

Abstract
Diphtheria toxin translocation (T) domain inserts in lipid bilayers upon acidification of the environment. Computational and experimental studies have suggested that low pH triggers a conformational change of the T-domain in solution preceding membrane binding. The refolded membrane-competent state was modeled to be compact and mostly retain globular structure. In the present work, we investigate how this refolded state interacts with membrane interfaces in the early steps of T-domain’s membrane association. Coarse-grained molecular dynamics (CG-MD) simulations suggest two distinct membrane-bound conformations of the T-domain in the presence of bilayers composed of a mixture of zwitteronic and anionic phospholipids (POPC:POPG with a 1:3 molar ratio). Both membrane-bound conformations show a common near parallel orientation of hydrophobic helices TH8-TH9 relative to the membrane plane. The most frequently observed membrane-bound conformation is stabilized by electrostatic interactions between the N-terminal segment of the protein and the membrane interface. The second membrane-bound conformation is stabilized by hydrophobic interactions between protein residues and lipid acyl chains, which facilitate deeper protein insertion in the membrane interface. A theoretical estimate of a free energy of binding of a membrane-competent T-domain to the membrane is provided.
Keywords: Diphtheria toxin, coarse-grained modeling, anionic lipids, insertion, low pH, histidine protonation
INTRODUCTION
Diphtheria toxin is a bacterial protein that penetrates eukaryotic cells, where it disrupts protein synthesis and results in cell death. The process of cell entry involves the internalization of the toxin bound to a cell-surface receptor followed by acidification of the endosome interior (Murphy, 2011). The full toxin consists of three domains, each associated with a specific function (Choe et al., 1992). The diphtheria toxin translocation (T) domain inserts into the membrane upon decrease of pH in the endosome interior. The T-domain aids membrane insertion and translocation of a toxin catalytic domain across the endosome membrane into the cytosol. In the cytosol, the catalytic domain facilitates cell death (Honjo, Nishizuka & Hayaishi, 1968). Thus, diphtheria toxin relies on a series of structural rearrangements of T-domain that enable delivery of the catalytic domain across endosomal membrane.
The stand-alone T-domain in solution adopts a monomeric globular form at neutral pH, comprised of ten α-helices (Bennett & Eisenberg, 1994). In the presence of anionic bilayers and acidic solution the protein undergoes major structural rearrangements, forming a membrane inserted state in which its most hydrophobic helices span the bilayer (Kyrychenko et al., 2009). Detailed understanding of T-domain membrane insertion has been precluded by the difficulty of applying high resolution structural methods (e.g. X-ray crystallography and NMR), primarily because of the protein tendency to aggregate at low pH, and the existence of multiple conformations among the membrane-associated states (Chenal et al., 2002; Kyrychenko et al., 2009; Ladokhin et al., 2004; Montagner et al., 2007; Palchevskyy et al., 2006; Vargas-Uribe et al., 2013a; Wang et al., 1997). Understanding the process of the T-domain membrane association will facilitate the initial steps towards complete characterization of its folding in membranes and its translocation function.
Spectroscopic experiments reveal that the T-domain’s insertion pathway contains several kinetic intermediates, some of which can be stabilized by the lipid composition of the target membrane and protein mutations (Kyrychenko et al., 2009; Rodnin et al., 2011; Vargas-Uribe et al., 2013a). Kinetic analysis of the T-domain membrane insertion process in low pH solution showed that the protein initially forms a membrane competent state in solution followed by its membrane association and a formation of an insertion competent intermediate (Kyrychenko et al., 2009). It has been suggested that the insertion competent state is characterized by an additional protonation of amino-acid side-chains at the membrane interface (Kyrychenko et al., 2009; Vargas-Uribe et al., 2013a). It has also been reported that increase of the molar fraction of anionic lipids promotes insertion of the hydrophobic helices TH8-9 into bilayers (Kyrychenko et al., 2009).
Protonation of T-domain histidine side-chains has been recognized to play an important role in various stages of the membrane insertion process, e.g. H257 and H223 were implicated to act as a molecular switch that triggers disruption of the native structure of T-domain in low pH solution (Flores-Canales, Simakov & Kurnikova; Kurnikov et al., 2013; Kyrychenko et al., 2009; Ladokhin, 2013; Perier et al., 2007; Rodnin et al., 2010). It has also been suggested that histidine protonation plays a role in the membrane binding (Perier et al., 2007), as well as in the final stages of the membrane insertion of the isolated T-domain (Rodnin et al., 2011; Vargas-Uribe et al., 2013a). Recently, we have performed atomistic MD simulations of T-domain destabilization in low pH solution. These simulations have directly demonstrated, for the first time, the role of N-terminal histidines in partial unfolding of the N-terminal helices, and a solvent exposure of the hydrophobic sites upon protein reorganization. The results of the simulations in conjunction with spectroscopic experiments (Kurnikov et al., 2013) suggest that the protein retains its compact structure while in solution. These features were interpreted as initial stages of formation of a membrane competent state of the T-domain in solution (Flores-Canales et al.; Kurnikov et al., 2013). Recent X-ray structures of diphtheria toxin distressed by exposure to low pH prior to crystallization process at neutral pH (Leka et al., 2014) have also indicated the possibility of refolding of the N-terminal helices as predicted by (Kurnikov et al., 2013).
No atomistic picture of the process of the T-domain membrane association and subsequent insertion is available thus far. In this work, we model initial association of the protein with the membrane to investigate whether the formation of the early intermediates in this process are affected by the protein structural and protonation states as well as by the lipid composition of the bilayer. All simulations are performed using a coarse-grained representation of the protein and the lipid. Coarse-grained molecular dynamics (CG-MD) simulations have been previously used to model protein-membrane association (Kalli, Campbell & Sansom, 2011; Kalli et al., 2010; Marrink & Tieleman, 2013; Stansfeld & Sansom, 2011). Such models successfully represent the role of electrostatic interactions between key residues and anionic lipids, as well as formation of hydrophobic contacts in membrane association of proteins (Kalli et al., 2011; Kalli et al., 2010). Coarse-graining the protein and the lipid structure allows one to perform efficient simulations on the microsecond time-scales of the mesoscopically sized systems (Marrink & Tieleman, 2013; Stansfeld & Sansom, 2011).
Two types of simulations are presented in this manuscript, 1) unrestrained CG-MD simulations of the T-domain association with the lipid bilayers, and 2) umbrella sampling (US) free energy CG-MD simulations to estimate a potential of mean force (PMF) of the protein-membrane association and a free energy of the protein-membrane interface binding in different conformations. A coarse-grained protein structure in this study is based on our previously generated atomistic model of the T-domain membrane competent state (Kurnikov et al., 2013).
RESULTS
1. Simulations of the T-Domain Approaching the Bilayer
To investigate the initial membrane association of the T-domain and the effects of anionic lipids, we modeled the interactions of the neutral pH and the low pH structures of the T-domain with pre-formed phospholipid bilayers composed of mixtures of POPC and POPG (see detailed description of protein structures in Fig. 1). The top and bottom panels in Fig. 2 display the center of mass (COM) distance (Z) between the neutral (top) or low pH protein structures (bottom), and bilayers of different compositions as a function of time, respectively. As can be seen, larger fractions of anionic lipids (POPC:POPG 1:3) promote steady association of either neutral or low pH T-domain structures to bilayer interfaces in ten CG-MD trajectories (see Fig. 2C, F). In these trajectories, the protein binds to the bilayer within 0.3 μs to 1 μs and remains attached to the bilayer interface over the rest of the simulation length. In both protein structures, the initial protein-membrane contacts are formed at residues located in the N-terminal helices. For example, the low pH T-domain structure initiates protein-membrane contacts with residues located in the loop between TH2 and TH3, the loop between TH3 and TH4, or in the protein terminals. The neutral pH structure forms the initial protein-membrane contacts with the residues located in the loop between TH2 and TH3, or with the residues of the helix TH2 and the loop TH3-TH4.
Fig. 1.

Neutral pH folded structure and a low pH unfolded structure of T-domain. (A) The neutral pH structure is obtained from the diphtheria multi-domain structure obtained under neutral pH conditions [PDB 1F0L] (Steere, 2001). All side-chains are set to their standard protonation state at pH 7.5. (B) The low pH partially unfolded T-domain structure is generated by fixing histidines in their protonated state while the other residues are set to their standard protonation state. Explicit solvent molecular dynamics simulations were performed over ca. 6.8 μs on ANTON supercomputer (Kurnikov et al., 2013). Helices TH1, TH2, TH4, and TH5 are shown as green, cyan, magenta and orange ribbons, respectively. Helices TH5′, TH6, TH7 are shown as grey ribbons. Helices TH8 and TH9 are shown as blue and brown ribbons, respectively. The axes of TH8 and TH9, used to calculate the orientation of the T-domain respect to the membrane normal axis, are drawn as arrows. All histidines are highlighted as red licorice representation.
Fig. 2.

Plots of the center of mass (COM) distance (Z) between T-domain and lipid bilayers of different composition along the bilayer normal axis obtained from all unrestrained CG-MD simulations. Distance between the neutral pH structure and bilayers of the following composition and molar ratios: (A) POPC; (B) POPC:POPG 3:1; and (C) POPC:POPG 1:3. Distance of the low pH unfolded T-domain structure and bilayers of the following composition: (D) POPC; (E) POPC:POPG 3:1; and (F) POPC:POPG 1:3. Each line color represents an independent CG-MD production simulation, which is initiated with the same protein orientation and different seed number. Accumulated CG-MD production simulation time is 60 μs.
Simulations of the T-domain in presence of bilayers containing a smaller fraction of anionic phospholipid (POPC:POPG 3:1) exhibit a different protein-lipid association behavior. Fig. 2E shows that the low pH T-domain structure rapidly associates with the bilayer and forms a stable membrane-associated state in a single trajectory. The neutral pH structure binds to the membrane interface over the last 1 μs in two trajectories (see Fig. 2B). Most of the initial protein-membrane contacts are formed in the N-terminal helices of the protein. For instance, the low pH structure has initial protein-membrane contacts in the loop TH2-TH3. The neutral pH structure forms initial lipid contacts with the residues of the helix TH2 or with the residues 295–296. In presence of pure POPC bilayers (see Figs. 2A, D) both the neutral pH and the low pH T-domain structures bind only transiently to the membrane interface. Therefore, addition of negatively charged phospholipids to bilayers promotes rapid and stable membrane association of T-domain.
To monitor the protein orientation dynamics, we calculated the angles between the axes of helices TH8 and TH9, and the normal axis to the membrane plane as a function of time (see Figs. S1, S2, and Fig. 1 for structure notation). Overall, the protein orientation relative to the membrane plane is stabilized as the fraction of anionic lipids is increased. Figs. S1F and S2F show that the low pH T-domain adopts two distinct membrane-associated conformations with bilayers containing POPC:POPG 1:3. These two orientations have helices TH8 and TH9 in a near parallel or at an oblique angle to the membrane plane. The neutral pH T-domain structure adopts two membrane-bound conformations in which helices TH8 and TH9 are near perpendicular to the membrane plane upon binding to the mostly anionic bilayer (see Figs. S1C and S2C). One of these orientations is observed in three of the five independent trajectories. In simulations with a small ratio of anionic lipids (POPC:POPG 3:1) or no anionic lipids (POPC) the protein orientation with respect to bilayer was highly dynamic (see Figs. S1A, D and S2A, D).
2. T-domain Equilibrated at the Bilayer Interface
To investigate the similarities and differences of membrane association of the neutral and low pH T-domain structures to bilayers containing anionic phospholipids, we analyzed T-domain’s binding to the bilayer interface using equilibrium fragments of the simulations (the last 500 ns of each trajectory, see Fig. S3). We then calculated histograms of the COM distances (Z) between the protein and bilayers, and histograms of the number of contacts between residues and head-group beads (see Fig. 3). Fig. 3A shows that anionic bilayers (POPC:POPG 1:3) promote deeper insertion of the low pH structure (major peak at Z = 37 Å) than that of the neutral pH structure (major peak at Z = 43 Å). This observation is supported by the significant number of contacts between the low pH structure and the membrane interface relative to that of the neutral pH structure (Fig. 3B). Bilayers composed of a smaller fraction of anionic lipids (POPC:POPG 3:1) induce similar insertion depths of both T-domain structures (major peaks at Z = 43 Å and 42 Å in Fig. 3C). Both T-domain structures have a predominant peak at zero protein-membrane interface contacts, which shows the decreased affinity of the protein to associate with membranes of lower fraction of negatively charged lipids (see Fig. 3D).
Fig. 3.

Histograms for the center of mass (COM) distances (Z) between the T-domain and lipid bilayers (left panels), and the number of contacts between the protein and membrane interface beads (right panels). Data obtained from simulations of neutral pH and low pH T-domain structures are represented by red and blue bars, respectively. (A) Histogram of the COM distance between T-domain bound to POPC:POPG 1:3. (B) Histogram of contacts between T-domain and the membrane interface containing POPC:POPG 1:3. (C) Histogram of the COM distance between T-domain and bilayers containing POPC:POPG 3:1. (D) Histogram of contacts between T-domain and bilayer interface containing POPC:POPG 3:1. Data is obtained from the last 500 ns segments of all unrestrained CG-MD simulations of anionic membranes.
3. Protein Orientations at the Bilayers with the Higher Anionic Content
Fig. 4 shows density profiles of cationic, hydrophobic and histidine residues and bilayer components for two distinct protein orientations of the low pH T-domain structure and bilayers of higher anionic content (POPC:POPG 1:3). These orientations are further referred to as conformations B1 and B2. Conformation B1 was observed in four of five independent trajectories and is characterized by insertion of positively charged residues at the membrane interface. B2 shows insertion of hydrophobic residues in the bilayer hydrophobic core. Conformation B1 shows that cationic residues reside between the lipid head-groups and water regions, and populate a pronounced peak close to the membrane interface at 30 Å in the normal membrane axis (see Fig. 4A). Protonated histidines have a predominant density peak between the lipid head-group and phosphate regions. Hydrophobic residues show less penetration in the membrane interface, compared to their distribution in conformation B2 (see Fig. 4B). Conformation B2 exhibits a decreased penetration of cationic and histidine residues and an increased insertion of hydrophobic residues in the lipid head-group and acyl chains in contrast to conformation B1 (see Fig. 4B). It also shows a shoulder in the density profile of hydrophobic residues located between the lipid head-group and water regions.
Fig. 4.

Normalized number densities of coarse-grained atoms as a function of their distance from the center of the bilayer along the normal to the bilayer. Data is obtained from the last 500 ns of representative unrestrained CG-MD simulations of T-domain structures with lipid bilayers containing POPC:POPG 1:3. (A) Membrane bound conformation B1 of low pH T-domain structure. (B) Membrane bound conformation B2 of low pH T-domain structure. (C) Neutral pH T-domain structure associated to a bilayer in three simulations of a set of five independent unrestrained CG-MD simulations. (D) Neutral pH T-domain structure attached to a lipid bilayer in two trajectories of a set of five independent unrestrained CG-MD simulations. Density profiles of headgroup atoms of POPC (choline) and POPG (glycerol) are shown in black and yellow lines, respectively. Density profiles of phosphate and acyl chains from all lipids are shown in red and green lines, respectively. Density profiles of water and sodium counterions (ions) are shown in blue and dark yellow, respectively. Profiles of cationic residues (Lys, Arg, and N-terminus), histidines and hydrophobic residues are shown in cyan, grey and magenta lines, respectively.
CG-MD simulations of the neutral pH T-domain structure and bilayers of mixture POPC:POPG 1:3 display two different membrane bound orientations (see Figs. S1C and S2C). The most frequently observed orientation displays penetration of cationic residues in the head-group region, as shown in Fig. 4C. The same figure shows that histidines and hydrophobic residues have no significant penetration in the membrane interface region. In contrast Fig. 4D shows that the less frequently observed orientation has hydrophobic and histidine residues residing in the head-group region.
In addition, five independent trajectories of the low pH T-domain structure with histidines in their neutral state and bilayers composed of POPC:POPG 1:3 results in the protein binding to the bilayer interface (data not shown). The protein adopts a stable conformation B2 in a single trajectory. The other four trajectories show less deep insertion of the protein. Two of these trajectories show exchange of conformations between B1 and similar orientations observed in the simulations of the neutral pH T-domain model and bilayers (POPC:POPG 1:3).
The low pH T-domain structure exhibits more membrane interacting residues than those of the neutral pH structure bound to the bilayers with high anionic content POPC:POPG 1:3 (see Figs. S4 and S5). Conformations B1 and B2 have two different patterns of the protein-membrane contacts. Conformation B1 has significant number of contacts via the residues located in the partially unfolded helices TH1 and TH2, the loop TH3-4, the N-terminus of TH8, and the C-terminus of TH9, as shown in Fig. S4A. Conformation B2 has protein-membrane contacts located in the helices TH3-5, TH6-7, N-terminus of TH8 and C-terminus of TH9 (Fig. S4A). B2 has a significant number of the protein-aliphatic tail contacts via the residues P271, I306, P308 and V371, while these contacts are not present in the orientation B1, as shown in Fig. S4B.
The neutral pH T-domain structure displays two different membrane bound orientations with respect to bilayers with the high anionic lipid content. Histograms of the protein-membrane interface contacts show that the most frequently observed orientation has the majority of contacts localized in the loop TH8-9 and helix TH3, shown in Fig. S5. The second membrane bound orientation shows contacts at the TH1 N-terminus, TH8 N-terminus and TH9 C-terminus, also shown in Fig. S5.
4. Protein Orientations at the Bilayers with the Lower Anionic Content
Decrease of anionic content (POPC:POPG 3:1) reduces the propensity of the membrane binding for both the neutral and low pH T-domain structures. Both neutral and low pH structures have the majority of the membrane interactions with the residues of the TH1 N-terminus, TH8 N-terminus and TH9 C-terminus (see Fig. S6). In general, both membrane bound conformations are similar to the one described above in the simulations of the neutral pH T-domain with highly anionic bilayers (POPC:POPG 1:3) (Compare to contact histograms in Fig. S5). There are only subtle differences in conformations between the neutral and low pH T-domains: The low pH T-domain structure shows histidine and cationic residues residing in the glycerol group region (see Fig. S7A). The neutral pH structure shows reduced penetration of histidines, and an increased density peak of cationic residues within the glycerol groups (Fig. S7B).
5. Potential of Mean Force Calculations
We performed umbrella sampling (US) calculations to determine the free energy profile of the low pH structure of the T-domain across a pre-formed bilayer of higher anionic content (POPC:POPG 1:3). To enhance the conformational sampling, we performed three sets of US simulations with three different initial protein orientations. These protein orientations are the binding modes B1, B2 and a conformation in a different orientation taken from a simulation of the low pH protein structure with bilayers containing POPC:POPG 3:1 (see Fig. S6B). US simulations of the latter initial orientation equilibrate towards protein conformations similar to B1. Fig. 5 shows the free energy profiles for the complete set of simulations, orientation B1, and B2. The free energy profile obtained for the entire set of US simulations has two minima; a global minimum located at 35 Å and an intermediate at 41 Å. The free energy difference between these minima is 1.6 kcal/mol. The free energy barrier from the intermediate to the relative global minimum is within the thermal noise. The calculated free energy of binding (−kBTln(K)) for this curve is −11.3 ± 0.2 kcal/mol, which is calculated as indicated in the Methods section.
Fig. 5.
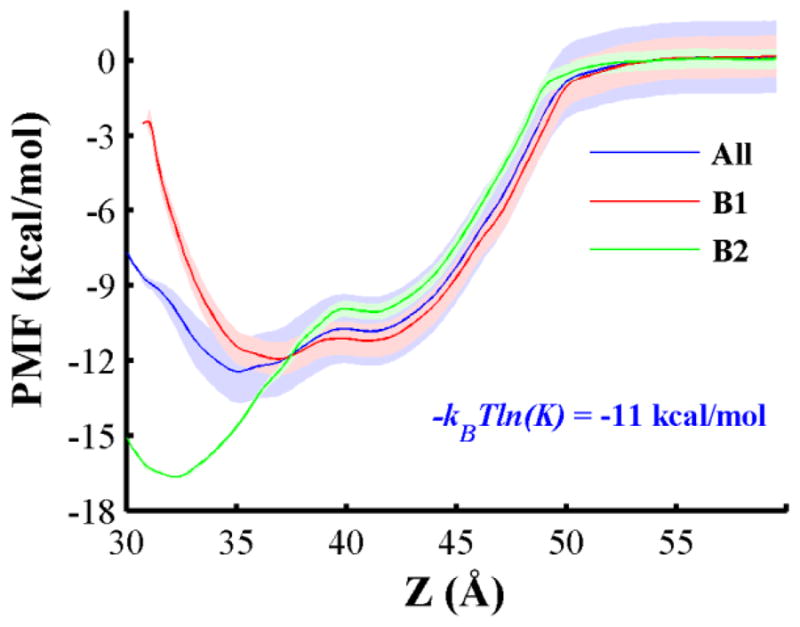
Potential of mean force (PMF) profile along the reaction coordinate Z. This reaction coordinate is the COM distance between the low pH T-domain structure and a pre-formed bilayer composed of POPC:POPG 1:3 along the normal axis to the membrane plane. US simulations start from protein orientations B1, B2 and an orientation observed at a lower fraction of POPG lipids. PMF curve and standard deviations calculated from the combined data of all US CG-MD simulations are shown by a blue line and shades, respectively. The corresponding free energy value is shown (see Methods section for description). PMF profiles of orientations B1 and B2 are shown in red and green lines, respectively. Standard deviations are shown by shades in their respective color. We performed 27 × 3 US window CG-MD simulations equally spaced every 1 Å over the reaction coordinate and the PMF profiles are calculated with WHAM method (Hub et al., 2010). Standard deviations are obtained from bootstrapping calculations. Additional computational details are described in methods.
Inspection of each set of umbrella simulations shows that orientation B1 and the non-preferable protein orientation have similar stabilized orientations and PMF profiles. Thus we combine their biased ensembles and calculate a single free energy profile, which is referred as PMF profile of conformation B1 (see red line in Fig. 5). Free energy profile of B1 has two minima, a global minimum located at Z = 37 Å and a local minimum located at Z = 41 Å. The local minimum represents an intermediate state when the protein initially touches the membrane, and the global minimum is observed upon subsequent reorientation of the protein on the membrane. B2 free energy profile also exhibits two energy minima. Its global minimum is observed at a more deeply inserted state (Z = 32 Å), its intermediate state is also at 41 Å. The free energy difference between the local and the global minima in each profile B1 and B2 is approximately 0.7 and 6.6 kcal/mol, respectively. Energy barriers between minima in both profiles B1 and B2 are located near 40 Å and are small. Overall, the lowest free energy states of the membrane bound conformations B1 and B2 are inserted at similar COM positions as observed in unrestrained CG-MD simulations, which sample the majority of conformations around Z = 37 Å and 34 Å, respectively.
The lowest minimum of conformation B2 is favored by 4.7 kcal/mol relative to the equivalent minimum of B1. However, a different trend is calculated for the intermediate and transition states of both conformations. B1 intermediate state is preferable by 1.2 kcal/mol, which is relative to the intermediate state of B2. Similarly, B1 transition state is lower by 1.2 kcal/mol than that of B2.
The convergence of protein orientation in our US simulations is monitored by the angles between helices TH8 and TH9, and the membrane normal. For example, Fig. S8 displays data from restrained simulations within the COM distance range of Z = 35 Å to 45 Å in which the protein is in contact with the bilayer interface. The protein orientation converges after the first 200 ns of each US window (see Fig. S8); however, some windows show some fluctuations of the angle values after this initial segment. Angle traces are shown for membrane-associated states with Z lower than 39 Å, at the transition state at 39 and for intermediate states with Z larger than 39 Å and lower than 45 Å. The latter restrained simulations sample protein conformations with helices TH8 and TH9 in a near perpendicular orientation to the membrane plane (see cyan lines in Fig. S8).
6. Membrane-associated Conformations at Equilibrium
Protein conformations associated with the bilayer are extracted from the global minima of PMF profiles B1 and B2. Fig. 6A shows that conformation B1 is characterized by protein-membrane interface contacts in the partially unfolded helix TH1 and the unfolded TH2. The majority of contacts in this region involve residues 223–227, which includes the protonated sidechain of H223. The N-terminus of helix TH8 forms contacts involving residues 322–327; residue E259 also forms contacts with the membrane interface. In Fig. 6B, conformation B2 shows that the partially unfolded helices TH1-2 are located away from the membrane interface in contrast to conformation B1. Helix TH5 is in a deeper inserted state and residues of helices TH3, TH4 and TH9 C-terminus form contacts with the membrane interface. Note that conformations B1 and B2 share a similar region of protein-membrane contacts, which is located around the N-terminus of helix TH8 (residues 320–322). H322 is a common protein-membrane contact in both conformations. The histograms of the protein-membrane contacts are similar with the ones obtained from the unrestrained CG-MD simulations.
Fig. 6.

Structural representation of the lowest free energy binding modes of the low pH T-domain structure associated to an anionic lipid bilayer composed of POPC:POPG 1:3. (A) Ribbon representation of membrane bound state B1 obtained from the last MD frame of a restrained US simulation at COM distance of Z = 37 Å. (B) Ribbon representation of membrane bound state B2 obtained from the last MD frame of a restrained US simulation at COM distance of Z = 32 Å. Helices TH1, TH2, TH4, TH5, TH8 and TH9 are shown in green, cyan, magenta, orange, blue and brown ribbons, respectively. Other helices are shown as grey ribbons. Residues with normalized number of contacts larger than 0.6 are highlighted in orange space filled representation. Residues 202, 222–227, 259, 322–327 are highlighted in B1 representation and residues 240–241, 244, 248, 264–265, 277, 280, 288, 299, 316, 318, 320–322, 373, 377 (not shown) are highlighted in B2 representation. Head-group beads of POPC and POPG are highlighted in grey and blue space filled representation. Phosphorous atoms are shown in dark brown space filled representation.
Protein structures at the free energy barrier of each PMF profile B1 and B2 are shown in Fig. 7. Overall, the protein orientations are slightly different from their respective global minima (see Fig. 6). At the free energy barrier of the conformation B1, the region near the N-termini of helices TH1 and TH8 forms the majority of contacts with the membrane interface (see Fig. 7A). In contrast, a transition structure of the conformation B2 displays the majority of the protein-membrane contacts in residues of the helices TH3, TH4, and near the N-terminus of the helix TH8 (as shown in Fig. 7B). We observe that protonated H322 forms the membrane interface contacts in both transition states. This observation suggests that H322 plays a role in the protein membrane association as well in the partial unfolding around this region. The histograms of the contacts for these transition states are shown in Fig. S9.
Fig. 7.

Structural representation of the intermediate membrane bound states along the free energy profile curves of the low pH T-domain structure in the presence of a pre-formed membrane containing POPC:POPG 1:3. Each representation corresponds to the last MD snapshot of restrained US simulations at COM distance of Z = 39 Å. (A) Ribbon representation of intermediate membrane bound state B1. (B) Ribbon representation of membrane bound state B2. Helices TH1, TH2, TH4, TH5, TH8 and TH9 are shown in green, cyan, magenta, orange, blue and brown ribbon representation, respectively. Other helices are shown in grey ribbons. Residues with normalized number of contacts larger than 0.6 are highlighted in orange space filled representation. Residues 202–204, 322, 326, 375 (not shown) are highlighted in B1 representation and residues 251–252, 264, 267, 321–322 are highlighted in B2 representation. Head-group beads of POPC and POPG are highlighted in grey and blue space filled representation. Phosphorous atoms are shown in dark brown space filled representation.
The protein structures at the intermediate states (the second minimum in each PMF profile) are similar to those observed in the unrestrained CG-MD simulations of the low pH structure with bilayers containing a small fraction of anionic lipids POPC:POPG 3:1 (compare Figs. S6B and S10). In this intermediate state, the residues 203–204 near the N-terminus of the helix TH1, the residues H322 and H323 near the N-terminus of TH8, the residue Y375 in the C-terminus of TH9 form stable contacts with the membrane interface, as shown in Fig. S10. Also, the residue E262 of the helix TH4 forms protein-membrane contacts, but less frequently.
7. Experimental Observation of T-domain Membrane Partitioning
To experimentally test whether the T-domain initially interacts with the membrane interface in a preferred orientation, we measured the membrane partitioning of the T-domain labeled with the fluorescence probe bimane at different positions. LUV (large unilamellar vesicles) containing the quencher LysoUB (to allow monitoring the binding process via quenching of the bimane probe (Ladokhin et al., 2002; Posokhov & Ladokhin, 2006; Posokhov et al., 2008b)) were composed of a mixture of POPC:POPG with a 9:1 molar ratio, which has been demonstrated to stabilize the interfacial intermediate state and prevent further insertion into the bilayer (Kyrychenko et al., 2009). In order to further exclude the possibility of transmembrane insertion and limit the observation to the interfacial intermediate, the measurements were performed at the mildly acidic pH of 6.0. As shown in Fig. 8, increasing concentration of lipids results in increasing partitioning of the W+-state (membrane competent state) to the membrane interface (more quenching of the probe) until reaching saturation. The total signal change at saturation appears to be dependent on the position of the label, which may indicate that the stable interfacial intermediate of the T-domain has a preferred orientation when interacting with the bilayer in which residue 369 is facing the bilayer. Fitting the data to a two state binding transition (Ladokhin, 2009) gives a similar free energy of partitioning for the T-domain labeled at any of the residues, suggesting that the labeling procedure did not affect the binding process.
Fig. 8.

Binding of the T domain to LUV composed of POPC:POPG 9:1 as a function of the lipid concentration. Each of the residues indicated in the figure were mutated to cysteine, one at the time, and then labeled with Bimane. The labeled protein was incubated with different concentrations of LUV containing 2% of LysoUB at pH 6.0, where the decrease in the emission intensity of bimane indicated interaction with the membrane. The free energies were calculated by fitting the data as previously described (Ladokhin, 2009). The data indicates that, although the free energy of binding of the T-domain to LUV is independent of the position of the label, the total signal change appears to be dependent on the position of labeling.
DISCUSSION
In our unrestrained CG-MD simulations T-domain associates with the membranes strongly and relatively rapidly in all simulations with bilayers predominantly composed of anionic lipids (POPC:POPG 1:3). In contrast, neutral lipids (pure POPC) alone allow for only transient binding of the protein.
The low pH T-domain model shows two predominant membrane-associated conformations B1 and B2 in bilayers composed of POPC:POPG 1:3 (see Fig. 6 and results for description). B1 is the most frequently observed membrane bound conformation, which forms stable protein-membrane contacts at residues of the partially unfolded helices TH1-2 and the loop between TH3-4 (see Fig. 6A). The conformation B2 has more protein-membrane interface contacts at residues of helices TH3, TH4-5, TH6-7 and is stabilized by hydrophobic interactions with the lipid tails (see Fig. 6B). Furthermore, B2 shows partially unfolded N-terminal helices exposed to the solvent. However, conformations B1 and B2 have a common binding surface composed by residues in the N-terminus of TH8 and C-terminus of TH9. Both conformations have similar orientations of helical axes of TH8-9, which are near parallel or at an oblique angle relative to the membrane plane. Finally, some trajectories show residues of TH2 or residues of the loop between TH2-3 as initial protein-membrane contacts. The neutral pH T-domain structure also repeatedly binds to anionic bilayers (POPC:POPG 1:3); however, no binding of the T-domain in neutral pH solution has been observed in experiments.
The above paragraph suggests that both neutral and low pH coarse-grained models bind to bilayers of large anionic fraction (POPC:POPG 1:3). However, the binding of the neutral pH T-domain model is probably due to the underlying simplifications of the coarse-grained force-field, which are described in the following paragraphs. Note that the low pH T-domain model forms larger number of protein-membrane contacts than those observed in the neutral pH model (see Fig. 3). The difference of protein-membrane contacts is the result of pre-refolding or formation of a membrane-competent state of the low pH T-domain model in explicit solvent (Kurnikov et al., 2013). This model is in qualitative agreement with the formation of a membrane-competent state of the T-domain in solution, which facilitates membrane-binding (Kyrychenko et al., 2009).
B1, B2 and a neutral pH T-domain bound conformation show H322 as a common protein-membrane contact in bilayers composed of POPC:POPG 1:3. It is possible that changes of protonation state of H322 can affect the formation and the orientation of these membrane bound conformations. Indeed, experiments have determined that mutations of H322 result in a misfolded state of the T-domain in membranes (Vargas-Uribe et al., 2013a).
CG-MD simulations of neutral and low pH T-domain structures and bilayers with low anionic content (POPC:POPG 3:1) resulted in similar membrane-bound conformations, namely, three of ten simulations resulted in formation of the long-lived protein-lipid complexes. For example, these conformations show similar protein-membrane interactions of cationic residues located in the protein terminals and residues near the N-terminus of the helix TH8 (residues H323 and E326). Note that the neutral pH T-domain structure has histidines in their neutral state, resulting in a decreased penetration in the membrane interface compared to the low pH T-domain structure. At the same time the membrane-associated neutral pH structure has a slightly larger fraction of its terminal cationic residues embedded between lipid head-groups (see Fig. S7). This observation suggests that the membrane association of T-domain with bilayers containing a small fraction of anionic POPG phospholipids depends on specific distribution of positive charge on the protein. Protonation of all histidines not only decreases the overall charge of the T-domain from −10 electron units to −4, letting it to insert deeper into a negatively charged bilayer, but also re-distributes the positive charges on the protein, thus promoting a slightly different orientation at the lipid interface.
Following the initial association of T-domain with the bilayer, helices TH8 and TH9 may require up to 1 μs to stabilize (e.g. see trajectory shown in black lines in Figs. 2F, S1F, and S2F). Note that the time-scales in the coarse-grained simulations are faster than those observed in atomistic simulations because, in part, of the lower viscosity of the simulated media (Marrink et al., 2007). A typical factor of 4 is used to scale time in such simulations (this factor corresponds to the ratio of the diffusion constant of the coarse-grained water relative to the real water). Using this scaling factor we estimate that the effective time-scale of the protein orientational dynamics in the membrane interface was within the 2.4 μs – 6 μs range (see stabilization of TH8 in Fig. S1F). Overall, these simulations indicate microsecond time-scales for the T-domain membrane association and reorientation in the membrane interface.
Equilibrium properties of the low pH T-domain structure as a function of the insertion depth in the bilayer (POPC:POPG 1:3) are best characterized using potential of mean force calculations. The estimated free energy of binding is about −11 kcal/mol. It is known that coarse-grained models over-estimate partitioning energies of peptides in lipid bilayers compared to atomistic models see e.g. (Wee, Ulmschneider & Sansom, 2010). Therefore, future atomistic modeling simulations based on the equilibrated CG models of the T-domain bound to the anionic bilayer may provide partition coefficient estimates quantitatively comparable to experimental values. The interfacial partitioning, which experimentally can be studied only for the membranes with lower content of anionic lipids (e.g., 10% POPG, Fig. 8) gives the estimates of approximately ΔGx = −7 kcal/mol. However, this value might be slightly underestimated due to potential aggregation of the protein in acidic solution (Palchevskyy et al., 2006; Posokhov et al., 2008a; Rodnin et al., 2008). It would be reasonable to assume that the free energy for interfacial partitioning for the 75% POPG LUV composition is somewhat higher than 7 kcal/mol but lower than the transmembrane insertion energy reported at 12 kcal/mol (Posokhov et al., 2008a; Vargas-Uribe et al., 2014). Indeed, a more accurate estimate for the partitioning of the T-domain into LUV composed of POPC:POPG 9:1 is −8.2 kcal/mol (Vargas-Uribe et al., 2014).
Based on our previous atomistic modeling of the membrane competent state of T-domain in explicit solvent followed by the present membrane binding study, we can propose the following mechanism of the initial stages of membrane association of T-domain. First, protonation of histidines induces solvent exposure of hydrophobic residues of the helixes TH8 N-terminus and TH9 C-terminus and a partial unfolding/refolding of N-terminal helices, which facilitates disruption of the inter-helical salt-bridges (Flores-Canales et al.; Kurnikov et al., 2013). Secondly, the low pH T-domain structure binds to the membrane with conformation B1, which is guided by electrostatic interactions between the exposed cationic residues located in the partially unfolded N-terminal helixes, and the anionic lipids. Note that the deeper inserted state B2 is only sampled by a single free CG-MD trajectory, which is probably due to its non-preferable transition state. However, at equilibrium, B2 is favored by hydrophobic interactions with the lipid and simultaneously a solvent exposure of the N-terminal helices. The CG-MD simulations of the low pH protein structure with histidines set in their neutral state provide further indication that conformation B1 is favored by protonation of histidines. The rate or probability of formation of B2 in our unrestrained CG-MD simulations were not affected by protonation of histidines, but was driven by conformational switching of the protein in solution. The predicted membrane-associated states share some similarities with reported pH-dependent conformational states of the T-domain by neutron reflectometry and solid state NMR experiments (Chenal et al., 2009). For example, the predicted conformation B2 has similar features to those observed in a protein bound state in pH 6 solution. Similarities such as solvation of the N-terminal helices and the near parallel orientation of the C-terminal helices relative to the bilayer plane (see Fig. 6B and Figs. S1F, S2F).
One of the questions that can be addressed by the comparison of the computational and experimental data is whether the intermediate state trapped at low content of anionic lipids (Fig. 8) is similar to the interfacial intermediates B1 and B2. As illustrated in Fig. 7, TH9 is not a part of the lipid-interacting interface, which is not surprising as it contains a negatively charged residue E362, which would disfavor the interaction with both negatively charged headgroups and hydrophobic core. Indeed, the experimental evidence collected with site-directed mutagenesis indicates that acidic residues located in the hydrophobic hairpin formed by TH8-9 also play a role in the process of the membrane association of T-Domain (Ghatak, Vargas-Uribe, Rodnin, Ladokhin, manuscript in preparation). We conclude that the B1 and B2 conformations represent early interfacial intermediates, formed prior to loss of negative charge on E362 which would also explain why in experiments we don’t see evidence for strong membrane interactions with 25% POPG composition. In experiments, conformations B1 and B2 are likely to represent early interfacial states that are kinetically populated prior to the state designated as I-state in Fig. 3 of a recent review (Ladokhin, 2013). Additional protonation in the membrane interface will trigger the formation of the insertion competent state (denominated I+-state), followed by the transmembrane insertion of helices TH8-9. In future studies, we will explore the role of protonation of the acidic side-chains in the predicted membrane bound conformations of T-domain.
The presented CG-MD simulations are possibly affected by several simplifications in the MARTINI force field. This force field is limited by the number and beads size representing the chemical groups and the simplified treatment of electrostatic interactions. The latter are limited to interactions between charged beads in the protein and lipid head-groups. As a result, the CG-MD simulations may not accurately model the binding dependency of the T-domain with anionic content in bilayers.
During the course of the CG-MD simulations, structures of the neutral and low pH T-domain models were restrained in order to model the initial membrane association of the protein. In this initial process, it is assumed that the protein structure will not change dramatically. Atomistic MD simulations of the membrane bound conformations B1 and B2 showed that the protein retains its overall structure over several microseconds (manuscript in preparation).
CONCLUSIONS
In summary, this work presents a coarse-grained modeling of the membrane association of T-domain models generated for neutral and mildly acidic pH. We show that T-domain associates more rapidly and steadily to bilayers as the fraction of anionic lipids increases. In particular, our simulations show that the low pH T-domain model binds deeply in the membrane interface of predominantly anionic bilayers and forms extensive protein-membrane contacts. Furthermore, combined approaches of unrestrained and umbrella sampling simulations suggest two membrane-associated states of the low pH T-domain structure, which are characterized by distinctive patterns of electrostatic and hydrophobic interactions between the protein and the bilayer. Based on these results, we propose an initial membrane association pathway of T-domain. This study supports previous findings of the role of protonation of histidines in the formation of a refolded state of T-domain in solution with properties of a membrane competent state. Our results indicate the micro-second time-scales are involved in the membrane insertion process of proteins of a few hundred of residues such as the T-domain. In future work, we will refine the identified membrane bound conformations by atomistic MD simulations. The role of neutralization of acidic side-chains in the membrane-associated states of T-domain will be also studied.
METHODS
1. Coarse-grained Models and Unrestrained CG-MD Simulations
Two coarse-grained models of T-domain were constructed using atomistic structures for the membrane incompetent and membrane competent states of T-domain, which are populated at neutral and mildly acidic pH, respectively (Kurnikov et al., 2013). The neutral pH T-domain model was built from a high resolution structure of diphtheria toxin at pH 7.5 (PDB ID code: 1F0L) and histidines were set to a neutral state (see Fig. 1A) (Steere, 2001). The mildly acidic pH (subsequently referred as low pH) T-domain model was constructed from the last MD generated structure of a 6.8 μs long all-atom MD simulation of a refolded state of T-domain, which was triggered by protonation of histidine side-chains (see Fig. 1B) (Kurnikov et al., 2013). The suggested conformational rearrangement of the all-atom T-domain model was supported by experimental observations (Kurnikov et al., 2013). All of the six histidines were also set to protonated state in the coarse-grained model of the low pH T-domain structure. All glutamate and aspartate side-chains were set in their standard protonation states in both structural models. Notice that the coarse-graining approach is a serial multi-scale modeling of the initial membrane association of the T-domain. MARTINI v2.1 force field was used to model both the protein and the lipid in this work (Monticelli et al., 2008). In order to maintain the tertiary structure of the CG protein models, an elastic network model ElneDyn was applied with a force constant 10 kJ mol−1 Å−2 and a cutoff radius of 9 Å (Periole et al., 2009).
To study T-domain membrane association with bilayers, our protein model was placed 100 Å away from an equilibrated lipid bilayer and solvated by approximately 11000 CG water beads. Preformed bilayers were composed of 256 phospholipids with different mixtures of anionic and neutral phospholipids and the total charge of the system was neutralized with sodium counterions. This computational setup corresponds to a low ionic strength of solution in experiments. We performed five independent MD runs for each T-domain folded state and lipid bilayers composed of 1-palmitoyl-2-oleoyl-phosphatidyl-choline (POPC) and 1-palmitoyl-2-oleoyl-phosphatidyl-glycerol (POPG) with the following ratios: POPC, POPC:POPG 3:1, and POPC:POPG 1:3. In total, 30 × 2 μs long CG-MD simulations were performed. In the following discussion, such equilibrium MD simulations performed using a typical protocol and in the absence of any biases will be referred to as unrestrained MD.
2. Potential of Mean Force Calculations
Umbrella sampling (US) calculations were performed to obtain biased distributions along a reaction coordinate z, which is the distance between the center of mass (COM) of the protein and a bilayer along the bilayer normal axis. The potential of mean force w(z) along the reaction coordinate z is defined:
| (Eq. 1) |
| (Eq. 2) |
where w(z*) and z* are defined as arbitrary, ρ(z) is the distribution function along z, R are the coordinates of the system, z′ (R) is a function that describes z as a function of R, U(R) is the potential energy as a function of R, kB is the Boltzmann’s constant, T is the temperature (Roux, 1995).
Three different initial orientations of the membrane competent state model of T-domain were set every 1 Å within the range of 32 Å to 58 Å along the reaction coordinate. Thus, a total of 81 restrained simulations were performed. To insert the protein in the pre-formed bilayer, the protein position and orientation were restrained relative to the membrane plane and protein atoms were gradually re-grown from the center of mass to their final positions in the bilayer interface. This growth process was carried out over ten stages. At each stage the protein atom coordinates were scaled by a factor and the whole system energy was minimized. The scaling factors were linearly scaled from 0.1 to 1.0. The protein and a pre-equilibrated bilayer containing 512 phospholipids were solvated by approximately 19800 CG water beads. The protein tertiary structure was restrained by an elastic network as indicated in subsection 2.1. Afterwards, neutralizing counterions were added. Each MD trajectory was equilibrated over 50 ps with time step of 1 fs with positional restraints applied to the entire protein, followed by 400 ns with time step of 20 fs with positional restraints on the protein backbone atoms. All positional restraints were removed for production simulations of length 800 ns and a time step of 20 fs. Total simulation time for PMF calculations accounts for a total of 64.8 μs. A harmonic restraint was applied to the center of mass distance between the protein and the bilayer with a force constant of K = 10 kJ mol−1 Å−2. The COM of the bilayer is calculated by using those lipids inside a cylinder of 38 Å. The weighted contribution of lipids was switched off between 33 Å and 38 Å. This cylinder was centered at the protein and was aligned in a parallel orientation relative to the membrane normal axis. MD frames were saved every 100 ps and position restraints data was saved every 1 ps. We constructed the PMF curves by combining and unbiasing the restrained CG-MD simulations using the weighted histogram analysis method (WHAM) (Kumar et al., 1992), as implemented in g_wham (Hub, de Groot & van der Spoel, 2010). Data was extracted from the last 600 ns of each restrained CG-MD trajectory. The standard deviations were calculated using the Bayesian bootstrap of complete histograms. The binding constant was calculated from the PMF curve as
| (Eq. 3) |
where w(z) = 0 is assumed for the unbound macrostate as described in detail by (Vivcharuk et al., 2008). The integral over the bound macrostate goes from l′ = 30 Å to l = 51 Å. The lower bound was chosen to study the initial membrane binding of T-domain in the bilayer-water interface. The upper bound was selected such that for z larger than l, the protein was free to reorient in the solvent. Integrals are approximated using the trapezoidal rule and the error is estimated from standard deviations along the PMF curve.
3. Simulation Protocols and Analysis
All CG-MD simulations were performed using GROMACS 4.5.3 (Hess et al., 2008). A semi-isotropic NPT ensemble was used for all simulations with Berendsen thermostat and barostat set to a reference of T = 310 K and P = 1 bar, respectively. The following constants were set to τT = 1 ps, τP = 1 ps and a compressibility value of 3.0 × 10−5 bar−1. The Coulomb interactions were shifted to zero between 0 and 12 Å and the Lennard-Jones interactions between 9 to 12 Å. The neighbor list was updated every 5 step with a cutoff radius of 14 Å. The dielectric constant was set to 15. Each protein-lipid system was first equilibrated for 50 ps with a time step of 1 fs and with the protein restrained in its position in space. This was followed by a 5 ns equilibration simulation with a time step of 20 fs and the protein still restricted in space. The protein was allowed to move freely in the production simulations that followed equilibration. A production trajectory was saved every 100 ps. Trajectory analysis was performed using GROMACS analysis tools and in-house scripts; visualization was performed using VMD 1.9.1 (Humphrey, Dalke & Schulten, 1996).
To analyze orientations of representative membrane bound conformations of T-domain, we computed the normalized density profiles of coarse-grained atoms as a function of their distance from the bilayer COM along the membrane normal axis. Density profiles were calculated over the last 500 ns of an unrestrained trajectory. Histograms of protein-lipids contacts were calculated over the last 200 ns of an unrestrained trajectory and the last 600 ns of a biased US trajectory. Coarse-grained atoms separated by a distance lower than 5 Å are considered to be in contact. The distance (Z) between the COM of the protein and a bilayer was measured along the bilayer normal axis for all unrestrained CG-MD simulations.
4. Experimental procedures
Diphtheria toxin T domain (amino acids 202–378) was cloned into NdeI-EcoRI-treated pET15b vector containing an N-terminal 6xHis-tag for purification (Rodnin et al., 2008), followed by site-directed mutagenesis to substitute specific residues to cysteine for site-directed labeling. The cysteine mutations were confirmed by DNA sequencing. The expression and purification of the proteins were performed using the previously described procedure (Rodnin et al., 2008; Zhan et al., 1994). Labeling with monoBrBimane was performed using a standard procedure for thiol-reactive derivatives (Haugland, 1996; Kyrychenko et al., 2009; Vargas-Uribe, Rodnin & Ladokhin, 2013b). LysoUB-labeled large unilamellar vesicles (LUV) were prepared by the addition of 2% of LysoUB to the lipid mixture during the LUV preparation. LUV (0.1μm of diameter) were composed of a molar mixture POPG:POPC of 1:9 and prepared by extrusion as previously described (Hope et al., 1986; Mayer, Hope & Cullis, 1986).
Bimane fluorescence quenching was followed by steady-state fluorescence using a SPEX Fluorolog FL3-22 steady-state fluorescence spectrometer (Jobin Yvon, Edison, NJ) equipped with double-grating excitation and emission monochromators. The cuvette, of dimensions 2×10 mm, was oriented perpendicular to the excitation beam and maintained at 25 °C using a Peltier device from Quantum Northwest (Spokane, WA). Emission spectra were collected in 1 nm steps from 400 nm to 700 nm using an excitation wavelength of 380 nm (slits were 3 nm for excitation and emission). The sample was acidified with a small aliquot of acetic buffer to reach pH 6, and the lipid titration was performed adding small amounts of LysoUB LUV to the mixture with 30 min of incubation for equilibration. The quenching of Bimane was quantified using the emission intensity at 470 nm in the absence of LUV as F0 and in the presence of LUV as F. The data was plotted as F0/F as function of the lipid concentration and fitted as previously described (Ladokhin, 2009) to obtain the partitioning coefficient Kx and the total change in intensity I. The free energy of partitioning was calculated as −kBTln(Kx).
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health grant GM-069783. Computational work was performed under XSEDE grant TGMCB040051N.
ABBREVIATIONS
- T-domain
Diphtheria Toxin Translocation domain
- CG
Coarse-grained
- MD
Molecular dynamics
- POPC
1-palmitoyl-2-oleoyl-phosphatidyl-choline
- POPG
1-palmitoyl-2-oleoyl-phosphatidyl-glycerol
- COM
Center of mass
- PMF
Potential of mean force
- K
Binding constant
- Kx
Partitioning coefficient
- ΔGx
Partitioning free energy
- LUV
Large unilamellar vesicles
- W+
Membrane competent state in low pH solution
References
- Bennett MJ, Eisenberg D. Refined structure of monomeric diphtheria toxin at 2.3 A resolution. Protein Sci. 1994;3:1464–75. doi: 10.1002/pro.5560030912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenal A, Prongidi-Fix L, Perier A, Aisenbrey C, Vernier G, Lambotte S, Haertlein M, Dauvergne MT, Fragneto G, Bechinger B, Gillet D, Forge V, Ferrand M. Deciphering membrane insertion of the diphtheria toxin T domain by specular neutron reflectometry and solid-state NMR spectroscopy. J Mol Biol. 2009;391:872–83. doi: 10.1016/j.jmb.2009.06.061. [DOI] [PubMed] [Google Scholar]
- Chenal A, Savarin P, Nizard P, Guillain F, Gillet D, Forge V. Membrane protein insertion regulated by bringing electrostatic and hydrophobic interactions into play. A case study with the translocation domain of diphtheria toxin. J Biol Chem. 2002;277:43425–32. doi: 10.1074/jbc.M204148200. [DOI] [PubMed] [Google Scholar]
- Choe S, Bennett MJ, Fujii G, Curmi PM, Kantardjieff KA, Collier RJ, Eisenberg D. The crystal structure of diphtheria toxin. Nature. 1992;357:216–22. doi: 10.1038/357216a0. [DOI] [PubMed] [Google Scholar]
- Flores-Canales JC, Simakov NA, Kurnikova M. In Preparation. [Google Scholar]
- Haugland RP. Handbook of Fluorescent Probes and Research Chemicals. Molecular Probes, Inc; Eugene, OR: 1996. [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Honjo T, Nishizuka Y, Hayaishi O. Diphtheria toxin-dependent adenosine diphosphate ribosylation of aminoacyl transferase II and inhibition of protein synthesis. J Biol Chem. 1968;243:3553–5. [PubMed] [Google Scholar]
- Hope MJ, Bally MB, Mayer LD, Janoff AS, Cullis PR. Generation of multilamellar and unilamellar phospholipid vesicles. Chem Phys Lipids. 1986;40:89–107. [Google Scholar]
- Hub JS, de Groot BL, van der Spoel D. g_wham-A Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. Journal of Chemical Theory and Computation. 2010;6:3713–3720. [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. Journal of Molecular Graphics & Modelling. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Kalli AC, Campbell ID, Sansom MS. Multiscale simulations suggest a mechanism for integrin inside-out activation. Proc Natl Acad Sci U S A. 2011;108:11890–5. doi: 10.1073/pnas.1104505108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli AC, Wegener KL, Goult BT, Anthis NJ, Campbell ID, Sansom MSP. The Structure of the Talin/Integrin Complex at a Lipid Bilayer: An NMR and MD Simulation Study. Structure. 2010;18:1280–1288. doi: 10.1016/j.str.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules .1. The Method. Journal of Computational Chemistry. 1992;13:1011–1021. [Google Scholar]
- Kurnikov IV, Kyrychenko A, Flores-Canales JC, Rodnin MV, Simakov N, Vargas-Uribe M, Posokhov YO, Kurnikova M, Ladokhin AS. pH-Triggered Conformational Switching of the Diphtheria Toxin T-Domain: The Roles of N-Terminal Histidines. J Mol Biol. 2013;425:2752–64. doi: 10.1016/j.jmb.2013.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrychenko A, Posokhov YO, Rodnin MV, Ladokhin AS. Kinetic Intermediate Reveals Staggered pH-Dependent Transitions along the Membrane Insertion Pathway of the Diphtheria Toxin T-Domain. Biochemistry. 2009;48:7584–7594. doi: 10.1021/bi9009264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladokhin AS. Fluorescence spectroscopy in thermodynamic and kinetic analysis of pH-dependent membrane protein insertion. Methods Enzymol. 2009;466:19–42. doi: 10.1016/S0076-6879(09)66002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladokhin AS. pH-Triggered Conformational Switching along the Membrane Insertion Pathway of the Diphtheria Toxin T-Domain. Toxins (Basel) 2013;5:1362–80. doi: 10.3390/toxins5081362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladokhin AS, Isas JM, Haigler HT, White SH. Determining the membrane topology of proteins: Insertion pathway of a transmembrane helix of annexin 12. Biochemistry. 2002;41:13617–13626. doi: 10.1021/bi0264418. [DOI] [PubMed] [Google Scholar]
- Ladokhin AS, Legmann R, Collier RJ, White SH. Reversible refolding of the diphtheria toxin T-domain on lipid membranes. Biochemistry. 2004;43:7451–8. doi: 10.1021/bi036157w. [DOI] [PubMed] [Google Scholar]
- Leka O, Vallese F, Pirazzini M, Berto P, Montecucco C, Zanotti G. Diphtheria toxin conformational switching at acidic pH. FEBS J. 2014;281:2115–22. doi: 10.1111/febs.12783. [DOI] [PubMed] [Google Scholar]
- Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B. 2007;111:7812–24. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Marrink SJ, Tieleman DP. Perspective on the Martini model. Chem Soc Rev. 2013;42:6801–22. doi: 10.1039/c3cs60093a. [DOI] [PubMed] [Google Scholar]
- Mayer LD, Hope MJ, Cullis PR. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim Biophys Acta. 1986;858:161–168. doi: 10.1016/0005-2736(86)90302-0. [DOI] [PubMed] [Google Scholar]
- Montagner C, Perier A, Pichard S, Vernier G, Menez A, Gillet D, Forge V, Chenal A. Behavior of the N-terminal helices of the diphtheria toxin T domain during the successive steps of membrane interaction. Biochemistry. 2007;46:1878–87. doi: 10.1021/bi602381z. [DOI] [PubMed] [Google Scholar]
- Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink SJ. The MARTINI coarse-grained force field: Extension to proteins. Journal of Chemical Theory and Computation. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- Murphy JR. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins (Basel) 2011;3:294–308. doi: 10.3390/toxins3030294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palchevskyy SS, Posokhov YO, Olivier B, Popot JL, Pucci B, Ladokhin AS. Chaperoning of insertion of membrane proteins into lipid bilayers by hemifluorinated surfactants: application to diphtheria toxin. Biochemistry. 2006;45:2629–35. doi: 10.1021/bi052257l. [DOI] [PubMed] [Google Scholar]
- Perier A, Chassaing A, Raffestin S, Pichard S, Masella M, Menez A, Forge V, Chenal A, Gillet D. Concerted protonation of key histidines triggers membrane interaction of the diphtheria toxin T domain. J Biol Chem. 2007;282:24239–45. doi: 10.1074/jbc.M703392200. [DOI] [PubMed] [Google Scholar]
- Periole X, Cavalli M, Marrink SJ, Ceruso MA. Combining an Elastic Network With a Coarse-Grained Molecular Force Field: Structure, Dynamics, and Intermolecular Recognition. Journal of Chemical Theory and Computation. 2009;5:2531–2543. doi: 10.1021/ct9002114. [DOI] [PubMed] [Google Scholar]
- Posokhov YO, Ladokhin AS. Lifetime fluorescence method for determining membrane topology of proteins. Anal Biochem. 2006;348:87–93. doi: 10.1016/j.ab.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Posokhov YO, Rodnin MV, Das SK, Pucci B, Ladokhin AS. FCS study of the thermodynamics of membrane protein insertion into the lipid bilayer chaperoned by fluorinated surfactants. Biophys J. 2008a;95:L54–6. doi: 10.1529/biophysj.108.141002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posokhov YO, Rodnin MV, Lu L, Ladokhin AS. Membrane insertion pathway of annexin B12: thermodynamic and kinetic characterization by fluorescence correlation spectroscopy and fluorescence quenching. Biochemistry. 2008b;47:5078–5087. doi: 10.1021/bi702223c. [DOI] [PubMed] [Google Scholar]
- Rodnin MV, Kyrychenko A, Kienker P, Sharma O, Posokhov YO, Collier RJ, Finkelstein A, Ladokhin AS. Conformational Switching of the Diphtheria Toxin T Domain. Journal of Molecular Biology. 2010;402:1–7. doi: 10.1016/j.jmb.2010.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnin MV, Kyrychenko A, Kienker P, Sharma O, Vargas-Uribe M, Collier RJ, Finkelstein A, Ladokhin AS. Replacement of C-terminal histidines uncouples membrane insertion and translocation in diphtheria toxin T-domain. Biophys J. 2011;101:L41–3. doi: 10.1016/j.bpj.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnin MV, Posokhov YO, Contino-Pepin C, Brettmann J, Kyrychenko A, Palchevskyy SS, Pucci B, Ladokhin AS. Interactions of fluorinated Surfactants with diphtheria toxin T-domain: Testing new media for studies of membrane proteins. Biophysical Journal. 2008;94:4348–4357. doi: 10.1529/biophysj.107.126235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux B. The Calculation of the Potential of Mean Force Using Computer-Simulations. Computer Physics Communications. 1995;91:275–282. [Google Scholar]
- Stansfeld PJ, Sansom MS. Molecular simulation approaches to membrane proteins. Structure. 2011;19:1562–72. doi: 10.1016/j.str.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Steere B. Characterization of high-order oligomerization and energetics in Diptheria Toxin. University of California; Los Angeles: 2001. [Google Scholar]
- Vargas-Uribe M, Rodnin MV, Kienker P, Finkelstein A, Ladokhin AS. Crucial role of H322 in folding of the diphtheria toxin T-domain into the open-channel state. Biochemistry. 2013a;52:3457–63. doi: 10.1021/bi400249f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Uribe M, Rodnin MV, Ladokhin AS. Comparison of membrane insertion pathways of the apoptotic regulator Bcl-xL and the diphtheria toxin translocation domain. Biochemistry. 2013b;52:7901–7909. doi: 10.1021/bi400926k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Uribe M, Rodnin MV, Ojemalm K, Holgado A, Kyrychenko A, Nilsson I, Posokhov YO, Makhatadze G, Von Heijne G, Ladokhin AS. Thermodynamics of membrane insertion and refolding of the diphtheria toxin T-domain. Journal of Membrane Biology. 2014 doi: 10.1007/s00232-014-9734-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivcharuk V, Tomberli B, Tolokh IS, Gray CG. Prediction of binding free energy for adsorption of antimicrobial peptide lactoferricin B on a POPC membrane. Physical Review E. 2008;77:031913. doi: 10.1103/PhysRevE.77.031913. [DOI] [PubMed] [Google Scholar]
- Wang Y, Malenbaum SE, Kachel K, Zhan H, Collier RJ, London E. Identification of shallow and deep membrane-penetrating forms of diphtheria toxin T domain that are regulated by protein concentration and bilayer width. J Biol Chem. 1997;272:25091–8. doi: 10.1074/jbc.272.40.25091. [DOI] [PubMed] [Google Scholar]
- Wee CL, Ulmschneider MB, Sansom MSP. Membrane/Toxin Interaction Energetics via Serial Multiscale Molecular Dynamics Simulations. Journal of Chemical Theory and Computation. 2010;6:966–976. doi: 10.1021/ct900652s. [DOI] [PubMed] [Google Scholar]
- Zhan HJ, Choe S, Huynh PD, Finkelstein A, Eisenberg D, Collier RJ. Dynamic Transitions of the Transmembrane Domain of Diphtheria-Toxin - Disulfide Trapping and Fluorescence Proximity Studies. Biochemistry. 1994;33:11254–11263. doi: 10.1021/bi00203a022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.