

Abstract
Transcriptome analysis is a widely used approach to study the molecular mechanisms underlying development and the responses of fungi to environmental cues. However, it is difficult to obtain cells with a homogeneous status from the sexually-induced culture of the plant pathogenic fungus Fusarium graminearum. In this study, we provided phenotypic and genetic evidence to show that the current conditions applied for perithecia induction inevitably highly induced asexual sporulation in this fungus. We also found that hundreds of genes under the control of the conidiation-specific gene ABAA were unnecessarily upregulated after perithecia induction. Deletion of ABAA specifically blocked conidia production in both the wild-type strain and sexually-defective mutants during sexual development. Taken together, our results suggest that the abaA strain could be used as a background strain for studies of the initial stages of perithecia production in F. graminearum. Further comparative transcriptome analysis between the abaA mutant and the sexually-defective transcription factor mutant carrying the ABAA deletion would contribute to the construction of the genetic networks involved in perithecia development in F. graminearum.
Introduction
Fusarium head blight (FHB) is an important and widespread disease of major small grains. Fusarium graminearum is the primary causative agent of FHB worldwide [1]. In addition to yield and quality losses, the importance of FHB lies in the accumulation of harmful mycotoxins [2]. FHB epidemics continue to occur throughout the world in accordance with recent emerging fungal diseases in animals and plants [3,4]. Because few resistant cultivars and fungicides with effective applications and competitive prices are available, FHB remains difficult to control [4]. Therefore, understanding the molecular mechanisms involved in the F. graminearum life cycle is required to develop effective strategies to control FHB.
F. graminearum uses conidia (asexual spores), ascospores (sexual spores), and hyphal fragments as disease inocula [5]. Whereas conidia and hyphal fragments are dispersed short distances by wind or rain-splash, ascospores in the perithecia are forcibly discharged into the air and can move several kilometers; therefore, ascospores function as the primary inocula instead of conidia [6,7]. Moreover, perithecia and perithecia-associated mycelia are known to play roles in the overwintering process [8]. Sexual reproduction ensures genetic diversity in the population, which provides a capacity for adaptability towards host plants [9,10].
Sexual reproduction of F. graminearum is accompanied by distinct alterations in cellular differentiation processes and metabolism [11,12]. Therefore, fine-tuned temporal and spatial gene regulation is necessary for proper fertilization, fruiting body formation, ascospore maturation and discharge. Because numerous metabolic and major signal transduction pathways are closely linked to these processes, disruption of the genes involved in these processes commonly results in defects in various steps of F. graminearum sexual development [13,14]. For instance, a genome-wide functional analysis of transcription factors and kinases identified hundreds of genes involved in sexual development and contributed to the comprehensive understanding of the genetic networks involved in F. graminearum sexual reproduction [15,16].
Transcriptome analysis utilizing the next-generation sequencing-based RNA-seq is a powerful approach that can be used to study the molecular mechanisms involved in various developmental stages and the responses of fungi to specific environmental stresses. Additionally, comparative transcriptome analysis between the wild-type strain and null mutants enables the characterization of novel genes or genetic pathways that are under the control of upstream signal transduction pathways or specific transcription factors. Recent advances in next-generation sequencing technologies enabled researchers to perform transcriptome analyses even in non-model filamentous fungi [17].
One of the critical factors that determines the success of transcriptome analysis is the extraction of mRNA from cells with a homogeneous status to exclude false-positive results. In filamentous fungi, there has been a limit to the ability to obtain fungal cells with the same physiological and/or developmental status because few conditions are available for the simultaneous induction of specific developmental stages. Recent advances in RNA-seq technology enabled transcriptome analysis with a minute amount of sample, such as microdissected tissues and even a single cell [18,19]. However, most high-tech methods are difficult to apply to filamentous fungi due to the limited accessibility of equipment and technological limitations.
It is also very important to obtain morphologically and/or physiologically distinct cells for the study of sexual development in filamentous fungi such as F. graminearum. However, mycelial growth and asexual sporulation commonly occur, and only a small portion of hyphal cells differentiate into fruiting bodies after sexual induction. Therefore, the development of specific conditions to suppress other developmental processes or dissecting methods to obtain specific cells/tissues are required to obtain a high quality transcriptome during sexual development in filamentous fungi.
AbaA is a well-known central regulator for conidiation and the distinct genetic pathway, BrlA-AbaA-WetA, orchestrates each step of conidiogenesis in Aspergillus nidulans and A. fumigatus [20,21]. In a previous study, we identified and functionally characterized the ABAA ortholog, which was previously thought to be absent in F. graminearum [22]. We found that AbaA is specifically required for phialide formation and function, and that AbaA exclusively localized to nuclei during conidiogenesis. Subsequent studies demonstrated that the AbaA-WetA pathway of A. nidulans is conserved in F. graminearum [23]. WetA is under control of AbaA and is required for phialide function and conidia maturation.
In this study, we found that the current condition for in vitro sexual induction highly induced asexual sporulation and resulted in the unnecessary expression of conidiation-related genes that disturbed proper transcriptome analysis in F. graminearum. Based on our previous genetic results and bioinformatics verification, we propose that the conidia nonproducing mutant, abaA, is useful for studies of F. graminearum sexual development.
Materials and Methods
Fungal strains and media
The wild-type strain Z-3639 and transgenic strains derived from this strain were used in this study (Table 1). The conidia nonproducing mutant ΔabaA and transcription factor mutants were described in previous studies [15,22]. All strains were stored as conidia and mycelia in a 30% glycerol solution at -80°C. Conidia production was induced on yeast malt agar (YMA) as previously described [24]. All other media used in this study were prepared as described in the Fusarium laboratory manual [25].
Table 1. F. graminearum strains used in this study.
| Strain | Genotype | Source or reference |
|---|---|---|
| Z-3639 | Fusarium graminearum wild-type | [26] |
| ΔabaA | ΔabaA::GEN | [22] |
| Δmat2 | ∆mat1-2::GFP-HYG | [27] |
| HK167 | Δmat1-2::HYG ΔabaA::GEN | Δmat2 × ΔabaA |
| Δgzp53l005 | Δgzp53l005::GEN | [15] |
| Δgzzc183 | Δgzzc183::GEN | [15] |
| Δgzzc258 | Δgzzc258::GEN | [15] |
| ΔFgnot3 | ΔFgnot3::GEN | [15,28] |
| Δgzzc302 | Δgzzc302::GEN | [15] |
| HK327 | Δgzp53l005::GEN ΔabaA::GEN | HK167 × Δgzp53l005 |
| HK328 | Δgzzc183::GEN ΔabaA::GEN | HK167 × Δgzzc183 |
| HK329 | Δgzzc258::GEN ΔabaA::GEN | HK167 × Δgzzc258 |
| HK330 | ΔFgnot3::GEN ΔabaA::GEN | HK167 × ΔFgnot3 |
| HK331 | Δgzzc302::GEN ΔabaA::GEN | HK167 × Δgzzc302 |
Sexual crosses
Sexual reproduction was induced by removing 5-day-old aerial mycelia with the back of the surgical blade (surgical blade #11; Feather Safety Razor, Osaka, Japan) and 2.5% sterilized Tween 60 solution for the self-cross [1]. For the outcrosses, the heterothallic ∆mat2 strain was fertilized with 1 ml of a conidial suspension (105 conidia/ml) from fertilizing parents [27]. All sexually-induced cultures were incubated under near UV light (365 nm wavelength; HKiv Import & Export Co., Ltd., Xiamen, China) at 25°C.
RNA-seq and bioinformatics analysis
The RNA-seq analysis was performed as previously described [22]. Total RNA was extracted using an Easy Spin Total RNA Extraction Kit (iNtRON Biotech, Seongnam, Korea). RNA-seq libraries were constructed using the Illumina TruSeqTM RNA sample prep kit (Illumina, San Diego, CA USA) with no modifications to the standard low-throughput protocol. Samples were run on an Illumina HiSeq2000 instrument using the reagents provided in the Illumina TruSeq paired-end (PE) cluster kit V3-cBot-HS and the TruSeq SBS kit v3-HS (200 cycles).
The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus [29] and are accessible through GEO Series accession number GSE46133 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE46133) and GSE79532 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE79532). Raw RNA-seq data from 2 h and 24 h after sexual induction (WT-2 h and WT-24 h) were obtained in a previous study [30] and realigned into the genome obtained from the Fusarium graminearum Genome Database [31] for the comparative transcriptome analysis.
Alignments were generated with BWA [32] using the F. graminearum genome [31] and the htseq-count script in the HTSeq package was used to compute the counts per gene [33]. Genome-wide transcript levels were quantified in reads per kilobase of exon per million mapped sequence reads (RPKM) [34]. Genes with maximum RPKM × 100 values below 10 were removed from the analysis. Three RNA-seq replicates were performed for each sample and mean values of them were used to calculate fold changes. Differentially expressed genes were identified based on fold change values. The time-course expression patterns during conidiation were clustered into 10 groups using the R package Mfuzz with default setting, which performs fuzzy c-means clustering [35].
Results and Discussion
Conidia production after sexual induction in F. graminearum
Sexual and asexual reproduction are commonly induced together in various Fusarium species, including F. graminearum [1]. To exclude the effects of conidiation from the transcriptome analysis during perithecia production in F. graminearum and F. verticillioides, in a previous study the surface of the agar medium was washed and visible perithecia were picked for RNA extraction [30]. Similarly, we observed that vigorous conidia production occurred in the current condition applied for F. graminearum sexual reproduction (Fig 1A and 1C). The conidia number peaked 2 days after sexual induction and was highly maintained during the sexual developmental stages. The results demonstrated that transcripts accumulated during conidiation; therefore, the transcripts stored within conidia should be included in the transcriptome of sexually-induced fungal cultures, especially during the initial stages (~ 2 days). Thousands of genes exhibit altered expression during F. graminearum conidiation [22,23], and dormant conidia have been known to store many transcripts in filamentous fungi [36,37].
Fig 1. Induction of asexual sporulation during perithecia production in F. graminearum.

(A) Conidia production after sexual induction. The number of conidia (conidia/mm2) was counted 0–5 days after sexual induction in the wild-type strain. (B) ABAA and WETA transcript levels during asexual and sexual reproduction. The transcript levels were quantified in reads per kilobase of exon per million mapped sequence reads (RPKM). (C) Mycelia and perithecia produced from the wild-type and abaA mutant strains. Scale bars represent 50 μm and 500 μm for the upper and below pictures, respectively. Arrows indicate mature conidia.
To analyze the transcriptome during conidiogenesis and the initial stage of perithecia production, we performed RNA-seq 3 and 9 h after conidia induction and 1 day after sexual induction in the F. graminearum wild-type strain Z-3639. The transcriptomes 6 and 12 h after conidia induction were obtained in a previous work [22]. Because all of the RNA-seq experiments were performed using the same platform (GEO platform ID, GPL17573) and normalized together, the RPKM values of each sample could be compared.
We investigated the expression of the conidiation-specific genes ABAA and WETA during conidiogenesis (Fig 1B). Our previous results showed that the AbaA-WetA pathway was specifically involved in phialide formation and function in F. graminearum [22,23]. In accordance with our previous results, ABAA transcripts began to increase 6 h after conidia induction and peaked at 12 h. WETA was expressed later than ABAA, confirming the involvement of the AbaA-WetA pathway for conidiation in this fungus. Transcript amounts of both ABAA and WETA 1 day after sexual induction were approximately half of those detected 12 h after conidia induction (Fig 1B). Taken together, we concluded that the present condition for sexual reproduction also highly induced asexual sporulation in F. grainearum.
Induction of conidiation-related genes during the initial stage of perithecia development
We investigated the extent of the distortion of the perithecia production transcriptome by unintended conidia induction in F. graminearum. ABAA deletion resulted in a complete lack of conidia production but normal sexual development (Fig 1C). Moreover, ABAA deletion did not affect any other fungal developmental stages in F. graminearum, including vegetative growth and virulence [22]. To analyze the abaA mutant transcriptome during the initial stage of perithecia development, we performed RNA-seq 1 day after sexual induction of the abaA mutant and compared the results with other transcriptome data (S1 Table). Overall, 933 and 472 genes were upregulated and downregulated, respectively, in the wild-type strain compared to the abaA mutant, showing that a profound alteration in gene expression occurred when conidiation was blocked during sexual development.
We compared genes that were differentially expressed due to the ABAA deletion (WT-1 d/abaA-1 d > 3 or < 3) with genes that exhibited altered expression during the initial stage of perithecia development in the wild-type strain (WT-24 h/WT-2 h > 2 or < 2). Raw RNA-seq data 2 h and 24 h after sexual induction were obtained from a previous study [30] and realigned for our analysis (S1 Table). Overall, 4200 and 1929 genes were upregulated and downregulated, respectively, 24 h after sexual induction compared to the uninduced condition (2 h). More than half of the upregulated genes in the wild-type strain compared to the abaA mutant exhibited simultaneous increases in expression during the initial stage of sexual induction (WT-1 d/abaA-1 d > 3 and WT-24 h/WT-2 h > 2; 570/933); however, a small portion of genes overlapped with genes that were downregulated during perithecia development (WT-1 d/abaA-1 d > 3 and WT-24 h/WT-2 h < 2; 98/933) (Fig 2A). The result suggests that approximately 14% (570/4200) of the sexually-induced genes may be related to conidiation but not perithecia development in F. graminearum. Among the downregulated genes in the wild-type strain compared to the abaA mutant, a relatively high proportion of genes were repressed during the initial stage of sexual induction (WT-1 d/abaA-1 d < 3 and WT-24 h/WT-2 h < 2; 102/472) (Fig 2B).
Fig 2. Induction of conidiation-related genes during the initial stage of sexual development.

(A) Venn diagrams illustrating the overlap between upregulated (A) or downregulated (B) wild-type-specific genes (blue circle, WT-1 d/abaA-1 d) and differentially expressed genes after sexual induction in the wild-type strain (green circle for downregulated genes, WT-24 h/WT-2 h < 2; orange circle for upregulated genes, WT-24 h/WT-2 h > 2). (C) Expression profiles of clustered groups including the conidiation-specific genes WETA and ABAA. Fuzzy clustering categorized 933 upregulated wild-type-specific genes into 10 groups. The genes included in groups 3 and 9 showed similar expression patterns during conidiation with WETA and ABAA, respectively. (D) The Venn diagram illustrating the overlap between the WETA- and ABAA-related genes (blue circle) and the upregulated genes after sexual induction in the wild-type strain (orange circle). Wild-type-specific genes (WT-1 d/abaA-1 d) represent differentially expressed genes in the wild-type strain compared to the abaA mutant 1 day after sexual induction. Genes that were differentially expressed 24 h after sexual induction compared to the uninduced condition (2 h) represent differentially expressed genes after sexual induction [30].
To dissect the relationship between the abaA-specific genes and conidiogenesis, we focused on downregulated genes in the abaA mutant (WT-1 d/abaA-1 d > 3, 933 genes) and categorized them into 10 groups depending on their expression patterns during conidiation (S2 Table and S1 Fig). Genes included in clusters 3 (239 genes) and 9 (125 genes) showed expression patterns similar to WETA and ABAA, respectively (Fig 2C). We hypothesized that genes under control of WetA or AbaA were members of these groups. Intriguingly, most genes in clusters 3 and 9 (264/364) were upregulated during the initial stage of perithecia development (Fig 2D). These results suggest that many of the genes required for asexual reproduction but not sexual development were highly expressed after sexual induction in F. graminearum.
Genome-wide functional analysis of transcription factors revealed the distinct positive correlation between the expression level and biological significance in F. graminearum [15]. We investigated the genetic correlation of fold changes between conidia-deficient and sexually-induced conditions based on the gene expression levels. First, correlations of fold changes between two experimental sets (WT-24 h/WT-2 h and WT-1 d/abaA-1 d) are shown as dot graphs (Fig 3). We focused on downregulated genes in the abaA mutant (WT-1 d/abaA-1 d > 3, 933 genes) for this analysis. As the cut-off RPKM values 12 h after conidia induction (0, 1, 10, and 100 for Fig 3A, 3B, 3C and 3D, respectively) were increased, fold changes of the two experimental sets showed stronger positive correlations. Additionally, highly expressed genes clearly tended to occur more frequently in clusters 3 and 9 (Fig 4). Taken together, we concluded that ABAA deletion abolished the expression of genes with a higher biological significance for conidiation.
Fig 3. Correlation analyses of transcriptomes depending on expression levels.

Genes with more than 0 (A), 1 (B), 10 (C), and 100 (D) RPKM values 12 h after conidia induction were selected for the analysis.
Fig 4. Percentages of genes included in clusters 3 and 9 based on their expression levels.
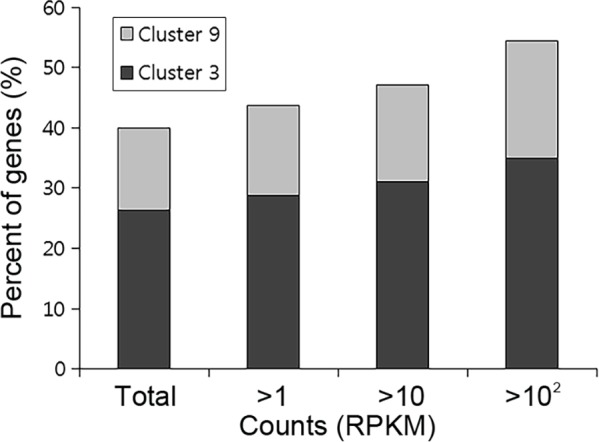
Genes with more than 1, 10, and 100 RPKM values 1 d after sexual induction were selected for the analysis.
Verification of the usefulness of the abaA mutant for sexual development studies
We investigated whether vigorous conidiation also occurred in F. graminearum perithecia-deficient mutants after sexual induction. We selected five representative transcription factor mutants for this analysis (Table 1). Three mutants (gzpl53l005, gzzc183, and gzzc258) had defects only in perithecia production but not in other phenotypes, whereas two mutants (Fgnot3 and gzzc302) showed pleiotropic developmental defects [15]. Among them, four mutants were normal in conidia production; only the Fgnot3 mutant produced significantly lower amounts of conidia compared to wild type in liquid medium [15,28]. Four mutants (gzpl53l005, gzzc183, gzzc258, and Fgnot3) highly produced conidia compared to wild type, whereas the gzzc302 mutant did not produce conidia after sexual induction (Fig 5A and 5B). In particular, a huge amount of conidia and conidiophores were observed in the gzzc183 and Fgnot3 mutants (Fig 5B). We thought that the variable conidiogenesis phenotypes during sexual development of the null mutants would be major obstacles to obtain quality transcriptomes. Therefore, transcriptome analyses of the perithecia-deficient mutants should be carefully performed to avoid false-positive signals caused by different conidiation phenotypes.
Fig 5. Phenotypic effects of ABAA deletion on representative transcription factor mutants.

(A) Conidia production. The number of conidia was measured 1 day after sexual induction. The number of conidia in the wild-type strain was arbitrarily set to one. (B) Mycelia and conidia of representative transcription factor mutants. Pictures were taken 1 day after sexual induction. Scale bar = 50 μm. (C) Sexual development of the transcription factor mutant with and without the ABAA deletion. Pictures were taken 7 days after sexual induction. Scale bar = 500 μm.
We questioned whether the conidia-deficient mutant abaA could be used as the alternative background strain to study sexual development in F. graminearum. To construct transcription factor mutants carrying the ABAA deletion, the Δmat2 strain was spermatized with hyphal fragments of ΔabaA to generate the HK167 (Δmat2 ΔabaA) strain (Table 1). Then, the HK167 strain was outcrossed with each transcription factor mutant. Subsequent genotyping confirmed the presence of double deletion mutants carrying deletions of both the transcription factor gene and ABAA. As expected, none of the double mutants produced conidia after sexual induction, suggesting that the effect of ABAA deletion was epistatic to the effects of the transcription factor genes (Fig 5A). We also examined whether the ABAA deletion affected the perithecia development phenotypes. The results confirmed that each single transcription factor mutant and corresponding double deletion mutant carrying the ABAA deletion showed indistinguishable perithecia production phenotypes (Fig 5C).
In a previous genome-wide functional analysis on transcription factor genes, we identified 105 mutants that were defective in multiple steps of sexual development in F. graminearum [15]. Among them, 44 mutants never produced mature perithecia and thus might be useful for studies of the mechanisms underlying the initial stages of perithecia development. Massive comparative transcriptome analyses using transcription factor mutants carrying the ABAA deletion will be performed to construct the genetic networks that orchestrate sexual reproduction in F. graminearum.
Conclusions
We provided phenotypic and genetic evidence that the current condition used for perithecia induction inevitably highly induced asexual sporulation in F. graminearum. Our comparative bioinformatics analysis revealed that many genes required for conidiation exhibited altered expression during the initial stages of sexual development. Moreover, deletion of ABAA specifically blocked conidia production in both the wild-type strain and sexually-defective mutants during sexual development. We strongly suggest that an alternative background strain (abaA) could be used to study the initial stages of perithecia production to obtain high quality transcriptome data.
Supporting Information
Fuzzy clustering categorized 933 upregulated wild-type-specific genes into 10 groups.
(TIF)
(XLS)
(XLS)
Data Availability
All raw RNA-seq files are available from the GEO database (accession numbers GSE46133, GSE79532).
Funding Statement
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (2013R1A6A3A04059121) and the Cooperative Research Program for Agricultural Science and Technology Development (Project PJ01085602), Rural Development Administration, Republic of Korea. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Leslie JF, Summerell BA (2006) The Fusarium laboratory manual Ames, I.A., USA: Blackwell Pub. [Google Scholar]
- 2.Desjardins AE, (2006) Fusarium mycotoxins: chemistry, genetics, and biology St. Paul, MN, USA: APS Press. [Google Scholar]
- 3.Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, et al. (2012) Emerging fungal threats to animal, plant and ecosystem health. Nature 484: 186–194. 10.1038/nature10947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goswami RS, Kistler HC (2004) Heading for disaster: Fusarium graminearum on cereal crops. Mol Plant Pathol 5: 515–525. 10.1111/j.1364-3703.2004.00252.x [DOI] [PubMed] [Google Scholar]
- 5.Sutton JC (1982) Epidemiology of wheat head blight and maize ear rot caused by Fusarium graminearum. Can J Plant Pathol 4: 195–209. [Google Scholar]
- 6.Maldonado-Ramirez SL, Schmale DG III, Shields EJ, Bergstrom GC (2005) The relative abundance of viable spores of Gibberella zeae in the planetary boundary layer suggests the role of long-distance transport in regional epidemics of Fusarium head blight. Agr Forest Meteorol 132: 20–27. [Google Scholar]
- 7.Parry DW, Jenkinson P, Mcleod L (1995) Fusarium ear blight (scab) in small grain cereals-a review. Plant Pathol 44: 207–238. [Google Scholar]
- 8.Dill-Macky R, Jones RK (2000) The effect of previous crop residues and tillage on Fusarium head blight of wheat. Plant Dis 84: 71–76. [DOI] [PubMed] [Google Scholar]
- 9.Zeller KA, Bowden RL, Leslie JF (2004) Population differentiation and recombination in wheat scab populations of Gibberella zeae from the United States. Mol Ecol 13: 563–571. [DOI] [PubMed] [Google Scholar]
- 10.Cuomo CA, Güldener U, Xu J-R, Trail F, Turgeon BG, Di Pietro A, et al. (2007) The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317: 1400–1402. [DOI] [PubMed] [Google Scholar]
- 11.Trail F, Common R (2000) Perithecial development by Gibberella zeae: a light microscopy study. Mycologia 92: 130–138. [Google Scholar]
- 12.Guenther JC, Hallen-Adams HE, Bücking H, Shachar-Hill Y, Trail F (2009) Triacylglyceride metabolism by Fusarium graminearum during colonization and sexual development on wheat. Mol Plant-Microbe Interact 22: 1492–1503. 10.1094/MPMI-22-12-1492 [DOI] [PubMed] [Google Scholar]
- 13.Geng Z, Zhu W, Su H, Zhao Y, Zhang K- Q, Yang J (2014) Recent advances in genes involved in secondary metabolite synthesis, hyphal development, energy metabolism and pathogenicity in Fusarium graminearum (teleomorph Gibberella zeae). Biotechnol Adv 32: 390–402. 10.1016/j.biotechadv.2013.12.007 [DOI] [PubMed] [Google Scholar]
- 14.Jia L-J, Tang W-H (2015) The omics era of Fusarium graminearum: opportunities and challenges. New Phytol 207: 1–3. 10.1111/nph.13457 [DOI] [PubMed] [Google Scholar]
- 15.Son H, Seo Y-S, Min K, Park AR, Lee J, Jin J-M, et al. (2011) A phenome-based functional analysis of transcription factors in the cereal head blight fungus, Fusarium graminearum. PLoS Pathog 7: e1002310 10.1371/journal.ppat.1002310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang C, Zhang S, Hou R, Zhao Z, Zheng Q, Xu Q, et al. (2011) Functional analysis of the kinome of the wheat scab fungus Fusarium graminearum. PLoS Pathog 7: e1002460 10.1371/journal.ppat.1002460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Dijk EL, Auger H, Jaszczyszyn Y, Thermes C (2014) Ten years of next-generation sequencing technology. Trends Genet 30: 418–426. 10.1016/j.tig.2014.07.001 [DOI] [PubMed] [Google Scholar]
- 18.Eberwine J, Sul J-Y, Bartfai T, Kim J (2014) The promise of single-cell sequencing. Nat Meth 11: 25–27. [DOI] [PubMed] [Google Scholar]
- 19.Ramsay K, Jones MGK, Wang Z (2006) Laser capture microdissection: a novel approach to microanalysis of plant–microbe interactions. Mol Plant Pathol 7: 429–435. 10.1111/j.1364-3703.2006.00348.x [DOI] [PubMed] [Google Scholar]
- 20.Park H-S, Yu J-H (2012) Genetic control of asexual sporulation in filamentous fungi. Curr Opin Microbiol 15: 669–677. 10.1016/j.mib.2012.09.006 [DOI] [PubMed] [Google Scholar]
- 21.Park H-S, Yu J-H (2016) Developmental regulators in Aspergillus fumigatus. J Microbiol 54: 223–231. 10.1007/s12275-016-5619-5 [DOI] [PubMed] [Google Scholar]
- 22.Son H, Kim M-G, Min K, Seo Y-S, Lim JY, Choi GJ, et al. (2013) AbaA regulates conidiogenesis in the ascomycete fungus Fusarium graminearum. PLoS One 8: e72915 10.1371/journal.pone.0072915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Son H, Kim M-G, Min K, Seo Y-S, Lim JY, Choi GJ, et al. (2014) WetA is required for conidiogenesis and conidia maturation in the ascomycete fungus Fusarium graminearum. Eukaryot Cell 13: 87–98. 10.1128/EC.00220-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris SD (2005) Morphogenesis in germinating Fusarium graminearum macroconidia. Mycologia 97: 880–887. [DOI] [PubMed] [Google Scholar]
- 25.Leslie JF, Summerell BA (2006) The Fusarium laboratory manual, 1st edn. Ames, IA, USA: Blackwell. [Google Scholar]
- 26.Bowden RL, Leslie JF (1999) Sexual recombination in Gibberella zeae. Phytopathology 89: 182–188. 10.1094/PHYTO.1999.89.2.182 [DOI] [PubMed] [Google Scholar]
- 27.Lee J, Lee T, Lee Y-W, Yun S-H, Turgeon BG (2003) Shifting fungal reproductive mode by manipulation of mating type genes: obligatory heterothallism of Gibberella zeae. Mol Microbiol 50: 145–152. [DOI] [PubMed] [Google Scholar]
- 28.Bui D-C, Son H, Shin JY, Kim J-C, Kim H, Choi GJ, et al. (2016) The FgNot3 subunit of the Ccr4-Not complex regulates vegetative growth, sporulation, and virulence in Fusarium graminearum. PLoS One 11: e0147481 10.1371/journal.pone.0147481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. (2013) NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res 41: D991–D995. 10.1093/nar/gks1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sikhakolli UR, López-Giráldez F, Li N, Common R, Townsend JP, Trail F (2012) Transcriptome analyses during fruiting body formation in Fusarium graminearum and Fusarium verticillioides reflect species life history and ecology. Fungal Genet Biol 49: 663–673. 10.1016/j.fgb.2012.05.009 [DOI] [PubMed] [Google Scholar]
- 31.Wong P, Walter M, Lee W, Mannhaupt G, Münsterkötter M, Mewes H-W, et al. (2011) FGDB: revisiting the genome annotation of the plant pathogen Fusarium graminearum. Nucleic Acids Res 39: D637–D639. 10.1093/nar/gkq1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anders S, Pyl PT, Huber W (2014) HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 35.Futschik M (2007) Mfuzz: Soft clustering of time series gene expression data. R package version 16.
- 36.Lamarre C, Sokol S, Debeaupuis J-P, Henry C, Lacroix C, Glaser P, et al. (2008) Transcriptomic analysis of the exit from dormancy of Aspergillus fumigatus conidia. BMC Genomics 9: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osherov N, May G (2000) Conidial germination in Aspergillus nidulans requires RAS signaling and protein synthesis. Genetics 155: 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fuzzy clustering categorized 933 upregulated wild-type-specific genes into 10 groups.
(TIF)
(XLS)
(XLS)
Data Availability Statement
All raw RNA-seq files are available from the GEO database (accession numbers GSE46133, GSE79532).