | 8.00e-05 | WP_005538080.1 |
| HLASA_2047 | 303 | no | ||
| HLASA_2048 | 3660 | hypothetical protein [Natrialba magadii] | 0 | WP_004217537.1 |
Another prophage 2 is ~43.5 kbp in length and containes 49 ORFs. One of the ORFs encodes a putative tape tail measure protein, which is a key feature of Siphoviridae morphological family. The closest homologues of ORFs of prophage 2 were related to several haloviruses: Bj1 (siphovirus that infects Halorubrum), phiCh1 (myovirus of Natrialba), HHTV1 (siphovirus of H. hispanica), HGTV1 (myovirus of Halogranum sp.), prophages in the Haloferax mucosum and Haloferax elongans genomes [42] and to environmental viral contigs (environmental halophages eHP-2, eHP-14, eHP-32, eHP-34, eHP-36). Interestingly, prophage 2 encodes an adenine-specific DNA methylase, which could be responsible for protection from host restriction endonucleases through methylation of the phage genome [43]. Prophage 2 is located next to a tRNA integration site and has a XerC/D integrase/recombinase gene on the opposite side of the genome: both features are always present as a part of a viable prophage. Archaeal tailed viral genomes integrated into cellular chromosomes have been identified before [44]. The fact that the closest host homologue in the database (H. sulfurireducens strain HSR2T, accession number CP008874) doesn’t contain a sequence of the prophage 2 suggests recent acquisition of this prophage. Viruses of Euryarchaeota encompass different morphologies, including spindle-shaped viruses (Salteproviridae), pleomorphic viruses (Pleolipoviridae), head-tailed viruses (Caudovirales), spherical viruses (Plasmaviridae), and unclassified icosahedral dsDNA viruses with an inner lipid layer [45]. Therefore, the presence of two different prophages in the same genome of H. sulfurireducens M27-SA2 suggests that archaea in hypersaline environemnts located at the bottom of the Mediterranean Sea are exposed to a constant threat of phage predation.
Genomic islands
Horizontally transferred genomic islands (GIs) in ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 genome were determined by the SeqWord Gene Island Sniffer program [46] and by the GOHTAM online tool [47]. The results of both GI identification methods are shown in Fig. 8. Six putative GIs characterized by alternative oligonucleotide usage patterns were detected by SWGIS, while GOHTAM search returned many short region (overall 52, see Fig. 8b) in addition to the six previously identified, probably due to a lower default sensitivity threshold of the latter method. Predicted GIs harbored mainly hypothetical proteins, transposases, glycosyltransferases (many in the third GI), and other enzyme-coding genes (transport and metabolism), a tRNA in the first GI, while the sixth GI covers the prophage 2 region (see above), not present in ‘Halanaeroarchaeum sulfurireducens’ HSR2T.
Fig. 8.
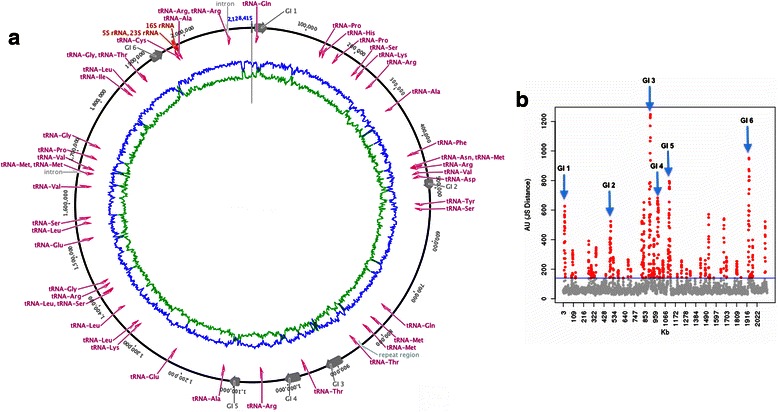
Localization of GIs on the chromosome of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2, as predicted by SWGIS [46] a (grey arrows on circular map) and GOTHAM [47] b (red dots). Common predicted regions of both methods are highlighted in b) (blue arrows)
Conclusions
In this manuscript we report on the complete genome sequence of ‘Halanaeroarchaeum sulfurireducens’ M27-SA2 which is composed of a 2,129,244-bp chromosome and a 124,256-bp plasmid. This is the first indication of the presence of obligate anaerobic sulfur-respiring haloarchaeon in deep-sea hypersaline anoxic lakes located on the seabed of Eastern Mediterranean Sea. This finding has significance for understanding of the biogeochemical functioning of these harsh ecosystems. Genome comparisons, analisys of amino acid biosynthesis pathways, CRISPR, phage-like elements and genomic islands was performed to understand the evolutionary success of Halanaeroarchaeum members in their adaptation to extreme biotopes around the world.
Acknowledgements
This work was supported by the European Commission’s program, under the MAMBA (FP7-KBBE-2009-2B-226977), MicroB3 (FP7-OCEAN.2011-2-287589) and INMARE (Horizon 2020–634486) Projects and under Russian Academy of Science Program “Molecular and Cellular Biology”. DS was supported by the RFBR grant 13-04-00049. We thank the master, crew, and participants to the cruises with research vessel RV Urania for their fruitful collaboration.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EM carried out the genome scaffolding, final assembling and annotations, and registration to GenBank, performed the genomic islands analysis and drafted the manuscript. DS, IK and ST carried out the genome raw and contig sequencing, and reviewed the overall manuscript. AL carried out the phage-like elements analisys and drafted the relative section. EA, GLS, and VLC participated in the organism growth, isolation and DNA preparation. FS carried out the CRISPR analisys, the amino acid biosynthesis pathways, and drafted the relative sections. MMY conceived the study, and participated in its design and coordination and helped to draft and review the manuscript. All authors read and approved the final manuscript.
References
- 1.Sorokin DY, Kublanov IV, Gavrilov SN, Rojo D, Roman P, Golyshin PN, Slepak VZ, Smedile F, Ferrer M, Messina E, La Cono V, Yakimov MM. Elemental sulfur and acetate can support life of a novel strictly anaerobic haloarchaeon. ISME J. 2016;10:240–52. doi:10.1371/journal.pone.0098229. PubMed. [DOI] [PMC free article] [PubMed]
- 2.Andrei AS, Banciu HL, Oren A. Living with salt: metabolic and phylogenetic diversity of archaea inhabiting saline ecosystems. FEMS Microbiol Lett. 2012;330:1–9. doi: 10.1111/j.1574-6968.2012.02526.x. [DOI] [PubMed] [Google Scholar]
- 3.Blum JS, Kulp TR, Han S, Lanoil B, Saltikov CW, Stolz JF, Miller LG, Oremland RS. Desulfohalophilus alkaliarsenatis gen. nov., sp. nov., an extremely halophilic sulfate- and arsenate-respiring bacterium from Searles Lake, California. Extremophiles. 2012;16:727–42. doi:10.1007/s00792-012-0468-6. PubMed. [DOI] [PMC free article] [PubMed]
- 4.Blum JS, Han S, Lanoil B, Saltikov C, Witte B, Tabita FR, Langley S, Beveridge TJ, Jahnke L, Oremland RS. Ecophysiology of “Halarsenatibacter silvermanii” strain SLAS-1T, gen. nov., sp. nov., a facultative chemoautotrophic arsenate respirer from salt-saturated Searles Lake, California. Appl Environ Microbiol. 2009;75:1950–60. doi:10.1128/AEM.02614-08. PubMed. [DOI] [PMC free article] [PubMed]
- 5.Sorokin DY, Detkova EN, Muyzer G. Sulfur-dependent respiration under extremely haloalkaline conditions in soda lake ‘acetogens’ and the description of Natroniella sulfidigena sp. nov. FEMS Microbiol Lett. 2011;319:88–95. doi: 10.1111/j.1574-6968.2011.02272.x. [DOI] [PubMed] [Google Scholar]
- 6.Antunes A, Taborda M, Huber R, Moissl C, Nobre MF, da Costa MS. Halorhabdus tiamatea sp. nov., a non-pigmented extremely halophilic archaeon from a deep-sea, hypersaline anoxic basin of the Red Sea, and emended description of the genus Halorhabdus. Int J Syst Evol Microbiol. 2008;58:215–220. doi: 10.1099/ijs.0.65316-0. [DOI] [PubMed] [Google Scholar]
- 7.Bonete MJ, Martínez-Espinosa RM, Pire C, Zafrilla B, Richardson DJ. Nitrogen metabolism in haloarchaea. Saline Systems. 2008;4:9. doi: 10.1186/1746-1448-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oren A, Trüper HG. Anaerobic growth of halophilic archaeobacteria by reduction of dimethylsulfoxide and trimethylamine N-oxide. FEMS Microbiol Lett. 1990;70:33–36. doi: 10.1111/j.1574-6968.1990.tb03772.x. [DOI] [Google Scholar]
- 9.Oren A. Anaerobic growth of archaeobacteria by reduction of fumarate. J Gen Microbiol. 1991;137:1387–1390. doi: 10.1099/00221287-137-6-1387. [DOI] [Google Scholar]
- 10.Yakimov MM, La Cono V, Slepak VZ, La Spada G, Arcadi E, Messina E, Borghini M, Monticelli LS, Rojo D, Barbas C, Golyshina OV, Ferrer M, Golyshin PN, Giuliano L. Microbial life in the Lake Medee, the largest deep-sea salt-saturated formation. Sci Rep. 2013;3:3554. doi: 10.1038/srep03554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Urakawa H, Martens-Habbena W, Stahl DA. High abundance of ammonia-oxidizing archaea in coastal waters, determined using a modified DNA extraction method. Appl Environ Microbiol. 2010;76:2129–2135. doi: 10.1128/AEM.02692-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lagesen K, Hallin PF, Rødland E, Stærfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi:10.1038/nature02340. PubMed. [DOI] [PubMed]
- 17.Toshchakov SV, Kublanov IV, Messina E, Yakimov MM, Golyshin PN. Genomic Analysis of Pure Cultures and Communities. In: Timmis KN, Nogales Fernández B, McGenity TJ, editors. Hydrocarbon and Lipid Microbiology Protocols: Cultivation. Springer Protocols Handbooks. 2015. pp. 1–23. [Google Scholar]
- 18.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2001;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 19.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi:10.1093/nar/25.17.3389. Pubmed. [DOI] [PMC free article] [PubMed]
- 20.Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- 21.Kiełbasa SM, Wan R, Sato K, Horton P, Frith MC. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011;21:487–493. doi: 10.1101/gr.113985.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crécy-Lagard V, Diaz N, Disz T, Edwards R, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar RC. PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics. 2007;8:18. doi: 10.1186/1471-2105-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pachiadaki MG, Yakimov MM, La Cono V, Leadbetter E, Edgcomb V. Unveiling microbial activities along the halocline of Thetis, a deep-sea hypersaline anoxic basin. ISME J. 2014;8:2478–2489. doi: 10.1038/ismej.2014.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narasingarao P, Podell S, Ugalde JA, Brochier-Armanet C, Emerson JB, Brocks JJ, Heidelberg KB, Banfield JF, Allen EE. De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J. 2012;6:81–93. doi:10.1038/ismej.2011.78. PubMed. [DOI] [PMC free article] [PubMed]
- 29.Garcia-Heredia I, Martin-Cuadrado AB, Mojica FJ, Santos F, Mira A, Antón J, Rodriguez-Valera F. Reconstructing viral genomes from the environment using fosmid clones: the case of haloviruses. PLoS One. 2012;7:e33802. doi:10.1371/journal.pone.0033802. PubMed. [DOI] [PMC free article] [PubMed]
- 30.Dinsdale EA, Edwards RA, Hall D, Angly F, Breitbart M, Brulc JM, Furlan M, Desnues C, Haynes M, Li L. Functional metagenomic profiling of nine biomes. Nature. 2008;452:629–32. doi:10.1038/nature06810. PubMed. [DOI] [PubMed]
- 31.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–77. doi:10.1038/nrmicro2577. PubMed. [DOI] [PMC free article] [PubMed]
- 32.Boujelben I, Yarza P, Almansa C, Villamor J, Maalej S, Antón J, Santos F. Virioplankton community structure in Tunisian solar salterns. Appl Environ Microbiol. 2012;78:7429–7437. doi: 10.1128/AEM.01793-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shah SA, Erdmann S, Mojica FJ, Garrett RA. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol. 2013;10:891–899. doi: 10.4161/rna.23764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fischer S, Maier LK, Stoll B, Brendel J, Fischer E, Pfeiffer F, Dyall-Smith M, Marchfelder A. An archaeal immune system can detect multiple protospacer adjacent motifs (PAMs) to target invader DNA. J Biol Chem. 2012;287:33351–63. doi:10.1074/jbc.M112.377002. PubMed. [DOI] [PMC free article] [PubMed]
- 35.Li M, Wang R, Xiang H. Haloarcula hispanica CRISPR authenticates PAM of a target sequence to prime discriminative adaptation. Nucleic Acids Res. 2014;42:7226–7235. doi: 10.1093/nar/gku389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Attar S, Westra ER, van der Oost J, Brouns SJ. Clustered regularly interspaced short palindromic repeats (CRISPRs): the hallmark of an ingenious antiviral defense mechanism in prokaryotes. Biol Chem. 2011;392:277–289. doi: 10.1515/bc.2011.042. [DOI] [PubMed] [Google Scholar]
- 37.Paul JH. Prophages in marine bacteria: dangerous molecular time bombs or the key to survival in the seas? ISME J. 2008;2:579–589. doi: 10.1038/ismej.2008.35. [DOI] [PubMed] [Google Scholar]
- 38.Fouts DE. Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006;34:5839–5851. doi: 10.1093/nar/gkl732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seed KD, Lazinski DW, Calderwood SB, Camilli A. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature. 2013;494:489–491. doi: 10.1038/nature11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pietilä MK, Atanasova NS, Manole V, Liljeroos L, Butcher SJ, Oksanen HM, Bamford DH. Virion architecture unifies globally distributed pleolipoviruses infecting halophilic archaea. J Virol. 2012;86:5067–5079. doi: 10.1128/JVI.06915-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abedon ST and Calendar RL (Eds.) The Bacteriophage, second edition, Oxford University Press, New York, 2005, Part 27, p. 409–447.
- 42.Maier LK, Dyall-Smith M, Marchfelder A. The Adaptive Immune System of Haloferax volcanii. Life (Basel) 2015;5:521–537. doi: 10.3390/life5010521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy J, Mahony J, Ainswoth S, Nauta A, van Sinderen D. Bacteriophage Orphan DNA Methyltransferases: Insights from Their Bacterial Origin, Function, and Occurrence. Appl Environ Microbiol. 2013;79:7547–7555. doi: 10.1128/AEM.02229-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krupovic M, Forterre P, Bamford DH. Comparative analysis of the mosaic genomes of tailed archaeal viruses and proviruses suggests common themes for virion architecture and assembly with tailed viruses of bacteria. J Mol Biol. 2010;397:144–160. doi: 10.1016/j.jmb.2010.01.037. [DOI] [PubMed] [Google Scholar]
- 45.Prangishvili The wonderful world of Archaeal viruses. Annu Rev Microbiol. 2013;67:565–585. doi: 10.1146/annurev-micro-092412-155633. [DOI] [PubMed] [Google Scholar]
- 46.Bezuidt O, Lima-Mendez G, Reva ON. SeqWord Gene Island Sniffer: a program to study the lateral genetic exchange among bacteria. World Acad Sci Eng Technol. 2009;58:1169–1174. [Google Scholar]
- 47.Ménigaud S, Mallet L, Picord G, Churlaud C, Borrel A, Deschavanne P. GOHTAM: a website for ‘Genomic Origin of Horizontal Transfers, Alignment and Metagenomics’. Bioinformatics. 2012;28:1270–1271. doi: 10.1093/bioinformatics/bts118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen M, Angiuoli SV. Towards a richer description of our complete collection of genomes and metagenomes: the “Minimum Information about a Genome Sequence” (MIGS) specification. Nat Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garrity GM, Phylum HJG, AII . Euryarchaeota phy. nov. In: Garrity GM, Boone DR, Castenholz RW, editors. Bergey’s Manual of Systematic Bacteriology. 2. New York: Springer; 2001. pp. 211–355. [Google Scholar]
- 51.List Editor Validation List no. 85. Validation of publication of new names and new combinations previously effectively published outside the IJSEM. Int J Syst Evol Microbiol. 2002;52:685–690. doi: 10.1099/00207713-52-3-685. [DOI] [PubMed] [Google Scholar]
- 52.Grant WD, Kamekura M, McGenity TJ, Class VA, III . Halobacteria class. nov. In: Garrity GM, Boone DR, Castenholz RW, editors. Bergey’s Manual of Systematic Bacteriology. 2. New York: Springer; 2001. p. 294. [Google Scholar]
- 53.Grant WD, Larsen H, Group III . Extremely halophilic archaeobacteria. Order Halobacteriales ord. nov. In: Holt JG, editor. Bergey’s Manual of Systematic Bacteriology, Volume 3. Baltimore: Williams & Wilkins; 1989. pp. 2216–2228. [Google Scholar]
- 54.The nomenclatural types of the orders Acholeplasmatales, Halanaerobiales, Halobacteriales, Methanobacteriales, Methanococcales, Methanomicrobiales, Planctomycetales, Prochlorales, Sulfolobales, Thermococcales, Thermoproteales and Verrucomicrobiales are the genera Acholeplasma, Halanaerobium, Halobacterium, Methanobacterium, Methanococcus, Methanomicrobium, Planctomyces, Prochloron, Sulfolobus, Thermococcus, Thermoproteus and Verrucomicrobium, respectively. Opinion 79. Int J Syst Evol Microbiol. 2005;55:517–518. PubMed doi:10.1099/ijs.0.63548-0. [DOI] [PubMed]
- 55.List Editor Validation List no. 31. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol. 1989;39:495–497. doi: 10.1099/00207713-39-4-495. [DOI] [Google Scholar]
- 56.Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol. 1980;30:225–420. doi: 10.1099/00207713-30-1-225. [DOI] [PubMed] [Google Scholar]
- 57.Gibbons NE, Family V. Halobacteriaceae Fam. nov. In: Buchanan RE, Gibbons NE, editors. Bergey’s Manual of Determinative Bacteriology. 8. Baltimore: The Williams and Wilkins Co.; 1974. pp. 269–273. [Google Scholar]
- 58.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swofford DL. PAUP*: Phylogenetic analysis using Parsimony (and other methods) 4.0 Beta. Sinauer Associates, Inc.; 2002. ISBN 978-0-87893-806-3
- 60.Carver TJ, Thomson N, Bleasby A, Berriman M, Parkhill J. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics. 2009;25:119–120. doi: 10.1093/bioinformatics/btn578. [DOI] [PMC free article] [PubMed] [Google Scholar]