

Abstract
Prior to 2011, the 1-year survival rates for patients suffering from advanced or metastatic melanoma was as low as 33%, with a median overall survival of about 9 months. Several chemotherapeutic regimens have been applied, either as monochemotherapy or as polychemotherapy, overall not resulting in an improvement of progression-free or overall survival. Novel insights into the epidemiology and biology of melanoma allowed the development of newer therapies. The discovery of mutations in BRAF, a part of the mitogen-activated protein kinase, allowed the development of two BRAF inhibitors, vemurafenib and dabrafenib, which significantly improved the outcome of metastatic melanoma treatment. This article reviews the mechanism of action, efficacy, and safety profile of dabrafenib. An in-depth knowledge of this medication will encourage clinicians to select the appropriate therapeutic strategy for each patient, as well as to prevent or adequately manage side effects, optimizing, thus, the drug’s applicability.
Keywords: melanoma, BRAF, target therapy, dabrafenib, melanoma survival
Introduction
Despite an increase in the incidence of advanced melanoma, little progress has been made over recent decades in addressing the poor prognosis of patients or the limited treatment options available.1–3 The “traditional” treatments for metastatic melanoma were associated with low response rates and complicated by severe toxicities. Dacarbazine was one of the first chemotherapies approved for metastatic melanoma, achieving a response rate of about 20% and a median response duration of 5–6 months. However, studies assessing the efficacy of dacarbazine revealed no benefit in overall survival (OS).4 High-dose interleukin (IL)-2 has been reported to achieve a 6%–16% response rate, with a progression-free survival (PFS) of 13.1 months.5,6 However, the response rate of IL-2 in patients with brain metastasis is only 5%.7 A slightly improved response rate and PFS has been reported with the combination of high-dose IL-2 therapy and the peptide vaccine gp-100. However, the use of high-dose IL-2 is restricted by its severe toxicity, consisting of capillary leak syndrome, arrhythmias, hypotension, and neurologic disturbances.5,6 Although lacking an OS benefit and associated with severe toxicity, IL-2 remained for years a first-line treatment for metastatic melanoma, on the basis of the prolonged PFS of responding patients.5 Temozolomide is an oral alkylating agent with a cytotoxic effect similar to dacarbazine. Based on its ability to penetrate the blood–brain barrier, temozolomide has been tested in previously untreated patients with brain metastases, achieving a response rate of ~7% and a median PFS of 1.2 months.8 Combination chemotherapy has also been tested in several studies, without showing any improvement in response rates.9
During the last years, the prognosis of metastatic melanoma substantially changed with the introduction of kinase inhibitors vemurafenib, dabrafenib, and trametinib and the immune checkpoint inhibitor ipilimumab, an anti-CTLA-4 antibody.10–13 More recently, clinical trials testing the efficacy of the programmed cell death-1 receptor inhibitors nivolumab and pembrolizumab showed a further improvement in OS of metastatic melanoma patients.7,8
Targeted therapy
Several key genetic mutations have been shown to contribute to melanoma development and progression. Approximately 40%–50% of melanomas harbor activating mutations in the BRAF oncogene, most of them found in exon 15, codon 600 (V600). The most frequent mutation event is the substitution of valine by glutamic acid (V600E), occurring in ~75% of the cases. Other, less frequent, substitutions include valine by lysine (V600K) and valine by arginine (V600R).
BRAF is a key molecule of the rat sarcoma gene (RAS), which activates several pathways, such as the mitogen-activated protein kinase (MAPK) pathway that induces cell growth and cell proliferation. Indeed, mutations in the MAPK signaling pathway may be detected in melanoma patients.14 Intracellular signaling is triggered by growth factors that enhance the binding of a GTP protein (RAS) to cell membranes, which, subsequently, promotes the formation of dimers leading to the activation of RAF kinases.15 RAF kinases stimulate the phosphorylation of MEK proteins, which in turn phosphorylate and activate the protein kinase ERK. ERK, finally, stimulates the signals for progrowth within the nucleus, leading to cell proliferation and differentiation and to an inhibitory feedback toward upstream components of the pathway.15–17 Therefore, the uncontrolled activation of the MAPK pathway is associated with the proliferation of malignant cells. This pathway is physiologically activated when extracellular signals bind to their cognate membrane receptor, typically a receptor tyrosine kinase.
BRAF mutations have been reported also in most of the melanocytic nevi, suggesting that the mutation is not responsible for malignancy in melanocytic proliferations. This indicates that BRAF mutations may contribute to an early increased proliferation of melanocytes, but not necessarily a malignant transformation.10 In fact, the formation of nevi might result from melanocytic proliferation driven by BRAF mutations and followed by oncogene-induced senescence. In contrast, melanoma formation requires that senescence does not occur.18–20 Most melanoma cells derive directly from transformed melanocytes, without a previous formation of a nevus, possibly resulting from other genetic alterations (eg, alterations in the p53 and Rb pathways) additional to the oncogenic BRAF mutations.
BRAF mutations in melanoma are significantly more frequent in younger patients, while BRAF mutational status has been shown to correlate to the anatomic site of primary melanoma, the histological subtype, the evidence of chronic sun damage and, partially, the geographic region (Table 1).21,22 For example, BRAF mutations are much less frequent in acral and mucosal melanoma, while they have never been documented in uveal melanoma.23,24
Table 1.
Frequency, type, and clinical characteristics associated to BRAF mutation
| Cancer type | Mutation frequency and type | Clinical characteristics |
|---|---|---|
| Melanoma | 46%–48%; V600E more common than V600K; other rare exon 15 mutations reported |
BRAFV600E mutations more common in younger persons and in tumors arising from intermittently sun-exposed skin. Mutually exclusive with NRAS |
Two different combinations of BRAF inhibitors have been developed and tested for advanced melanoma: type 1 BRAF kinase inhibitors, which bind and inhibit the effect of BRAF mutation, and type 2 BRAF inhibitors, binding to the inactive kinase.17 Wild-type BRAF status represents an absolute contraindication for such compounds, due to paradoxical activation of MAPK.15
Sorafenib, a nonselective BRAF inhibitor, acts as a pan-inhibitor of BRAF and has largely failed in melanoma treatment. In contrast, drugs that selectively target a mutated and activated form of the BRAF kinase have been shown to be appropriate for BRAF mutant melanoma treatment.
Vemurafenib (Zelboraf®) was the first molecular agent targeting the mutated BRAF kinase that demonstrated an improved OS in a Phase III randomized trial. Vemurafenib is an orally administered small-molecule showing a remarkable antitumor activity against BRAFV600E mutant melanoma cell lines. On the basis of the documented efficacy of vemurafenib in Phase I and II studies,17,25 a Phase III randomized clinical trial (BRIM-3) compared vemurafenib to dacarbazine in patients with unresectable stage III or IV melanoma.26 In this trial, 675 previously untreated patients with BRAFV600E mutation-positive advanced melanoma were randomized to receive either 960 mg of vemurafenib orally twice a day or 1 g/m2 of dacarbazine intravenously every 3 weeks. The endpoints of the study were PFS and OS. After a median follow-up period of 3.8 months for patients treated with vemurafenib and 2.3 months for those receiving dacarbazine, vemurafenib was associated with a relative reduction of 63% in the risk of death and of 74% in the relative risk of disease progression, as compared with dacarbazine (P<0.001). Vemurafenib was also associated with a higher disease control rate and a higher response rate. The safety and efficacy of vemurafenib and dacarbazine in this Phase III study were updated at a median follow-up of 12.5 and 9.5 months, respectively, with median OS reaching 13.6 months with vemurafenib compared to 9.7 months with dacarbazine. The hazard ratio (HR) for death in the vemurafenib group was 0.70 (95% confidence interval [CI]: 0.57–0.87; P=0.0008).10 The vemurafenib group showed significantly longer PFS than the dacarbazine group (6.9 vs 1.6 months; HR 0.38 (95% CI: 0.32–0.46); P<0.0001). The relative impact of vemurafenib with respect to mutated BRAF subtypes was also assessed by the updated analysis, showing comparable efficacy and toxicity in patients with BRAFV600E and BRAFV600K mutation.10
The recommended dose of vemurafenib is 960 mg to be taken orally twice each day. The most common adverse events (AEs) recorded in the BRIM-3 registration trial included arthralgia, fatigue, nausea, rash, photosensitivity, and development of cutaneous squamous cell carcinoma (cSCC) or keratoacanthoma (KA) (25).10 The most frequent grade 3 or 4 AEs were cSCC/KA, transaminitis, and rash.14 The dose of vemurafenib was modified or interrupted due to AEs in 38% of patients,26 while the drug was permanently discontinued in only 7% of the patients treated.10
The second selective BRAF inhibitor approved for treatment of BRAF-mutated metastatic melanoma is dabrafenib (Tafinlar®), which is a highly potent adenosine triphosphate-competitive inhibitor of BRAFV600E kinase with proven antitumor activity within the brain and systemically. The recommended dose is 150 mg twice a day.
Dabrafenib monotherapy
The approval of dabrafenib was mainly based on the BREAK trial, which began in 2009; the initial results of the Phase I trial were presented in 2010 and the final results in 2012.19,20 The trial in Phase I included 184 patients, 156 suffering from melanoma and 28 from other solid tumors, with an Eastern Cooperative Oncology Group performance status of 0 or 1. The main aim of the study was to evaluate the safety and tolerability of dabrafenib, as well as to determine the recommended dose for Phase II. Secondary aims were to investigate the pharmacokinetic/pharmacodynamic profile of the drug and the tumor response. The treatment was continued until disease progression, intolerable toxic events, or withdrawal of consent. The tumor response was assessed by the Response Evaluation Criteria in Solid Tumours version 1.0. The most common AEs included development of cSCC (20 patients, 11%), fatigue (14, 8%), and pyrexia (11, 6%). A reduction in drug dose was required in 13 (7%) patients, while no deaths or discontinuations due to AEs occurred. On the basis of safety and the pharmacokinetic profile of dabrafenib, the dose of 150 mg twice daily was determined as the recommended dose for Phase II. Among the 36 patients with BRAFV600-mutant melanoma receiving the recommended dose, 18 (50%, 32.9–67.1 CI 95%) had confirmed response, while the response rate for patients with BRAFV600E was 56% (15 out of 27, 56%, 35.3–74.5).27 The median duration of response was 6.2 months (95% CI, 4.2–7.7) and the PFS was similar for patients with BRAFV600E and V600K mutations (5.5 and 5.6 months, respectively).
After establishment of the optimal dose, three expanded cohorts were added; one with metastatic melanoma, one with asymptomatic untreated brain metastases (3 mm or larger), and one with nonmelanoma solid tumors. In the BREAK-2, single-arm, open-label, Phase II trial, 76 patients with melanoma and BRAFV600E and 16 with BRAFV600K mutation were enrolled.28 The response rate was much better in the V600E group (59% with 7% complete response) than the V600K group (13%). PFS and OS were also longer in the V600E group (6.3 and 13.1 months, respectively), compared to patients with V600K (4.5 and 12.9 months, respectively).
Although the different response between patients harboring the V600E and those with V600K mutation cannot be adequately explained, it provides evidence supporting that these genotypes correspond to biologically distinct subtypes of melanoma, with V600K mutation associated with a significantly shorter disease-free interval but no difference in survival thereafter.22
In the Phase I study, a reduction in the size of brain metastases was reported in nine of ten patients, four of whom experienced a complete remission.19 Based on this observation, the BREAK-MB Phase II study was designed to assess the efficacy of dabrafenib in patients with BRAFV600E-mutant melanoma with untreated or recurrent/progressing after local treatments on brain metastases. The study found dabrafenib to be effective in both groups of patients (with previously treated brain metastasis and untreated ones). The reported survival was approximately three times longer compared to the temozolomide study (patients were not BRAF genotyped).8,29 The safety profile was acceptable; the three most frequent serious AEs were pyrexia (6%), intracranial hemorrhage (6%, one treatment related), and development of cSCC (6%).
The pivotal Phase III trial11 (BREAK-3) compared dabrafenib with dacarbazine (DTIC) in patients with stage IV or unresectable stage IIIC BRAFV600E melanoma with an Eastern Cooperative Oncology Group performance status of 0 or 1. The trial enrolled patients with a 3:1 randomization to receive either dabrafenib 150 mg or DTIC (1,000 mg/m2 every 3 weeks). Patients receiving DTIC crossed over to dabrafenib in case of disease progression. The primary endpoint of the study was PFS as assessed by a local investigator. Secondary endpoints were the following: PFS assessed by an independent review committee (IRC); OS; objective response rate (ORR) according to revised Response Evaluation Criteria in Solid Tumours guidelines, version 1.1,30 as assessed by an investigator and IRC; PFS after crossover from DTIC to dabrafenib, response duration, quality of life, safety, and tolerability. Confirmed responses by an IRC were recorded in 50% of dabrafenib patients (3% complete, 47% partial) and 6% of DTIC patients (2% complete, 4% partial). The median time to response in the dabrafenib group was 6.2 weeks. Similarly to the previous vemurafenib Phase III trial, a median PFS of 6.9 months was found for dabrafenib treatment versus 2.7 months for DTIC.
The most recent update at 16.9 months median follow-up31 reported a median OS of 20.0 months for dabrafenib versus 15.6 months for DTIC (59% of DTIC patients crossed over to dabrafenib arm). Of note, 18 patients (10%) remain on dabrafenib without disease progression.
Toxic AEs and cSCC are reported less frequently in patients treated with dabrafenib compared to those treated with vemurafenib, but a direct comparison has never been conducted.
Dabrafenib in combination
Although BRAFi were the first agents achieving a significant efficacy in metastatic melanoma, their beneficial effect is limited by the frequent development of acquired resistance, while ~15% of patients do not respond at all. Therefore, several challenges remain to be addressed to optimize the efficacy of these drugs and minimize treatment failures. Resistance to targeted therapy with BRAFi is a result of reactivation of the MAPK pathway, limiting the PFS benefit to 6–8 months. BRAF inhibitors have also been suggested to induce secondary primary tumor development through a paradoxical activation of the MAPK pathway in cells lacking BRAF mutations.
The estimated crucial role of the MAPK pathway in the development of resistance to BRAFi generated the hypothesis that its blockage by an MEK inhibitor might improve the efficacy of BRAFi. This led to the first combination treatment of a BRAF inhibitor (dabrafenib) with an MEK inhibitor (trametinib). Trametinib, an MEK1/2 inhibitor targeting the kinase downstream of BRAF in the MAPK pathway, is also active in monotherapy in BRAF-mutated melanomas but with lower efficacy than BRAF inhibitors. As opposed to dabrafenib, the metabolism of trametinib is predominantly nonhepatic, involving deacetylation as well as secondary modifications including oxidation and glucuronidation. Trametinib does not appear to have significant inhibitory activity toward CYP isozyme or transport proteins, limiting, thus, the possibility of interactions with dabrafenib.
The efficacy of the combination treatment has been tested in different schemes, including synchronous and sequential initiation of the two agents. It has been shown that inhibition of MEK by a single agent has limited value in patients with melanoma after progression on a BRAF inhibitor.32 Instead, the response rate to MEK inhibition was higher when this agent was given first, followed by the initiation of dabrafenib.12 However, all sequential regimens were shown to be inferior to the synchronous initiation of the two drugs.
Analytically, a Phase I/II study explored the combination of dabrafenib and trametinib in patients affected by advanced melanoma harboring mutations in BRAFV600E/K. Combination treatment with 150 mg of dabrafenib twice per day and 2 mg of trametinib daily was compared with monotherapy with 150 mg of dabrafenib twice per day. A significantly higher response rate (76% vs 54%, P=0.03) and a prolonged PFS was observed in the combination arm, compared to dabrafenib monotherapy.33 Furthermore, combination therapy was associated with a lower rate of cSCC development (7% vs 19%), whereas pyrexia was more frequent in the combination arm (71% vs 26%). Based on these promising data, US Food and Drug Administration approved the combination treatment of dabrafenib and trametinib for metastatic melanoma. Subsequently, the superiority of the combination regimen was further documented by the results of two Phase III trials. In COMBI-V study,34 dabrafenib plus trametinib were compared to vemurafenib plus placebo as first-line treatment for BRAF-mutant metastatic melanoma. The study was terminated early since, at the preplanned interim analysis, the combination treatment was assessed as significantly superior. In detail, patients receiving the combination treatment had a longer OS (median OS not reached vs 17.2 months), a longer PFS (11.4 vs 7.3 months; HR 0.56 [95% CI 0.46–0.69]; P<0.001), a prolonged response duration (13.8 vs 7.5 months) and a higher response rate (64% vs 51%; P<0.001).35 The last data cutoff was then performed at 349 events, with the combination treatment associated with a higher 2-year OS compared to vemurafenib (51% and 38%, respectively), a higher median OS (25.6 vs 18.0 months (HR 0.66; P<0.001), a prolonged PFS (12.6 vs 7.3 months, HR 0.61; P<0.001), and improved ORR and deep of response. Good prognostic features at baseline, associated with durable response and prolonged OS, were the following: lactate dehydrogenase, with a 2-year OS rate of 66% and a median PFS of 17.5 months, earlier-stage melanoma, and fewer metastatic sites.36
Furthermore, this study showed that treatment with the combination of dabrafenib plus trametinib does not result in deterioration of quality of life, adding a clear benefit over monotherapy with the BRAF inhibitor vemurafenib.34 The latter finding is highly relevant in clinical practice, since, in addition to the survival benefit, the minimization of disease-associated symptoms and drug-associated AEs also represent pursued goals.
In the second Phase III study, COMBI-D, untreated BRAF-mutant patients were randomized to receive either dabrafenib and trametinib or dabrafenib plus placebo. The median OS for the combination group was 25.1 months (95% CI 19.2 to not reached) versus 18.7 months (15.2–23.7 95% CI) for the monotherapy group, while 1- and 2-year survival rates were also higher in the combination group. Among patients receiving the combination of dabrafenib, 87% experienced treatment-related AEs, compared to 90% of patients in the dabrafenib group. Fever was the most common AE (52%) in the combination group and hyperkeratosis (33%) in the monotherapy group, while grade 3 or 4 AEs were similar in the two groups (32% and 31%).37
Finally, a Phase III randomized, double-blind and placebo-controlled trial (COMBI-AD) is underway, assessing the efficacy of the combination of dabrafenib plus trametinib as an adjuvant therapy, following surgical excision, of high-risk BRAF mutation-positive melanoma (NCT0909453).
Since targeted therapy has an important effect on the immune system, the possibility of combining a BRAF or MEK inhibitor with immunotherapy is an interesting approach. However, Phase I data showed that combined administration of vemurafenib and ipilimumab increases liver toxicity (although this was not reported with dabrafenib plus ipilimumab),38 while the triple combination of ipilimumab plus dabrafenib and trametinib was reported to increase the risk of bowel perforation. The development of anti-PD-1/PD-L1 agents, which appear to be more effective and less toxic than ipilimumab, reintroduces the possibility of a combined approach with BRAF or MEK inhibitor. Indeed, a Phase I study reported data on the combination of the anti-PD-L1 antibody, MEDI14736 (durvolumab) with dabrafenib and trametinib in patients with clinical stage IIIC or stage IV melanoma. The triple combination resulted in an ORR of 69% and disease control rate of 100%, showing also a manageable safety profile. However, longer follow-up will be necessary to determine the efficacy and safety of the triple drug combination.39
Safety evaluation
Cumulative experience with the two BRAFi, vemurafenib and dabrafenib, has shown that, although some toxicities such as skin toxicity, joint pain, and fever are common with both inhibitors, their type and severity vary considerably and may influence the choice of the drug (Table 2). For example, photosensitivity is common in patients treated with vemurafenib and much less frequent in patients treated with dabrafenib, whereas fever and chills are much more frequent with dabrafenib treatment. Skin toxicities are the most common AEs associated with BRAFi, experienced by up to 57% of patients, and appearing within days after therapy initiation (Figures 1–4).
Table 2.
Skin toxicity
| Vemurafenib | Dabrafenib |
|---|---|
| Rash: 49% | Hyperkeratosis: 39% |
| Photosensitivity: 31% | Photosensitivity: rare |
| SCC: 19% | SCC: 10% |
| Alopecia: 26% | Alopecia: rare |
| Others (pruritus, dry skin, papillomas): frequent | Others (pruritus, dry skin, papillomas): not frequent |
Abbreviation: SCC, squamous cell carcinoma including keratoacanthoma.
Figure 1.
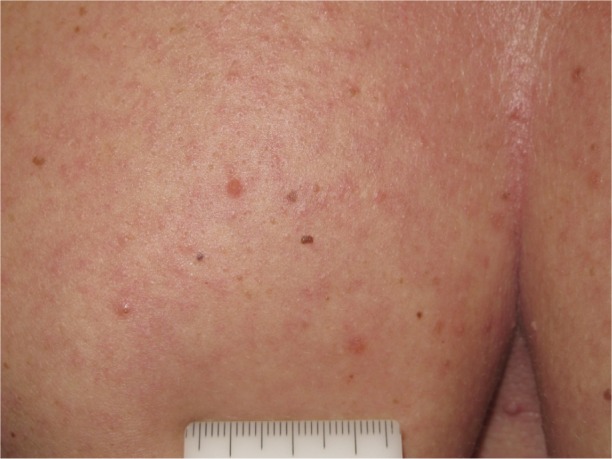
Acantholytic dyskeratosis.
Figure 2.
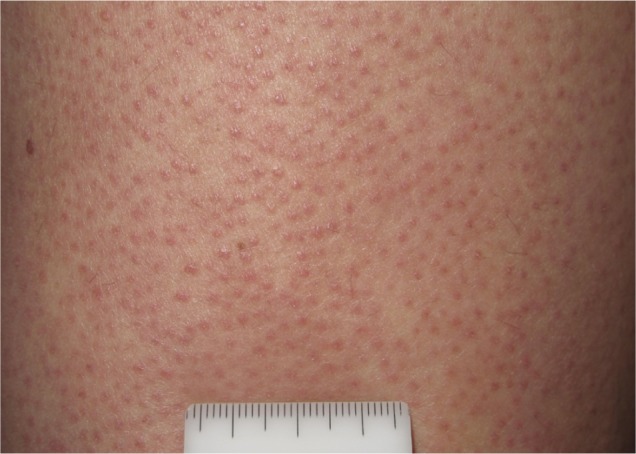
Keratosis pilaris.
Figure 3.
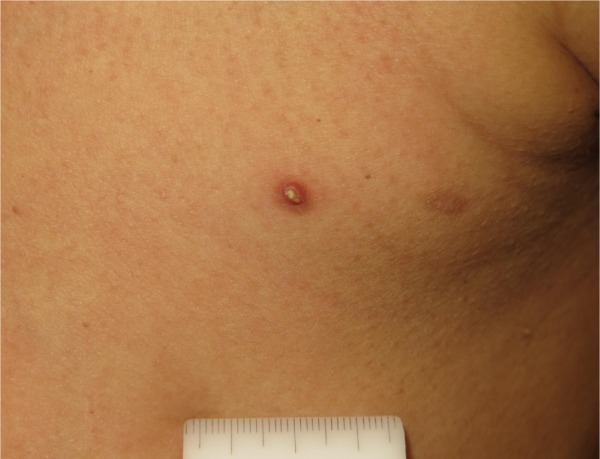
Verrucous keratosis.
Figure 4.
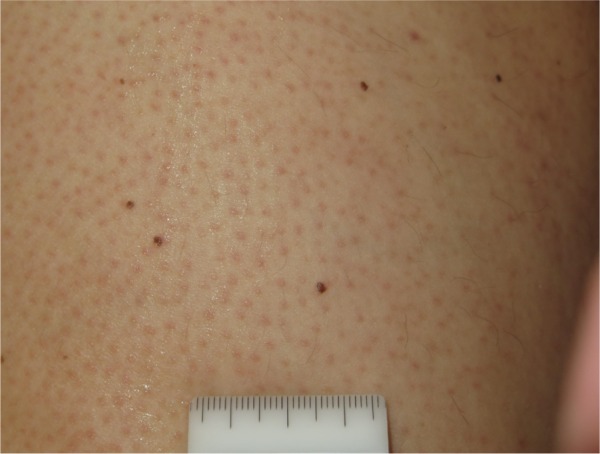
New and enlarging melanocytic nevi.
Overall, dabrafenib is well tolerated by the patients, including those with brain metastases, since its side effects are common but usually manageable. The toxicities in the Phase III trial were similar to those observed in early-phase trials.11 The most common grade 2 or higher AEs were cutaneous manifestations (hyperkeratosis, papillomas, palma-plantar erythrodysesthesia), pyrexia, fatigue, headache, and arthralgia. In the BREAK-3 trial,11 7% of patients developed cSCC/KA, three patients (2%) developed new primary melanomas, while phototoxicity was rare (3%), as were grade 3 AEs. Dose reductions were necessary in 28% of patients, and therapy was permanently discontinued due to toxicity in only 3% of patients. In the BREAK-MB study, 82% of patients experienced at least one grade 2 or higher side effect and 22% had grade 3 or higher AE. However, only 2% discontinued dabrafenib due to toxicity.
Although vemurafenib is considered to be well tolerated as well, adverse effects are frequent, with skin toxicity representing the most common problem. Among several skin AEs reported in the literature, the most common are alopecia, photosensitivity, pruritus, hand–foot skin reactions (HFSR), cutaneous manifestations resembling hyperkeratotic and dyskeratotic diseases (Figure 1), follicular-centered eruption resembling keratosis pilaris (Figure 2),40 seborrheic dermatitis-like eruptions, and Darier or Grover-like eruptions. Less frequent events include pyogenic granuloma, gingival hyperplasia, and lupus erythematosus-like skin eruption.41–43
The development of keratosis pilaris-like eruptions associated with facial erythema and HFSR in patients treated with vemurafenib validate the association of facial erythema with the BRAF pathway and indicate VEGF inhibition as the molecular mechanism responsible for HFSR. The specificity of vemurafenib for BRAF suggests that inhibition of the BRAF pathway alone is sufficient to induce HFSR.40 The occurrence of malignant and benign hyperproliferative skin lesions like cSCC, KA, warty dyskeratoma, and verrucous keratosis (Figure 3) has also been documented. The paradoxical phenomenon of vemurafenib-induced SCCs suggests that transformation of sensitive cells, harboring clinically silent RAS mutations, is regulated by BRAF-inhibitor-induced MAPK signaling via noninhibited RAF isoforms, highlighting the complexity and redundancy of kinase signaling.44 Notably, cSCCs and KAs associated with vemurafenib therapy are easily treated by simple excision, without requiring discontinuation of vemurafenib. Another peculiar side effect of vemurafenib is the darkening of existing nevi and the appearance of new nevi within 2 months after drug initiation (Figure 4).45–47 Skin toxicities generally cannot be prevented, but rarely require permanent discontinuation of the treatment, being usually adequately managed with dose modification and appropriate therapy.48,49
The absence of photosensitivity with dabrafenib and the lower frequency of cutaneous AEs suggest dabrafenib as an appropriate alternative treatment option for patients who are intolerant to vemurafenib due to skin toxicity.
The combination of dabrafenib plus trametinib and vemurafenib plus cobimetinib resulted in similar clinical efficacy. Therefore, the selection of combination regimen might mainly be based on its expected toxicity. Pyrexia is the most frequent AE reported with the combination of dabrafenib and trametinib (51%–53%; G3: 4%–6%), representing the most common reason for dose interruptions, dose reductions, and permanent discontinuation.34,35,37 No baseline features have been identified to predict pyrexia, and it does not seem to be associated with the clinical outcome.50 Other frequent AEs include fatigue (35%), nausea (30%–35%), headache (30%), chills (30%–31%), diarrhea (24%–32%), arthralgia (24%), rash (22%–23%), and hypertension (22%).34,35,37
Cardiac-related AEs may occur when trametinib is administered as a single agent or in combination with dabrafenib. Specifically, trametinib has been reported to decrease LVEF. In clinical trials, the median time to the first occurrence of left ventricular dysfunction, cardiac failure, and LVEF decrease was between 2 and 5 months. Integrated safety data from COMBI-D (N=209) and COMBI-V studies (N=559) suggest a decreased ejection fraction as a common AE, whereas LVEF dysfunction and cardiac failure were not reported during COMBI-D, but were noted as uncommon events in the integrated safety analysis.
Pyrexia was reported also in patients treated with the combination of vemurafenib and cobimetinib, although to a much lesser extent as regarding frequency and severity (26%; G3: 2%).51 On the other hand, photosensitivity reaction (28%), diarrhea (56%; G3: 6%), increased aspartate/alanine aminotransferase (22%–23%; G3: 8%–11%) and increased creatinine kinase (27%; G3: 7%) are more frequent and severe with vemurafenib plus cobimetinib than dabrafenib plus trametinib. Ocular toxicity is also more commonly reported in patients treated with vemurafenib and cobimetinib (chorioretinopathy: 1%), though most of these events were low-grade and reversible without any treatment, or with dose reduction/withdrawal of cobimetinib.52
The most striking safety difference between combination therapy and monotherapy is the decreased incidence of new skin cancers and other hyperproliferative skin lesions. As discussed earlier, this finding is consistent with the suggested pathogenesis of these tumors, which includes a paradoxical activation of the MAPK pathway with upstream activation of signaling by preexisting RAS mutation.53 The addition of an MEK inhibitor leads to a block of RAS signaling along the MAPK pathway and prevents the cellular proliferation.
Conclusion
BRAF inhibitors (vemurafenib and dabrafenib) have become worldwide standards of care for patients with BRAF-mutant metastatic melanoma, especially those with high tumor burden or progression after immunotherapy. Both agents improve survival, compared with chemotherapy, and have acceptable toxicity profiles. Combined BRAF and MEK inhibition achieves a statistically significant further improvement in response rate, PFS, and OS compared to monotherapy. However, the majority of the patients develop resistance and tumor progression similar to that observed with BRAF inhibitor monotherapy. Preclinical evidence, a Phase I trial,45 reported promising data indicating that a multitargeted upfront approach, including immunotherapy, might have the potential to achieve the greatest survival benefit for patients with metastatic melanoma.
Definitely, the appropriate selection of systemic therapy for metastatic melanoma remains an evolving field and requires further elucidation. For instance, long-term data on the combination therapy with dabrafenib and trametinib revealed that long-term survival and durable responses are associated with good prognostic features at baseline, including factors related to low-volume disease.54 However, such baseline factors are classically considered a reason to choose front-line immunotherapy. The need to develop specific predictive molecular markers for each therapy is now more important than ever. Another issue requiring further clarification is the determination of the optimal sequence of administered therapeutic agents, since it remains unknown whether treating patients with ipilimumab and nivolumab followed by dabrafenib and trametinib is more effective than treatment with dabrafenib and trametinib followed by ipilimumab and nivolumab (ongoing trial NCT02224781).
Finally, neoadjuvant BRAF-targeted therapy in advanced locoregional BRAFV600 mutant melanoma patients represents a novel challenge to be addressed, with a relevant clinical trial urgently required.
Acknowledgments
Study supported in part by the Italian Ministry of Health (RF-2010-2316524).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Ferlay J, Parkin DM, Steliarova-Foucher E. Estimates of cancer incidence and mortality in Europe in 2008. Eur J Cancer. 2010;46:765–781. doi: 10.1016/j.ejca.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.Garbe C, Eigentler TK, Keilholz U, Hauschild A, Kirkwood JM. Systematic review of medical treatment in melanoma: current status and future prospects. Oncologist. 2011;16:5–24. doi: 10.1634/theoncologist.2010-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serrone L, Zeuli M, Sega FM, Cognetti F. Dacarbazine-based chemotherapy for metastatic melanoma: thirty-year experience overview. J Exp Clin Cancer Res. 2000;19(1):21–34. [PubMed] [Google Scholar]
- 5.Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 6.Schwartzentruber DJ, Lawson DH, Richards JM, et al. Gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–2127. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guirguis LM, Yang JC, White DE, et al. Safety and efficacy of high-dose interleukin-2 therapy in patients with brain metastases. J Immunother. 2002;25(1):82–87. doi: 10.1097/00002371-200201000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agarwala SS, Kirkwood JM, Gore M, et al. Temozolomide for the treatment of brain metastases associated with metastatic melanoma: a phase II study. J Clin Oncol. 2004;22(11):2101–2107. doi: 10.1200/JCO.2004.11.044. [DOI] [PubMed] [Google Scholar]
- 9.Seigler HF, Lucas VS, Pickett NJ, Huang AT. DTIC, CCNU, bleomycin, and vincristine in metastatic melanoma. Cancer. 1980;46(11):2346–2348. doi: 10.1002/1097-0142(19801201)46:11<2346::aid-cncr2820461104>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 10.McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–332. doi: 10.1016/S1470-2045(14)70012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauschild A, Grob J-J, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 12.Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 13.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signaling pathways in cancer. Oncogene. 2007;26(22):3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 15.Carnahan J, Beltran PJ, Babij C, et al. Selective and potent Raf inhibitors paradoxically stimulate normal cell proliferation and tumor growth. Mol Cancer Ther. 2010;9:2399–2410. doi: 10.1158/1535-7163.MCT-10-0181. [DOI] [PubMed] [Google Scholar]
- 16.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19(11):1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 17.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kefford R, Arkenau H, Brown MP, et al. Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors. J Clin Oncol. 2010;28(15 Suppl):S611. Abstract 850. [Google Scholar]
- 19.Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumors: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893–1901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trefzer U, Minor DR, Ribas A, et al. BREAK-2: a phase IIA trial of the selective BRAF kinase inhibitor GSK2118436 in patients with BRAF (V600E/K)-positive metastatic melanoma [abstract] Pigment Cell Melanoma Res. 2011;24:1020. Abstract LBA 1021–1021. [Google Scholar]
- 21.Bauer J, Büttner P, Murali R, et al. BRAF mutations in cutaneous melanoma are independently associated with age, anatomic site of the primary tumor, and the degree of solar elastosis at the primary tumor site. Pigment Cell Melanoma Res. 2011;24(2):345–351. doi: 10.1111/j.1755-148X.2011.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menzies AM, Haydu LE, Visintin L, et al. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin Cancer Res. 2012;18(12):3242–3249. doi: 10.1158/1078-0432.CCR-12-0052. [DOI] [PubMed] [Google Scholar]
- 23.Si L, Kong Y, Xu X, et al. Prevalence of BRAF V600E mutation in Chinese melanoma patients: large scale analysis of BRAF and NRAS mutations in a 432-case cohort. Eur J Cancer. 2012;48(1):94–100. doi: 10.1016/j.ejca.2011.06.056. [DOI] [PubMed] [Google Scholar]
- 24.Rimoldi D, Salvi S, Liénard D, et al. Lack of BRAF mutations in uveal melanoma. Cancer Res. 2003;63(18):5712–5715. [PubMed] [Google Scholar]
- 25.Sosman JA, Kim KB, Schuter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davies MA, Liu P, McIntyre S, et al. Prognostic factors for survival in melanoma patients with brain metastases. Cancer. 2011;117(8):1687–1696. doi: 10.1002/cncr.25634. [DOI] [PubMed] [Google Scholar]
- 28.Ascierto PA, Minor D, Ribas A, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol. 2013;31(26):3205–3211. doi: 10.1200/JCO.2013.49.8691. [DOI] [PubMed] [Google Scholar]
- 29.Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 study. Lancet Oncol. 2012;13(11):1087–1095. doi: 10.1016/S1470-2045(12)70431-X. [DOI] [PubMed] [Google Scholar]
- 30.Carlino MS, Fogarty GB, Long GV. Treatment of melanoma brain metastases: a new paradigm. Cancer J. 2012;18(2):208–212. doi: 10.1097/PPO.0b013e31824b2890. [DOI] [PubMed] [Google Scholar]
- 31.Hauschild A, Grob JJ, Demidov LV, et al. An Update on Overall Survival (OS) and Follow-On Therapies in BREAK-3, a Phase III, Randomized Trial: Dabrafenib (D) vs. Dacarbazine (DTIC) in Patients (pts) with BRAF V600E Mutation-Positive Metastatic Melanoma (MM); Poster presented at ESMO Annual Meeting; 29 September, 2014; Madrid. [Google Scholar]
- 32.Kim KB, Kefford R, Pavlick AC, et al. Phase II study of the MEK1/MEK2 inhibitor trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31:482–489. doi: 10.1200/JCO.2012.43.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grob JJ, Amonkar MM, Karaszewska B, et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val 600-mutation-positive melanoma (COMBI-V): results of a phase 3, open label, randomized trial. Lancet Oncol. 2015;16:1389–1398. doi: 10.1016/S1470-2045(15)00087-X. [DOI] [PubMed] [Google Scholar]
- 35.Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 36.Robert C, Karaszewska B, Schachter J, et al. Two year estimate of overall survival in COMBI-v, a randomized, open-label, phase III study comparing the combination of dabrafenib and trametinib with vemurafenib as first-line therapy in patients with unresectable or metastatic BRAF V600E/K mutation-positive cutaneous melanoma; ECC 2015; 28 September, 2015; Vienna. Abstract 3301. [Google Scholar]
- 37.Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomized controlled trial. Lancet. 2015;386(9992):444–451. doi: 10.1016/S0140-6736(15)60898-4. [DOI] [PubMed] [Google Scholar]
- 38.Puzanov I, Callahan MK, Linette GP, et al. Phase I study of the BRAF inhibitor dabrafenib with or without the MEK inhibitor trametinib in combination with ipilimumab for V600E/K mutation-positive unresectable or metastatic melanoma. J Clin Oncol. 32(suppl):5s. abstr 2511. [Google Scholar]
- 39.Ribas A, Butler M, Lutzky J, et al. Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and MEK (trametinib) inhibitors in advanced melanoma. J Clin Oncol. 33(suppl) abstr 3003. [Google Scholar]
- 40.Huang V, Hepper D, Anadkat M, Cornelius L. Cutaneous toxic effects associated with vemurafenib and inhibition of the BRAF pathway. Arch Dermatol. 2012;148:628–633. doi: 10.1001/archdermatol.2012.125. [DOI] [PubMed] [Google Scholar]
- 41.Sammut SJ, Tomson N, Corrie P. Pyogenic granuloma as a cutaneous adverse effect of vemurafenib. N Engl J Med. 2014;371(13):1265–1267. doi: 10.1056/NEJMc1407683. [DOI] [PubMed] [Google Scholar]
- 42.Mangold AR, Bryce A, Sekulic A. Vemurafenib-associated gingival hyperplasia in patient with metastatic melanoma. J Am Acad Dermatol. 2014;71(5):e205–e206. doi: 10.1016/j.jaad.2014.03.043. [DOI] [PubMed] [Google Scholar]
- 43.Reinholz M, Berking C, Hermans C, Ruzicka T, Braun-Falco M. Lupus erythematosus-like skin eruption after vemurafenib therapy. J Am Acad Dermatol. 2014;71(4):e159–e160. doi: 10.1016/j.jaad.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 44.Wang CM, Fleming KF, Hsu S. A case of vemurafenib-induced keratosis pilaris-like eruption. Dermatol Online J. 2012;18:7. [PubMed] [Google Scholar]
- 45.Menzies AM, Kefford RF, Long GV. Paradoxical oncogenesis: are all BRAF inhibitors equal? Pigment Cell Melanoma Res. 2013;26:611–615. doi: 10.1111/pcmr.12132. [DOI] [PubMed] [Google Scholar]
- 46.Göppner D, Müller J, Krüger S, Franke I, Gollnick H, Quist SR. High incidence of naevi-associated BRAF wild-type melanoma and dysplastic naevi under treatment with the class I BRAF inhibitor vemurafenib. Acta Derm Venereol. 2014;94(5):517–520. doi: 10.2340/00015555-1813. [DOI] [PubMed] [Google Scholar]
- 47.Perier-Muzet M, Thomas L, Poulalhon N, et al. Melanoma patients under vemurafenib: prospective follow-up of melanocytic lesions by digital dermoscopy. J Invest Dermatol. 2014;134(5):1351–1358. doi: 10.1038/jid.2013.462. [DOI] [PubMed] [Google Scholar]
- 48.Chu EY, Wanat KA, Miller CJ, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: A clinicopathologic study. J Am Acad Dermatol. 2012;67(6):1265–1272. doi: 10.1016/j.jaad.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sinha R, Edmonds K, Newton-Bishop JA, Gore ME, Larkin J, Fearfield L. Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: practical advice on diagnosis, prevention and management of the main treatment-related skin toxicities. Br J Dermatol. 2012;167:987–994. doi: 10.1111/bjd.12010. [DOI] [PubMed] [Google Scholar]
- 50.Dummer R, Rinderknecht J, Goldinger SM. Ultraviolet A and photosensitivity during vemurafenib therapy. N Engl J Med. 2012;366:480–481. doi: 10.1056/NEJMc1113752. [DOI] [PubMed] [Google Scholar]
- 51.Menzies AM, Ashworth MT, Swann S, et al. Characteristics of pyrexia in BRAFV600E/K metastatic melanoma patients treated with combined dabrafenib and trametinib in a phase I/II clinical trial. Ann Oncol. 2014;26:415–421. doi: 10.1093/annonc/mdu529. [DOI] [PubMed] [Google Scholar]
- 52.Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 53.Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–215. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Long VG, Weber JS, Infante JR, et al. Overall survival and durable responses in patients with BRAF V600_mutant metastatic melanoma receiving dabrafenib combined with trametinib. J Clin Oncol. 2016 Jan 25; doi: 10.1200/JCO.2015.62.9345. Epub. [DOI] [PubMed] [Google Scholar]