

Abstract
Herein, we report a Zn-ProPhenol catalyzed Mannich reaction using α-branched ketones as nucleophilic partners for the direct enantio- and diastereoselective construction of quaternary carbon stereocenters. The reaction can be run on a gram-scale with a low catalyst loading without impacting its efficiency. Moreover, the Mannich adducts can be further elaborated with complete diastereocontrol to access molecules possessing complex stereotriads.
Graphical abstract
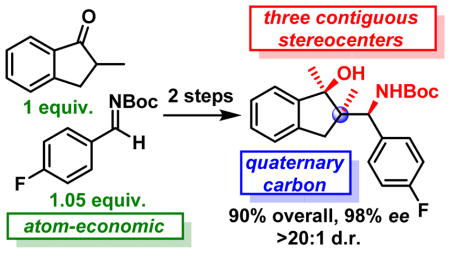
One of the modern challenges for the synthetic chemist is the fast, selective and atom-economic access to molecular complexity starting from simple and widely available starting materials.1 The Mannich reaction is an important tool for C-C bond formation.2 Moreover, it is also one of the most robust ways to produce nitrogen-containing compounds which are ubiquitous in nature. All-carbon quaternary stereocenters are also a common feature of natural products and their construction still represents a synthetic challenge, especially in a catalytic enantioselective fashion.3 In this context, we report herein the first direct asymmetric Mannich reaction that allow the use of α-branched ketone donors. This atom-economic transformation provides an efficient enantio- and diastereoselective access to chiral β-amino ketones decorated with a quaternary carbon stereocenter.
The Mannich reaction was discovered in the early 20th century and has received a lot of attention, especially in the context of the total synthesis of alkaloids.4 Despite its synthetic importance, the first examples of catalytic enantioselective Mannich reaction only appeared in the late 90’s.5 These reactions require preformed enolates such as silyl enol ethers that are reacted with an imine activated by a chiral Lewis acid catalyst. One serious drawback of this approach is the preparation and the instability of the preformed enolates. A more effective and atom-economic approach was later introduced by directly using an unmodified carbonyl donor.6 This strategy really became popular with the advent of organocatalysis.7 Bimetallic catalysis has also emerged as a powerful way to perform direct enantioselective Mannich reactions.8 However, the use of ketones as donors for such reactions is so far limited to methyl ketones, cyclohexanones or α-hydroxyketones.6–9
The formation of α-quaternary carbonyl compounds using a direct Mannich reaction has been limited to the use of easily enolizable ketones10 such as 1,3-dicarbonyl compounds11 or 3-substituted oxindoles12 (Scheme 1a). To the best of our knowledge, the lone exception is a proline-catalyzed reaction using branched aldehydes developed by Barbas and coworkers13 (Scheme 1b). However, this reaction proceeds with poor to moderate diastereoselectivities and is restricted to the use of N-PMP-protected glyoxylate ethyl ester as acceptor. In this respect, we wished to develop the first direct catalytic enantioselective Mannich reaction allowing the use of a broad array of α-branched aromatic ketones together with a broad range of imine acceptors.
Scheme 1.
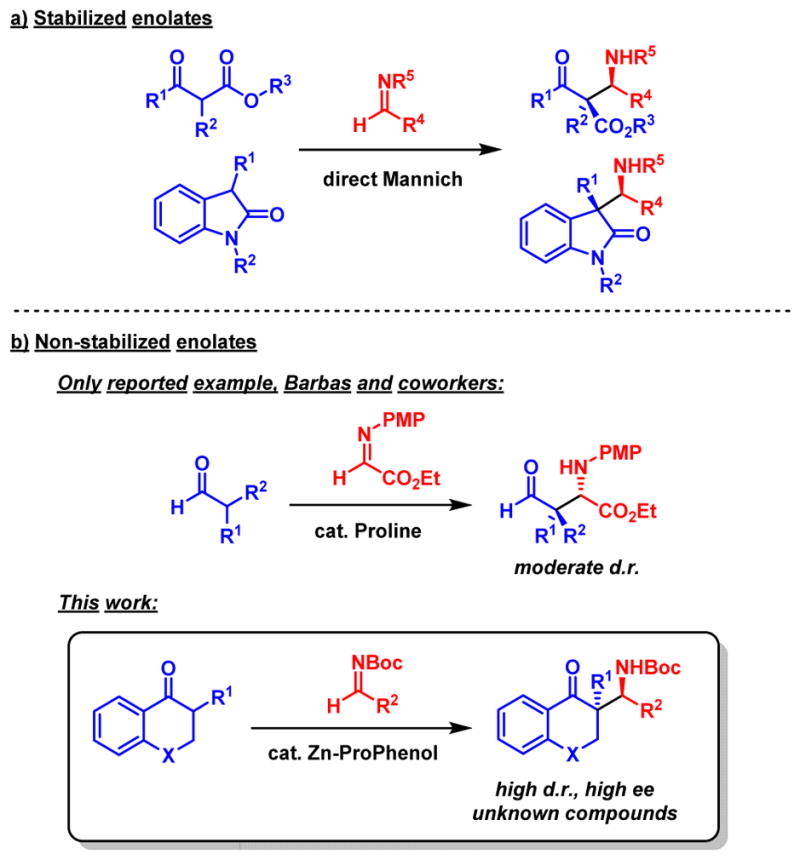
Direct Mannich reactions forming quaternary carbon stereocenters
The ProPhenol ligand is part of the aza-hemicrown family of ligands and forms dinuclear main group metal catalysts when treated with alkyl metal reagents such as Et2Zn. The Zn-ProPhenol catalytic system has proven to be particularly efficient for a range of enantioselective aldol and Mannich reactions.8c However, the scope of donors that is compatible with this catalytic system is mostly limited to methyl ketones (acetophenones, acetone, methyl vinyl ketone, methyl ynones) or unsubstituted α-hydroxy carbonyl compounds. Very recently, we reported the use of more substituted ketones14 for Mannich-type reactions but so far the formation of quaternary stereogenic centers using this strategy has remained elusive.
Our recent work with fluorinated aromatic ketones14a led us to wonder about the special importance of the fluorine atom concerning the reactivity/selectivity for this Mannich reaction. Could α-branched ketones substituted with simple alkyl substituents be suitable for related processes using our Zn-ProPhenol catalytic system? A priori, such alkyl substituents should deactivate the ketone partner for both steric and electronic reasons thus significantly impacting the outcome of the reaction. With these challenges in mind, we initiated our studies with 2-methyl tetralone 1a to explore the feasibility of such a transformation (Table 1). Pleasingly, when 1a was reacted with Boc-protected aldimine 3a in the presence of 20 mol% of the Zn-ProPhenol catalyst in THF at 60°C, the desired Mannich product 4aa was obtained with very promising selectivities albeit in low yield due to a poor conversion (entry 1). When the reaction temperature was raised to 80°C, a better conversion was observed and remarkably, the selectivities were not affected (entry 2). Increasing the concentration of the reaction was beneficial too (entry 3) and the catalyst loading could be reduced to 10 mol% to afford 4aa with only a slightly reduced yield (entry 4). Under similar reaction conditions, 2-methyl indanone 2a proved to be more reactive than 1a (entry 5–7) and 5aa was also obtained with excellent selectivities. Pleasingly, the catalyst loading could even be lowered to 5 mol% without impacting the efficiency of the reaction (entry 8).
Table 1.
Optimization of the reaction conditionsa
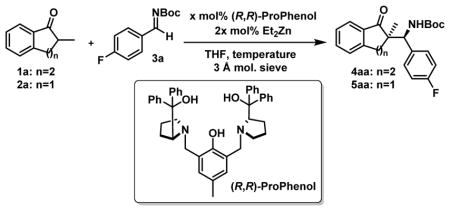
| ||||||
|---|---|---|---|---|---|---|
| entry | 1 | x | [conc.], T | yieldb | d.r.c | eed |
| 1 | 1a | 20 | (0.4 M), 60°C | 28% | >20:1 | 97% |
| 2 | 1a | 20 | (0.4 M), 80°C | 76% | >20:1 | 96% |
| 3e | 1a | 20 | (1.0 M), 80°C | 93% | >20:1 | 99% |
| 4e | 1a | 10 | (1.0 M), 80°C | 75% | >20:1 | 95% |
| 5 | 2a | 20 | (0.4 M), 60°C | 99% | >20:1 | 99% |
| 6 | 2a | 10 | (0.4 M), 60°C | 91% | >20:1 | 99% |
| 7e | 2a | 10 | (0.4 M), 80°C | 99% | >20:1 | 99% |
| 8e | 2a | 5 | (0.4 M), 80°C | 95% | >20:1 | 99% |
Reaction conditions: 0.20 mmol 1a or 2a, 0.24 mmol 3a, x mol% (R,R)-ProPhenol, 2x mol% Et2Zn (1M in hexanes), 3 A molecular sieves (5 mg), in THF for 40 h at the indicated temperature and concentration.
Isolated yield.
Determined by 1H-NMR analysis.
Determined by HPLC on a chiral stationary phase.
Reaction time is 16h.
We selected 2a to evaluate the generality of the reaction concerning the imine partner using our optimized conditions. A variety of aromatic Boc-aldimines were successfully reacted under our optimized reaction conditions to afford highly functionalized β-amino ketones 5ab–5ag (Scheme 2). Both electron-withdrawing and electron-donating groups were tolerated on the aromatic ring as well as heteroaromatic imines. The reaction is not restricted to aromatic imines; vinyl imine 3f and alkyl imine 3g gave the Mannich products 5af and 5ag in high yields and selectivities. Finally, Cbz-protected imines can be used with similar efficiency which allows the use of orthogonal protecting group strategies.
Scheme 2.
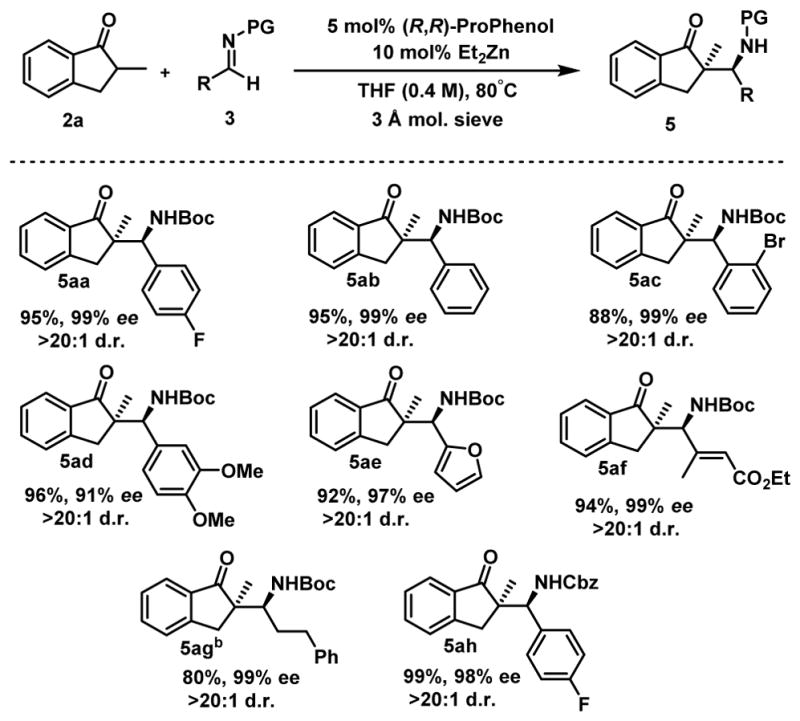
Scope of the reactiona
aReaction conditions: 0.20 mmol 2a, 0.24 mmol 3, 5 mol% (R,R)-ProPhenol, 10 mol% Et2Zn (1M in hexanes), 3 A molecular sieves (5 mg), in THF (0.4 M) at 80°C for 16 h. bReaction run at 70°C.
We then investigated several 5-membered aromatic ketones for this direct Mannich reaction (Scheme 3). Indanones 2a–e which are substituted with linear alkyl groups are competent partners and the Mannich products 5aa–5eh were obtained in good yields together with perfect selectivities. Notably, an ethyl ester and a free terminal alkyne are well tolerated under these reaction conditions. Substitution with a branched alkyl group did not impact the efficiency of the reaction as shown with 5fa. Importantly, even a tert-butyl substituent could be used to afford 5ga possessing two adjacent quaternary carbons. 3-Coumaranones are important heterocyclic compounds that are found in numerous natural products and bioactive compound.15 In this context, we were pleased to see that 3-coumaranone 2h could be used for this direct Mannich reaction.
Scheme 3.
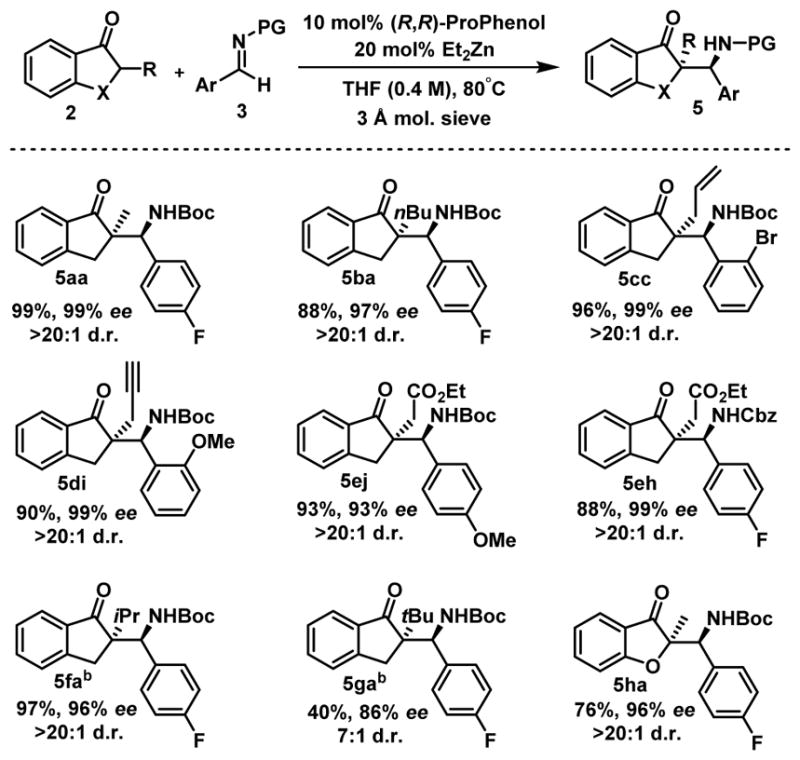
Scope of the reactiona
aReaction conditions: 0.20 mmol 2, 0.24 mmol 3, 10 mol% (R,R)-ProPhenol, 20 mol% Et2Zn (1M in hexanes), 3 A molecular sieves (5 mg), in THF (0.4 M) at 80°C for 16 h. bReaction run in THF (1.0 M).
Moreover, this method was extended to other important aromatic ketones (Scheme 4) such as tetralone 1a–b and 4-chromanone 1c which afforded the Mannich products 4aa–4cf with similar levels of efficiency.16 Thiochromanone 4d, possessing a coordinating sulfur atom that could potentially poison the Zn-catalyst, was also successfully used but in this case the innate diastereoselectivity of the reaction was 2.6:1. However, the two diastereoisomers could be easily separated by chromatography and the major diastereoisomer 4da was isolated in 72% yield together with excellent selectivities. Benzosuberone 4e was also a competent partner for this Mannich reaction. Finally, challenging acyclic substrates were attempted for this reaction. Pleasingly, isobutyrophenone afforded β-amino ketone 4fa in good yield and high enantioselectivity. However, only traces of the Mannich product were detected when the highly hindered phenone 4g was reacted under these reaction conditions, the remainder being unreacted starting material.
Scheme 4.
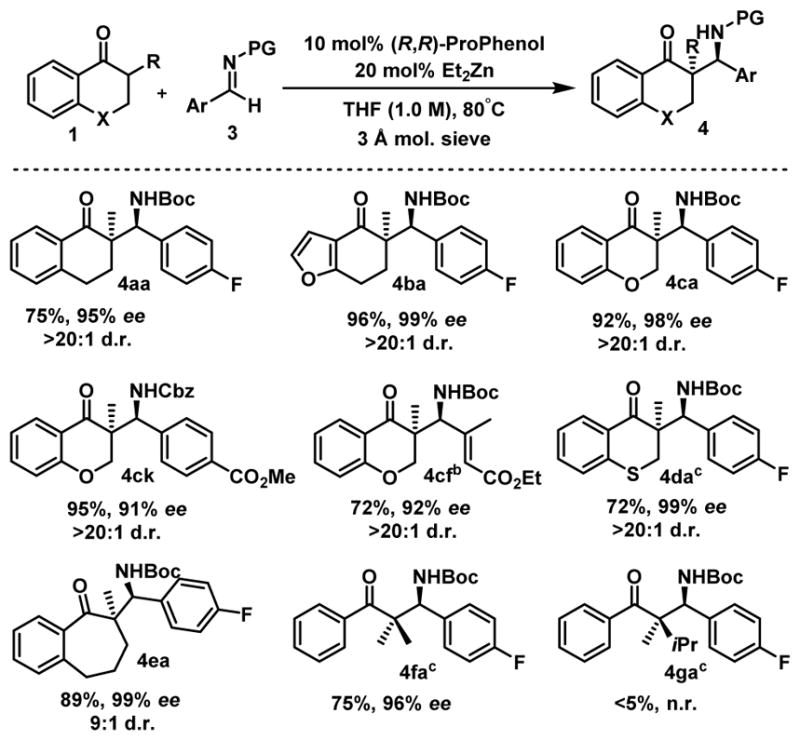
Scope of the reactiona
aReaction conditions: 0.20 mmol 1, 0.24 mmol 3, 10 mol% (R,R)-ProPhenol, 20 mol% Et2Zn (1M in hexanes), 3 A molecular sieves (5 mg), in THF (1.0 M) at 80°C for 16 h. b8:1 d.r. before chromatography. c2.6:1 d.r. before chromatography. cReaction run in Et2O (1.0 M).
To showcase the scalability and the practicality of the process, we performed a gram-scale reaction using 2a and 1.05 equivalent of imine 3a (Scheme 5). Pleasingly, the catalyst loading could be reduced to only 2 mol% without impacting the outcome of the reaction and 5aa was obtained with comparable excellent yield and selectivities.
Scheme 5.
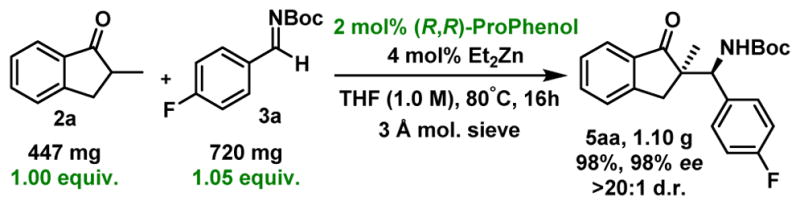
Gram-scale reaction
We then attempted to further elaborate the Mannich products 5 to demonstrate their synthetic utility (Scheme 6).
Scheme 6.
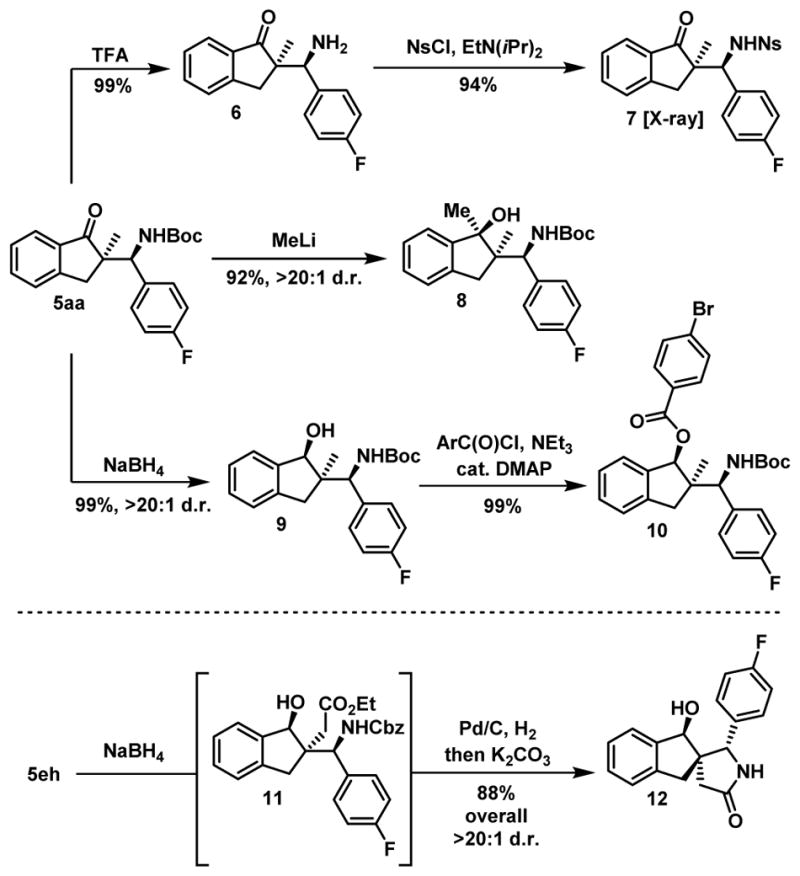
Synthetic applications
First, the Boc protecting group was quantitatively removed with trifluoroacetic acid in dichloromethane at room temperature. Of note, this type of reaction is usually plagued by retro-Mannich processes that were not observed in this case. Then, primary amine 6 can be further functionalized and a protecting group switch to a para-nosyl group afforded compound 7 from which crystals were grown. The relative and absolute configuration of 7 derived from 5aa was unambiguously established by X-ray crystallographic analysis and by analogy the same configuration was assigned to all compounds 4–5. To account for the stereochemical outcome of the reaction, we propose a transition state where the Boc-imine is engaged via two-point binding with the dinuclear catalyst as shown in scheme 7. When 5aa was reacted with MeLi at −78°C in THF, a diastereoselective addition of the methyl group was achieved to form 8 possessing three contiguous stereocenters and featuring two vicinal tetrasubstituted carbons. The use of MeMgCl instead of MeLi led to a sluggish reaction under similar conditions and to decomposition when the reaction temperature was increased. Additionally, a diastereoselective reduction of 5aa with NaBH4/methanol at 0°C was also developed. The seemingly hindered neopentylic secondary alcohol 9 could then be acylated under standard conditions using DMAP as a catalyst. A diastereoselective reduction of ketone 5eg was also performed using our optimized reaction conditions and 11 was used for the next step without chromatography purification. Removal of the Cbz protecting group under hydrogenolysis in methanol directly followed by the addition of K2CO3 led to the complex spirolactam 12 in excellent yield.
Scheme 7.
Proposed transition-state for the formation of 5aa
In summary, we have developed the first direct asymmetric Mannich reaction using α-branched ketones. This strategy allows an enantio- and diastereoselective access to a range of functionalized β-amino ketones featuring an all-carbon quaternary stereocenter. The reaction exhibits many interesting features: the pre-activation of the reaction partners is not required; the reaction is highly atom-economic and can be run on a gram-scale with a low catalyst loading. Moreover, two convenient and orthogonal protecting groups can be used on the imines with similar efficiency. Finally, the Mannich adducts can be further elaborated to complex molecules possessing three contiguous stereogenic centers with complete control of diastereoselectivity. The extension of this strategy to the synthesis of quaternary carbon in acyclic systems is a major goal of our future efforts and will be reported in due course.
Supplementary Material
Acknowledgments
We thank the National Science Foundation (CHE-1360634) and the National Institute of health (GM-033049) for their generous support of our programs. We thank Dr. Jana Maclaren (Stanford University) for X-ray crystallographic analysis and Jacob S. Tracy (Stanford University) for conducting NOE experiments. T.S. is extremely grateful to the Swiss National Science Foundation for a fellowship.
Footnotes
The authors declare no competing financial interest.
Experiment details, compound characterization data, and spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Trost BM. Science. 1991;254:1471. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- 2.Tramontini M, Angiolini L. Tetrahedron. 1990;46:1791. [Google Scholar]
- 3.(a) Corey EJ, Guzman-Perez A. Angew Chem Int Ed. 1998;37:388. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; (b) Douglas CJ, Overman LE. Proc Nat Acad Sci USA. 2004;101:5363. doi: 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Das JP, Marek I. Chem Commun. 2011;47:4593. doi: 10.1039/c0cc05222a. [DOI] [PubMed] [Google Scholar]; (d) Quasdorf KW, Overman LE. Nature. 2014;516:181. doi: 10.1038/nature14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arend M, Westermann B, Rish N. Angew Chem Int Ed. 1998;37:1044. doi: 10.1002/(SICI)1521-3773(19980504)37:8<1044::AID-ANIE1044>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 5.Ishitani H, Ueno M, Kobayashi S. J Am Chem Soc. 1997;119:7153.For an early review, see: Kobayashi S, Ishitani H. Chem Rev. 1999;99:1069. doi: 10.1021/cr980414z.
- 6.(a) Cordova A. Acc Chem Res. 2004;37:102. doi: 10.1021/ar030231l. [DOI] [PubMed] [Google Scholar]; (b) Arrayas RG, Carretero JC. Chem Soc Rev. 2009;38:1940. doi: 10.1039/b820303b. [DOI] [PubMed] [Google Scholar]
- 7.For the first organocatalytic direct asymmetric Mannich reaction, see: List B. J Am Chem Soc. 2000;122:9336.. For general reviews, see: Notz W, Tanaka F, Barbas CF., III Acc Chem Res. 2004;37:580. doi: 10.1021/ar0300468.Mukherjee S, Yang JW, List B. Chem Rev. 2007;107:5471. doi: 10.1021/cr0684016.Verkade JMM, Van Hemert LJC, Quaedflieg PJLM, Rutjes FPJT. Chem Soc Rev. 2008;37:29. doi: 10.1039/b713885g.
- 8.(a) Shibasaki M, Yoshikawa N. Chem Rev. 2002;102:2187. doi: 10.1021/cr010297z. [DOI] [PubMed] [Google Scholar]; (b) Shibasaki M, Kanai M, Matsunaga S, Kumagai N. Acc Chem Res. 2009;42:1117. doi: 10.1021/ar9000108. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Bartlett MJ. Acc Chem Res. 2015;48:688. doi: 10.1021/ar500374r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Matsunaga S, Yoshida T, Morimoto H, Kumagai N, Shibasaki M. J Am Chem Soc. 2004;126:8777. doi: 10.1021/ja0482435. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Jaratjaroonphong J, Reutrakul V. J Am Chem Soc. 2006;128:2778. doi: 10.1021/ja057498v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For a general review, see: Kobayashi S, Mori Y, Fossey JS, Salter MM. Chem Rev. 2011;111:2626. doi: 10.1021/cr100204f.
- 11.For selected examples, see: Hamashima Y, Sasamoto N, Hotta D, Somei H, Umebayashi N, Sodeoka M. Angew Chem Int Ed. 2005;44:1525. doi: 10.1002/anie.200462202.Ting A, Lou S, Schaus SE. Org Lett. 2006;8:2003. doi: 10.1021/ol060304b.Kang YK, Kim DY. J Org Chem. 2009;74:5734. doi: 10.1021/jo900880t.Hatano M, Horibe T, Ishihara K. J Am Chem Soc. 2010;132:56. doi: 10.1021/ja909874b.
- 12.(a) Tian X, Jiang K, Peng J, Du W, Chen YC. Org Lett. 2008;10:3583. doi: 10.1021/ol801351j. [DOI] [PubMed] [Google Scholar]; (b) Cheng L, Liu L, Jia H, Wang D, Chen YJ. J Org Chem. 2009;74:4650. doi: 10.1021/jo9006688. [DOI] [PubMed] [Google Scholar]; (c) He R, Ding C, Maruoka K. Angew Chem Int Ed. 2009;48:4559. doi: 10.1002/anie.200901277. [DOI] [PubMed] [Google Scholar]; (d) Shimizu S, Tsubogo T, Xu P, Kobayashi S. Org Lett. 2015;17:2006. doi: 10.1021/acs.orglett.5b00749. [DOI] [PubMed] [Google Scholar]
- 13.Chowdari NS, Suri JT, Barbas CF., III Org Lett. 2004;6:2507. doi: 10.1021/ol049248+.. For a single example using a branched ketone, see: Yang X, Toste FD. J Am Chem Soc. 2015;137:3205. doi: 10.1021/jacs.5b00229.
- 14.(a) Trost BM, Saget T, Lerchen A, Hung C-I. Angew Chem Int Ed. 2016;55:781. doi: 10.1002/anie.201509719. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, Hung C-I. J Am Chem Soc. 2015;137:15940. doi: 10.1021/jacs.5b11248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Boumendjel A. Curr Med Chem. 2003;10:2621. doi: 10.2174/0929867033456468. [DOI] [PubMed] [Google Scholar]; (b) Seabra RM, Andrade PB, Ferreres F, Moreira MM. Phytochemistry. 1997;45:839. [Google Scholar]; (c) Lee CY, Chew EH, Go ML. Eur J Med Chem. 2010;45:2957. doi: 10.1016/j.ejmech.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 16.The utilization of saturated alkyl imines with 4c and 4e has proven to be problematic due to competitive isomerization to the corresponding enecarbamates and will be a focus for future efforts.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.