

Recent work by Choi et al. proposes a new protonation site (N7) and associated pKa for one-electron oxidized guanine.1 Oxidation of guanine is the first step toward the production of a whole series of biologically important DNA damage products (e.g., 8-oxo-G) some with mutagenic effects. Various groups have published extensively on these radical species, their resulting products, as well as their significance.2–6 Consequently, studies on characterization and chemistry of the guanine cation radical (G•+ (Figure 1)) are especially important and need careful evaluation.6
Figure 1.
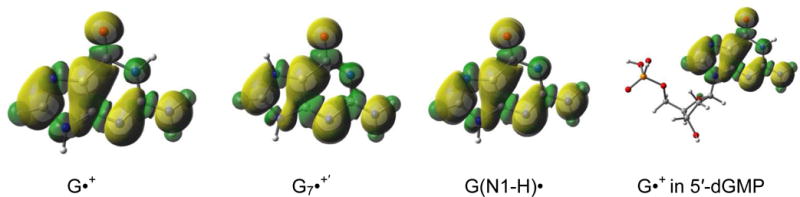
The spin density distribution plots for G•+, G7•+′(protonated at N7), and G(N1-H)• in guanine and that for G•+ in 5′-dGMP obtained by employing B3LYP-PCM/6-31++G(d,p) method. Yellow represents positive spin densities, whereas the green shows regions of small negative spin densities. Numerical spin values are given in the supporting information Table S2. All geometries are fully optimized.
Choi et al. report that the neutral guanyl radical, G(N1-H)• (Figure 1), formed by N1-deprotonation of the guanine cation radical (G•+), undergoes rapid reprotonation (rate constant = 8.1 × 106 s−1) at N7 of the guanine ring to form a new guanine cation radical species, G7•+′ (Figure 1).1 The assignment to G7•+′ is based on time-resolved resonance Raman (TRRR) study.1 According to their interpretation of the TRRR result, G7•+′ shows a C-O stretching mode at 1266 cm−1 owing to a C6-O6 single bond with a localized spin on O6 (i.e., on the exocyclic oxygen at C6).1
Evidence is presented below that the Raman spectral assignments, the localization of spin at O6, and the reprotonation at N7 are in error.
The authors incorrectly associate the 1266 cm−1 resonance they observe with an oxygen-localized radical at O6 which is asserted to be similar in nature to the neutral phenoxyl radical. This is suggested to be the evidence for the single bonded C-O nature of these radical species. According to literature, the 1266 cm−1 resonance is closer to that of the parent phenol C-OH stretch which is reported as 1255 cm−1.7 In fact, the neutral phenoxyl radical is reported to have a 1505 cm−1 C-O stretch in the literature.7 To verify the previous experimental assignments,7 we have calculated the Raman C-O stretching resonances for the phenoxyl radical (1504 cm−1) and parent phenol (1270 cm−1) at the B3LYP-PCM/6-31++G(d,p) level; we report the calculated Raman spectra in the supporting information. The agreement of the Raman C-O stretching resonances between theory and experiment for the phenol and the phenoxyl radical confirms that the 1266 cm−1 resonance cannot be associated with a phenoxyl-type C-O radical.
The question then arises about the Raman spectral assignments of the various one-electron oxidized guanyl species, i.e., G•+, G7•+′ and G(N1-H)•. The authors state that they observe no C6-O stretching mode at >1600 cm−1 in G7•+′ but find a 1266 cm−1 resonance. They suggest that this 1266 cm−1 resonance corresponds to the decrease of the C-O double-bond character. To verify these assignments, we have calculated the Raman modes of various guanyl species at the B3LYP-PCM/6-31++G(d,p) level which are shown in supporting information and discussed below. We first calculated the C6-O Raman stretch for the parent 5′-dGMP and the guanine base itself. We obtain 1716 cm−1 for the parent 5′-dGMP and 1714 cm−1 for the guanine base. We note that the experimental value for the Raman C6-O stretch for the parent 5′-dGMP is 1688 cm−1.1 These results show the guanine base alone is sufficient to verify the C6-O stretch frequency and the theoretical results agree well with experiment. Therefore, employing the guanine base as a model, we have calculated the Raman spectra of G7•+′ as well as of G•+ and G(N1-H)• at the B3LYP-PCM/6-31++G(d,p) level. Our theoretical calculations show that for G7•+′ the C6-O Raman stretch is at 1668 cm−1. The other guanyl radicals considered here using the guanine base model show Raman active C6-O stretches of 1608-1654 cm−1 (G(N1-H)•) and 1747 cm−1 (G•+). We note that G•+ in 5′-dGMP shows the same C6-O stretch frequency of 1747 cm−1 as found for G•+ in the guanine base, again demonstrating no significant effect of the sugar-phosphate group. These results indicate the largely double bonded character of C6-O in all these guanyl radicals.
The authors also argue that the low level semi-empirical INDO calculations by Rakvin et al.8 support spin localization (i.e., localization of the unpaired electron) at O6 in G•+. Actually, the calculations by Ravkin et al. report only ca. 24% spin at O6 and the remainder of the spin density was reported to be spread over the other sites in the guanine base (see supporting information Table S1).8 To clarify this, we have calculated the spin density distributions for each of the guanine radicals G•+, G7•+′, and G(N1-H)• by employing the more reliable B3LYP-PCM/6-31++G(d,p) method. The results are presented in Figure 1 below and the actual numerical values are provided in the supporting information Table S2.
The spin density distribution plots in Figure 1 clearly show that there is no evidence for localization of the spin at O6 in the guanyl radicals (G•+, G7•+′, and G(N1-H)•) and the spin is fully delocalized as is typical for pi-radicals.9 Furthermore, the spin density distribution plots (for numerical spin densities, consult Table S2 in supporting information) show that the protonation site does not significantly affect the spin density distribution. In fact, the deprotonated guanine cation radical (G(N1-H)•) has an almost identical spin distribution to that of G•+ or G7•+′ .The calculated spin density at O6 is 0.13 for G•+, 0.13 for G(N1-H)•, and 0.14 for G7•+′. Addition of the sugar-phosphate to N9 in G•+ (5′-dGMP•+) is found to have no effect on the spin density at O6 which remains at 0.13. These spin density values are, therefore, inconsistent with the authors’ claim of spin localization at O6 in G7•+′, i.e., the species suggested to be formed by reprotonation of G(N1-H)• at N7.
The question may then arise as to the accuracy of the prediction of these spin densities by DFT calculations. For these pi-radicals, the isotropic hyperfine coupling constant (HFCC) of a nucleus is directly proportional to the spin density at the nitrogen atom site or for C-H proton couplings at the carbon pz orbital.10,11 These isotropic HFCC values predicted by DFT employing the B3LYP functional match well with the experimental values (see Table S2, supporting information and also ref. 6, 10, 11). The successful prediction of hyperfine couplings is strong evidence that the spin densities are also well-predicted.
Finally, the question arises whether the guanine cation radical, after its deprotonation from N1, is favored energetically to re-protonate at N7. We have performed calculations at the thermodynamically reliable G4 level as well as at B3LYP-PCM/6-31++G(d,p) for the free energy of G•+ and G7•+′ (see supporting information Table S3). At the G4 level, we find that the structure with protonation at N1, i.e. G•+, is favored by 11.2 kcal/mol (11.4 kcal/mol for B3LYP) over that with protonation at N7, i.e. G7•+′. This 11.2 kcal/mol difference eliminates the possibility of any significant reprotonation of G(N1-H)• at N7. In addition, we have considered the protonation at O6 vs. the protonation at N1. Again, protonation at N1 is favored by 8.4 kcal/mol (B3LYP) over O6. Clearly, the favored site of protonation in the guanine cation radical is at N1 not N7.
In summary, the discussion presented above of Raman C-O modes, spin density distributions and relative stabilities all strongly indicate that the authors have not shown evidence for the formation of G7•+′.
From the response of the authors to our comment, we see that their DFT results reported in their comment agree well with our DFT results. In spite of this, the authors claim that DFT fails to predict the experiment. We note that our highly accurate G4 results agree well with both sets of DFT results. Additionally, in the supporting information our DFT calculations for several CO bond stretches in radicals and parent molecules also agree well with experiment.
Supplementary Material
Acknowledgments
We thank the National Cancer Institute of the National Institutes of Health (Grant R01CA045424) for support.
Footnotes
Supporting Information Available: (i) Theoretical prediction (B3LYP-PCM/6-31++G(d,p)) of Raman spectra of various radicals and parent molecules (geometry optimized); (ii) B3LYP-PCM/6-31++G(d,p) calculated spin densities (Tables S1 and S2) and hyperfine coupling constants of various guanyl radicals (Table S2); (iii) B3LYP-PCM/6-31++G(d,p) and G4-PCM calculated relative stability (ΔG (kcal/mol)) of the guanine cation radicals with protonation at N1, at N7, and O6 (Table S3).
This information is available free of charge via the internet at http://pubs.acs.org/.
References
- 1.Choi J, Yang C, Fujitsuka M, Tojo S, Ihee H, Majima T. Proton Transfer of Guanine Radical Cations Studied by Time-Resolved Resonance Raman Spectroscopy Combined with Pulse Radiolysis. J Phys Chem Lett. 2015;6:5045–5050. doi: 10.1021/acs.jpclett.5b02313. [DOI] [PubMed] [Google Scholar]
- 2.von Sonntag C. Free-radical-induced DNA Damage and Its Repair. Springer-Verlag; Berlin, Heidelberg: 2006. pp. 213–482. [Google Scholar]
- 3.Shukla LI, Adhikary A, Pazdro R, Becker D, Sevilla MD. Formation of 8-oxo-7,8-dihydroguanine-radicals in gamma-irradiated DNA by multiple one-electron oxidations. Nucleic Acids Res. 2004;32:6565–6574. doi: 10.1093/nar/gkh989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenberg MM. The formamidopyrimidines: purine lesions formed in competition with 8-oxopurines from oxidative stress. Acc Chem Res. 2012;45:588–597. doi: 10.1021/ar2002182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fleming AM, Alshykhly O, Zhu J, Muller JG, Burrows CJ. Rates of chemical cleavage of DNA and RNA oligomers containing guanine oxidation products. Chem Res Toxicol. 2015;28:1292–1300. doi: 10.1021/acs.chemrestox.5b00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adhikary A, Kumar A, Becker D, Sevilla MD. The Guanine Cation Radical: Investigation of Deprotonation States by ESR and DFT. J Phys Chem B. 2006;110:24171–24180. doi: 10.1021/jp064361y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tripathi GNR, Schuler RH. The resonance Raman spectrum of phenoxyl radical. J Chem Phys. 1984;81:113–121. [Google Scholar]
- 8.Rakvin B, Herak JN, Voit K, Hüttermann J. Free radicals from single crystals of deoxyguanosine 5′-monophosphate (Na salt) irradiated at low temperatures. Radiat Environ Biophys. 1987;26:1–12. doi: 10.1007/BF01211360. [DOI] [PubMed] [Google Scholar]
- 9.Kumar A, Sevilla MD. π- vs σ-radical states of one-electron-oxidized DNA/RNA bases: a density functional theory study. J Phys Chem B. 2013;117:11623–11632. doi: 10.1021/jp407897n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adhikary A, Kumar A, Becker D, Sevilla MD. Theory and ESR Spectral Studies of DNA-radicals. In: Chatgilialoglu C, Struder A, editors. Encyclopedia of Radicals in Chemistry, Biology and Materials. John Wiley & Sons Ltd; Chichester, UK: 2012. pp. 1371–1396. [Google Scholar]
- 11.Adhikary A, Becker D, Sevilla MD. Electron Spin Resonance of Radicals in Irradiated DNA. In: Lund A, Shiotani, editors. Applications of EPR in radiation research. Springer-Verlag; Berlin, Heidelberg: 2014. pp. 299–352. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.