

Abstract
A challenge in the structure-based design of specificity is modeling the negative states, i.e. the complexes that you do not want to form. This is a difficult problem because mutations predicted to destabilize the negative state might be accommodated by small conformational rearrangements. To overcome this challenge, we employ an iterative strategy that cycles between sequence design and protein docking in order to build up an ensemble of alternative negative state conformations for use in specificity prediction. We have applied our technique to the design of heterodimeric CH3 interfaces in the Fc region of antibodies. Combining computationally- and rationally-designed mutations produced unique designs with heterodimer purities greater than 90%. Asymmetric Fc crystallization was able to resolve the interface mutations; the heterodimer structures confirmed that the interfaces formed as designed. With these CH3 mutations, and those made at the heavy-/light-chain interface, we demonstrate one-step synthesis of four fully IgG bispecific antibodies.
Graphical Abstract
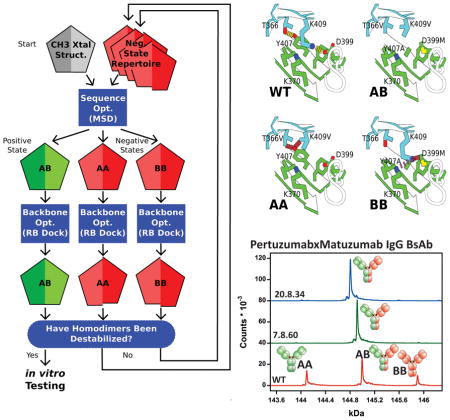
Introduction
Advances in computational protein design have led to a variety of design successes. Protein designers have stabilized existing proteins, designed new proteins de novo, created and manipulated protein complexes, and introduced catalytic active sites into proteins (Pantazes et al., 2011; Stranges and Kuhlman, 2013; Woolfson et al., 2015; Zanghellini, 2014). In these cases, the design problems challenge the designer in similar but slightly different ways: Does the protein have a low energy? Is the interface tightly packed? Does the protein form favorable contacts with the transition state? What these challenges share is a common narrow focus, each allowing the designer to develop a protocol that at any one time concentrates on only a single model of the protein being designed. These protocols often start from a conformation of the protein backbone and then optimize the side-chain identities and rotamers (Ponder and Richards, 1987) on that backbone. Most protocols then allow the backbone to relax in response to the new sequence and may iterate between sequence design and backbone movement (Mandell and Kortemme, 2009). There are other design problems, however, that cannot be solved by modeling a sequence in only a single context.
A specificity design problem, for example, might challenge the design protocol: do the mutations to protein A allow it to bind protein B but prevent it from binding protein C? It involves aspects of both positive design, as the AB interaction must be favored, and negative design (Hecht et al., 1990), as the AC interaction must be disfavored. Multistate design (MSD) (Davey and Chica, 2012), which designs for multiple protein states simultaneously, has proven itself useful in designing a single protein sequence to adopt multiple conformations (Ambroggio and Kuhlman, 2006; Fromer et al., 2009), in understanding what kind of sequences can adopt multiple conformations (Babor et al., 2011; Humphris and Kortemme, 2007; Willis et al., 2013), and in designing specificity such as when designing a protein to bind one target but to avoid another (Ashworth et al., 2010; Grigoryan et al., 2009; Zheng et al., 2014) or when organizing multimeric assemblies (Fallas and Hartgerink, 2012; Havranek and Harbury, 2003; Lewis et al., 2014).
The requirement that a single sequence be shared between multiple states has meant that most MSD protocols are very similar. In an outer loop, they employ a search algorithm to explore sequence space, picking a single sequence in each iteration, and then in an inner loop, they thread that sequence onto each of the states to compute an energy for each one. MSD then aggregates the state energies to compute a fitness for the sequence, and this fitness guides the outer-loop search through sequence space.
Three features distinguish the various MSD efforts: the fitness function, the search algorithm used in the outer loop, and – the focus of this work – the energy function and associated conformational sampling routine used in the inner loop. The choice of energy function is tied intimately to the kind of sampling that is required. For an atomic-resolution energy function, the atomic coordinates have to be predicted, which means that after the new sequence is placed onto the backbone, at a minimum its rotamers have to be optimized (and rotamer optimization is computationally challenging (Pierce and Winfree, 2002)). When negative design is included, MSD will often design collisions into the negative states. However, there are two kinds of collisions: there are “true collisions” which will disrupt a negative state, and there are “false collisions” that can be relaxed away by moving the backbone. The problem for MSD is that, when it looks at a single fixed backbone, it cannot distinguish between true and false collisions. A potential solution to this problem is to use an ensemble of backbones to represent how the complexes might adjust to accommodate mutations. Davey and Chica found that considering many near-native conformations improved ΔΔG predictions (Davey and Chica, 2014). Here, we build on this result by using negative-state repertoires (NSRs) during the sequence optimization process, which involves sampling a large sequence space (~1940, i.e. 19 amino acids considered at 40 residue positions). We apply our method to an important problem in antibody engineering: the design of fully IgG bispecific antibodies (BsAbs).
The most common form of circulating antibodies, IgGs, are homodimers of heterodimers where two heavy chains (HCs) homodimerize with one another and form intermolecular disulfide bonds, and a light chain (LC) binds to each HC with a disulfide bond forming across the HC/LC interface. The HC is made of four domains: the VH, CH1, CH2, and CH3 domains; the LC is made of two domains: VL and CL. A symmetric interface between the two HCs forms between the CH3 domains, and the disulfide bonds between the HCs form in the hinge region between CH1 and CH2. IgGs are often conceptualized as a capital letter “Y,” where the stem (aka Fc) represents the interface between the two HCs (each half stem comprising CH2 and CH3), and the two arms represent the HC/LC portion with each arm capable of binding an antigen with their VH/VL domains.
The alluring fact that each antibody has two arms has drawn many scientists to consider whether a single antibody could be engineered to bind two different antigens. Uses for such BsAbs include cancer and autoimmune therapy (Baeuerle and Reinhardt, 2009; Chan and Carter, 2010; Scott et al., 2012), tumor detection (Barbet et al., 1998), T cell redirection (Lum et al., 2006), and broadly, as affinity reagents to bring two proteins together. IgG antibodies and BsAbs make attractive therapeutics due to their long serum half-lives (mediated by Fc/Neonatal-receptor interactions (Chan and Carter, 2010)), their ability to recruit the patient’s own immune system (mediated by CH2/FcγR interactions (Nimmerjahn and Ravetch, 2008)), and their flexibility to bind a wide variety of targets with exquisite specificity. However, actually constructing IgG BsAbs is not easy. A naïve mixture of two LCs and two HCs will result in the formation of ten different species, with the desired BsAb representing only 1/8th of the desired product (Suresh et al., 1986). To properly orchestrate the desired assembly between an A and B pair of light- and heavy chains in a single cell, LC-A must preferentially bind HC-A and not HC-B and vice versa (the light-chain pairing problem), and HC-A must heterodimerize with HC-B without homodimerizing with itself and vice versa (the heavy-chain pairing problem) (Klein et al., 2012; Spiess et al., 2015).
The heavy-chain pairing problem has been solved previously, through rational design using sterics (Atwell et al., 1997; Ridgway et al., 1996), electrostatics (Gunasekaran et al., 2010; Mimoto et al., 2014), or both (Choi et al., 2013) to disfavor homodimer formation; it has been solved by mixing IgG and IgA residues (Davis et al., 2010); and two groups approached it computationally (von Kreudenstein et al., 2013; Moore et al., 2011). It has not, to our knowledge, been solved using MSD.
This paper presents the use of NSRs in MSD to design CH3 mutations that when refined with additional rationally chosen mutations, produced designs yielding 93% pure heterodimer, comparable to previously published designs. It also includes the first asymmetrically crystallized CH3 interface, allowing direct observation of the mutant residues without averaging between the two chains. Finally, it demonstrates the utility of these CH3 mutations in the one-step generation of BsAbs when combined with Fab mutations that control light-chain pairing.
Results
Negative-State Repertoires
The mpi_msd application in Rosetta implements an MSD protocol that, in its inner loop, threads a sequence on each of several states and performs a fixed-backbone rotamer optimization on each one (Leaver-Fay et al., 2011). By using fixed backbones, the protocol is able to reuse rotamer energies and thus run quickly. However, negative design across protein/protein interfaces often produces false collisions because most collisions across an interface can be resolved by pulling the chains slightly apart or by sheering them. Rigid-body docking would unmask false collisions but docking would be too slow to run in MSD’s inner loop.
To let MSD see through false collisions, we instead use a repertoire of different backbone conformations for the negative states – a negative-state repertoire (NSR). Each sequence for one of the negative species (AA, or BB) is threaded onto each of the backbone conformations in the NSR, and then the lowest energy produced over the set is the one taken for that species. This means a collision that produces a high energy for one backbone conformation but a low energy in another will correctly be seen by MSD as a false collision.
We build each NSR iteratively, running MSD starting from a crystal structure and then docking the output homodimers. If docking identifies a false collision, then we add the backbone conformation that relieved the collision to the NSR (Figure 1). The efficacy of NSRs at weeding out false collisions can be seen in comparing the differences in binding energies for the homodimers as measured before docking and after docking with and without NSRs (Figure S1). The improved agreement in these energies shows the value of NSRs and confirms our previous in silico observations in a different system (Leaver-Fay et al., 2011). Although NSRs do not avoid all false collisions, they improve over previous MSD approaches where negative states were modeled only onto the backbones of existing structures (Havranek and Harbury, 2003), or where the extra backbone conformations were generated using only the wild-type sequence (Allen et al., 2010; Davey and Chica, 2014).
Figure 1. Negative State Repertoires (NSRs).

A) The iterative design process begins by running the multistate design (MSD) sequence optimization executable, feeding in the wild type CH3 homodimer backbone from the 1L6X crystal structure (grey pentagon; each chain of the homodimer is represented by half a pentagon). At its completion, MSD outputs models of the AB heterodimer positive state (green pentagon), and the AA and BB homodimer negative states (red pentagons), threaded onto the backbone of the input structures that produce the lowest-energy. MSD, in trying to destabilize the homodimers, typically introduces collisions across their interfaces; however, these collisions often disappear if the interfaces are allowed to relax. Such false collisions dupe MSD into outputting sub-optimal sequences. Next, backbone optimization through rigid-body docking (RB dock) identifies nearby, low-energy conformations. If the homodimer-backbone conformations it finds (shifted red pentagons) show favorable binding energies, then these structures are added to a NSR for use in subsequent iterations. B) The seven CH3 conformations in the NSR of design 20.8, aligned on chain A (green). Alternate rigid-body orientations of chain B (cyan) show the conformational variability within the space of structures that gave favorable binding energies.
Notation
This paper refers to CH3 mutations using a leading chain identifier; e.g. if chain A includes Y407A, and chain B includes K409V, it will refer to this pair as A_Y407A and B_K409V. Additionally, when describing unfavorable interactions in a homodimer, it will use lowercase letters to denote which side of the interface another residue is on, e.g. in the BB homodimer, B_K409V collides with a_Y407; both residues are on the same “B” species, but the collision is across the interface. The name “7.2” for this design designates it as a member of design family 7. The inverted design, design 7.2−1, swaps 7.2’s chain A and chain B mutations, to produce A_K409V and B_Y407A.
Round 1 Testing
We employed a multi-round design strategy. We sought from MSD a list of small, recurring subsets of mutations that would be unlikely to destabilize the heterodimer, yet would significantly destabilize one of the two homodimers. In the first round of experimental screening, we identified designs that destabilized their intended homodimer without destabilizing the heterodimer, and in the second round, combined those designs to eliminate both homodimers.
Starting from the 1L6X crystal structure of the CH3 interface (Idusogie et al., 2000), we built an NSR, and then performed several hundred MSD trajectories with this NSR followed by rigid-body docking. We examined the designs that produced the greatest binding-energy separation between the heterodimers and the homodimers, pruning away extraneous mutations. Design families were gathered as variations on common sets of mutations. Sixty-six designs, falling into thirteen families (Table S1), were tested experimentally in the first round.
We screened the designs using two assays: 1) a gel-filtration assay and 2) a Förster resonance energy transfer (FRET) assay. The first assay expressed A chain mutations on a full HC (alongside a separate LC), and expressed B chain mutations on an Fc. Purified protein was passed through an Ultra-performance liquid chromatography (UPLC) sizing column, and eluted protein was measured by absorption at 280 nm. We transfected mammalian cells using A:B DNA ratios of either 1:1 or 4:1 (Figure S2A). In the second assay, chain A’s Fc was expressed with an N-terminally fused EGFR domain 3, and chain B’s Fc with an N-terminally fused VEGFR1 domain 3 (Figure S2B). Probing with fluorescently labeled anti-EGFR and anti-VEGFR Fabs gave an approximation to the concentration of each species that qualitatively matched our expectation; WT homodimers showed more FRET signal than those of the positive controls (Figure S2C). The main benefit of using two assays was to filter out designs that performed well in one assay and poorly in the other; we favored those designs that performed well in both.
Of the thirteen families, eleven contained members that shifted the observed homodimer:heterodimer ratio in the intended direction, though some more effectively than others (Table S2). The majority of the designs tested in this first round (37 of 66) performed well in at least one of the two assays; however, we could not carry all of them forward into round 2. Several of the designs gave nearly 90% heterodimer when expressed at a 1:1 ratio, in particular, those from families 7 and 11, whose mutations are shown in Figure 2.
Figure 2. CH3 Interface Designs.

A) Cartoons of the residues at the CH3 interface. B) Design 7.8 and how its mutations destabilize the two homodimers (red dashes = collision; purple dashes = electrostatic repulsion). Design 7.4 differs from design 7.8 only in the absence of A_D399M. Design 7.7 differs from design 7.8 only in having A_K409I instead of A_K409V. C) Design 11.2. Collisions between B_E357D and a_Y349 and between B_S364Q and a_K370 are predicted to destabilize the BB homodimer; the AA homodimer was not predicted to be destabilized.
We sought and obtained crystal structures for nine of these designs to determine the extent of structural rearrangement induced by the mutations. Previous structures of CH3 heterodimers, however, did not resolve their interfaces. Although the center of each CH3 interface is asymmetric, the rest of the Fc is not, so the crystal lattice formed without placing the A and B chains consistently. As a result, the residues at the interface were averaged, rendering the most intriguing part of the structure invisible (Elliott et al., 2014; Gunasekaran et al., 2010; Strop et al., 2012). We sought to avoid this averaging by crystallizing the two chains with chain A bound to the Fc-III peptide (DeLano, 2000) at the hinge between CH2 and CH3, but where chain B’s ability to bind Fc-III had been ablated by three mutations: M252E, I253A, & H435A.
With this asymmetric crystallization strategy, we solved eleven crystal structures of our designed heterodimers (Table S3) and two of the previously published heterodimers: the knobs-into-holes (KH) design (Atwell et al., 1997), PDB ID 5DI8, and Amgen’s charge swap design (DD-KK) (Gunasekaran et al., 2010), PDB ID 5DK2. The CH3 interfaces showed very little structural rearrangement so that the crystal structures very closely resembled both the design models and the starting crystal structure. The RMSDs between the crystal structures and the design models for the backbone heavy-atoms of the 60 interface residues (see Supplemental Methods) ranged between 0.24 and 0.33 Å, with a mean of 0.27 Å. Of the 20 non-alanine, non-glycine mutations in the nine crystal structures, Rosetta correctly predicted both the χ1 and χ2 dihedrals to within 20° for 14 of them. Figures 3 and 4 compare the design models and crystal structures for two of the more important designs to emerge from this round, designs 7.8 and 11.2 (PDB IDs 5DJZ and 5DJ0).
Figure 3. 1.9Å Crystal structure of design 7.8 (green=chain A & cyan=chain B) compared against the design model (gray).

PDB ID 5DJZ. Rosetta mispredicted the rotamer adopted by A_D399M, but even the crystal rotamer would be close enough to b_K409 to collide, either pushing it unfavorably towards a_K370, or pushing it away from aK370 but still disrupting the solvation shell surrounding b_K409. In any case, A_D399M consistently increased heterodimer purity when paired with other family 7 mutations. Rosetta correctly identified the rotamers for B_T366V and B_K409V, which both adopt the most commonly seen β-sheet rotamer. Surprisingly, the backbone for B_T366V moved away from the center of the interface by 0.5Å, enlarging the cavity left by A_Y407A, which is incompletely filled by B_K409V. The interface-residue heavy-atom RMSD between the two structures is 0.86 Å.
Figure 4. Design Model vs 2.28Å Crystal Structure of Design 11.2.

PDB ID 5DJ0. The crystal structure of design 11.2 (green: chain A; cyan: chain B) showed an unexpected rearrangement with the Cα-Cβ bond vector of A_K370Y changing by 7°. This is accompanied by a 0.1Å movement of its Cα, and together these motions swing the tyrosine’s terminal hydroxyl by 1.4Å from its predicted location; though it still adopted the predicted rotamer, the contact it formed with B_E357D was quite different. A_K370Y’s movement was accompanied by a comparatively large movement of the B-chain helix from residues 354 to 359, where the backbone atoms moved by about 0.5Å. This helix showed the greatest variability within our crystal structures. The motion of this helix allowed B_E357D to form an ideal hydrogen bond to A_K370Y where the donor hydrogen lies in the plane of the phenol ring and also in the sp2 plane of the carboxylate group, and furthermore the CG-OD1 - - OH angle is 118°. The design model for 11.2 (gray) contained a hydrogen bond between this pair, but the geometry was significantly worse. The interface-residue, heavy-atom RMSD between the two structures is 0.65 Å.
As a final test before advancing designs into round 2, we performed differential scanning calorimetry (DSC) to determine the mutations’ effect on stability (Table S2). Most of the designs tested decreased the midpoint of thermal unfolding (Tm) of the CH3 domain by about 12° C which appeared to be linked to the mutation of K409, though a handful produced destabilizations in the range between 3 and 8° C. None of the designs resulted in an increased melting temperature. Twenty four designs were selected for combination in round 2.
Round 2 Testing
We combined pairs of designs selected from round 1 and screened them computationally by running rigid-body docking on the three dimers. From these simulations, we selected an additional 44 combinations for testing in the UPLC and FRET assays. Surprisingly, very few of the design combinations formed heterodimers with purities we had anticipated. Four combinations of round 1 designs showed high heterodimer yields and seemed promising (Table S4); repeated assays showed that design 20.8 (composed from designs 11.2 & 7.4−1) expressed well with the greatest consistency, so we looked to refine this design.
Design refinement
We first attempted to refine design 20.8. We grafted 7.4−1’s mutations onto the crystal structure of design 11.2 to generate a model of 20.8 and fed this model back into MSD to look for additional mutations to improve the total energy of the heterodimer while continuing to disfavor the homodimers. We synthesized an additional 24 designs (Table S5). Of these additional designs, most showed worsened heterodimer purity. Rosetta had suggested a reversion to wild type at A_K409 (A_K409V in the parental 7.4−1 design), which it predicted would improve the total energy of the heterodimer; however, all of the designs that contained this mutation displayed a significant increase in the formation of the AA homodimer, indicating that the collision between A_K409V and b_Y407 was critical in preventing homodimer formation.
After obtaining the crystal structure of 20.8 (PDB ID 5DJY), we rationally designed three more mutations: a) A_T366M (A_T366V in the parental 7.4−1 design) to fill much of the cavity vacated by B_Y407A, b) B_E356G to relieve tension between A_Y349S and b_E356, and c) B_S364R (B_S364Q in the parental 11.2 design) to repel a_K370 in the BB homodimer. These mutations lead to designs 20.8.34, which included A_T366M and B_E356G, and 20.8.37, which included A_T366M and B_S364R. We were able to obtain a crystal structure of 20.8.34 (Figure 5, PDB ID 5DK0), which very closely resembled the crystal structures of 20.8 and 11.2 before it. We also obtained crystal structures of the AA and BB homodimers for 20.8.37 (Figure S3, Table S6). These structures very closely resemble the recently published KH homodimers (Elliott et al., 2014) where the CH2 and CH3 are paired in a head-to-tail orientation. Because those homodimers were expressed in E. coli, it was not clear whether the CH2/CH3 interface was real or an artifact of missing CH2-domain glycosylation. As our homodimers were expressed in mammalian cells, this head-to-tail arrangement appears to be real. We also observed this head-to-tail arrangement in our UPLC assay in the formation of a “long dimer” (Figure 6), with the long dimer forming even for WT.
Figure 5. Comparison of design 20.8.34’s crystal structure with WT.

The crystal structure of design 20.8.34 (chain A: green, chain B: cyan) is aligned by its interface residues with the WT crystal structure 1L6X (grey). A) The mutations from 7.4−1 and A_T366M. The rotamer adopted by A_T366M helps explain why Rosetta never predicted this mutation to improve 20.8: at −148°, its χ2 angle is 30° off from the nearest rotamer Rosetta would have sampled. This conformation appears to relieve the collision it would otherwise have had with B_L351. Additionally, the A_T366M’s Cα-Cβ bond vector swings by 4 degrees, and Cα shifts 0.2A, both motions separating the methionine’s sulfur from BL351. Rosetta did however suggest T366M in design 6.1, the crystal structure of which matched the design model closely (Figure S5). B) The mutations from 11.2 and B_E356G. The largest changes in this structure were seen in the small helix from 355 to 358 on the B chain so B_E357D’s side chain ended up unresolved in the structure and seems to no longer contact A_K370Y. The interface-residue, heavy-atom RMSD between the two structures is Å.
Figure 6. Comparison of our best designs against positive controls.

The UPLC experiments in 1:1 and 4:1 A:B transfection ratios allowed observation of several species. The wild type (WT) sequence showed four prominent peaks corresponding, from left to right, to the AA homodimer, the AB heterodimer, and two BB homodimer peaks. Visible to the left of the AA homodimer peak in the 4:1 experiment is a trace amount of an AA “long dimer” (inset), a species formed by a head-to-tail arrangement brought about by a CH2/CH3 interface which we and others have observed in several crystal structures. This species would project its variable domains in opposite directions, decreasing its elution time. After extinction coefficient normalization, the WT experiments showed an average AA:AB:BB ratio of 1.2:2:0.6. The deviation from the theoretical 1:2:1 ratio is likely due to the full heavy chain (the A chain) expressing better than the Fc alone (the B chain). Designs 20.8.34 and 20.8.37, though they improved upon 20.8, both showed accumulation of a left shoulder (pink) on the AB heterodimer elution peak. We were not able to isolate this species, but believe it to be related to the AB heterodimer. A similar shoulder is also observed in the 4:1 assay of the DD-KK control. Curves: hexp – the experimentally observed absorption, sum – the sum of the fitted-curve heights, and individual peaks (earlier to later): AAl3 – AAl1 (the three AA longdimer peaks), AA, ABsh (the unidentified left shoulder on the AB peak), AB, A monomer, Aplz (proteolized A monomer), BB1 & BB2 (the two BB homodimer peaks), and B monomer.
In parallel, we explored refinements on design 7.8 from round 1. Design 7.8 showed high heterodimer purity but still formed more BB homodimer than desired, which was especially apparent in the 4:1 UPLC experiments of 7.8−1 (Figure S4). We manually searched for mutations that could introduce an unfavorable electrostatic interaction between the BB partners, and identified a mutation, B_Q347R, that could be paired with A_K360D to form a salt bridge across the heterodimeric interface; B_Q347R in the BB homodimer, however, would interact unfavorably with the wild type a_K360. We modeled these mutations in Rosetta using the FastRelax protocol (Khatib et al., 2011) and found that B_Q347R could escape its unfavorable interaction with a_K360 by forming a salt bridge with the neighboring b_E345; Rosetta predicted that B_E345R would prevent B_Q347R’s escape. The design containing these mutations, 7.8.60, showed decreased BB homodimer formation by UPLC. In the 1:1 assay of 7.8.60−1, the formation of the standard AA dimer decreased to 0.4%, and the formation of the AA-long dimer also decreased compared to 7.8−1.
We compared our designs against a set of existing CH3 heterodimer designs: the KH design, the DD-KK charge-swap design, and the sterics-based design published by Zymeworks (ZW1) (von Kreudenstein et al., 2013). In the UPLC assays, these three designs formed heterodimers with between 91 and 94% purity (Table 1, Figure 6), whereas our design 7.8.60 achieved 93% purity. The DD-KK, KH, and ZW1 designs also showed significant AA homodimer formation in the 4:1::A:B assay in both the regular and inverted forms, with the exception of the (non-inverted) ZW1 design, which formed very little.
Table 1. Summary of our top designs.
The WT control shows that both the UPLC and FRET experiments underreport the amount of BB homodimer formed, compared to the theoretical 25% if both the A and B chains expressed equally well. The percentages reported for the UPLC experiments represent the fraction of the dimeric species; free monomer species are not included in the tally, as they could be more easily purified away and/or adjusted by changing the transfection ratio of the two chains. Standard errors are reported in parentheses for experiments performed more than once. In the 1:1 UPLC experiments, our best designs, 7.8.60 and 20.8.34, perform roughly as well as the positive controls (KH, DD-KK, and ZW1). For each design, only chain A mutations are given, but looking at each design and its inverse gives mutations for both chains.
| DSC | UPLC 1:1 | UPLC 4:1 | FRET 1:1 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Design | A Chain Muts | N | Tm | N | AAL | AA | AB | BB | N | AAL | AA | AB | BB | N | AA | AB | BB |
| WT | - | 1 | 82.9 | 12 | 1.6 (0.5) | 31.3 (1.7) | 51.3 (0.6) | 15.9 (1.5) | 3 | 2.3 (0.7) | 65.0 (2.3) | 27.4 (0.9) | 2.9 (0.3) | 13 | 24.2 (1.4) | 61.2 (1.5) | 14.6 (2.5) |
| DD-KK | K409D K392D | 1 | 68.8 | 2 | 1.4 (0.5) | 0.5 (0.2) | 91.7 (0.4) | 6.3 (0.2) | 6 | 3.6 (0.6) | 19.9 (3.3) | 37.5 (4.6) | 2.6 (0.4) | 4 | 2.3 (0.5) | 96.2 (1.0) | 1.6 (0.6) |
| DD-KK−1 | D399K E356K | 2 | 1.8 (0.1) | 1.2 (0.4) | 93.6 (0.2) | 3.3 (0.1) | 2 | 9.6 (1.3) | 11.3 (1.2) | 29.4 (2.3) | 3.5 (0.6) | ||||||
| KH | T366W | 1 | 69.9 | 4 | 3.2 (0.8) | 0.8 (0.2) | 92.4 (1.0) | 3.6 (0.2) | 3 | 14.2 (3.5) | 15.7 (4.0) | 35.0 (7.5) | 2.2 (1.0) | 4 | 2.0 (0.2) | 96.2 (0.7) | 1.7 (0.6) |
| KH−1 | T366S L358V Y407A | 2 | 2.6 (0.2) | 0.8 (0.1) | 94.1 (0.2) | 2.4 (0.1) | 1 | 6.4 | 4.2 | 33.1 | 6.5 | ||||||
| ZW1 | T350V T366L K392L T394W | 1 | 81.2 | 3 | 1.3 (0.4) | 0.6 (0.3) | 94.3 (0.7) | 3.8 (0.4) | 3 | 2.1 (0.8) | 5.1 (2.5) | 18.3 (0.7) | 3.5 (1.3) | 2 | 3.5 (0.2) | 94.8 (0.1) | 1.6 (0.3) |
| ZW1−1 | T350V L351Y F405A Y407V | 2 | 2.4 (0.7) | 1.4 (0.4) | 87.6 (5.2) | 8.6 (5.4) | 2 | 8.6 (2.0) | 9.0 (1.6) | 25.6 (1.3) | 19.4* (18.0) | ||||||
| 7.8 | D399M Y407A | 1 | 71 | 6 | 1.5 (0.2) | 0.5 (0.1) | 89.9 (1.9) | 8.2 (1.9) | 1 | 4.9 (2.6) | 9.4 (3.7) | 42.3 (8.1) | 1.9 (0.5) | 4 | 5.8 (2.1) | 89.0 (2.0) | 5.5 (1.9) |
| 7.8−1 | T366V K409V | 6 | 3.8 (0.4) | 4.1 (1.2) | 88.6 (1.0) | 3.5 (0.4) | 2 | 24.1 (14.6) | 28.6 (7.6) | 26.9 (6.1) | 2.4 (0.6) | ||||||
| 7.8.60 | K360D D399M Y407A | 1 | 70.4 | 1 | 3.9 | 2.3 | 92.9 | 0.9 | 1 | 9.8 | 16.8 | 46.2 | 0.8 | ||||
| 7.8.60−1 | E345R Q347R T366V K409V | 3 | 2.0 (0.3) | 0.4 (0.0) | 93.3 (1.3) | 4.3 (1.4) | 2 | 5.4 (3.6) | 26.4 (6.0) | 32.5 (4.9) | 3.2 (0.4) | ||||||
| 20.8 | Y349S K370Y T366V K409V | 4 | 70.0 | 6 | 4.0 (1.1) | 2.2 (1.0) | 84.9 (1.4) | 6.0 (2.0) | 1 | 1 | 8.4 | 11.7 | 12 | 5 | 3 (2.3) | 93.3 (3.2) | 3.7 (1) |
| 20.8−1 | E357D S364Q Y407A | 2 | 0.2 (0.1) | 0.3 (0.1) | 92.5 (0.1) | 7.0 (0.0) | 1 | 1 | 3.4 | 4.7 | 38.6* | ||||||
| 20.8.34 | Y349S K370Y T366M K409V | 1 | 68.9 | 4 | 3.0 (0.4) | 1.3 (0.2) | 83.8 (3.5) | 3.5 (0.8) | 3 | 9.5 | 12.0 | 23.6 | 30.3* | 4 | 2.3 (0.7) | 93.9 (1.5) | 3.7 (1.6) |
| 20.8.34−1 | E356G E357D S364Q Y407A | 1 | 1.3 | 1.2 | 94.2 | 3.3 | 6 | 3.8 | 4.3 | 31.8 | 3.8 | ||||||
| 20.8.37 | Y349S K370Y T366M K409V | 1 | 68.9 | 2 | 2.3 (0.3) | 0.2 (0.2) | 83.5 (1.7) | 4.8 (1.3) | 1 | 7.9 | 11.9 | 34.7 | 1.5 | ||||
| 20.8.37−1 | E357D S364R Y407A | 1 | 2.3 | 0.7 | 85.5 | 4.6 | 4 | 10.4 | 1.8 | 29.9 | 3.6 | ||||||
In some 4:1 UPLC assays, a prominent peak appeared where the second of the two BB homodimer peaks ought to have been; this likely represents a proteolyzed A monomer species whose peak has shifted significantly later.
BsAb Production
We tested how well our CH3 designs correctly assembled into IgG BsAbs in mammalian cells (the norm for antibody manufacture). We chose four different IgG1 mAbs – Pertuzumab (anti-HER-2), Matuzumab (anti-EGFR), BHA10 (anti-LTβR) and MetMAb (anti-cMet) – and paired them to form four possible BsAbs: Pertuzumab with BHA10, Pertuzumab with Matuzumab, MetMab with BHA10, and MetMab with Matuzumab (Figure 7, Table S7). We combined our previously published orthogonal Fab interface mutations (Lewis et al., 2014) with designs 7.8.60 and with 20.8.34 and compared their BsAb assembly against designs with a wild-type CH3 sequence. Each of the four putative BsAbs were expressed transiently in 293F cells, affinity purified, and characterized for assembly using liquid chromatography with in-line mass spectrometry (LCMS). LCMS gave a readout on the percentage of correctly assembled BsAb and the percentage and identity of misassembled byproducts. Both 20.8.34 and 7.8.60 designs formed very high purity BsAbs, each averaging 93% purity over the four target BsAbs. In three of the four cases, at least one of the two designs formed > 95% pure BsAb. Our previous work showed the LCMS experiment accurately predicted BsAb binding behavior (Lewis et al., 2014).
Figure 7. LCMS Traces for four BsAbs constructed using designs 20.8.34 and 7.8.60, compared against the WT CH3 sequence.

All three sequences relied on our previously reported orthogonal Fab mutations (Lewis et al., 2014). Design 20.8.34 yielded 90.3, 96.9, 89.0, and 95.8% of the correctly formed BsAb, whereas design 7.8.60 yielded 95.2, 98.1, 89.3, and 90.8% of the correctly formed BsAb (See Table S7).
Discussion
This paper has presented a new technique for performing negative design at protein/protein interfaces – that of using negative-state repertoires. The extra fixed-backbone conformations mimicked real docking trajectories and allowed MSD to better see the structural effect of a mutation on a negative state. As a result, eleven of the thirteen design families tested in the first round destabilized their intended homodimer. Our top computationally designed heterodimers achieved between 80 and 90% pure heterodimer. To make them comparable to other published CH3 heterodimers, however, required additional rationally designed mutations based on X-ray structures to minimize residual homodimer formation. This paper has also presented a novel crystallization technique that was able to resolve the asymmetry at the CH3 interface; the crystal structures of our heterodimers matched the design models closely, and the structures of the previously-published KH and DD-KK heterodimers represent the first fully-resolved view of their interfaces.
It is likely that allowing slight internal backbone flexibility would further improve MSD. Though the use of NSRs mimics some degree of flexibility – rigid-body flexibility – the NSRs do not incorporate internal-geometry flexibility and there is evidence that this flexibility is valuable. The crystal structures for 11.2 and 20.8.34, for instance, both showed sufficient variability in their Cα-Cβ bond vectors to fit larger residues than otherwise appeared possible or to form better hydrogen bonds with polar residues. However, naïvely opening up backbone flexibility without aggressively sampling backbone conformation space is unlikely to improve predictions since the space is enormous and the energy landscape is rugged: the noise arising from energy minima in regions far away from the CH3 interface would likely overpower any signal from new low-energy sequences. Aggressive sampling, however, would dramatically increase running time, which is already long. Perhaps incorporating additional low-energy backbone conformations that are held fixed throughout simulation could sufficiently mimic internal backbone flexibility (Davey and Chica, 2014).
Since the 1980s, the idea of producing fully IgG BsAbs has been a paramount goal for those in the field of antibody engineering (Milstein and Cuello, 1983). Chimeric or humanized/fully human IgG antibodies have demonstrated their utility in the treatment of many different diseases with >40 approved and 100s in clinical trials (Ecker et al., 2015) and their advantageous properties (stability, solubility, manufacturability, pharmacokinetics) are well documented. Fully IgG BsAbs should maintain these attributes, which may pose an advantage over non-native BsAb formats such as tandem scFvs, diabodies, and even IgG-like formats such as IgG-scFvs and dual variable domain Igs (DVD-Igs) (Spiess et al., 2015). However, fully IgG BsAbs have been challenging to manufacture compared to some of the antibody fragment (Fv, scFv, or diabody) approaches due to the complex heterotetrameric assembly of the native IgG architecture. Nonetheless, several new strategies have been described in the past few years for generating IgG BsAbs either through post expression/purification biochemical methods (Labrijn et al., 2013; Spiess et al., 2013; Strop et al., 2012) or by the direct expression of correctly assembled IgG BsAbs (Bostrom et al., 2009; Lewis et al., 2014; Liu et al., 2015; Mazor et al., 2015; Schaefer et al., 2011). Here we demonstrate the ability to express and assemble fully IgG BsAbs using a single cell mammalian process amenable for transfer to standard industry manufacturing protocols using both Fab designs (Lewis et al., 2014) and CH3 heterodimer designs generated exclusively by our MSD design processes.
Experimental Procedures
Computational Methods
MSD (sequence optimization) and rigid-body docking (backbone optimization) were iterated between to build up an NSR (Figure 1). Conformations for the homodimers generated by docking with a large difference in binding energies as measured by MSD and by the InterfaceAnalyzer application following docking were gathered. Those conformations that by eye appeared dissimilar to other conformations from the same iteration were chosen for the the next iteration’s NSR. Prior to the first iteration, rigid-body docking was performed on the 1L6X crystal structure so that the homodimer conformations that comprised the NSR would not be drastically lower in energy than the conformation used for the positive state. The lowest energy structure of 20 docking trajectories was then used in subsequent MSD simulations; from the 1L6X crystal structure, this docked structure had a heavy-atom RMSD of 0.315Å over all four domains, and a backbone heavy-atom RMSD of 0.069Å over the CH3 interface residues.
UPLC
For each design, three plasmids (0.25 μg MetMab heavy chain + 0.25 μg Fc + 1.5 μg MetMab light chain) were transiently transfected into 2mL of HEK293F cells. Transfected cells were grown at 37 °C in a 5% CO2 incubator while shaking at 125 rpm for 5 days. Secreted protein was harvested by centrifugation at 2K rpm for 5 min and the supernatant recovered. Antibody was protein-G purified. Eluted samples were neutralized with 1M Tris pH9.0 (Sigma) and filtered with an Ultrafree-MC-GV centrifugal filter (Millipore). A 30 μL sample was added to Waters UPLC tube, from which 10 μL is injected into a Waters Acquity UPLC with a BEH200 SEC column, equilibrated in PBS and run at 0.3 mL/min. A dilution series of purified MetMab was also run as a standard. Peaks from the UPLC traces were deconvoluted and integrated using a custom set of Octave scripts. Molar percentages were computed by normalizing each species by its predicted extinction coefficient.
FRET
Chain A’s plasmid contained an Fc appended to EGFR; chain B’s plasmid contained an Fc appended to VEGFR1. For protein production, the two plasmids were transfected (1:1) into HEK293F cells using Freestyle transfection reagents (Life Technologies). Transfectants were grown and secreted protein harvested as described above. Supernatants were purified using 2 μm filters.
Europium(Eu)-labeled MF1 (anti-mVEGFR1 D3) or Eu-labeled Matuzumab FAb (anti-hEGFR D3) were mixed with Cy5-labeled MF1 or Cy5-labeled Matuzumab Fab to determine heterodimer and homodimer ratios (Figure S2). 96-1/2 well microtiter plates (black from Costar) were incubated for approximately 30 minutes at room temperature. Fluorescence measurements were carried out on a Wallac Envision 2103 Multilabel Reader with a dual mirror (PerkinElmer Life Sciences) with the laser excitation of the Europium at wavelength at 340 nm and the emission filters Europium 615 and APC 665. Delay between excitation and emission was 20μs.
DSC
A- and B-chain mutations were incorporated into plasmids containing Fc and HA-tagged FCs. These plasmids were used to transfect cells as above, and secreted protein was harvested as above. Supernatants were passed through 2 μm filters for purification. Purification was performed using protein A chromatography (Lewis et al., 2014). DSC measurements were carried out as described previously (Clark et al., 2014) with the exception that the scan rate was 1.5 deg. C/min. All DSC thermograms were fit using analysis software provided by the manufacturer (GE Healthcare).
Crystallography
The designs’ A chains were cloned into a standard-length IgG1 Fc construct. Their B chains contained an N-terminal Histidine tag (8xHis) and the three Fc-III related mutations. Plasmids harboring the chain A and chain B DNA sequences were transfected into cells, the cells grown, and protein harvested as above. Supernatants were passed through 2 μm filters. Purification was performed using a two-step Protein A/His-tag process.
The purified proteins were screened using vapor diffusion in 96-well format (Intelli-plates, Art Robins Instrument) using commercially available screens: PEGs, PEGs II, ComPAS, Classics, Classics II Suites (Qiagen). A Phoenix robot (Art Robins Instrument) performed the initial setup, combining 0.3 μL of protein with 0.3 μL of well solution. When necessary, crystal growth was optimized with additional screens based on the best conditions observed in the commercial screens. Optimization screens utilized streak seeding from the original crystals.
The structure of the first asymmetrically crystallized heterodimer was solved by molecular replacement using the published homodimeric Fc complex with the Fc-III peptide (PDB ID 1DN2) as a search model in PHASER (McCoy et al., 2007). The resulting solution with only one bound Fc-III peptide was refined using xtalview/xfit and the ccp4 family of programs (Winn et al., 2011). Subsequent structures utilized the most similar previously solved structure for molecular replacement and were similarly refined.
LCMS
To generate IgG BsAb protein, four plasmids, each containing either an HC or an LC from two separate MAbs, were transfected into cells, the cells grown, and the protein harvested as above. The cell culture supernatants were collected and passed through 0.2 μm filters. The supernatants were purified, prepared, and analyzed by high pressure liquid chromatography/mass spectrometry (LCMS) as described previously (Lewis et al., 2014), except that the proteins were enzymatically deglycosylated after purification and neutralization to approximately pH 8.0 using 1 M Tris, pH 8.5–9.0. Proteins were deglycosylated by the addition of 1 μL N-Glycanase (Prozyme) for 3–14 hrs at 37 °C prior to being submitted for LCMS.
Supplementary Material
Highlights.
Protein specificity design improves with the use of repertoires of negative states.
This paper presents several novel CH3 heterodimers, resulting from multistate design.
A novel xtal technique revealed the heterodimeric interfaces, otherwise unresolved.
LCMS showed 2 CH3 interfaces both averaged 93% purity across 4 bispecific antibodies.
Acknowledgments
We would like to thank Mr. Benjamin Gutierrez for assistance with transient transfections. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility.
Footnotes
Author Contributions
ALF & BK designed and ran computational experiments; KJF, HA, & SHD made the constructs; FH & RY purified the proteins; KJF & SJD designed the FRET assay; KJF performed FRET assays; HA designed the UPLC assay; HA, KJF, & SHD performed the UPLC assays; SA & ALF deconvoluted the UPLC traces; SA & AKC designed the asymmetric crystallization technique; SA, AP, & FL crystallized the structures; SJD & JRF performed LCMS experiments; SJD analyzed the LCMS data; SA, HA, KJF, SD, BK, and ALF suggested rational mutations; ALF, SA, KJF, SJD and BK wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Allen BD, Nisthal A, Mayo SL. Experimental library screening demonstrates the successful application of computational protein design to large structural ensembles. Proc Natl Acad Sci. 2010;107:19838–19843. doi: 10.1073/pnas.1012985107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambroggio XI, Kuhlman B. Computational Design of a Single Amino Acid Sequence that Can Switch between Two Distinct Protein Folds. J Am Chem Soc. 2006;128:1154–1161. doi: 10.1021/ja054718w. [DOI] [PubMed] [Google Scholar]
- Ashworth J, Taylor GK, Havranek JJ, Quadri SA, Stoddard BL, Baker D. Computational reprogramming of homing endonuclease specificity at multiple adjacent base pairs. Nucleic Acids Res. 2010;38:5601–5608. doi: 10.1093/nar/gkq283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwell S, Ridgway JBB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol. 1997;270:26–35. doi: 10.1006/jmbi.1997.1116. [DOI] [PubMed] [Google Scholar]
- Babor M, Mandell DJ, Kortemme T. Assessment of flexible backbone protein design methods for sequence library prediction in the therapeutic antibody herceptin--HER2 interface. Protein Sci. 2011;20:1082–1089. doi: 10.1002/pro.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009;69:4941–4944. doi: 10.1158/0008-5472.CAN-09-0547. [DOI] [PubMed] [Google Scholar]
- Barbet J, Peltier P, Bardet S, Vuillez JP, Bachelot I, Denet S, Olivier P, Leccia F, Corcuff B, Huglo D, et al. Radioimmunodetection of medullary thyroid carcinoma using indium-111 bivalent hapten and anti-CEA x anti-DTPA-indium bispecific antibody. J Nucl Med Off Publ Soc Nucl Med. 1998;39:1172–1178. [PubMed] [Google Scholar]
- Bostrom J, Yu SF, Kan D, Appleton BA, Lee CV, Billeci K, Man W, Peale F, Ross S, Wiesmann C, et al. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science. 2009;323:1610–1614. doi: 10.1126/science.1165480. [DOI] [PubMed] [Google Scholar]
- Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10:301–316. doi: 10.1038/nri2761. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Kim YJ, Lee S, Kim YS. A heterodimeric Fc-based bispecific antibody simultaneously targeting VEGFR-2 and Met exhibits potent antitumor activity. Mol Cancer Ther. 2013;12:2748–2759. doi: 10.1158/1535-7163.MCT-13-0628. [DOI] [PubMed] [Google Scholar]
- Clark LA, Demarest SJ, Eldredge J, Jarpe MB, Li Y, Simon K, van Vlijmen HWT. Influence of canonical structure determining residues on antibody affinity and stability. J Struct Biol. 2014;185:223–227. doi: 10.1016/j.jsb.2013.08.009. [DOI] [PubMed] [Google Scholar]
- Davey JA, Chica RA. Multistate approaches in computational protein design. Protein Sci. 2012;21:1241–1252. doi: 10.1002/pro.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey JA, Chica RA. Improving the accuracy of protein stability predictions with multistate design using a variety of backbone ensembles. Proteins. 2014;82:771–784. doi: 10.1002/prot.24457. [DOI] [PubMed] [Google Scholar]
- Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo KM, Huston JS. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel. 2010;23:195–202. doi: 10.1093/protein/gzp094. [DOI] [PubMed] [Google Scholar]
- DeLano WL. Convergent Solutions to Binding at a Protein-Protein Interface. Science. 2000;287:1279–1283. doi: 10.1126/science.287.5456.1279. [DOI] [PubMed] [Google Scholar]
- Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs. 2015;7:9–14. doi: 10.4161/19420862.2015.989042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JM, Ultsch M, Lee J, Tong R, Takeda K, Spiess C, Eigenbrot C, Scheer JM. Antiparallel Conformation of Knob and Hole Aglycosylated Half-Antibody Homodimers Is Mediated by a CH2--CH3 Hydrophobic Interaction. J Mol Biol. 2014;426:1947–1957. doi: 10.1016/j.jmb.2014.02.015. [DOI] [PubMed] [Google Scholar]
- Fallas JA, Hartgerink JD. Computational design of self-assembling register-specific collagen heterotrimers. Nat Commun. 2012;3:1087. doi: 10.1038/ncomms2084. [DOI] [PubMed] [Google Scholar]
- Fromer M, Yanover C, Linial M. Design of multispecific protein sequences using probabilistic graphical modeling. Proteins Struct Funct Genet. 2009;78:530–547. doi: 10.1002/prot.22575. [DOI] [PubMed] [Google Scholar]
- Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding proteins. Nature. 2009;458:859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, Ng S, Bin, Born T, Retter M, Manchulenko K, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285:19637–19646. doi: 10.1074/jbc.M110.117382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havranek JJ, Harbury PB. Automated design of specificity in molecular recognition. Nat Struct Biol. 2003;10:45–52. doi: 10.1038/nsb877. [DOI] [PubMed] [Google Scholar]
- Hecht MH, Richardson JS, Richardson DC, Ogden RC. De novo design, expression and characterization of {F}elix: a four-helix bundle protein of native-like structure. Science. 1990;249:884–891. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- Humphris EL, Kortemme T. Design of multi-specificity in protein interfaces. PLoS Comput Biol. 2007;3:e164. doi: 10.1371/journal.pcbi.0030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idusogie EE, Presta LG, Gazzano-Santoro H, Totpal K, Wong PY, Ultsch M, Meng YG, Mulkerrin MG. Mapping of the C1q Binding Site on Rituxan, a Chimeric Antibody with a Human IgG1 Fc. J Immunol. 2000;164:4178–4184. doi: 10.4049/jimmunol.164.8.4178. [DOI] [PubMed] [Google Scholar]
- Khatib F, Cooper S, Tyka MD, Xu K, Makedon I, Popović Z, Baker D, Players F. Algorithm discovery by protein folding game players. Proc Natl Acad Sci. 2011;108:18949–18953. doi: 10.1073/pnas.1115898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Sustmann C, Thomas M, Stubenrauch K, Croasdale R, Schanzer J, Brinkmann U, Kettenberger H, Regula JT, Schaefer W. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. MAbs. 2012:653–663. doi: 10.4161/mabs.21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Kreudenstein TS, Escobar E, Lario PI, D’Angelo I, Brault K, Kelly J, Durocher Y, Baardsnes J, Woods JR, Xie MH, et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: Quality by molecular design. MAbs. 2013;5 doi: 10.4161/mabs.25632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrijn AF, Meesters JI, de Goeij BECG, van den Bremer ETJ, Neijssen J, van Kampen MD, Strumane K, Verploegen S, Kundu A, Gramer MJ, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci U S A. 2013;110:5145–5150. doi: 10.1073/pnas.1220145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A, Jacak R, Stranges PB, Kuhlman B. A Generic Program for Multistate Protein Design. PLoS One. 2011;6:e20937. doi: 10.1371/journal.pone.0020937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, Guntas G, Leaver-Fay A, Smith EM, Ho C, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotechnol. 2014;32:191–198. doi: 10.1038/nbt.2797. [DOI] [PubMed] [Google Scholar]
- Liu Z, Leng EC, Gunasekaran K, Pentony M, Shen M, Howard M, Stoops J, Manchulenko K, Razinkov V, Liu H, et al. A Novel Antibody Engineering Strategy for Making Monovalent Bispecific Heterodimeric IgG Antibodies by Electrostatic Steering Mechanism. J Biol Chem. 2015:M114.620260. doi: 10.1074/jbc.M114.620260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum LG, Davol PA, Lee RJ. The new face of bispecific antibodies: targeting cancer and much more. Exp Hematol. 2006;34:1–6. doi: 10.1016/j.exphem.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Mandell DJ, Kortemme T. Backbone flexibility in computational protein design. Curr Opin Biotechnol. 2009;20:420–428. doi: 10.1016/j.copbio.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, Sachsenmeier K, Carlson M, Gadre DV, Borrok MJ, et al. Improving target cell specificity using a novel monovalent bispecific IgG design. 2015 doi: 10.1080/19420862.2015.1007816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein C, Cuello AC. Hybrid hybridomas and their use in immunohistochemistry. Nature. 1983;305:537–540. doi: 10.1038/305537a0. [DOI] [PubMed] [Google Scholar]
- Mimoto F, Kadono S, Katada H, Igawa T, Kamikawa T, Hattori K. Crystal structure of a novel asymmetrically engineered Fc variant with improved affinity for FcγRs. Mol Immunol. 2014;58:132–138. doi: 10.1016/j.molimm.2013.11.017. [DOI] [PubMed] [Google Scholar]
- Moore GL, Bautista C, Pong E, Nguyen DHT, Jacinto J, Eivazi A, Muchhal US, Karki S, Chu SY, Lazar GA. A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. MAbs. 2011;3:546–557. doi: 10.4161/mabs.3.6.18123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- Pantazes RJ, Grisewood MJ, Maranas CD. Recent advances in computational protein design. Curr Opin Struct Biol. 2011;21:467–472. doi: 10.1016/j.sbi.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Pierce NA, Winfree E. Protein design is NP-hard. Protein Eng. 2002;15:779–782. doi: 10.1093/protein/15.10.779. [DOI] [PubMed] [Google Scholar]
- Ponder JW, Richards FM. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- Ridgway JBB, Presta LG, Carter P. “Knobs-into-holes” engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9:617–621. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A. 2011;108:11187–11192. doi: 10.1073/pnas.1019002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AM, Allison JP, Wolchok JD. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012;12 [PMC free article] [PubMed] [Google Scholar]
- Spiess C, Merchant M, Huang A, Zheng Z, Yang NY, Peng J, Ellerman D, Shatz W, Reilly D, Yansura DG, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat Biotechnol. 2013;31:753–758. doi: 10.1038/nbt.2621. [DOI] [PubMed] [Google Scholar]
- Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015 doi: 10.1016/j.molimm.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Stranges PB, Kuhlman B. A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. 2013;22:74–82. doi: 10.1002/pro.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strop P, Ho WH, Boustany LM, Abdiche YN, Lindquist KC, Farias SE, Rickert M, Appah CT, Pascua E, Radcliffe T, et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J Mol Biol. 2012;420:204–219. doi: 10.1016/j.jmb.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Suresh MR, Cuello AC, Milstein C. Immunochemical Techniques Part I: Hybridoma Technology and Monoclonal Antibodies. Elsevier; 1986. [Google Scholar]
- Willis JR, Briney BS, DeLuca SL, Crowe JE, Jr, Meiler J. Human germline antibody gene segments encode polyspecific antibodies. PLoS Comput Biol. 2013;9:e1003045. doi: 10.1371/journal.pcbi.1003045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfson DN, Bartlett GJ, Burton AJ, Heal JW, Niitsu A, Thomson AR, Wood CW. De novo protein design: how do we expand into the universe of possible protein structures? Curr Opin Struct Biol. 2015;33:16–26. doi: 10.1016/j.sbi.2015.05.009. [DOI] [PubMed] [Google Scholar]
- Zanghellini A. de novo computational enzyme design. Curr Opin Biotechnol. 2014;29:132–138. doi: 10.1016/j.copbio.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Zheng F, Jewell H, Fitzpatrick J, Zhang J, Mierke DF, Grigoryan G. Computational Design of Selective Peptides to Discriminate between Similar PDZ Domains in an Oncogenic Pathway. J Mol Biol. 2014 doi: 10.1016/j.jmb.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.