

Abstract
Genetic heterogeneity is a common problem for genome-wide association studies of complex human diseases. Ordered-subset analysis (OSA) reduces genetic heterogeneity and optimizes the use of phenotypic information, thus improving power under some disease models. We hypothesized that in a genetically heterogeneous disorder such as Alzheimer’s disease (AD), utilizing OSA by age at onset (AAO) of AD may increase the power to detect relevant loci. Using this approach, 8 loci were detected, including the chr15: 30,44 region harboring CHRFAM7A. The association was replicated in the NIA-LOAD Familial Study dataset. CHRFAM7A is a dominant negative regulator of CHRNA7 function, the receptor that facilitates amyloid-β1–42 internalization through endocytosis and has been implicated in AD. OSA, using AAO as a quantitative trait, optimized power and detected replicable signals suggesting that AD is genetically heterogeneous between AAO subsets.
Keywords: Age at onset, Alzheimer’s disease, copy number variation
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder affecting approximately 5.4 million individuals in the US [1–3]. AD and age at onset (AAO) of AD have high heritability [4, 5]. Rare Mendelian forms confirmed and elucidated pathways involved in amyloid accumulation, but only account for a small percentage of AD [6]. Genome-wide association studies (GWAS) using single-nucleotide polymorphisms (SNPs) elucidated 10 loci contributing to the heritability of AD [7, 8].
Copy number variation (CNV) is a specific form of genetic variation representing extra or missing copies of normal sequence, such as deletions, duplications, and inversions [9]. CNVs have properties that are distinct from SNPs: they are often multiallelic and have a higher de novo mutation rate, and therefore they are often not adequately tagged by SNPs [10]. CNVs influence gene expression and phenotypic variation by altering either gene dosage or genome organization [11, 12]. As the mutational load by variant type is locus specific, CNVs serve as an alternative genetic marker map representing a supplementary approach to SNP association [13].
Genetic heterogeneity is a common challenge for GWAS of complex human diseases. Genetic heterogeneity at the population level may manifest as variable risk allele frequencies in subsets of patients. Ordered subset analysis (OSA) is a statistical method that is able to identify a more homogeneous subset of patients [14]. OSA is designed to test genotypic differences between cases and controls in the case-only quantitative trait situation [15]. If the quantitative trait is important in the genotype-phenotype relationship, then one end of the distribution of trait values will contribute disproportionately to the association signal and offset the penalty for multiple testing. The power of OSA for case-control studies was particularly superior to the Cochran-Armitage trend test (OSA power 83.4% versus trend test power 39.45 at α = 0.3) when the subset of interest was about 15–30% of the available dataset. Furthermore OSA allows the selection of the most informative subset of individuals for deeper genotyping [15].
This genome-wide CNV association study applying ordered subset by AAO analysis was undertaken to identify loci that confer risk of AD in a subset of AD subjects defined by AAO of the disease.
MATERIALS AND METHODS
Discovery set
781 subjects participated in the study. Probable AD was diagnosed based on NINCDS-ADRDA criteria [16]. The methodology of the Texas Alzheimer Research and Care Consortium project has been described [17]. Exclusion criteria included a Hachinski score >4 and clinical or imaging evidence of a stroke.
The control group consisted of non-demented subjects; inclusion criteria were the following: unrelated to cases, age over 55 years, normal performance on activities of daily living, and Clinical Dementia Rating (CDR) global score 0 (by surrogate historian). Control subjects underwent neuropsychological testing after enrollment in the study which included the assessment of global cognitive functioning/status (Mini-Mental State Exam and CDR), attention (Digit Span and Trails A), executive function (Trails B and Clock Drawing; Texas Card Sorting), memory (Wechsler Memory Scale (WMS) Logical Memory I and WMS Logical Memory II), language (Boston Naming and FAS Verbal Fluency), premorbid IQ (American National Adult Reading Test), visuospatial memory (WMS-Visual Reproduction I and II), psychiatric symptoms (Geriatric Depression Scale; Neuropsychiatric Inventory- Questionnaire), and functional assessment (Lawton-Brody Activities of Daily Living: PSMS, IADL). Subjects with impairment (Z-score < −1.5 on any measure) were excluded from the control cohort following consensus review.
IRB at each site approved the study. Informed consent was obtained. Genomic DNA was extracted from whole blood with the Puregene DNA isolation kit (Qiagen).
Replication set
For replication of the results we used the NIA-LOAD Familial Study dataset probands (n = 866) and unrelated controls (n = 906) deposited in dbGAP (http://www.ncbi.nlm.nih.gov/gap). Selection criteria for cases were probands only, Caucasian, non-Hispanic, and AAO data available. For controls, the selection criteria included unrelated to cases, Caucasian, and non-Hispanic.
Gene expression set
We obtained 22 pathologically confirmed AD temporal lobe tissue samples (mean age at death: 80 years, range 61–93) and 15 control temporal lobes (mean age at death: 65 years, range 41–93) from two institutions (Alzheimer’s Disease and Memory Disorders Center (ADMDC) tissue collection, New York Brain Bank). Board-certified neuropathologists assigned the diagnosis based on plaque and tangle assessment and Braak staging. De-identified brain samples were exempt from IRB.
AAO phenotyping
AAO was determined with two standardized methods in the TARC cohort according to previously published data [18, 19]: i) caregiver estimate of onset of symptoms and ii) physician estimate of duration of illness using a structured interview with landmark event to facilitate recall [20]. AAO in the LOAD cohort was determined by a standard question inquiring when the symptoms started.
APOE genotyping
APOE genotyping was performed using real-time PCR according to the manufacturer’s instructions. The amplifications were done on an ABI 7900HT thermal cycler (Applied Biosystems, Inc.). The custom Taq-Man probes (Applied Biosystems, Inc) were unique to SNPs at nucleotide positions 112 (rs7412) and 158 (rs429358) of the APOE gene, respectively. The combination of alleles at the two polymorphisms determined the APOE genotype. APOE status for the LOAD was not available and imputation was not possible due to lack of linkage disequilibrium between SNPs on the array and the APOE polymorphisms.
CNV genotyping by the genome-wide human SNP array 6.0
Array based genotyping was performed on the Genome-Wide Human SNP Array 6.0 (Affymetrix) according to the manufacturer’s instructions. Standard QC measures included contrast QC (>0.4) and Median of the Absolute values of all Pairwise Differences (MAPD) <0.4 and number of calls more than 2 SD from the mean.
CNV genotyping in LOAD
The NIA-LOAD Familial Study genotyping was performed on the Illumina Human610 Quad_array. The logR ratio was calculated in the GenomeStudio (Illumina) software. Candidate locus specific data was extracted and OSA was performed on the logR numeric data.
Detection of copy number variation and test of association
The Affymetrix intensity data was corrected for 16 principal components to avoid inflation of the test statistics (Supplementary Figure 1). The PCA corrected data was segmented by the CNAM algorithm implemented in GoldenHelix (GoldenHelix).
The univariate method is sensitive for larger, rare CNVs by scanning the data subject by subject. Algorithm settings included a moving window of 20000 probes, could combine maximum number of segments and minimum number of markers per segment set at 1, maximum pairwise permuted p-value 0.005, applying 2000 permutations per pair to achieve high sensitivity. The multivariate method is sensitive for common CNVs with superior resolution. Algorithm settings were similar to the univariate method, except we did not apply a moving window. Both algorithms reduce the dataset to regions where CNV events occur, thus limiting the number of tests of association.
For test of association, OSA was performed using the covariates from the univariate and multivariate segmentation algorithms. The subsets were added sequentially by 20 subjects from youngest AAO to oldest. The OSA script was created in R (script available upon request). We used the false discovery rate (FDR) approach considering all comparisons within the univariate or multivariate tests and the criteria for significance was set at p-value of <0.05. As the univariate and multivariate segments have substantial overlap, considering both sets would have been overly conservative. Ultimately, we required locus specific replication in the replication set. To account for the APOE effect on AAO in a way that does not change the fundamental concept of the ordered subset analysis, the following strategy was applied: a derived AAO matrix was generated where the APOE effect was accounted for by adding 5 years for one APOE4 allele, and 10 years for two APOE4 alleles. The statistics are added to Tables 2 and 3. All except one association remained significant.
Table 2.
Ordered subset analysis adding multivariate segmentation data by AAO groups in ascending order with bins of 20
| Start probe | Chr | Position | traitval | Pval | lowerCI | upperCI | meanCtrl | meanCase | nCtrl | nCase | qval | APOE pval | Nearest gene |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CN_857286 | 2 | 4213852 | 60 | 0.000281 | −0.189 | −0.058 | −0.030 | 0.093 | 190 | 50 | 0.021 | 3.84E-05 | None |
| CN_847338 | 2 | 208355166 | 86 | 0.000523 | 0.045 | 0.160 | −0.135 | −0.237 | 190 | 364 | 0.032 | 0.0006221 | CREB1 |
| SNP_A-8602968 | 5 | 97048466 | 55 | 0.000415 | −0.072 | −0.021 | −0.011 | 0.035 | 190 | 24 | 0.029 | None | |
| CN_1196516 | 7 | 109437080 | 56.47 | 0.000679 | −0.180 | −0.051 | −0.022 | 0.094 | 190 | 29 | 0.037 | 0.0152776 | EIF3IP1 |
| SNP_A-8363651 | 13 | 69248299 | 56 | 0.000148 | −0.080 | −0.026 | −0.017 | 0.036 | 190 | 28 | 0.014 | LINC00550 | |
| CN_119094 | 15 | 30444264 | 74.77 | 0.000726 | 0.016 | 0.060 | −0.014 | −0.052 | 190 | 226 | 0.038 | 0.00085 | CHRFAM7A |
Chr, chromosome; traitval, age at onset when maximum p value was detected; pval, p value; lowerCI, lower confidence Interval; upperCI, upper confidence interval; meanCtrl, mean logR of controls; meanCase, mean logR of cases; nCtrl, number of controls; nCase, number of cases; qval, False Discovery Rate (FDR) corrected p value; APOE pval, APO-adjusted p value; Affy N, number of probes covering the region on the Genome-Wide Human SNP Array 6.0 array; Illumina N, number of probes covering the region on the Illumina Human610_Quad Array.
Table 3.
Ordered subset analysis adding univariate segmentation data by AAO groups in ascending order with bins of 20
| Start probe | Chr | Position | traival | pval | lowerCI | upperCI | meanCtrl | meanCase | nCtrl | nCase | qval | APOE pval | Nearest gene |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CN_847338 | 2 | 208355166 | 84 | 6.55E-06 | 0.069 | 0.174 | −0.117 | −0.238 | 190 | 355 | 0.030 | 7.42E-06 | CREB1 |
| CN_1066295 | 4 | 173424935 | 55 | 4.33E-08 | 0.074 | 0.152 | 0.105 | −0.008 | 190 | 24 | 0.001 | 0.1047305 | GALNTL6 |
| CN_641648 | 13 | 69248299 | 55 | 1.37E-05 | −0.054 | −0.021 | −0.050 | −0.012 | 190 | 24 | 0.042 | 7.30E-06 | LINC00550 |
| CN_690430 | 14 | 41607622 | 55 | 1.97E-06 | −0.136 | −0.058 | −0.108 | −0.011 | 190 | 24 | 0.013 | 0.0003186 | BX248273 |
Chr, chromosome; traitval, age at onset when maximum p value was detected; pval, p value; lowerCI, lower confidence interval; upperCI, upper confidence interval; meanCtrl, mean logR of controls; meanCase, mean logR of cases; nCtrl, number of controls; nCase, number of cases; qval, False Discovery Rate (FDR) corrected p value; APOE pval, APOE-adjusted p value.
TaqMan assay for the CNV encompassing the CHRFAM7A fusion gene
TaqMan Copy Number Assay was performed to validate the copy number calls of CHRFAM7A. Primers (forward primer: GTAATAGTGTAATACTGTAACTTTAAAATGTGTTACTTGT, reverse primer: AGCCGGGATGGTCTCGAT) and probe (TCCTGACTGTACACATAAAA) were supplied by FAM dye-labeled assay targeted to CHRFAM7A and the VIC dye-labeled RNaseP Applied Biosystems. The duplex real-time PCR assays were performed using a FAM dye-labeled assay targeted to CHRFAM7A and the VIC dye-labeled RNaseP (TaqMan copy number reference assay, part #4403326) as a reference gene. Each sample was assayed in quadruplicate by using 10 ng DNA in each reaction. Real-time PCR was performed using the CFX384 Real-time PCR Detection System (Bio-Rad). Threshold cycle (Ct) values were determined for CHRFAM7A and compared with Ct values for RNase P. Relative quantity was determined by the DD Ct method [21].
Gene expression analysis of CHRFAM7A and CHRNA7 in postmortem human brain tissue
Microarray expression profiling was performed by the Microarray Core Facility of The University of Texas Health Science Center at Houston, Houston, TX. RNA QC included 260/280 nm 1.9–2.0, 260/230 nm >1.5 for RNA by Nanodrop 1000. RNA quality was further assessed by calculating RNA integrity number (RIN) with Agilent 2100 Bioanalyzer (Microarray core facility, Baylor College Medicine). Samples were entered to an expression array experiment if RIN > 4 (range 4.3–6.9) [22]. The RNA was amplified into cRNA and biotinylated by in vitro transcription using the Illumina® TotalPrep RNA Amplification Kit (Ambion, Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol. Biotinylated cRNAs were purified, fragmented, and subsequently hybridized to an Illumina Human-6 V3 BeadChip (Illumina, San Diego, CA).
RESULTS
756 samples passed the contrast QC and were adequate for copy number analysis. Forty samples failed MAPD cutoff of 0.4 and 143 samples failed due to number of CNV calls more than 2 SD of the mean. 573 samples passed QC, 381 AD subjects and 192 normal controls. Six AD subjects had missing AAO, thus 375 AD subjects entered the OSA AAO analysis. Demographics of the 375 subjects are depicted in Table 1.
Table 1.
Cohort characteristics
| N | % Female | AAO mean | AAO SD | AAO range | Age mean | Age SD | Age range | APOE 4/4 | APOE N/4 | APOEN/N | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AD | 375 | 59 | 71.4 | 8.6 | 45–90 | 79.3 | 8.5 | 56-1-5 | 52 (13.9%) | 180 (48%) | 143 (38.1%) |
| Control | 192 | 74 | NA | NA | NA | 73.3 | 8.8 | 57–96 | 4 (2.1%) | 56 (29.1%) | 130 (67.7%) |
AAO, age at onset; SD, age at onset standard deviation.
The OSA using the multivariate covariates identified six chromosomal regions (Table 2), while the univariate segmented data resulted in four chromosomal regions (Table 3). Two chromosomal regions (chr2: 208,35 and chr13: 69,24) were detected by both segmentation methods. The association of the chr15: 30,44 chromosomal region was replicated in the NIA-LOAD Familial Study dataset (Bonferroni corrected p-value <0.05, Supplementary Table 1). A subset of samples (n = 499) was validated by TaqMan assay for the chr15: 30,44 region (genotyping accuracy: 93.3%) (Fig. 1). We detected 0.73% homozygous deletion, 18.12% heterozygous deletion, and 79.21% diploid genotypes. In addition, we detected rare three copies of the fusion gene (1.87%) confirmed by Taq-Man assay, consistent with previous studies using locus specific genotyping (in contrast to array based genome wide genotyping) [23]. Expression of CHRFAM7A correlated with CNV state in the postmortem human temporal lobe tissue (Fig. 2).
Fig. 1.
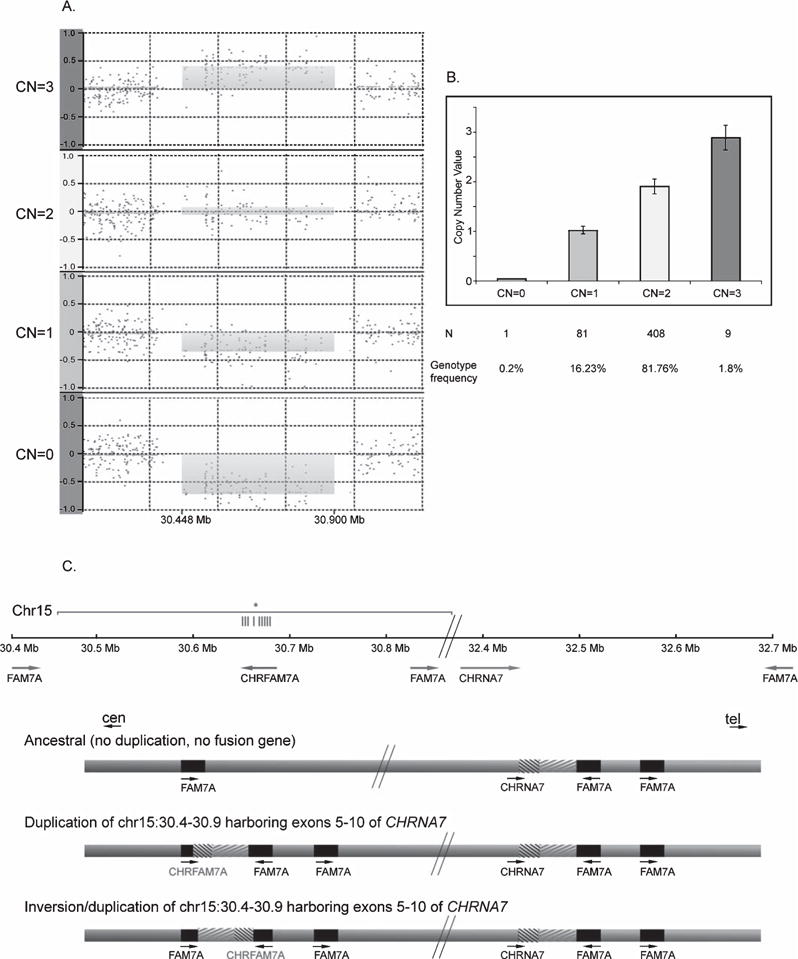
Genotyping, validation, and genomic context of the CHRFAM7A fusion gene. (a) Genome-Wide Human SNP Array 6.0 is presented as a plot of normalized probewise log2ratio data in the four panels for duplication, diploid, heterozygous and homozygous deletion carriers (CN = 3,2,1,0). (b) Validation of the 4 CN states using TaqMan Copy Number Assay. (c) Genomic context of the structure of the CHRFAM7A region. Top: Chromosomal location and orientation of genes based on the human reference genome. The horizontal lines indicate the 9 CNV probes located in the CHRNA7 exons of the fusion gene (grey line: duplicated sequence, *: the site of the TaqMan probe). Bottom: The three structures of the CHRFAM7A allele. The ancestral allele contains both the CHRNA7 and FAM7A but no hybrid gene. Partial duplication and fusion of CHRNA7 and FAM7A generated the hybrid gene, CHRFAM7A (CHRNA7 and CHRFAM7A in direct orientation). Inversion of the fusion gene results in opposite orientation of CHRNA7 and CHRFAM7A.
Fig. 2.
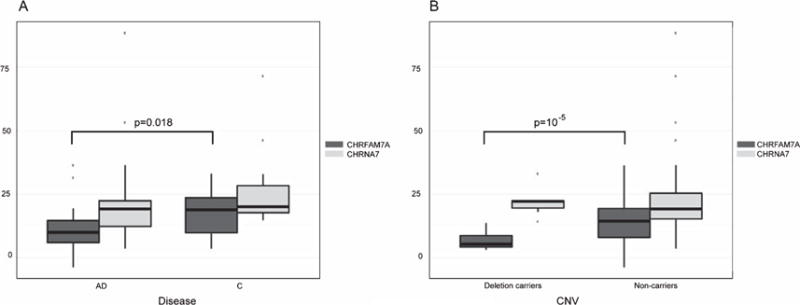
CHRFAM7A and CHRNA7 gene expression. Expression level of CHRFAM7A correlates with the (A) disease state (p = 0.018) and (B) CNV state (p = 10−5) in human temporal lobe samples. Expression of CHRNA7 is similar in AD versus controls (A) or in deletion carriers and non-carriers (B) consistent with in vitro experiments suggesting that the presence of the fusion gene decreases the number of functional receptors reaching the cell membrane without changes in CHRNA7 expression levels.
DISCUSSION
All six regions associated with AD from the multivariate segmented dataset are known CNV loci and are significant after accounting for the APOE effect on AAO. The chr2: 4,21 and chr5: 97,04 regions do not harbor genes within 500 kb. Two of the CNVs are near noncoding RNA sequences. The chr7: 109,43 CNV is in close proximity of EIF3IP1, a pseudogene encoding Eukaryotic Translation Initiation Factor 3, Subunit I Pseudogene 1. The chr13:69,24 region encompasses Long Intergenic Non-Protein Coding RNA 550 (LINC00550). LINC00550 is of unknown function; however, it is overexpressed in neurofibrillary tangles as compared to neurons in subjects with AD (GEO: GDS2795). The chr2: 208,35 deletion upstream from CREB1 has been reported previously from the same dataset using alternative statistical approach incorporating gene expression as a quantitative trait and CNV into the model [24]. The chr15: 30,44 region was reported to be associated with AD [23]. Inspection of the kernel distribution of the logR data suggested that the previous reports grossly underestimated the deletion frequency on chr15: 30,44 likely due to over-stringent algorithm settings and detected only the homozygous deletion. We confirmed the CNV calls with TaqMan assay and demonstrated that the deletion allele frequency is 18.85%. Locus specific association studies in schizophrenia and bipolar disorder reported similar allele frequencies [25–27].
The four associations detected by the univariate segmented data included the chr2: 208,35 and the chr13:69,24 regions also detected by the multivariate segmentation. The univariate and multivariate segmentation methods are sensitive to larger, rarer and smaller, more common CNV events, respectively, with overlap between the two. These two regions were large enough for the univariate segmentation and frequent enough for the multivariate method, thus detected by both. Due to this overlap, correcting for multiple testing using Bonferroni correction for the two methods would be overly conservative. The chr4: 173,42 region is intragenic in GALNTL6. GALNTL6 has been associated with lipid metabolism [28], body mass index [29], and hypertension, raising the possibility of its association with AD through a vascular mechanism; this association lost its significance after correcting for the APOE effect on AAO. The nearest gene to the chr14: 41,60 region is BX248273 of unknown function.
The association of the chr15: 30,44 chromosomal region was replicated in the NIA-LOAD Familial Study dataset. The lack of replication for the other regions with the NIA-LOAD Familial Study dataset is inconclusive due to limited coverage of these regions on the Illumina Human610 Quad array (Table 2 and 3). Gene expression of CHRFAM7A was lower in deletion carrier status versus non-carriers in postmortem human temporal lobes. CHRFAM7A is a fusion gene derived through a presumptive series of duplication, deletion, and inversion events [25–27]. The ancestral allele does not possess the fusion gene; as the most frequent genotype is one copy of the fusion gene on each chromosome (2 copy of the fusion gene), and the CNV calls are relative to the most frequent variant, thus the ancestral allele is the homozygous deletion (0 copy of the fusion gene) (Fig. 1). CHRFAM7A association has been reported previously; however the segmentation algorithm failed to detect the heterozygous deletion in this study [30]. It has been implicated in schizophrenia [31, 32], bipolar disorder [33], and episodic memory impairment [34]. Large deletions of approximately 2 MB affecting the chromosomal region between two low copy repeats from CHRFAM7A to CHRNA7 are more frequent in idiopathic epilepsies and developmental disorders [31, 32, 35, 36].
The fusion gene is derived from the neuronal acetylcholine receptor subunit alpha-7 (CHRNA7) and family with sequence similarity 7A (FAM7A or ULK4) genes. The presence of the fusion gene reduces the nicotine-elicited alpha 7 current in Xenopus laevis oocytes [37, 38]. The reduction is mainly caused by the decreased number of functional receptors reaching the oocyte membrane [37, 38]. These findings suggest that CHRFAM7A acts as a dominant negative modulator of CHRNA7 function and is critical for receptor regulation in humans.
Amyloid-β (Aβ)1−42 and α7 nicotinic acetylcholine receptor are co-expressed in AD brains [39]. Aβ1–42 binds with high affinity to the α7 nicotinic acetylcholine receptor and the receptor facilitates internalization of Aβ1–42 through endocytosis in cell culture [39] supporting the observation that neurons expressing the α7 nicotinic acetylcholine receptor are selectively vulnerable in AD. α7 nicotinic acetylcholine receptor expressed in vascular smooth muscle cells is implicated in cerebral amyloid angiopathy, one of the pathological hallmarks of AD [40].
Memantine, one of the drugs used in the treatment of AD, is a low molecular weight antagonist of α7 nicotinic acetylcholine receptor. As lower copy number and lower expression levels of the fusion gene (the negative regulator of α7 nicotinic acetylcholine receptor) are associated with AD, memantine could be particularly effective in this subgroup of AD, allowing molecular based therapy.
This genome-wide CNV association study applying OSA by AAO analysis was undertaken to identify loci that confer risk of AD in a subset of AD subjects defined by AAO of the disease. AAO was determined with two methods with high correlation as reported previously [18, 19]. The power of OSA for case-control studies is superior to trend tests when the subset of interest is about 15–30% of the available dataset, corresponding to the allele frequencies we detected for the replicated locus. Additional advantages include the method’s nonparametric nature: through the ranking procedure it is robust to outliers; and the identification of a case subset that is enriched for a particular susceptibility allele. The major limitation of OSA is the evaluation of a single covariate in the model.
CNV GWAS studies face multiple challenges that impact the power of traditional case control design: i) CNVs are often multiallelic in contrast with the biallelic SNPs, ii) CNVs cannot be computed, iii) compiling datasets is inefficient due to batch effects and variable coverage, thus limiting sample size and power, and iv) CNV calls cannot be optimized at the whole genome level as the frequency and contribution of deletions and duplications at each locus is different resulting in variable references and dynamic ranges which are locus specific. OSA as an alternative statistical approach with increased power is useful as it can detect a signal in a smaller, homogeneous dataset and subsequently these signals can be followed up from other datasets in a locus specific manner allowing optimization of CNV calls for that specific locus.
The contribution of CNVs to the heritability of AD has not been fully explored due to these limitations and larger scale studies with high resolution and high dynamic range assays are needed.
Supplementary Material
Acknowledgments
This study was supported by the Texas Alzheimer’s Research and Care Consortium (TARCC) funded by the state of Texas through the Texas Council on Alzheimer’s Disease and Related Disorders and an Alzheimer Association New Investigator Research Grant to KS.
Investigators from the Texas Alzheimer’s Research and Care Consortium: Baylor College of Medicine: Susan Rountree MD, Valory Pavlik PhD, Wen Chan PhD, Paul Massman PhD, Eveleen Darby, Tracy Evans RN, Aisha Khaleeq; Texas Tech University Health Science Center: Benjamin Williams, MD, Gregory Schrimsher, PhD, Andrew Dentino, MD, Ronnie Orozco; University of North Texas Health Science Center: Thomas Fairchild, PhD, Janice Knebl, DO, Sid E. O’Bryant, PhD, James R. Hall, PhD, Robert C. Barber, PhD, Douglas Mains, Lisa Alvarez; University of Texas Southwestern Medical Center: Perrie Adams, PhD, Roger Rosenberg, MD, Myron Weiner, MD, Mary Quiceno, MD, Joan Reisch, PhD, Ryan Huebinger, PhD, Guanghua Xiao, PhD, Doris Svetlik, Amy Werry, Janet Smith; University of Texas Health Science Center – San Antonio: Donald Royall, MD, Raymond Palmer, PhD, Marsha Polk.
Authors’ disclosures available online (http://www.j-alz.com/disclosures/view.php?id=2193).
Footnotes
Handling Associate Editor: Israel Ampuero
SUPPLEMENTARY MATERIAL
The supplementary table and figure are available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-132693.
References
- 1.Ebly EM, Parhad IM, Hogan DB, Fung TS. Prevalence and types of dementia in the very old: Results from the Canadian Study of Health and Aging. Neurology. 1994;44:1593–1600. doi: 10.1212/wnl.44.9.1593. [DOI] [PubMed] [Google Scholar]
- 2.Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD, van Belle G, Jolley L, Larson EB. Dementia and Alzheimer disease incidence: A prospective cohort study. Arch Neurol. 2002;59:1737–1746. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- 3.Rocca WA, Hofman A, Brayne C, Breteler MM, Clarke M, Copeland JR, Dartigues JF, Engedal K, Hagnell O, Heeren TJ, et al. Frequency and distribution of Alzheimer’s disease in Europe: A collaborative study of 1980–1990 prevalence findings. The EURODEM-Prevalence Research Group. Ann Neurol. 1991;30:381–390. doi: 10.1002/ana.410300310. [DOI] [PubMed] [Google Scholar]
- 4.Daw EW, Heath SC, Wijsman EM. Multipoint oligogenic analysis of age-at-onset data with applications to Alzheimer disease pedigrees. Am J Hum Genet. 1999;64:839–851. doi: 10.1086/302276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daw EW, Payami H, Nemens EJ, Nochlin D, Bird TD, Schellenberg GD, Wijsman EM. The number of trait loci in late-onset Alzheimer disease. Am J Hum Genet. 2000;66:196–204. doi: 10.1086/302710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T. Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65:664–670. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li YJ, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Allen FA, Jr, Goetz CG, Mastaglia F, Stajich JM, Gibson RA, Middleton LT, Saunders AM, Scott BL, Small GW, Nicodemus KK, Reed AD, Schmechel DE, Welsh-Bohmer KA, Conneally PM, Roses AD, Gilbert JR, Vance JM, Haines JL, Pericak-Vance MA. Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet. 2002;70:985–993. doi: 10.1086/339815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conrad DF, Bird C, Blackburne B, Lindsay S, Mamanova L, Lee C, Turner DJ, Hurles ME. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet. 2010;42:385–391. doi: 10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, Tyler-Smith C, Carter N, Scherer SW, Tavare S, Deloukas P, Hurles ME, Dermitzakis ET. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conrad DF, Hurles ME. The population genetics of structural variation. Nat Genet. 2007;39:S30–S36. doi: 10.1038/ng2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCarroll SA. Extending genome-wide association studies to copy-number variation. Hum Mol Genet. 2008;17:R135–R142. doi: 10.1093/hmg/ddn282. [DOI] [PubMed] [Google Scholar]
- 14.Hauser ER, Watanabe RM, Duren WL, Bass MP, Langefeld CD, Boehnke M. Ordered subset analysis in genetic linkage mapping of complex traits. Genet Epidemiol. 2004;27:53–63. doi: 10.1002/gepi.20000. [DOI] [PubMed] [Google Scholar]
- 15.Qin X, Hauser ER, Schmidt S. Ordered subset analysis for case-control studies. Genet Epidemiol. 2010;34:407–417. doi: 10.1002/gepi.20489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blacker D, Albert MS, Bassett SS, Go RC, Harrell LE, Folstein MF. Reliability and validity of NINCDS-ADRDA criteria for Alzheimer’s disease. The National Institute of Mental Health Genetics Initiative. Arch Neurol. 1994;51:1198–1204. doi: 10.1001/archneur.1994.00540240042014. [DOI] [PubMed] [Google Scholar]
- 17.O’Bryant SE, Waring SC, Cullum CM, Hall J, Lacritz L, Massman PJ, Lupo PJ, Reisch JS, Doody R. Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: A Texas Alzheimer’s research consortium study. Arch Neurol. 2008;65:1091–1095. doi: 10.1001/archneur.65.8.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw CA, Li Y, Wiszniewska J, Chasse S, Zaidi SN, Jin W, Dawson B, Wilhelmsen K, Lupski JR, Belmont JW, Doody RS, Szigeti K. Olfactory copy number association with age at onset of Alzheimer disease. Neurology. 2011;76:1302–1309. doi: 10.1212/WNL.0b013e3182166df5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szigeti K, Lal D, Li Y, Doody RS, Wilhelmsen K, Yan L, Liu S, Ma C, Texas Alzheimer R, Care C. Genome-wide scan for copy number variation association with age at onset of Alzheimer’s disease. J Alzheimers Dis. 2013;33:517–523. doi: 10.3233/JAD-2012-121285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doody RS, Dunn JK, Huang E, Azher S, Kataki M. A method for estimating duration of illness in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2004;17:1–4. doi: 10.1159/000074078. [DOI] [PubMed] [Google Scholar]
- 21.Aldhous MC, Abu Bakar S, Prescott NJ, Palla R, Soo K, Mansfield JC, Mathew CG, Satsangi J, Armour JA. Measurement methods and accuracy in copy number variation: Failure to replicate associations of beta-defensin copy number with Crohn’s disease. Hum Mol Genet. 2010;19:4930–4938. doi: 10.1093/hmg/ddq411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weis S, Llenos IC, Dulay JR, Elashoff M, Martinez-Murillo F, Miller CL. Quality control for microarray analysis of human brain samples: The impact of postmortem factors, RNA characteristics, and histopathology. J Neurosci Methods. 2007;165:198–209. doi: 10.1016/j.jneumeth.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 23.Swaminathan S, Huentelman MJ, Corneveaux JJ, Myers AJ, Faber KM, Foroud T, Mayeux R, Shen L, Kim S, Turk M, Hardy J, Reiman EM, Saykin AJ, Alzheimer’s Disease Neuroimaging I, Group N-LNFS Analysis of copy number variation in Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. PLoS One. 2012;7:e50640. doi: 10.1371/journal.pone.0050640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Shaw CA, Sheffer I, Sule N, Powell SZ, Dawson B, Zaidi SN, Bucasas KL, Lupski JR, Wilhelmsen KC, Doody R, Szigeti K. Integrated copy number and gene expression analysis detects a CREB1 association with Alzheimer’s disease. Transl Psychiatry. 2012;2:e192. doi: 10.1038/tp.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flomen RH, Collier DA, Osborne S, Munro J, Breen G, St Clair D, Makoff AJ. Association study of CHRFAM7A copy number and 2 bp deletion polymorphisms with schizophrenia and bipolar affective disorder. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:571–575. doi: 10.1002/ajmg.b.30306. [DOI] [PubMed] [Google Scholar]
- 26.Flomen RH, Davies AF, Di Forti M, La Cascia C, Mackie-Ogilvie C, Murray R, Makoff AJ. The copy number variant involving part of the alpha7 nicotinic receptor gene contains a polymorphic inversion. Eur J Hum Genet. 2008;16:1364–1371. doi: 10.1038/ejhg.2008.112. [DOI] [PubMed] [Google Scholar]
- 27.Flomen RH, Shaikh M, Walshe M, Schulze K, Hall MH, Picchioni M, Rijsdijk F, Toulopoulou T, Kravariti E, Murray RM, Asherson P, Makoff AJ, Bramon E. Association between the 2-bp deletion polymorphism in the duplicated version of the alpha7 nicotinic receptor gene and P50 sensory gating. Eur J Hum Genet. 2013;21:76–81. doi: 10.1038/ejhg.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kathiresan S, Manning AK, Demissie S, D’Agostino RB, Surti A, Guiducci C, Gianniny L, Burtt NP, Melander O, Orho-Melander M, Arnett DK, Peloso GM, Ordovas JM, Cupples LA. A genome-wide association study for blood lipid phenotypes in the Framingham Heart Study. BMC Med Genet. 2007;8(Suppl 1):S17. doi: 10.1186/1471-2350-8-S1-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fox CS, Heard-Costa N, Cupples LA, Dupuis J, Vasan RS, Atwood LD. Genome-wide association to body mass index and waist circumference: The Framingham Heart Study 100K project. BMC Med Genet. 2007;8(Suppl 1):S18. doi: 10.1186/1471-2350-8-S1-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heinzen EL, Need AC, Hayden KM, Chiba-Falek O, Roses AD, Strittmatter WJ, Burke JR, Hulette CM, Welsh-Bohmer KA, Goldstein DB. Genome-wide scan of copy number variation in late-onset Alzheimer’s disease. J Alzheimers Dis. 2010;19:69–77. doi: 10.3233/JAD-2010-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Moller HJ, Hartmann A, Shianna KV, Ge D, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Muhleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, Group. Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nothen MM, Peltonen L, Collier DA, St Clair D, Stefansson K. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.International Schizophrenia C. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong CJ, Lai IC, Liou LL, Tsai SJ. Association study of the human partially duplicated alpha7 nicotinic acetylcholine receptor genetic variant with bipolar disorder. Neurosci Lett. 2004;355:69–72. doi: 10.1016/j.neulet.2003.10.043. [DOI] [PubMed] [Google Scholar]
- 34.Dempster EL, Toulopoulou T, McDonald C, Bramon E, Walshe M, Wickham H, Sham PC, Murray RM, Collier DA. Episodic memory performance predicted by the 2bp deletion in exon 6 of the “alpha 7-like” nicotinic receptor subunit gene. Am J Psychiatry. 2006;163:1832–1834. doi: 10.1176/ajp.2006.163.10.1832. [DOI] [PubMed] [Google Scholar]
- 35.Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, Muhle H, de Kovel C, Baker C, von Spiczak S, Kron KL, Steinich I, Kleefuss-Lie AA, Leu C, Gaus V, Schmitz B, Klein KM, Reif PS, Rosenow F, Weber Y, Lerche H, Zimprich F, Urak L, Fuchs K, Feucht M, Genton P, Thomas P, Visscher F, de Haan GJ, Moller RS, Hjalgrim H, Luciano D, Wittig M, Nothnagel M, Elger CE, Nurnberg P, Romano C, Malafosse A, Koeleman BP, Lindhout D, Stephani U, Schreiber S, Eichler EE, Sander T. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Bon B, Mefford HC, Menten B, Koolen DA, Sharp AJ, Nillesen WM, Innis JW, de Ravel TJ, Mercer CL, Fichera M, Stewart H, Connell LE, Ounap K, Lachlan K, Castle B, Van der Aa N, van Ravenswaaij C, Nobrega MA, Serra-Juhé C, Simonic I, de Leeuw N, Pfundt R, Bongers EM, Baker C, Finnemore P, Huang S, Maloney VK, Crolla JA, van Kalmthout M, Elia M, Vandeweyer G, Fryns JP, Janssens S, Foulds N, Reitano S, Smith K, Parkel S, Loeys B, Woods CG, Oostra A, Speleman F, Pereira AC, Kurg A, Willatt L, Knight SJ, Vermeesch JR, Romano C, Barber JC, Mortier G, Pérez-Jurado LA, Kooy F, Brunner HG, Eichler EE, Kleefstra T, de Vries BB. Further delineation of the 15q13 microdeletion and duplication syndromes: A clinical spectrum varying from non-pathogenic to a severe outcome. J Med Genet. 2009;46:511–523. doi: 10.1136/jmg.2008.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Lucas-Cerrillo AM, Maldifassi MC, Arnalich F, Renart J, Atienza G, Serantes R, Cruces J, Sanchez-Pacheco A, Andres-Mateos E, Montiel C. Function of partially duplicated human alpha77 nicotinic receptor subunit CHRFAM7A gene: Potential implications for the cholinergic anti-inflammatory response. J Biol Chem. 2011;286:594–606. doi: 10.1074/jbc.M110.180067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Araud T, Graw S, Berger R, Lee M, Neveu E, Bertrand D, Leonard S. The chimeric gene CHRFAM7A, a partial duplication of the CHRNA7 gene, is a dominant negative regulator of alpha7*nAChR function. Biochem Pharmacol. 2011;82:904–914. doi: 10.1016/j.bcp.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagele RG, D’Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1–42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience. 2002;110:199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- 40.Clifford PM, Siu G, Kosciuk M, Levin EC, Venkataraman V, D’Andrea MR, Nagele RG. Alpha7 nicotinic acetylcholine receptor expression by vascular smooth muscle cells facilitates the deposition of Abeta peptides and promotes cerebrovascular amyloid angiopathy. Brain Res. 2008;1234:158–171. doi: 10.1016/j.brainres.2008.07.092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.