

Abstract
Alkynyl ketones are attractive but challenging nucleophiles in enolate chemistry. Their susceptibility to other reactions such as Michael additions and the difficulty of controlling the enolate geometry make them difficult substrates. Mannich-type reactions, which previously have not been reported using N-carbamoyl-imines with simple ketone enolates became our objective. In this report, we describe the first direct catalytic Mannich-type reaction between various ynones and N-Boc imines, whose stereocontrol presumably derives from catalyst control of enolate geometry. This method produces α-substituted β-amino ynones with excellent chemo-, diastereo- and enantioselectivity. The products can be readily transformed into a broad range of molecular scaffolds upon further one-step transformations, demonstrating the utility of ynones as masked synthetic equivalents for a variety of unsymmetrically substituted acyclic ketones. In particular, alkynyl alkyl ketones resolve the longstanding problem of the inability to use the enolates of unsymmetrical dialkyl ketones lacking α-branching for regio- and stereoselective reactions.
Graphical Abstract

INTRODUCTION
C-C bond formation through enolate chemistry is one of the most fundamental and broadly utilized transformations in modern synthetic organic chemistry. Over the past few decades, significant progress in this field has been made not only in achieving high levels of control in additions to diverse electrophiles, but also with processes that no longer require stoichiometric generation of the enolate intermediates.1 Alkynyl ketones are valuable yet underexplored nucleophiles in this active area of research. Their propensity to act as Michael acceptors and the difficulty of controlling the enolate geometry due to lack of steric bulk on the acetylene moiety render them challenging substrates.2 Nonetheless, ynones are versatile building blocks for structural proliferation into various synthetic targets.3 The high metallophilicity of the acetylene moiety can be employed in chemoselective C-C bond formations such as [2+2+2] cycloadditions,4 alkyne-alkyne or alkene-alkyne coupling,5 and reductive coupling reactions.6 The adjacent carbonyl group also allows rapid construction of various heterocycles.7 The valuable ability of ynone enolates to act as a broad spectrum enolate equivalent is significant, especially in a lead generation program in drug discovery, where rapid access to a broad diversity of skeletal classes is required.
Our initial motivation derived from the fact that unsymmetrical acyclic ketones lacking functionality in proximity of the carbonyl group, such as 3-hexanone, do not participate in reactions proceeding through enol or enolates with high chemo-, regio-, diastereo-, and enantioselectivity. As shown in eq. 1 (Scheme 1), the source of these difficulties lies in the inability to control the regio- and geometric selectivity of the enolization process. Thus, in attempting to develop the addition of such a ketone with an electrophile under catalytic conditions, up to eight total isomers are possible (eq. 2).
Scheme 1.

Problems and Known Strategy to Selectively Functionalize Unsymmetrical Dialkyl Ketones
One way to generate regio-defined acyclic dialkyl ketone enolates involves the metal-catalyzed reductive coupling of α,β-unsaturated ketones via hydrogenation (eq. 3).8 In such processes, the selected enone first undergoes 1,4-addition by hydride to generate the reactive enolate intermediate, which then is trapped by the desired electrophile.9,10 Although effective, this strategy is generally applied to ketones bearing a simple vinyl group. Since an alkyne can function as a masked alkane, the use of ynones also provides a potential route to direct regio-, diastereo-, and enantioselective modification on nonsymmetrical dialkyl ketones (Scheme 2). This particular strategy relies on an acetylene moiety on one side of the ketone, preventing enolization on that side. After performing the enolate chemistry, the acetylene moiety can be transformed into a variety of compounds, including fully saturated unsymmetrical ketones via hydrogenation.
Scheme 2.

Ynones as Advantageous Acyclic Donors in Enolate Chemistry
Reports of direct catalytic enolization of ynones for enantioselective additions are rare.11 Previous examples in the literature mainly focus on aldol reactions between aldehydes and methyl ynones, and good diastereocontrol was only observed in the presence of an α-hydroxy group. More importantly, the use of ynones in Mannich-type reactions is unknown. The lack of methods for diastereo- and enantioselective Mannich-type reactions of ynones and the valuable α-substituted β-amino ynone products provided a fitting challenge and opportunity for the further development of our metal-ProPhenol system.12 The commercially available ProPhenol ligand is a member of the aza-crown family that was mainly inspired by the work of Cram on chiral crown compounds.13 When treated with an alkyl metal reagent such as Et2Zn or Bu2Mg, this proline-derived ligand spontaneously assembles into a chiral dinuclear metal complex via deprotonation of the three hydroxyl groups. The resulting complex contains a Lewis acidic site to activate an electrophile and a Brønsted basic site to deprotonate a pronucleophile.
Despite previous success in an array of direct asymmetric aldol and Mannich-type reactions using zinc-ProPhenol catalysis, the scope of donors has been restricted to methyl ketones (acetophenones,12a acetone,12e methyl vinyl ketone,12f methyl ynones11a) or unsubstituted α-hydroxy carbonyl compounds.12f–g To demonstrate the difficulty of using higher alkyl ketones, a control experiment using 3-hexanone and imine A was conducted. As shown in eq. 4 (Scheme 3), no desired Mannich product was observed using the Zn-ProPhenol catalyst. We therefore anticipated the possibility of ynones to overcome this observed limitation due to the unique steric and electronic properties of the alkyne group. The excellent properties of Boc as a nitrogen protecting group make the use of N-Boc imines preferentially attractive. However, curiously, according to a recent review, there are no examples of simple ketone enolates serving as suitable donors for such imines.14 To illustrate the difficulty of controlling the enolate geometry via traditional approaches even in the case of alkynyl ketones, a control experiment using imine A and ynone B was conducted under standard enolate conditions (eq. 5). As expected, poor diastereoselectivity of 1.25 : 1 was obtained, suggesting the lack of steric differentiation between the oxygen and the acetylene moiety. In this respect, the catalyst must exercise control of the enolate geometry.
Scheme 3.

Control Experiments
RESULTS AND DISCUSSION
We initiated our studies with silyl-protected ynone 2a as the model substrate since it is less likely to undergo undesired Michael addition due to steric bulk (Table 1). Benzyldimethylsilyl group was chosen due to the potential subsequent functionalization via Hiyama cross-coupling.15 Moreover, Boc-protected aldimine 1a was selected for initial optimization process due to its stability and ease of handling. Astonishingly, the first experiment using 2 equivalents of 2a and aldimine 1a in the presence of 20 mol% of Zn-ProPhenol catalyst produced a near perfect result (entry 1), in which the desired Mannich adduct 3a was obtained as a single diastereomer in 99% yield and 97% ee. This result suggested that our catalyst system did generate one enolate isomer preferentially over the other. Decreasing the catalyst loading to 10 mol% did not show any detrimental effect. Although excellent selectivity was retained by further decreasing the catalyst loading to 5 mol%, the reaction never proceeded to full conversion even at room temperature (entry 3). Subsequent optimization showed that ynone 2a can be reduced to 1.2 equivalents to give 3a in 99% yield and excellent diastereo- and enantioselectivity (entry 5). To our delight, reactions with alkyl-substituted ynone 2g, which is more prone to undergo undesired Michael additions, gave comparable results despite minor erosion of the diastereoselectivity (entry 7), and further adjustment of the reaction temperature did not give significant improvement (entry 8 & 9). Eventually, by reducing the catalyst loading to 5 mol%, 3h was obtained as a single diastereomer in 91% yield with excellent ee (entry 10). The high conversion of 2g with only 5 mol% of catalyst in contrast to 2a might potentially be due to the absence of the bulky silyl-protecting group directly attached to the acetylene moiety. Finally, it should be noted that the ligand can be easily recovered and recycled via chromatography (80% recovery), hence enhancing the effective turnover number.
Table 1.
Optimization of the reaction conditionsa
 | ||||||
|---|---|---|---|---|---|---|
| entry | 3 | x | equiv. of 2, T (°C) |
yieldb | drc | eed |
| 1 | 3a | 20 | 2, 4 | 99% | >20 : 1 | 97% |
| 2 | 3a | 10 | 2, 4 | 95% | >20 : 1 | 99% |
| 3e | 3a | 5 | 2, rt | 78% | >20 : 1 | 98% |
| 4 | 3a | 10 | 1.5, 4 | 96% | >20 : 1 | 97% |
| 5 | 3a | 10 | 1.2, 4 | 99% | >20 : 1 | 99% |
| 6 | 3a | 10 | 1.0, 4 | 85% | >20 : 1 | 99% |
| 7 | 3h | 10 | 1.2, 4 | 93% | 15 : 1 | 99% |
| 8 | 3h | 10 | 1.2, −10 | 70% | 16 : 1 | 99% |
| 9 | 3h | 10 | 1.2, rt | 95% | 15 : 1 | 95% |
| 10e | 3h | 5 | 1.2, 4 | 91% | >20 : 1 | 99% |
Reaction conditions: : 0.24 mmol 1, x mol% L1, 2x mol% Et2Zn (1.0 M in hexanes), 4 Å molecular sieves (10 mg), in THF (0.3 M) at the indicated temperature and concentration.
Isolated yield.
Determined by 1H-NMR analysis.
Determined by HPLC on a chiral stationary phase.
Reaction was done in 0.6 M THF.
With the optimized conditions in hand, we then evaluated the generality of this catalytic process (Table 2). On the acceptor side, a variety of aromatic N-Boc aldimines was successfully reacted with ynone donors. Both electron-withdrawing and electron-donating groups were tolerated in ortho, meta, and para positions. Heteroaromatic imines also gave the product in high yields and selectivities without poisoning the Zn catalyst. On the donor side, a variety of functional groups, including alkenes, unprotected terminal acetylenes, esters, protected alcohols, arenes, and bulky alkyl chains were tolerated without significant perturbation of the reactivity and selectivities, demonstrating the high chemoselectivity of this catalytic process. Moreover, as described in the optimization table, reactions with alkyl-substituted ynones were performed using 5 mol% loading of catalyst (3h–3k).
Table 2.
Scope of the Reaction with Aromatic Iminesa
 | ||||
|---|---|---|---|---|
| entry | 3 | Structure | yieldb | dr/ee |
| 1 | a | 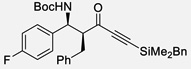 |
99% | >20 : 1, 99% |
| 2 | b |  |
95% | >20 : 1, 91% |
| 3 | c | 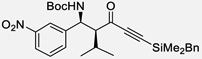 |
88% | >20 : 1, 99% |
| 4 | d | 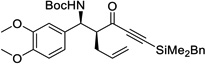 |
86% | >20 : 1, 98% |
| 5 | e | 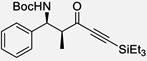 |
75% | >20 : 1, 99% |
| 6 | f | 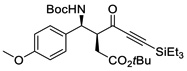 |
95% | >20 : 1, 92% |
| 7 | g | 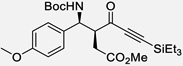 |
89% | 9.5 : 1, 99% |
| 8b | h | 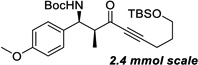 |
91% | >20 : 1, 99% |
| 9b | i | 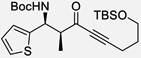 |
81% | 13 : 1, 92% |
| 10b | j | 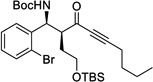 |
85% | 17 : 1, 96% |
| 11b | k | 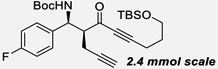 |
91% | 16 : 1, 99% |
| 12c | l | 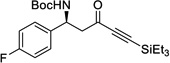 |
85% | NA, 97% |
| 13c | m | 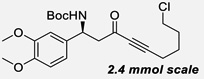 |
85% | NA, 94% |
| 14c | n | 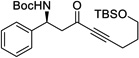 |
83% | NA, 92% |
| 15c | o | 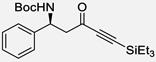 |
80% | NA, 95% |
Reaction conditions: 0.24 mmol 1, 0.29 mmol 2, 10 mol% L1, 20 mol% Et2Zn, 4 Å molecular sieves (10 mg), in THF (0.3 M) at 4 °C for 16 h.
Reaction run with 5 mol% catalyst in THF (0.6 M).
Reaction run at −10 °C for 48 h.
This reaction can also be performed using simple methyl ynones as donors (3l–3o) to give high yields and excellent selectivity at −10 °C. To show the scalability and practicality of the method, several examples (3h, 3k, 3m) were performed at 2.4 mmol scale, giving comparable results to the small-scale reactions. In contrast to the result from the control experiment (Scheme 3), our new approach can allow the formation of ynone 3j in 85% yield with excellent ee and dr. In addition, N-Dpp protected imines were also examined. However, low conversions and selectivities were observed (See SI Table S5).
The catalytic process was also applicable to nonaromatic imines. Using the standard conditions with L1, α,β-unsaturated imine 1j gave high product yields and diastereoselectivity (entry 1, Table 3). To enhance the enantioselectivity of this process, several ligands were examined, especially the recently developed non-C2-symmetric ProPhenol ligands, which have shown to improve the enantioselectivity substantially in the vinylation reaction previously described.12h To our delight, the combination of p-trifluoromethylphenyl and phenyl in the prolinol backbone L2 was found to be optimal without perturbing the conversion and diastereoselectivity (entry 4). Interestingly, when switching to the more electron-rich ligand L3 (entry 5), the enantioselectivity was similar to the parent ProPhenol ligand L1. The differential enantioinduction of L2 was further demonstrated by performing the reaction with the C2-symmetric analogous L5, in which significant decreases in yield and enantioselectivity were observed (entry 7). Curiously, using a donor-acceptor substituted as in L4 also showed a decrease in ee as well as yield (entry 6). ProPhenol L6, which has shown to improve the enantioselectivity of aldol reaction of glycinate benzophenone imines, did not result in any beneficial effect.11i
Table 3.
Optimization with α,β-unsaturated imine 1j a
 | ||||||
|---|---|---|---|---|---|---|
| entry | L | x | T (°C) | yieldb | drc | eed |
| 1 | L1 | 10 | 4 | 88% | 9 : 1 | 75% |
| 2 | L1 | 10 | −10 | 68% | 9 : 1 | 77% |
| 3 | L1 | 5 | 4 | 60% | 10 : 1 | 74% |
| 4 | L2 | 10 | 4 | 96% | 9 : 1 | 88% |
| 5 | L3 | 10 | 4 | 75% | 7 : 1 | 77% |
| 6 | L4 | 10 | 4 | 85% | 8 : 1 | 82% |
| 7 | L5 | 10 | 4 | 63% | 8 : 1 | 65% |
| 8 | L6 | 10 | 4 | 80% | 9 : 1 | 70% |
Reaction conditions: : 0.24 mmol 1, x mol% L, 2x mol% Et2Zn (1.0 M in hexanes), 4 Å molecular sieves (10 mg), in THF (0.3 M) at the indicated temperature for 8 h.
Isolated yield.
Determined by 1H-NMR analysis.
Determined by HPLC on a chiral stationary phase.
Initial examination using aliphatic imine 1k, on the other hand, led to complete isomerization to the corresponding enecarbamate under typical reaction conditions (entry 1, Table 4). This observation suggested the α-proton of imine 2k is comparably or even more acidic than the α-protons of ynone 2a under the reaction conditions. Fortunately, we were able to overcome this undesirable process via slow addition of the imine. Subsequent adjustments of the addition rate as well as the equivalence of imine gave 84% yield and 85% ee as a single diastereomer (entry 5). Finally, further ligand optimization showed that the same non-C2-symmetric ligand L2 was optimal for the enantioselectivity (entry 6).
Table 4.
Optimization with aliphatic imine 1ka
 | ||||||
|---|---|---|---|---|---|---|
| entry | L | equiv. of 1k |
slow add. (h) |
yieldc | drd | eee |
| 1b | L1 | 1.0 | 0 | n.p. | NA | NA |
| 2 | L1 | 2.0 | 3 | 32% | >20 : 1 | 82% |
| 3 | L1 | 2.0 | 6 | 55% | >20 : 1 | 80% |
| 4 | L1 | 2.0 | 12 | 88% | >20 : 1 | 85% |
| 5 | L1 | 1.5 | 12 | 84% | >20 : 1 | 85% |
| 6 | L2 | 1.5 | 12 | 89% | >20 : 1 | 95% |
| 7 | L3 | 1.5 | 12 | 75% | >20 : 1 | 80% |
| 8 | L4 | 1.5 | 12 | 86% | >20 : 1 | 88% |
Reaction conditions: : 0.24 mmol 2a, x mol% L, 2x mol% Et2Zn (1.0 M in hexanes), 4 Å molecular sieves (10 mg), in THF (0.3 M) at rt and stirred for 12 h after imine addition.
No desired product.
Isolated yield.
Determined by 1H-NMR analysis.
Determined by HPLC on a chiral stationary phase.
With the optimized conditions, different nonaromatic N-Boc imines were examined (Table 5). In general, good to excellent selectivities were observed. More importantly, imines bearing two enolizable α-protons were also compatible substrates in this process (entry 6 & 7), giving the corresponding ynones in excellent yields and selectivities. The incorporation of these highly enolizable imines not only considerably enhances the scope of this reaction, but also serves as a rare occasion where simple ketones are used as successful donors.14
Table 5.
Reaction with Nonaromatic Imines
 | ||||
|---|---|---|---|---|
| entry | 3 | Structure | yieldb | dr/ee |
| 1a | p | 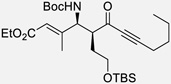 |
86% | 9 : 1, 88% |
| 2a | q | 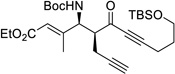 |
90% | 9 : 1, 91% |
| 3a | r | 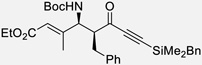 |
86% | >20 : 1, 99% |
| 4b | s | 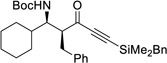 |
89% | >20 : 1, 95% |
| 5b | t | 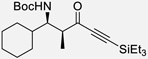 |
85% | >20 : 1, 94% |
| 6b | u | 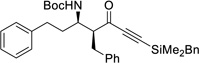 |
95% | >20 : 1, 98% |
| 7b | v | 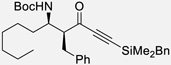 |
95% | >20 : 1, 97% |
Reaction conditions: 0.24 mmol 1, 0.29 mmol 2, 10 mol% L2, 20 mol% Et2Zn (1.0 M in hexanes), 4 Å molecular sieves (10 mg), in THF (0.3 M) at 4 °C for 8 h.
Reaction run with dropwise addition of imines (1.5 equiv) over 12 h at rt.
We then further elaborated the obtained products 3 to demonstrate their synthetic utility, especially with respect to library design in which each Mannich adduct can be converted into a wide diversity of scaffolds via a single step (Scheme 4). These transformations also validate the utility of ynones as a masked synthetic equivalent for a variety of unsymmetrically substituted acyclic ketones. Ketones with different degrees of unsaturation can be readily prepared. Pd-C catalyzed hydrogenation gave the fully saturated ketone 3ha in 95% yield, demonstrating an efficient route to products equivalent to direct functionalization of unsymmetrical dialkyl ketones via two effective atom-economic catalytic addition processes. Our previously developed Ru-catalyzed hydrosilylation and subsequent same-pot desilylation with CsF gave (E)-enones 3hb and 3ma in good yields,16 whereas (Z)-enone 3ja was generated upon partial hydrogenation using Lindlar’s catalyst with quinoline. Heterocycles, including pyrazole 3ka, pyrimidine 3na, and triazole 3hc, were obtained in excellent yields using literature single-step processes.7a–c Upon NaBH4 reduction, chiral propargyl alcohol 3da was prepared in high yield with 8 : 1 dr, and the relative configuration was determined by NOESY after cyclization to the cyclic carbamate 3db using NaH. The benzyldimethylsilyl group was removed during the reaction as well due to in situ-generated tert-butoxide.
Scheme 4.

Exemplified One-Step Structural Diversification of β-Amino Ynones
A 6-endo-dig cyclization catalyzed by in situ-generated PPh3AuSbF6 gave pyridinone 3mb in 96% yield,17 which is an important intermediate for a later total synthesis (Scheme 5). α-Substituted ynones, which have never been employed in this type of transformation, reacted sluggishly with significant epimerization at the α-position using the same catalyst, but changing to a more σ-donating NHC ligand gave 3hd and 3jb in high yields without any side reaction. The relative configurations of the α-substituted β-amino ynones were unambiguously determined by comparing the vicinal homonuclear 1H-1H coupling constants upon Au-catalyzed 6-endo-dig cyclizations, which are consistent with other results reported in the literature.18 Using our reported Pd-catalyzed alkyne-alkyne coupling conditions, heavily functionalized enyne 3pa was obtained in 82% yield with excellent stereo- and chemoselectivity.5c Finally, Ru-catalyzed [2+2+2] cycloaddition19 with 1-hexyne gave β-amino indanone 3kb, a member of a class of molecule that can modify muscarinic receptor activity for treating or alleviating diseases,20 in 93% yield as a single regioisomer.
Scheme 5.

Total Synthesis of (−)-Lasubine II
Pyridinone 3mb was readily converted into (−)-lasubine II 6 (Scheme 5), which was originally isolated from the leaves of Lagerstroemia subcostata koehne by Fuji et al.21 The quinolizidine moiety is widely encountered in drug-like compounds and natural products of potential biological activities.22 After Pd-C catalyzed hydrogenation, ZnBr2-mediated Boc deprotection followed by a same-pot intramolecular SN2 displacement under basic condition furnished quinolizidine 5 in 85% yield. Other typical Boc removal conditions, including TFA, TMSOTf, HCl in organic solvents, thermolysis, and other metallic Lewis acids, generally resulted in ring-opening via elimination of the C-N bond due to the adjacent electron-rich aromatic group. Subsequent reduction using L-selectride gave (−)-lasubine II 6 in 93% yield as a single diastereomer.23 Alkaloid 6 is also an essential fragment in many bioactive compounds from the Lythraceae alkaloid family,24 one noticeable example being (−)-decinine 7, which can be readily synthesized from 6.25 The total synthesis of 6 further demonstrated the exceptional synthetic utility of our methodology incorporating ynones, in which cyclic tertiary alkaloids can be readily constructed from the common backbone 3.
The absolute configuration of the C-N bond of 3 was further confirmed using O-methylmandelic amides26 9 and 9’ derived from 3da after Boc removal using TFA and the subsequent same-pot EDC coupling (Scheme 6). Substantial anisotropic magnetic effects were observed on H1 and H2 of 9 due to the proximal phenyl ring on the O-methylmandelic acid moiety in the most stable conformation. Similar shielding effect was also observed on the 3,4-dimethoxyphenyl group of 9’ when the opposite enantiomer of O-methylmandelic acid was applied.
Scheme 6.

Determination of Absolute Stereochemistry using O-Methylmandelic Acid
The proposed catalytic cycle is depicted in Scheme 7. After the formation of the dinuclear Zn-ProPhenol complex 10, deprotonation of ynone 2 occurs and places the nucleophile in a chiral environment (11 and 11’), in which the enolate can adopt either the (E)- or (Z)-geometry with reversible interconversion. N-Boc imine 1 then enters the reaction cycle, and binds preferentially to the catalyst system bearing the (Z)-enolate via a bidentate coordination to both zinc atoms (12). This explains the high diastereoselectivity of the reaction even when the electrophiles were introduced in slow addition as shown in the case of aliphatic imines. The particular arrangement of 12 minimizes the steric interactions of notably R and R1 of both substrates and the corresponding prolinol diphenyl ligand substituents. The enolate then attacks the electrophile to give intermediate 13 with the observed stereochemistry. Finally, deprotonation of ynone 2 liberates the Mannich-adduct 3, and turnovers the catalytic cycle.
Scheme 7.

Proposed Catalytic Cycle
CONCLUSION
The present study provides a highly chemo-, diastereo-, and enantioselective Mannich-type reaction using ynones as the donors catalyzed by the Zn-ProPhenol complex. This method tolerates a variety of functional groups on the donors and acceptors, allowing rapid construction of chiral and heavily functionalized α-substituted β-amino ynones. This process provides the broadest spectrum of Mannich products in a catalytic fully selective manner. More importantly, the products offer rapid access to a broad spectrum of chiral-amine-containing skeletal types utilizing the synthetic versatility of the ynone moiety, and also provide an effective route to selectively functionalize unsymmetrical dialkyl ketones. This Mannich-ynone approach can facilitate lead discovery in medicinal chemistry and the preparation of pharmaceutical analogues and complex natural products. Considering the broad scope and functional group compatibility of this methodology along with the high diastereo- and enantioselectivity, we anticipate that this process can be expanded to other electrophiles, such as aldehydes, ketones, or ketimines, offering new classes of chiral ynones ready for structural diversification.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM-033049) and the National Science Foundation (CHE-1360634) for their generous support of our program. We also thank Dr. Stephen R. Lynch (Stanford University) for conducting NOE experiments.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experiment details, compound characterization data, and spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Experimental procedures and compound characterization description (PDF)
Compound spectra (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.For selected reviews, see (a) Mahrwald R. Modern methods in stereoselective aldol reactions. Weinheim: Wiley-VCH; 2013. Notz W, Tanaka F, Barbas CF., III Acc. Chem. Res. 2004;37:580–591. doi: 10.1021/ar0300468. Mukherjee S, Yang JW, Hoffmann S, List B. Chem. Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016. MacMillan DWC. Nature. 2008;455:304–308. doi: 10.1038/nature07367. Trost BM, Brindle CS. The direct catalytic asymmetric aldol reaction. Chem. Soc. Rev. 2010;39:1600–1632. doi: 10.1039/b923537j.
- 2.A control experiment was conducted by exposing ynone 2g to catalytic potassium tert-butoxide (5 mol%) in anhydrous THF. The resulting complicated mixture due to decomposition suggested the intrinsic instability of ynones under basic conditions.
- 3.For selected applications and reviews of ynones in synthesis, see Shin Y, Fournier J-H, Fukui Y, Brückner AM, Curran DP. Angew. Chem. Int. Ed. 2004;116:4734–4737. Zinzalla G, Milroy L-G, Ley SV. Org. Biomol. Chem. 2006;4:1977. doi: 10.1039/b603015g. Müller TJJ. Top. Heterocycl. Chem. 2010;25:25–94. Bagley MC, Glover C, Merritt EA. Synthlett. 2007:2459–2482. Tamber UK, Kano T, Stoltz BM. Org. Lett. 2005;7:2413–2416. doi: 10.1021/ol050705b. Trost BM, Frederiksen MU, Papillon JPN, Harrington PE, Shin S, Shireman BT. J. Am. Chem. Soc. 2005;127:3666–3667. doi: 10.1021/ja042435i. Trost BM, Stivala CE, Hull KL, Huang A, Fandrick DR. J. Am. Chem. Soc. 2014;136:88–91. doi: 10.1021/ja411270d. Trost BM, Biannic B, Bridle CS, O’Keefe BM, Hunter TJ. J. Am. Chem. Soc. 2015;137:11594–11597. doi: 10.1021/jacs.5b07438. Fujii K, Maki K, Kanai M, Shibasaki M. Org. Lett. 2003;5:733–736. doi: 10.1021/ol027528o.
- 4.Saito S, Yamamoto Y. Chem. Rev. 2000;100:2901–2916. doi: 10.1021/cr990281x. [DOI] [PubMed] [Google Scholar]
- 5.(a) Trost BM, Toste FD, Pinkerton AB. Chem. Rev. 2001;101:2067–2096. doi: 10.1021/cr000666b. [DOI] [PubMed] [Google Scholar]; (b) Trost BM. Chem. Eur. J. 1998;4:2405–2412. [Google Scholar]; (c) Trost BM, Sorum MT, Chan C, Harms AE, Rühter G. J. Am. Chem. Soc. 1997;119:698–708. [Google Scholar]
- 6.Jang H, Krische MJ. Acc. Chem. Res. 2004;37:653–661. doi: 10.1021/ar020108e. [DOI] [PubMed] [Google Scholar]
- 7.(a) Kirkham JD, Edeson SJ, Stokes S, Harrity JPA. Org. Lett. 2012;14:5354–5357. doi: 10.1021/ol302418b. [DOI] [PubMed] [Google Scholar]; (b) Karpov AS, Müller TJJ. Org. Lett. 2003;5:3451–3454. doi: 10.1021/ol035212q. [DOI] [PubMed] [Google Scholar]; (c) Lie Ken Jie MSF, Pasha MK, Alam MS. Chem. Phys. Lipids. 1998;91:71–78. [Google Scholar]; (d) Kel'i AV, Gevorgyan V. J. Org. Chem. 2002;67:95–98. doi: 10.1021/jo010832v. [DOI] [PubMed] [Google Scholar]; (e) Van den Hoven BG, Ali BE, Alper H. J. Org. Chem. 2000;65:4131–4137. doi: 10.1021/jo000230w. [DOI] [PubMed] [Google Scholar]; (f) Awuah E, Capretta A. Org. Lett. 2009;11:3210–3213. doi: 10.1021/ol901043q. [DOI] [PubMed] [Google Scholar]; (g) Wang Z, Li L, Huang Y. J. Am. Chem. Soc. 2014;136:12233–12236. doi: 10.1021/ja506352b. [DOI] [PubMed] [Google Scholar]
- 8.For selected reviews, see Nishiyama H, Shiomi T. Top. Curr. Chem. 2007:105–137. Garner SA, Han S-B, Krische MJ. Metal-Catalyzed Reductive Aldol Coupling. In: Andersson PG, Munslow IJ, editors. Modern Reduction Methods. Weinheim, Germany: Wiley-VCH; 2008. pp. 387–417. Krische MJ, Han S, Hassan A. Synthesis. 2008:2669–2679. doi: 10.1055/s-2008-1067220.
- 9.Bee C, Han S, Hassan A, Iida H, Krische MJ. J. Am. Chem. Soc. 2008;130:2746–2747. doi: 10.1021/ja710862u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garner SA, Krische MJ. J. Org. Chem. 2007;72:5843–5846. doi: 10.1021/jo070779w. [DOI] [PubMed] [Google Scholar]
- 11.(a) Trost BM, Fettes A, Shireman BT. J. Am. Chem. Soc. 2004;126:2660–2661. doi: 10.1021/ja038666r. [DOI] [PubMed] [Google Scholar]; (b) Maki K, Motoki R, Fujii K, Kanai M, Kobayashi T, Tamura S, Shibasaki M. J. Am. Chem. Soc. 2005;127:17111–17117. doi: 10.1021/ja0562043. [DOI] [PubMed] [Google Scholar]; (c) Silva F, Sawicki M, Gouverneur V. Org. Lett. 2006;8:5417–5419. doi: 10.1021/ol0624225. [DOI] [PubMed] [Google Scholar]; (d) Shi S, Kanai M, Shibasaki M. Angew. Chem. Int. Ed. 2012;51:3932–3935. doi: 10.1002/anie.201109209. (2012) [DOI] [PubMed] [Google Scholar]
- 12.(a) Trost BM, Bartlett MJ. Acc. Chem. Res. 2015;48:688–701. doi: 10.1021/ar500374r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, Ito H. J. Am. Chem. Soc. 2000;122:12003–12004. [Google Scholar]; (c) Trost BM, Silcoff ER, Ito H. Org. Lett. 2001;3:2497–2500. doi: 10.1021/ol0161211. (2001) [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Shin S, Sclafani JA. J. Am. Chem. Soc. 2005;127:8602–8603. doi: 10.1021/ja051526s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Trost BM, Ito H, Silcoff ER. J. Am. Chem. Soc. 2001;123:3367–3368. doi: 10.1021/ja003871h. [DOI] [PubMed] [Google Scholar]; (f) Trost BM, Jaratjaroonphong J, Reutrakul V. J. Am. Chem. Soc. 2006;128:2778–2779. doi: 10.1021/ja057498v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Trost BM, Michaelis DJ, Truica MI. Org. Lett. 2013;15:4516–4519. doi: 10.1021/ol402081p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Trost BM, Hung C-I, Koester DC, Miller Y. Org. Lett. 2015;17:3778–3781. doi: 10.1021/acs.orglett.5b01755. [DOI] [PubMed] [Google Scholar]; (i) Trost BM, Miege F. J. Am. Chem. Soc. 2014;136:3016–3019. doi: 10.1021/ja4129394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Cram DJ. Angew. Chem. Int. Ed. 1988;27:1009–1020. [Google Scholar]; (b) Kyba EB, Koga K, Sousa LR, Siegel MG, Cram DJ. J. Am. Chem. Soc. 1973;95:2692–2693. [Google Scholar]
- 14.All examples mentioned involve easily enolizable ketones, such as 1,3-dicarbonyl and hydroxylmethyl ketones. Vesely J, Rios R. Chem. Soc. Rev. 2014;43:611–630. doi: 10.1039/c3cs60321k.
- 15.Trost BM, Ball ZT. J. Am. Chem. Soc. 2005;127:17644–17655. doi: 10.1021/ja0528580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Trost BM, Ball ZT, Jöge TA. J. Am. Chem. Soc. 2002;124:7922–7923. doi: 10.1021/ja026457l. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Crawley ML. J. Am. Chem. Soc. 2002;124:9328–9329. doi: 10.1021/ja026438b. [DOI] [PubMed] [Google Scholar]
- 17.Gouault N, Le Roch M, Cheignon A, Uriac P, David M. Org. Lett. 2011;13:4371–4373. doi: 10.1021/ol201698m. [DOI] [PubMed] [Google Scholar]
- 18.Etayo P, Badorrey R, Díaz-de-Villegas MD, Gálvez JA. Eur. J. Org. Chem. 2008;2008:6008–6014. doi: 10.1021/jo801515k. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto Y, Arakawa T, Ogawa R, Itoh K. J. Am. Chem. Soc. 2003;125:12143–12160. doi: 10.1021/ja0358697. [DOI] [PubMed] [Google Scholar]
- 20.Brann M, Messier T, Currier E, Duggento K, Spalding T, Friberg M, Skjaerbaek N. Compounds with Activity on Muscarinic Receptors. 6528529 B1. USA: Acadia Pharmaceuticals Inc; US Patent. 2003 Mar 4;
- 21.Fuji K, Yamada T, Fujita E, Murata H. Chem. Pharm. Bull. 1978;26:2515–2521. [Google Scholar]
- 22.Michael JP. Nat. Prod. Rep. 2008;25:139–165. doi: 10.1039/b612166g. [DOI] [PubMed] [Google Scholar]
- 23.Garcia Mancheño O, Gómez Arrayás R, Adrio J, Carretero JC. J. Org. Chem. 2007;72:10294–10297. doi: 10.1021/jo702076j. [DOI] [PubMed] [Google Scholar]
- 24.Wright H, Clardy J, Ferris JP. J. Am. Chem. Soc. 1973;95:6467–6468. [Google Scholar]
- 25.Shan Z-H, Liu J, Xu L-M, Tang Y-F, Chen J-H, Yang Z. Org. Lett. 2012;14:3712–3715. doi: 10.1021/ol3015573. [DOI] [PubMed] [Google Scholar]; (b) Cui L, Li C, Zhang L. Angew. Chem., Int. Ed. 2010;49:9178–9181. doi: 10.1002/anie.201004712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trost BM, Bunt RC, Pulley SR. J. Org. Chem. 1994;59:4202–4205. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.