

Pulmonary arterial hypertension (PAH) is a complex and progressive disorder, which almost always leads to right heart failure and death1. PAH is invariably associated with a spectrum of structural changes in the pulmonary arteries: increased adventitial and medial thickness, eccentric and concentric intimal thickening, the obliteration and recanalization of arteries and the appearance of dilation lesions. Virtually all of these changes are characterized, to a greater or lesser degree, by increased numbers of cells expressing α-smooth muscle (SM)-actin. However, neither the origins of these cells nor the molecular mechanisms operating to cause their accumulation have been fully elucidated. Traditionally it has been thought that the α-SM-actin expressing (SM)-like cells that accumulate in the above-mentioned vascular lesions were derived from proliferative expansion of resident vascular media SM cells (SMCs) or adventitial fibroblasts through the processes of de-differentiation of the former or differentiation of the latter. Through the years, however, this concept has been challenged by experimental data in not only the lung but also in the heart, kidney, and liver, demonstrating many possible sources of α-SM-actin expressing cells, including differentiation of resident vascular progenitor cells, recruitment of circulating progenitors or multi-functional inflammatory cells (fibrocytes), and finally the possibility that endothelial cells can transition into mesenchymal SM-like phenotype in a process recapitulating their developmental capabilities2, 3. Studies by Ranchoux et al.4, and now by Hopper et al. in this issue of Circulation5, provide convincing experimental evidence that endothelial-to-mesenchymal transition (EndMT) occurs in the setting of PAH/PH in both humans and animal models and potentially constitutes a target against which specific therapeutic agents could be utilized to abrogate the process and improve the status of the patient with PAH.
The potential of endothelial cells to undergo transition toward a mesenchymal-like phenotype is similar to that of epithelial cells, which have been long known to undergo epithelial-to-mesenchymal transition (EMT)6. EMT and EndMT have many regulatory pathways in common and both yield to a cell type capable of producing and remodeling extracellular matrix (ECM). In the process of EndMT, initially endothelial cells in a monolayer detach from to their surrounding cells and develop the ability to migrate (Figure 1). As cells migrate away from the monolayer, their cortical cytoskeleton is rearranged to enable cell motility via developing actin-rich projections such as lamellipodia or filopodia. Loss of endothelial markers, such as vascular endothelial cadherin (VE-cadherin), platelet endothelial cell adhesion molecule (PECAM-1 or CD31), corresponds to loss of cell-cell adhesion, increased migration, and “de-differentiation” of the endothelial cell2, 3, 7. It is well described that as the cells move away from the monolayer and into an ECM rich environment, they develop the ability to not only produce but to remodel existing ECM. Important functional indicators of EndMT are increased expression of collagens and elastin, as well as production of ECM-degrading enzymes, such as matrix metalloproteinase-2 (MMP)-2 and MMP-9 (Figure 1). The gain of myofibroblast markers, such as α-SM-actin and type-1 collagen, correspond to a newly acquired ability to produce and remodel ECM. The pathways, through which EndMT is induced, can be many in number as is carefully pointed out by Hopper et al., including vascular stresses on top of a potentially abnormal genetic background, and converge on several signaling proteins, such as Snail, Twist, Slug, and zinc finger E-box protein homeobox-1 (ZEB1) (Figure 1)8. These transcription factors, commonly used as markers of EndMT, play an essential role in downregulating expression of the proteins maintaining cell-cell adhesion, such as integrins and focal adhesion kinase (FAK), and upregulation of proteins involved in cell migration and ECM production. Some studies have suggested that besides these transcription factors, EndMT also requires downregulation of glycogen synthase kinase 3-β (GSK3β to decrease Snail degradation9. This distinct control of EndMT appears to be consistent with the current study of Hopper et al., and may differ from the EMT, where Snail alone has been suggested as sufficient for induction9. Findings of Hopper et al., raise the possibility that Slug activation alone in the context of BMPR2 knockdown, leads to EndMT.
Figure 1.
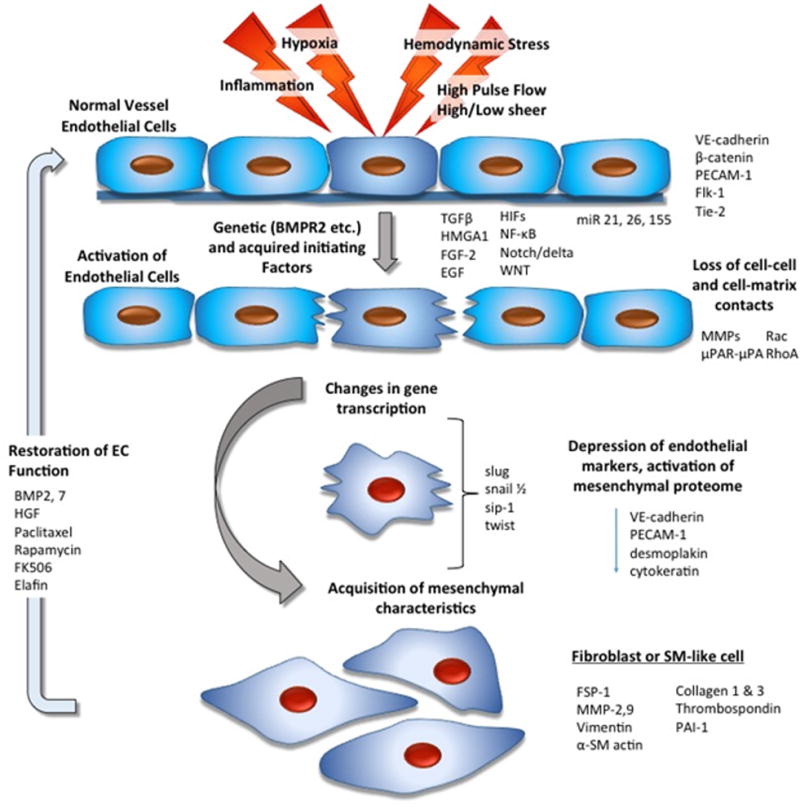
Schematic diagram illustrating potential mechanisms involved in endothelial-mesenchymal transition. Many initiating factors, all of which can lead to loss of cell-cell contacts, have been described. These factors are capable of activating metalloproteinase and serine protease family members and are important in loss of cell-cell contact. Following this interruption of cell-cell or cell-matrix contact, there is activation of transcription factors, which are actively involved in suppression of E-cadherins and other endothelial-specific proteins. Upregulation of a mesenchymal gene program follows. Importantly, in some situations, this process may be reversible leading to restoration of endothelial cells (EC) function.
Regulation of EndMT can involve changes in growth factors, inflammatory signaling, and the mechanical in situ environment. At present, the best described inducers of EndMT are members of the TGFβ super-family10. High levels of TGFβ in the microenvironment lead to the phosphorylation of SMAD2 and SMAD3 by ALK5, which appears critical at least in some forms of EndMT11. Yet, low levels of TGFβ will activate SMAD1, SMAD5, and/or SMAD8 through ALK1, which can then attenuate ALK5 signaling and therefore inhibit EndMT11. Hopper et al. now clearly demonstrate that high mobility Group AT-hook-1 (HMGA1), a member of a family of architectural factors that bind AT-rich regions of DNA and alter the chromatin structure to influence transcriptional activity, is a critical intracellular molecule regulating the ability of the cells to undergo EndMT5. It will therefore be essential to understand the environmental stimuli or microenvironmental cues that control HMGA1 expression in endothelial cells. In this light, it is important to note that microRNAs, which are clearly under the control of TGFβ signaling and potentially under the control HMGA1, have been implicated in the induction or inhibition of EndMT. For instance, miR21, which has been reported to be overexpressed in the setting of vascular remodeling associated with PAH/PH, has been shown to be involved in TGFβ-induced EndMT via PTEN/Akt-dependent pathway12. In vivo, the antifibrotic effects of miR21 antagonists are partly mediated through blocking EndMT12. A number of other miRNAs, including miR155, miR29 family members, and miR195, have also been implicated in this process3. However, consistent with the complexities of the EndMT process, Hopper et al. did not find that miR21 or miR26a regulated expression of HMGA1 in human pulmonary artery endothelial cells.
PAH is also consistently characterized by early and persistent increases in inflammatory signaling and oxidative stress, as well as significant changes in the mechanical factors imposed on the blood vessel. The inflammatory microenvironment observed in PAH contains a myriad of signaling proteins such as tumor necrosis factor-α (TNFα), interleukin1-β (IL-1β), IL-6, and reactive oxygen species (ROS). Each may have individual effects on the EndMT process, but ultimately the combination of factors will dictate the outcome. For example, oxidative stress can increase the expression and secretion of TGFβ1 and β2. Recent work has also demonstrated that H2O2 can induce EndMT potentially independent of the effects of oxidative stress or TGFβ signaling13. Interestingly, hypoxia, which is often associated with inflammatory signaling, has also been shown to induce Snail expression through HIF1α, at least in mice14. Hypoxia has also been shown to induce other transcription factors such as STAT3, NFKβ, interferon regulatory factor-1 (IRF-1), and β-catenin, that can interact with HIF to drive changes in endothelial function and EndMT.
PAH progression is associated with changes in the shear stresses imposed on the vascular wall, which can include high shear, high pulse intensity and disturbed flow. Endothelial cells are capable of responding to different patterns of flow in a number of ways as they sense flow through adhesion molecules, cytoskeletal deformation, nuclear displacement, and cilia. Interestingly both embryonic endothelium and adult endothelium are capable of responding to high shear patterns with EndMT through what looks principally like TGFβ/ALK5 signaling3. This supports the idea that high flow may play a major role in vascular remodeling and potentially, in part, through EndMT. Recent studies have demonstrated that high pulsatile flow, which has been shown to exist in the pulmonary circulation secondary to stiffening of the large pulmonary arteries, can promote vascular fibrosis by triggering EndMT, as well as fibroblast activation15.
Since decreased expression and abnormal function of bone morphogenetic protein receptor (BMPR)-2 is observed in PAH16, Hopper et al. analyzed the link between BMPR2 deficiency and EndMT. They demonstrated that both events are linked through an HMGA1-Slug axis. Ranchoux et al. also analyzed the relationship between BMPR2 deficiency and EndMT, using the first-ever Bmpr2 mutant rats4. These rats, spontaneously displayed pulmonary vascular remodeling associated with pulmonary neoexpression of Twist-1 and phosphorylation of vimentin4. They also demonstrated that the decrease in pulmonary BMPR2 expression preceded Twist-1 related EndMT and severe vascular remodeling in MCT-induced PH. Noteworthy is the fact that both EndMT and BMPR2 deficiency have previously been implicated in the vascular remodeling observed in experimental models of PH induced by either MCT or chronic hypoxic exposure17, 18. In one instance, the EndMT process observed in MCT-treated mice was reverted in IKBα mutant MCT-treated mice, suggesting an important role of NFKβ in the process17. In lung microvascular endothelial cells, IKBα mutant plasmid restored the decreased BMPR2 protein levels and reversed the EndMT process induced by TGFβ117. In another study, EndMT was partially ameliorated by stimulating BMPR2 signaling with rh-BMP2 and rh-BMP7 even in the presence of TGFβ118. Collectively, these studies support a strong link between abnormalities in BMP signaling and EndMT.
On the other hand, it could be argued that EndMT is an epiphenomenon in the PAH-remodeled pulmonary vasculature, without a significant biological relevance. For instance, histologic assessment of lungs from patients with systemic sclerosis-associated PAH and from Sugen+hypoxia murine model identified the presence of von Willebrand factor/α-SM-actin double-positive “transitional” endothelial cells in about 5% of pulmonary vessels19. Though this is a low prevalence, it must be kept in mind that the analyzed samples are human lungs at the end stage of disease evolution. Actually, even vascular cell proliferation, that is one of the paradigms of PAH, is a rare event in late-stage human lungs, with the exception of plexiform lesions (PLs). PLs are angio-proliferative glomeruloid-like vascular structures pathognomonic of PAH, whose hemodynamic effect is still debated. An immunohistochemical analysis of human PAH lungs for the proliferation nuclear marker Ki67, revealed that even though there is marked cell proliferation in PLs, only single endothelial cells stained positive for Ki67 in the adjacent arteries20. And last but not least, one must also keep in mind that EndMT can be detected only in a narrow time window, during the transitional stage when endothelial cells express both endothelial and mesenchymal markers. Once endothelial cell markers are lost, the cells are hardly distinguishable from other mesenchymal-like cells.
In conclusion, the recognition of EndMT as a new paradigm in PAH pathobiology will open exciting opportunities for anti-remodeling therapeutic interventions summarized in Figure 1 with a potentially limited effect on adult healthy tissues.
Acknowledgments
Funding Sources: This work was supported by NIH/NHLBI Axis Grant (HL114887-03), Program Project Grant (HL014985-40), Department of Defense Grant (PR140977), and ANR Grant (ANR-13-JSV1-0011-01).
Footnotes
Disclosures: None.
References
- 1.McLaughlin VV, Shah SJ, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardiol. 2015;65:1976–97. doi: 10.1016/j.jacc.2015.03.540. [DOI] [PubMed] [Google Scholar]
- 2.Arciniegas E, Frid MG, Douglas IS, Stenmark KR. Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1–8. doi: 10.1152/ajplung.00378.2006. [DOI] [PubMed] [Google Scholar]
- 3.Muylaert DE, de Jong OG, Slaats GG, Nieuweboer FE, Fledderus JO, Goumans MJ, Hierck BP, Verhaar MC. Environmental Influences on Endothelial to Mesenchymal Transition in Developing Implanted Cardiovascular Tissue-Engineered Grafts. Tissue Eng Part B Rev. 2015 Oct 8; doi: 10.1089/ten.TEB.2015.0167. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 4.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Pechoux C, Bogaard HJ, Dorfmuller P, Remy S, Lecerf F, Plante S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen-Kaminsky S, Perros F. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–18. doi: 10.1161/CIRCULATIONAHA.114.008750. [DOI] [PubMed] [Google Scholar]
- 5.Hopper R, Moonen JR, Diebold I, Cao A, Rhodes C, Ferreira Tojais N, Hennigs J, Gu M, Wang L, Rabinovitch M. In Pulmonary Arterial Hypertension, Reduced BMPR2 Promotes Endothelial-to-Mesenchymal Transition Via HMGA1 and its Target Slug. Circulation. 2016;133:XX–XXX. doi: 10.1161/CIRCULATIONAHA.115.020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–96. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Welch-Reardon KM, Wu N, Hughes CC. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler Thromb Vasc Biol. 2015;35:303–8. doi: 10.1161/ATVBAHA.114.303220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boutet A, De Frutos CA, Maxwell PH, Mayol MJ, Romero J, Nieto MA. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J. 2006;25:5603–13. doi: 10.1038/sj.emboj.7601421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20:556–67. doi: 10.1016/j.tcb.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Goumans MJ, Lebrin F, Valdimarsdottir G. Controlling the angiogenic switch: a balance between two distinct TGF-b receptor signaling pathways. Trends Cardiovasc Med. 2003;13:301–7. doi: 10.1016/s1050-1738(03)00142-7. [DOI] [PubMed] [Google Scholar]
- 12.Kumarswamy R, Volkmann I, Jazbutyte V, Dangwal S, Park DH, Thum T. Transforming growth factor-beta-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler Thromb Vasc Biol. 2012;32:361–9. doi: 10.1161/ATVBAHA.111.234286. [DOI] [PubMed] [Google Scholar]
- 13.Montorfano I, Becerra A, Cerro R, Echeverria C, Saez E, Morales MG, Fernandez R, Cabello-Verrugio C, Simon F. Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-beta1 and TGF-beta2-dependent pathway. Lab Invest. 2014;94:1068–82. doi: 10.1038/labinvest.2014.100. [DOI] [PubMed] [Google Scholar]
- 14.Luo D, Wang J, Li J, Post M. Mouse snail is a target gene for HIF. Mol Cancer Res. 2011;9:234–45. doi: 10.1158/1541-7786.MCR-10-0214. [DOI] [PubMed] [Google Scholar]
- 15.Elliott WH, Tan Y, Li M, Tan W. High Pulsatility Flow Promotes Vascular Fibrosis by Triggering Endothelial EndMT and Fibroblast Activation. Cell Mol Bioeng. 2015;8:285–295. doi: 10.1007/s12195-015-0386-7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–8. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- 17.Li L, Wei C, Kim IK, Janssen-Heininger Y, Gupta S. Inhibition of nuclear factor-kappaB in the lungs prevents monocrotaline-induced pulmonary hypertension in mice. Hypertension. 2014;63:1260–9. doi: 10.1161/HYPERTENSIONAHA.114.03220. [DOI] [PubMed] [Google Scholar]
- 18.Reynolds AM, Holmes MD, Danilov SM, Reynolds PN. Targeted gene delivery of BMPR2 attenuates pulmonary hypertension. Eur Respir J. 2012;39:329–43. doi: 10.1183/09031936.00187310. [DOI] [PubMed] [Google Scholar]
- 19.Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ, Holmes AM. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am J Pathol. 2015;185:1850–8. doi: 10.1016/j.ajpath.2015.03.019. [DOI] [PubMed] [Google Scholar]
- 20.Jonigk D, Golpon H, Bockmeyer CL, Maegel L, Hoeper MM, Gottlieb J, Nickel N, Hussein K, Maus U, Lehmann U, Janciauskiene S, Welte T, Haverich A, Rische J, Kreipe H, Laenger F. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am J Pathol. 2011;179:167–79. doi: 10.1016/j.ajpath.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]