

To initiate infection, enveloped viruses must fuse with a cell membrane, a process mediated by a dedicated viral fusion protein. To date, these proteins group into three basic structural classes. Most require priming (via a protease) to prepare them to respond to a fusion‐triggering signal. Known fusion triggers include receptors, low pH and proteases (and combinations thereof). Here, we provide an update on viral fusion protein priming and triggering, with a focus on virus fusion in endosomes.
Keywords: enveloped virus, fuse, low pH, membrane, prime, proteases, trigger, viral fusion protein, virus entry, virus receptors
Abstract
Ari Helenius launched the field of enveloped virus fusion in endosomes with a seminal paper in the Journal of Cell Biology in 1980. In the intervening years, a great deal has been learned about the structures and mechanisms of viral membrane fusion proteins as well as about the endosomes in which different enveloped viruses fuse and the endosomal cues that trigger fusion. We now recognize three classes of viral membrane fusion proteins based on structural criteria and four mechanisms of fusion triggering. After reviewing general features of viral membrane fusion proteins and viral fusion in endosomes, we delve into three characterized mechanisms for viral fusion triggering in endosomes: by low pH, by receptor binding plus low pH and by receptor binding plus the action of a protease. We end with a discussion of viruses that may employ novel endosomal fusion‐triggering mechanisms. A key take‐home message is that enveloped viruses that enter cells by fusing in endosomes traverse the endocytic pathway until they reach an endosome that has all of the environmental conditions (pH, proteases, ions, intracellular receptors and lipid composition) to (if needed) prime and (in all cases) trigger the fusion protein and to support membrane fusion.
All enveloped viruses deliver their genomes into the cytoplasm of their host cell by fusing with a cellular membrane. Ari Helenius inaugurated and has had a continual impact on this field in three major ways. He and his coworkers provided the first evidence that viruses can productively enter cells following endocytosis and transport to endosomes 1, 2, 3, 4, 5. His group was among the first to demonstrate that low pH is necessary and sufficient to trigger the fusion activity of certain enveloped viruses that enter cells through endosomes 1, 2, 6, 7, 8, 9, 10. And, extensive work led by Helenius demonstrated that not only can enveloped viruses productively enter cells through endosomes (Figure 1) but that most do so [for recent reviews, see 14, 15, 16]. The focus of this review is on enveloped virus fusion in endosomes, in particular on the diversity of endosomal cues that trigger virus fusion.
Figure 1.
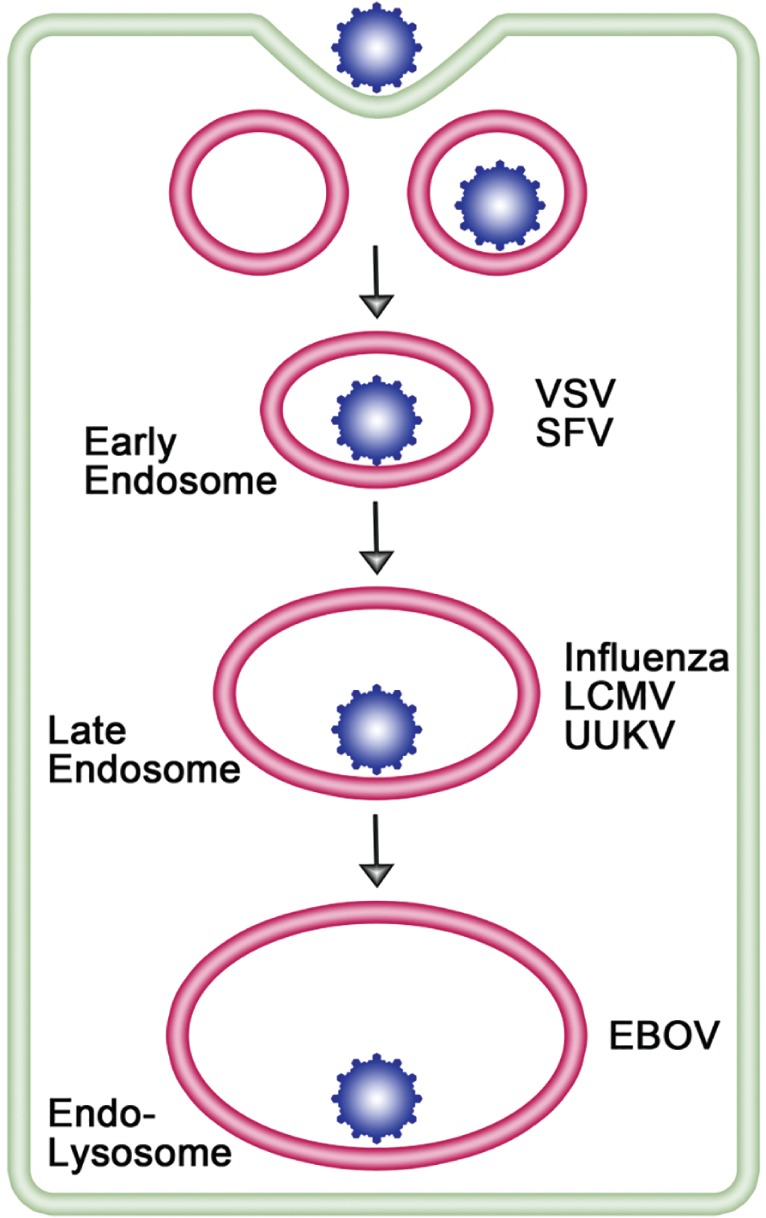
Enveloped virus entry through different endosomal compartments. Most enveloped viruses that enter the cell via endocytosis traverse the normal endocytic pathway (early endosome to late endosomes to endolysosome) and exit, by membrane fusion, where the conditions are sufficient to trigger the viral fusion protein; in some cases, the viral fusion protein is also proteolytically primed in the endocytic pathway as a prerequisite to fusion. [LCMV particles were found, however, to bypass early endosomes and traffic directly to, and fuse in, late endosomes 11, 12.] Examples of enveloped viruses that exit through early endosomes, late endosomes and endolysosomes are indicated. Viruses that enter through late endosomes or endolysosomes are termed ‘late penetrating viruses’ 13. See text and table legends for abbreviations.
Virus Fusion in Endosomes: General Considerations
Enveloped viruses that enter cells through endosomes begin their journey after binding to attachment factors and/or receptors on the cell surface followed by internalization through a variety of endocytic processes 14, 15, 16, 17, 18 (see Table 1 for definitions of terms). The major routes of virus internalization are clathrin‐mediated endocytosis, used by Semliki Forest virus (SFV) 1 and vesicular stomatitis virus (VSV) 19, and macropinocytosis, used by vaccinia virus 20, 21, Ebola virus (EBOV) 22, 23, 24 and others 25, 26. Some viruses, notably influenza, can use either mode of internalization depending on which pathway is functional in a given cell under given conditions 27, 28, 29, 30. Other means of internalization are caveolar endocytosis, used by certain non‐enveloped viruses, as well as clathrin‐ and caveolin‐independent endocytosis.
Table 1.
Definition of terms pertinent to viral membrane fusion proteins
| Fusion protein | The transmembrane protein on the surface of an enveloped virus that engages the target bilayer to mediate virus–cell membrane fusion. Examples: influenza HA, HIV Env, Dengue E. All characterized viral fusion proteins contain both a fusion peptide (or fusion loop) that engages the target membrane and a transmembrane domain that anchors the protein in the viral membrane. |
| Fusion subunit | Certain viral fusion proteins (e.g. influenza HA and HIV Env) are trimers of heterodimers that consist of a receptor binding and a fusion subunit, held together by either a disulfide bond (influenza HA) or non‐covalent interactions (HIV Env). In all of these cases the fusion subunit contains both the fusion peptide (or fusion loop) and a transmembrane domain. |
| Class I, II or III fusion proteins | All characterized viral fusion proteins fall into one of three classes based on the structure of their fusion protein/subunit: class I, largely α‐helical; class II, largely β‐structures; class III, contains both α‐helical and β‐structures. See Table 3. |
| Fusion peptide | A fusion peptide is a relatively hydrophobic sequence found at the N‐terminal end of a fusion subunit. It is the portion of the fusion protein that engages the target membrane. See Table 3. |
| Fusion loop | A fusion loop is a relatively hydrophobic sequence found internal to the fusion protein/subunit. Like a fusion peptide, it is the region of the fusion protein that engages the target membrane. See Table 3. |
| Prefusion conformation | The conformation of the viral fusion protein as it appears on the viral membrane after priming, but before fusion triggering. See Figure 2, panel ii. |
| Priming | All characterized class I and class II viral fusion proteins are primed to a state capable of responding to a fusion trigger. This involves a proteolytic cleavage event in the fusion protein precursor or in a companion protein. See Table 3. |
| Triggering | All viral fusion proteins must be triggered for fusion. Triggering converts the prefusion conformation to a postfusion conformation through a series of structural changes (Figure 2), and is induced by an environmental cue (Figure 3) at the cellular fusion site (e.g. low pH in endosomes). In most cases, a single trigger is sufficient, but in some cases (e.g. for ASLV Env), two triggers (receptor and low pH) are required: for ASLV Env, one to convert the protein to a prehairpin and a second to convert the prehairpin to a hairpin. |
| Refolding | Refolding encompasses all of the conformational changes in the fusion protein/subunit during fusion. The fold‐back stage of refolding encompasses the changes that convert the prehairpin to the hairpin (see Figure 2). |
| Postfusion conformation | The conformation of the fusion protein/subunit after the fusion reaction has been executed. For all characterized fusion proteins, this state is a trimer‐of‐hairpins (Figure 2). |
| Fusion cascade | The fusion cascade encompasses all changes in the viral fusion protein as well as in the target and viral membranes during the fusion reaction (panels ii to vi in Figure 2). |
Despite the various modes of internalization, most endocytosed enveloped viruses traverse the canonical endocytic pathway and enter the cytoplasm through either early endosomes, late endosomes or endolysosomes depending on which compartment has the proper environmental cue(s) to trigger and support fusion (Figure 1). While entry through endosomes does not necessarily imply a requirement for low pH for fusion 31, 32, 33, for most endocytosed enveloped viruses a major determinant of the entry site is the pH dependence of the viral fusion reaction (Table 2). The pH dependence of fusion varies among enveloped viruses 7, and these differences can correlate with the endosomal site of fusion 41. Hence, viruses with relatively high (∼pH 6) pH dependencies, such as SFV 41 and VSV 42, generally fuse in early endosomes, whereas those with lower pH dependencies (∼pH 5), including most strains of influenza, generally fuse in late endosomes 13. The pH dependence for influenza virus fusion varies by ∼0.7 pH units among different strains, with human influenza viruses generally requiring lower pH than avian ones. This variation in pH triggering has been proposed as an ‘acid stability marker’ as part of risk‐assessment analyses designed to predict the human transmissibility of influenza virus 43, 44, 45, 46, 47, 48. However, single‐particle tracking analyses of several clinical and laboratory‐adapted H3 influenza viruses show that the typical acid stability assay (pH onset of syncytia formation) may not provide enough information about the rates of fusion and fusion inactivation to predict viral tropism in all cases 49.
Table 2.
Site and fusion‐triggering mechanism for representative enveloped viruses
| Family | Virus | Site | Trigger |
|---|---|---|---|
| Retroviridae | MLV | Plasma membrane | Receptor |
| Paramyxoviridae | PIV5 | Plasma membrane | Receptor |
| Herpesviridae | HSV‐1 | Plasma membrane | Receptor |
| Coronaviridae | SARS | Plasma membrane or late endosome | Receptor + protease |
| Rhabdoviridae | VSV | Early endosome | Low pH |
| Togaviridae | SFV | Early endosome | Low pH |
| Bornaviridae | BDV | Early endosome | Low pH |
| Flaviviridae | TBE | Endosome | Low pH |
| Orthomyxoviridae | Influenza | Late endosome | Low pH |
| Arenaviridae | LCMV | Late endosome | Low pH |
| Bunyaviridae | UUKV | Late endosome | Low pH |
| Filoviridae | EBOV | Endolysosome | Low pH + additional cue(s) |
| Asfarviridae | ASFV | Late endosome | Low pH + additional cue(s) |
| Poxviridae | VV | Late endosome | Low pH [+ additional cue(s)] |
| Arteriviridae | PRRSV | Early endosome | Low pH [+ additional cue(s)] |
| Hepadnaviridae | HBV | Late endosome |
Information is for the specific virus listed (viruses of invertebrates only not included). Variations on entry sites and triggers exist for different family members (see main text). ‘Endosome’ denotes that the specific endosomal entry site is not yet known. ‘Low pH + additional cue(s)’ denotes that low pH is necessary but not sufficient; ‘low pH [+ additional cue(s)]’ indicates that low pH is needed, but it is unclear if it is sufficient. Blank indicates insufficient information.
PIV5, parainfluenza virus 5; TBE, tick‐borne encephalitis virus; LCMV, lymphocytic choriomenengitis virus; UUKV, Uukuniemi virus; VV, vaccinia virus (data are for the mature form, WR strain); ASFV, African swine fever virus; SHFV, simian hemorrhagic fever virus; BDV, Borna disease virus; HBV, hepatitis B virus. Additional information for the table can be found in references 34, 35, 36, 37, 38, 39, 40. For HBV, entry appears independent of late endosomal pH, but may require redox potential 38.
SFV, VSV and influenza represent relatively simple cases for which low pH is sufficient to trigger fusion. For these, the pH of fusion correlates with the pH needed to induce fusion‐activating conformational changes in the viral fusion protein, which in turn generally correlates with fusion in an early or a late endosome. However, even these simple cases have modifiers, such as special target membrane lipid requirements 6, 50, 51, 52, 53, 54. In other cases, low pH may be necessary, but not sufficient, to trigger fusion. This applies for endosomal entry of certain retroviruses and coronaviruses.
Viral Membrane Fusion Proteins: General Considerations
Enveloped viruses vary in the number of different types of glycoproteins that protrude from their membranes. For example, retroviruses display a single transmembrane glycoprotein (Env), while most influenza viruses display two: a hemagglutinin (HA) and a neuraminidase. Other enveloped viruses display more than two surface proteins. Nonetheless, for all characterized enveloped viruses one glycoprotein is the fusion protein, the protein that actually merges the viral and cellular membranes (Table 3). For reviews on the structures and mechanisms of viral fusion proteins, see references 60, 61, 65, 66, 67, 68, 69 and primary citations within. For viruses that encode a single transmembrane glycoprotein (e.g. retroviruses and filoviruses) that glycoprotein is the fusion protein. For viruses with two transmembrane glycoproteins (e.g. orthomyxoviruses and paramyxoviruses), the fusion protein is one of the two (e.g. HA of influenza virus). As reviewed in references 59, 60, 70, 71, large DNA viruses such as herpesviruses and poxviruses employ fusion machines, which consist of four (herpesviruses) or more (poxviruses) proteins associated with the virus membrane. However, at least for herpesviruses, one transmembrane glycoprotein, gB, is the fusion protein. Another important point is that despite their differing prefusion and postfusion structures, all characterized viral fusion proteins share a common architecture in their postfusion forms, a trimer‐of‐hairpins.
Table 3.
Examples of viral membrane fusion proteins
| Family | Virus example | Fusion protein (subunit) | Fusion protein class | Protein primed | Priming protease(s)a | Metastable | Fusion peptide or fusion loop | Fusion trigger |
|---|---|---|---|---|---|---|---|---|
| Orthomyxoviridae | Influenza | HA (HA2) | I | HA0 | Furin, trypsin | Yes | Peptide | Low pH |
| Paramyxoviridae | PIV5 | F (F1) | I | F0 | Furin, trypsin | Yes | Peptide | Receptor |
| Retroviridae | HIV | Env (gp41) | I | Env (gp160) | Furin | Yes | Peptide | Receptors |
| Coronaviridae | SARS | S (S2) | I | S0 | Trypsin, cathepsin | Yes | Peptide | Receptor + protease |
| Arenaviridae | LCMV | GP (GP2 + SSP) | I | GPC | SKI/S1P | Yes | Peptide and loop | Low pH |
| Filoviridae | EBOV | GP (GP2) | I | GP | Cathepsinsb | Yes | Loop | Low pH + additional cue(s)c |
| Togaviridae | SFV | E1 | II | pE2 | Furin | Yes | Loop | Low pH |
| Flaviviridae | TBE | E | II | prM | Furin | Yes | Loop | Low pH |
| Bunyaviridae | UUKV | Gc | II | GPC | Signal peptidase | Yes | Loop | Low pH |
| Rhabdoviridae | VSV | G | III | N/A | N/A | Nod | Loops | Low pH |
| Herpesviridae | HSV‐1 | gB | III | N/A | N/A | (Yes)e | Loops | Receptor |
| Bornaviridae | BDV | GP (GP2)f | GP | Furin | Low pH + [additional cue(s)]c | |||
| Poxviridae | VV | EFC | Low pH [+ additional cue(s)]c | |||||
| Asfarviridae | ASFV | Low pH + additional cue(s)c | ||||||
| Arteriviridae | PRRSV | Low pH [+ additional cue(s)]c | ||||||
| Hepadnaviridae | HBVg |
Information is for the specific virus listed. Fusion proteins from most family members share structural class, metastability and presence of a fusion peptide or loops (but see text: pesti‐ and hepaciviruses). Within families, differences exist in the priming proteases and triggering mechanisms (see text). Information is not included for viruses that only infect invertebrates [but note that Baculovirus gp64 is a class III fusion protein 55], nor for cell–cell fusion proteins from J paramyxovirus 56 or encoded by reoviruses 57. Blank indicates insufficient information.
HIV, human immunodeficiency virus; N/A, not applicable; SSP, stable signal peptide 58; EFC, entry fusion complex 59.
Denotes furin‐ and trypsin‐like proteases; SKI/S1P is a furin‐family member.
EBOV GP is cleaved to GP1 and GP2 by furin, but requires cathepsins for priming (see text).
See Table 2.
VSV G undergoes pH‐reversible conformational changes..
The fusion mechanism of BDV is not clear. Its single glycoprotein (GP) is cleaved by furin (to GP1 and GP2), this cleavage is important for fusion 34 and GP2 is postulated to possess fusion activity 35. However, high‐resolution structural information is not available for either GP1 or GP2.
HBV encodes a preS surface protein that is processed to large (L), medium (M) and small (S) proteins, but which is/are the major player(s) in fusion and how HBV fusion is activated remain to be clarified 64.
Priming and Triggering Viral Membrane Fusion Proteins
The field has converged on a model for how viral fusion proteins function [Figure 2; also see 60, 61, 65, 66, 67, 68, 69, 70]. All characterized class I and II viral fusion proteins must be primed before they can be triggered to induce fusion. Priming entails a proteolytic event that converts the fusion protein from a fusion‐incompetent to a fusion‐competent state (Figure 2). For class I fusion proteins the cleavage occurs in the fusion protein (precursor), whereas for class II fusion proteins, it occurs in a companion protein (Table 3). Depending on the viral fusion protein and the cells infected, priming can occur in the Golgi, at a cell surface or in an endosome. For example, for most strains of influenza virus, cleavage of the HA precursor (HA0) occurs in the extracellular space or at the cell surface by trypsin‐like proteases, but highly pathogenic avian influenza viruses such as H5N1 are cleaved in the Golgi by furin‐like proteases based on the presence of a multibasic cleavage site 73. Proteolytic priming separates HA0 into a receptor binding (HA1) and a fusion (HA2) subunit. Consequently, the fusion peptide is found at the amino terminus of HA2. Aspects of HA priming, for example accessibility of the cleavage site within HA0 (including obstruction by carbohydrates) as well as the host cells and tissues where priming occurs and the proteases involved, are thought to be critical determinants of influenza pathogenesis 43, 74, 75. Other priming events, as for the EBOV glycoprotein (GP) 76, 77 and the F protein of henipaviruses 78, 79, 80, occur in endosomes, mediated by cathepsins or other proteases. Most importantly, irrespective of the cellular site where it occurs, the protein (fusion or companion) cleaved or the proteases involved, priming converts the fusion protein to a fusion‐competent state (Figure 2).
Figure 2.
Model for how viral fusion proteins function. The model shown is for a class I fusion protein, but related models apply to class II and III fusion proteins. The term for the state of the protein is given above each image. For most class I fusion proteins [see 67 for paramyxovirus F proteins], prior to triggering (i and ii), the receptor‐binding subunit (deep purple, rb) clamps the fusion subunit (dark blue, f). Upon triggering, the receptor‐binding subunit moves out of the way unclamping the fusion subunit so that it can form a prehairpin embedded in the target membrane via the fusion peptide (red). The prehairpin then folds back causing the N‐ and C‐α‐helical heptad repeats to form a six‐helix bundle (6HB) and progressively pulling the target (pink) and viral (light blue) membranes through stages of close apposition (iv), hemifusion (v) and fusion pore formation (vi). In some cases (e.g. for influenza HA), membrane coalescence is aided by further packing of sequences C‐terminal to the C‐heptad in the grooves of the central N‐heptad coiled coil 72. Importantly, for all characterized viral fusion proteins, the final (postfusion) conformation (vi) is a trimer‐of‐hairpins.
The next stage of fusion is triggering, which for viruses that enter through endosomes is induced by an endosomal cue(s). An early consequence of triggering is exposure and repositioning of the fusion peptide (or loop) to engage the target bilayer; before triggering, the fusion peptide/loop is buried or tacked down in the fusion protein structure (or, for VSV G, points to the viral membrane) and therefore inaccessible to the target membrane. Upon triggering the fusion peptide/loop is exposed and inserts into the target membrane forming an intermediate termed a prehairpin (Figure 2). The prehairpin is a unique biological structure in which a single protein, the viral fusion protein, is simultaneously anchored in, and therefore bridges, two distinct membranes: the target membrane via the fusion peptide/loop and its own (viral) membrane through its transmembrane domain (TMD). Class I fusion proteins are trimers in their prefusion and postfusion states. Interestingly, while all characterized class II fusion proteins are dimers (that sit low on the viral surface) in their prefusion states, they convert, through a monomeric intermediate, to homotrimers once their fusion loops lodge in the target membrane 68, 81, 82. After prehairpin formation, the fusion protein folds back upon itself, drawing the attached target and viral membranes closer and closer together. The final postfusion state of all characterized viral fusion proteins is, therefore, a trimer‐of‐hairpins. Although there is debate 83, it is commonly thought that multiple fusion protein trimers act in concert 84, 85, 86, 87, 88, 89, 90, 91 to induce the later stages of fusion: close membrane apposition, formation of a lipid stalk, opening of a small fusion pore and expansion of the pore 92, 93, 94, 95, to create a passage for the viral nucleocapsid.
Classes of Viral Fusion Proteins
The diagram in Figure 2 is for a class I viral fusion protein (Table 3) such as the influenza HA 96, 97. For these, the fusion subunit is largely α‐helical, containing an N‐ and a C‐helical heptad repeat. In the prehairpin, the N‐heptad is thought to sit atop the C‐heptad in contiguous alignment. During the fold‐back stages, the three C‐heptads bind in the grooves of the trimeric N‐heptad coiled coil creating a six‐helix bundle‐containing trimer‐of‐hairpins (Figure 2). Since the N‐ and C‐heptads connect, respectively, to the fusion peptide and the TMD, hairpin formation pulls the attached membranes (cell and viral) together. Final interactions between the fusion peptide and TMD are thought to complete the event 98, 99, 100, 101, possibly aided in some cases by a ‘membrane proximal external region’, located upstream of the TMD 102, 103, 104, 105, 106.
Based on structural criteria there are two other recognized classes of viral fusion proteins 60, 61, 65, 66, 67, 68, 69, 70. Class II fusion proteins are largely composed of β‐strands and β‐sheets, with a fusion loop(s) at the tip of an extended β‐sheet domain 68. Class III fusion proteins contain both α‐helical and β‐sheet regions, with fusion loops at the tips of an extended β‐sheet 55, 60, 61, 70, 107. Most importantly, as for class I fusion proteins, the final ‘postfusion’ structure for characterized class II and III proteins is a trimer‐of‐hairpins (in which previously separated fusion peptides/loops and TMDs, and their attached target and viral membranes, have been brought together).
Fusion Peptides and Fusion Loops
The segments that engage the target membrane are termed ‘fusion peptides’ if at the N‐terminus of the fusion subunit and ‘fusion loops’ if internal to the polypeptide chain. Most class I fusion proteins contain fusion peptides, but those of avian sarcoma leukosis virus (ASLV) and filoviruses contain a fusion loop. Arenavirus GPs may contain both a fusion peptide and a fusion loop 58. The situation for coronaviruses is not fully resolved, with most evidence for a fusion peptide 108, 109, 110 (see below).
In membranes or membrane mimetics, the fusion peptide of influenza HA forms a kinked or hairpin α‐helical structure 111, 112 while that of HIV Env forms α‐helical or β‐structures depending on the target lipid composition 113, 114, 115. These structures maximize interactions of hydrophobic side chains with the hydrocarbon portion of the bilayer. The EBOV GP, a class I fusion protein, contains an internal disulfide‐bonded fusion loop. At neutral pH it adopts a relatively flat structure, as seen in the prefusion trimer 116, 117. At low pH the structure bends to more firmly grasp the target membrane 117; low pH‐induced changes also occur in the influenza HA fusion peptide 118, 119. Where studied, specific residues in fusion peptides and loops affect their structure, membrane insertion and function [for example, see 119, 120, 121]. Hence, the sequences and structures of fusion peptides/loops are important; their noted structural plasticity may be relevant at different stages of fusion.
Class II and III fusion proteins contain fusion loops at the tips of extended β‐sheet domains. While some class II fusion proteins have a single fusion loop 87, 122, 123, 124, 125, others, as well as all characterized class III fusion proteins, possess two fusion loops (per monomer) at the tip of a β‐sheet domain 126, 127, 128, 129, 130, 131, 132.
Fusion Triggering: General Considerations
There are four known ways by which a viral fusion protein can be triggered: by binding to a receptor(s), by exposure to low pH, by binding to a receptor followed by exposure to low pH and by a binding to a receptor followed by proteolytic cleavage (Figure 3). Interaction(s) with receptors is sufficient to trigger fusion for most retroviruses, paramyxoviruses and herpesviruses (Table 2), reflecting their predominant fusion at the cell surface at neutral pH [for reviews, see 60, 67, 70, 133, 134]. Most enveloped viruses, however, fuse in endosomes reflecting a requirement for low pH. Low pH may be sufficient, or it may work in concert with, or after binding to, a receptor. Alternatively, following receptor engagement, low pH may be needed for the action of an endosomal protease (Table 4).
Figure 3.
Different mechanisms by which class I, II and III fusion proteins are triggered. The three known classes of viral fusion proteins and the four confirmed mechanisms for fusion protein activation are shown on the left and right sides, respectively. Among fusion‐triggering mechanisms (right), blue denotes events that occur at neutral, and pink denotes ones that require low, pH. Some receptor + protease mechanisms do, whereas others do not, require low pH. Lines join ways in which specific viral fusion proteins, from different structural classes, are triggered. See text for details and abbreviations.
Table 4.
Examples of endosomal viral fusion triggers
| Trigger | Virus | Fusion protein | Additional facilitating factors/other comments |
|---|---|---|---|
| Low pH | Influenza | HA | One study suggests a role for cathepsin W for influenza entry, but the substrate (viral or cell) is not known. Events initiating nucleocapsid uncoating require K+. |
| SFV | E1 | SFV fusion requires cholesterol and sphingomyelin in the target membrane. | |
| Rubella | E1 | Rubella virus fusion is enhanced by Ca++ ions. | |
| VSV | G | VSV fusion is enhanced by anionic lipids such as LBPA. | |
| Dengue | E | Dengue virus fusion is enhanced by anionic lipids such as LBPA. | |
| Andes | Gc | Andes virus fusion requires high levels of cholesterol in the target membrane. | |
| UUKV | Gc | UUKV fusion is enhanced by anionic lipids such as LBPA. | |
| LCMV | GP | Low pH appears sufficient to trigger LCMV fusion (no known enhancing factors). | |
| Receptor + low pH | ASLV | Env | ASLV Env is the best‐characterized fusion protein activated in two sequential steps by receptor (Tva) binding followed by exposure to low pH. |
| JSRV | Env | JSRV Env appears to require both interaction with its receptor (Hyal2) and low pH, but details remain to be clarified. | |
| HCV | E1/E2 | Fusion and entry mediated by HCV E1/E2 appears to require both binding to cell surface receptor(s) and low pH, but details remain to be clarified. | |
| LASV | GP | LASV fusion is reported to require its intracellular receptor (LAMP1) and low pH. The exact roles of LAMP1 and low pH remain to be clarified. | |
| EBOV | GP | After priming GP1 to ∼19 kDa, EBOV GP requires its intracellular receptor (NPC1) and low pH for fusion, but the exact roles of NPC1, low pH and additional factor(s) remain to be clarified. | |
| Receptor + endosomal protease | SARS | S | SARS requires binding to its surface receptor (ACE2) plus cathepsin L for endosomal entry. Low pH is not needed for fusion per se (rather for cathepsin activity); trypsin can trigger ACE2‐bound SARS at the cell surface at neutral pH. |
| MERS | S | MERS fusion requires binding to its cell surface receptor (DPP4) followed by proteolytic activation at the cell surface (by trypsin‐like proteases) or in endosomes (by furin or cathepsins). Fusion may be triggered at neutral pH or under low pH conditions. | |
| RSV | F | RSV fusion is triggered in a pH‐independent manner in early endosomes following two distinct furin‐mediated cleavage events. |
See text for references and details.
JSRV, Jaagsiekte sheep retrovirus. See text and other tables for other abbreviations.
For some virus families, the fusion trigger is common to all members. For examples, low pH is sufficient to trigger the fusion proteins of influenza, alpha‐, flavi‐ and rhabdoviruses. For other families, different members employ different triggers. For example, the coronavirus infectious bronchitis virus (IBV) is triggered by simple exposure to low pH 135, while others, including severe acute respiratory syndrome coronavirus (SARS‐CoV), Middle East respiratory syndrome coronavirus (MERS‐CoV) and feline coronavirus (FCoV), are activated by binding to a receptor followed by protease action. Similarly, while paramyxoviruses of the paramyxovirinae subfamily (e.g. measles and mumps) fuse at neutral pH in response to receptor binding 67, some metapneumoviruses (pneumovirinae subfamily) enter the cytoplasm following endocytosis, and for some this correlates with low pH enhancement of fusion activity 32, 136, 137. For respiratory syncytial virus (pneumovirus genus; pneumovirinae subfamily), endocytosis appears to be followed by pH‐independent proteolytic activation of the fusion protein 25. Differences in neutral pH/cell surface versus low pH‐dependent/endosomal entry apply among retroviruses 133, 134, 138, 139, 140, 141, 142 and to herpesviruses in some cell types 143.
An analysis of the ways in which different viral fusion proteins are activated also reveals that the mechanism of fusion triggering does not necessarily correlate with the structural class of the fusion protein (Table 3, Figure 3). Different class I fusion proteins can be activated by each of the four characterized fusion‐triggering mechanisms (Table 3, Figure 3), while all characterized class II fusion proteins are activated by exposure to low pH alone. And, different class III fusion proteins can be activated either by low pH alone (VSV G), by binding to a receptor at neutral pH (e.g. most herpesviruses gB proteins in most cells) or by binding to a receptor followed by exposure to low pH (e.g. certain herpesviruses gB proteins in certain cells).
A major, perhaps the major, consideration about enveloped virus fusion in endosomes is that the specific endosome that serves as the fusion site (Figure 1, Table 2) is the endosome that possesses all of the necessary environmental cues to trigger the fusion protein. In some cases, this, or an upstream, endosome must also possess prefusion triggering priming factors. The endosomal fusion site will therefore be dictated by where the pH, receptors, ions, lipid composition, proteases and other factors are all present to (prime), trigger and support fusion.
Triggering by Simple Exposure to Low pH
Details of the fusion process were first elaborated for the influenza HA (Figure 2), for which low pH is sufficient to trigger all of the steps in the fusion cascade 7, 8, 9, 96, 97, 144, 145, 146, 147, 148. Unsurprisingly then, the pH dependence for key conformational changes in HA correlates with the pH dependence for fusion 43, 149, which, as mentioned previously, varies for different influenza strains. Low pH is also sufficient to trigger alpha‐, arena‐, bunya‐, flavi‐ and rhabdovirus fusion proteins (Table 3). For each of these, encompassing class I 150, 151, 152, 153, class II 36, 154, 155, 156, 157, 158, 159 and class III 55, 126, 160, 161 fusion proteins, protonation of multiple residues 147, 149, 162, 163, in particular histidines (pKa ∼ 6), is known or thought to be involved 164. For viruses activated solely by low pH, fusion in early or late endosomes is generally dictated by the pH dependence for key conformational changes in the viral fusion protein; those with higher pH thresholds generally fuse in earlier endosomes than ones requiring lower pH 7, 41. Exposure of certain enveloped viruses to low pH can also elicit changes to the matrix layer underlying the virus membrane. For influenza, protons can access the matrix layer through the M2 channel in the viral envelope 165, 166. Exposure of the matrix to H+ and K+, which can also enter via the M2 channel, likely aids later stages of fusion and/or uncoating 167, 168, 169. A related process may occur for other endosomally entering enveloped viruses 170, 171.
Among viruses whose fusion activity is activated solely by low pH, several have been shown to have specific lipid or ionic requirements that can influence the specific endosomal site of fusion (Table 4). Where studied, these requirements reside at the level of either the initial interaction between the exposed fusion peptide/loop and the target membrane or at a later step in the fusion process. For example, SFV requires ∼33 mol % cholesterol and ∼1–2% sphingomyelin in the target membrane for optimal fusion 6, 50, 51, 172. The cholesterol requirement is for stable insertion of the fusion loop (within its fusion protein, E1) into the target membrane prior to the fold‐back steps 68, 173. Similarly, hantaviruses such as Andes virus require high levels of cholesterol in the target membrane (in addition to low pH) for fusion 52.
Another intriguing case is dengue virus, which requires anionic lipids such as lysobisphosphatidic acid (LBPA) in the target membrane for fusion. Although dengue virus E protein displays conformational changes at a pH compatible with fusion in early endosomes, it traffics to late endosomes for fusion and entry 53, 174, which was attributed to higher concentration of anionic lipids in later endocytic organelles 53. Other flaviviruses 175, 176 as well as the rhabdovirus VSV 54, 177, 178 and the bunyavirus Uukuniemi (UUKV) 179 show higher rates and/or efficiencies of fusion when anionic lipids (including LBPA) are present in the target membrane. However, the physiological roles of specific anionic lipids in specific endosomal compartments for specific stages of fusion and/or capsid release are not fully understood. Some have argued that LBPA is needed for back‐fusion of intraluminal vesicles laden with already fused virus particles, and hence capsid release 175, 178.
In another twist, a recent study showed that in addition to low pH, rubella virus (RV) requires Ca++ for fusion 128. The RV fusion protein (E1) contains two fusion loops, which are closely associated through a metal ion‐binding site in the postfusion structure 129. Ca++ appears to be needed to properly position the two fusion loops for coordinated stable target membrane interaction (Figure 2) and hence for RV fusion 128.
Triggering by Binding to a Receptor Followed by Exposure to Low pH or the Action of a Protease
Below, we will discuss how engagement of a host cell receptor can play an active, albeit not exclusive, role in triggering certain viral fusion proteins that function in endosomes, As a prelude, we review how viruses that fuse at neutral pH are activated by their host cell receptors.
Paradigms from viruses that fuse at neutral pH following receptor binding
Most enveloped viruses that fuse at neutral pH are triggered solely by binding to a host cell receptor(s) (Figure 3, Tables 2 and 3). Some neutral pH fusing viruses (most retroviruses) possess a single glycoprotein (Env) whereas others (e.g. herpesviruses) have more. Retroviral Envs are class I fusion proteins. Binding of the receptor(s), to the receptor‐binding subunit of Env, unclamps the fusion subunit, which then proceeds through conformational changes that mediate fusion (Figure 2). An interesting case is Env from murine leukemia virus (MLV), a γ‐retrovirus. MLV Env contains a thiol exchange motif (CXXC) in its receptor‐binding subunit. Engagement of the host cell surface receptor activates this motif, which then isomerizes a disulfide bond between the CXXC motif and a CX6CC motif in the fusion subunit. By breaking this critical disulfide bond, the fusion subunit is unclamped and executes the fusion cascade 180. A similar isomerization occurs in Env of human T‐cell leukemia virus, a δ‐retrovirus 181. Human immunodeficiency virus (HIV), a lentivirus within the retrovirus family, presents a different case. The two subunits of HIV Env are not disulfide‐bonded and HIV Env does not contain a thiol exchange motif. For most HIV strains Env engagement with its primary receptor (CD4) elicits a change in its receptor‐binding subunit that reveals a binding site for a co‐receptor (CXCR4 or CCR5). When the co‐receptor binds it unclamps the fusion subunit allowing the fusion cascade to follow 138, 182, 183, 184. While it had been thought that all of these events happen at the plasma membrane, some findings suggest that the (posthemifusion) content mixing stage of HIV fusion requires endocytosis and dynamin (but not low pH) 31. However, the exact site of fusion for specific strains of HIV in specific (physiologically relevant) cell types is currently under study 133, 134.
Most, but not all, paramyxoviruses and herpesviruses fuse at neutral pH at the plasma membrane. In most of these cases, binding of a host cell receptor to a viral receptor‐binding protein induces conformational changes (in the viral receptor‐binding protein) that are relayed to the fusion protein, thereby sparking the fusion cascade. For paramyxoviruses, the relay is generally from the binding protein (HN, H or G) to the fusion protein (F) 67, with findings converging on a (receptor‐binding protein) ‘stalk exposure’ model for fusion triggering 67, 185, 186. For herpesviruses, the relay goes from the binding subunit (gD for HSV‐1) to an intermediate complex (gH/gL for HSV‐1) to the fusion protein (gB) 60, 70.
Triggering by binding to receptors followed by exposure to low pH
Unlike the cases described so far where either low pH alone or binding of a receptor(s) alone triggers the viral fusion protein, some viruses, notably the retrovirus ASLV, utilize a hybrid, two‐step process. The fusion protein of ASLV (Env) is a class I fusion protein. Unlike MLV Env, ASLV Env does not contain a thiol exchange motif in its receptor‐binding subunit (SU). In this case, binding of the ASLV receptor (Tva) to SU triggers conformational changes in SU 187, 188 that release the clamp on the fusion subunit, thereby allowing Env to form a prehairpin embedded in the target membrane via its fusion loop 189, 190, 191. Subsequent exposure to low pH induces the fold‐back events that generate a six helix bundle‐containing trimer‐of‐hairpins (Figure 2) and hence fusion and viral entry 139, 140, 141, 142, 152, 192, 193, 194, 195. A His residue in the chain reversal region (between the N‐ and C‐heptads) of the fusion subunit influences the pH dependence of fusion 152. This and a nearby His also influence the stability of the ASLV six‐helix bundle at low pH 196. Interestingly, differences among six‐helix bundles in terms of their dipole moments, stability at low versus neutral pH as well as the types of stabilizing interactions employed correlate with the pH dependence of fusion for certain retroviral (and filoviral) fusion proteins 196. ASLV Env is the only viral fusion protein currently known to use a clear‐cut two‐step mechanism where receptor binding triggers prehairpin formation and low pH triggers the fold‐back steps leading to hemifusion and fusion pore formation. However, the Env glycoprotein of the Jaagsiekte sheep retrovirus appears to use a two‐step process involving its receptor and low pH 197.
EBOV (discussed below) and Lassa virus (LASV) have recently been shown to employ intracellular receptors, located in late endocytic compartments, for productive entry. For EBOV the intracellular receptor is Niemann‐Pick C1 (NPC1); for LASV it is LAMP1 197, 198, 199, 200. Both viruses require low pH for entry. In the case of LASV, both LAMP1 and low pH appear to be needed to induce LASV GP‐mediated cell–cell fusion 199. A recent study employing electron cryotomography reported small changes in the membrane distal region of LASV GP1 at pH 5.0 and upon binding LAMP1 at pH 5.0, as well as shedding of GP1 at pH 3.0 201. However, future work is needed to determine if LAMP1 and physiological low pH elicit clear‐cut sequential conformational changes in LASV GP akin to those induced by Tva and low pH (pH 5.0) in ASLV Env.
Triggering by binding to receptors followed by the action of a (low pH‐dependent) protease
In some cases, viral fusion proteins are triggered by binding to a receptor followed by the action of a protease, which may or may not be low pH‐dependent. Such triggering mechanisms are employed by some coronaviruses and also by the paramyxovirus respiratory syncytial virus (RSV). SARS‐CoV has been studied extensively in this regard. Following its interaction with the ACE2 receptor, SARS‐CoV is activated by proteolytic cleavage; cathepsin L activates the virus in late endosomes in a pH‐dependent manner 202, 203, 204, but the virus can also be activated by trypsin‐like proteases at the cell surface in a pH‐independent manner 205, 206, 207. As first identified with SARS‐CoV 208, coronaviruses are somewhat unusual in that they have two distinct cleavage sites within their spike proteins 209 termed S1/S2 and S2′. In the case of SARS‐CoV either cathepsin L or trypsin‐like enzymes cleave at both positions but likely in a sequential manner (S1/S2 followed by S2′) 208. The use of each protease may be different in different cell types (e.g. Vero cells versus respiratory epithelial cells).
The currently emerging MERS‐CoV shares many features with SARS‐CoV with regard to membrane fusion 210, 211, 212, but has wider cell tropism. While both cathepsin L and trypsin‐like enzymes can activate MERS‐CoV following binding to its receptor (DPP4), there is an additional ability to use furin as an activating enzyme 213, which may explain its broader tropism. As with SARS‐CoV, the use of a given activating protease may be different in distinct cell types. Notably, MERS‐CoV is activated at the S2′ site by endosomal furin in a pH‐dependent manner 213, 214. It is unclear if the effect of low pH reflects a role in activation of furin or for conformational changes in the spike protein. Given that coronavirus fusion can function under neutral pH conditions, it is possible that the pH dependency is related to the level of proteolytic cleavage, with decreased cleavage of S compensated by fusion at low(er) pH. If so, this would be reminiscent of findings with a p62 cleavage site‐defective mutant of SFV 215.
While SARS‐CoV does not appear to be able to use furin for cleavage activation, furin is commonly used by many other coronaviruses. In most cases, furin appears to prime the S precursor (S0) at the S1/S2 location likely during biosynthesis, as first shown with mouse hepatitis virus (MHV) 216. In general, coronaviruses appear to be quite flexible with regard to their postreceptor means of fusion activation, and this can profoundly affect viral pathogenesis. For instance, feline coronavirus and the human coronavirus HCoV‐OC43 show distinct modulation of their pathogenesis based on sequence alterations in their furin cleavage sites 217, 218, 219. The second (endosomal or cell surface) protease likely acts at S2′, in close proximity to the fusion peptide 109, 110, to drive the fusion reaction. While many details remain to be resolved, studies of MHV have provided firm evidence for an endosomal activation pathway 214 driving membrane fusion following proteolytic activation at a fusion peptide‐proximal position, i.e. S2′ 214, 220. Another coronavirus that has been used to demonstrate postreceptor triggering of fusion is porcine epidemic diarrhea virus (PEDV), in this case in a trypsin‐dependent manner at S2′ 221 following interaction with the APN receptor. The action of trypsin on PEDV is considered to be pH‐independent.
As mentioned previously, the avian coronavirus IBV appears to be an exception, with fusion based solely on low pH triggering 135. In this case, S may be cleaved at S2′ during biosynthesis 208, 222, but any role of a yet‐to‐be‐identified receptor is unknown.
RSV is a paramyxovirus whose fusion protein (F) has two cleavage activation sites. RSV F shares many features with MERS‐CoV S, with furin activating at both cleavage positions and with the fusion peptide‐proximal cleavage event occurring in early endosomes 25. However, the second RSV cleavage event appears to be pH‐independent and is selective for furin, unlike for MERS‐CoV.
Other endosomal proteases that have been implicated in virus entry include cathepsin E for porcine reproductive and respiratory syndrome virus (PRRSV) 37 and cathepsin W for influenza virus 223. In these cases, it is not yet known if the proteases act on the virus or on a host factor within the endosomal network.
Cases for Which the Triggering Mechanism Is Not Yet Clear
There are several viruses that enter cells through endosomes for which the mechanism of fusion triggering is unclear. Three, touched on above, are why certain herpesviruses require endocytosis and low pH for entry into certain cell types, whether an endosomal factor plays an active role in HIV fusion in all cell types and the precise roles of LAMP1 and low pH in triggering LASV fusion. Here, we elaborate on two important viruses that may use novel fusion‐triggering mechanisms: EBOV and hepatitis C virus (HCV).
Triggering EBOV GP for fusion
EBOV fusion and entry are mediated by its sole GP, a trimer of a heterodimer containing a receptor‐binding subunit (GP1) disulfide bonded to a fusion subunit (GP2) 224, 225, 226, 227. Following internalization by macropinocytosis 22, 23, EBOV traffics to and fuses in endolysosomes (Figure 1) that contain its intracellular receptor, NPC1, and two‐pore channel 2 (TPC2), which is also required for EBOV entry 198, 228, 229, 230, 231, 232, 233. Before or upon arrival in endolysosomes, low pH‐dependent cathepsins remove the mucin‐like domain and glycan cap from GP1 76, 77, 116, 234, 235. This priming step converts EBOV GP1 (formerly 130 kDa) to a 19‐kDa species. Importantly, the fusion subunit (GP2) is still clamped in 19 kDa‐GP1‐S‐S‐GP2 236, 237. Priming (to 19‐kDa GP1) has two important consequences: it exposes residues in GP1 critical for binding to NPC1 228, 235 and it appears to potentiate 19 kDa‐GP1‐S‐S‐GP2 for fusion triggering 236. An important observation about primed 19 kDa‐GP1‐S‐S‐GP2 is that it still requires low pH and a factor(s) thwarted by the cysteine protease inhibitor E64d to mediate entry 77, 230, 233, 238. Concordantly, low pH and binding to NPC1 are necessary, but apparently not sufficient, to trigger productive 19 kDa‐GP1‐S‐S‐GP2‐mediated virus–cell fusion 228.
Three lines of evidence indicate that low pH plays a role in triggering 19 kDa‐GP1‐S‐S‐GP2 beyond its role for optimal cathepsin activity. The first is that the fusion loop (in GP2) undergoes a low pH‐dependent conformational change contingent on residues important for GP‐mediated entry 117, 121. The second is that low pH stabilizes the postfusion (six‐helix bundle) form of EBOV GP2 239, 240, 241. The third is that in vitro‐induced conformational changes in primed EBOV GP occur more readily at low pH 236. While a recent study detected cell–cell fusion mediated by 19 kDa‐GP1‐S‐S‐GP2 at neutral pH, the fusion pores formed were small and non‐expanding 242, likely not large enough to pass a viral nucleocapsid. Moreover, the latter findings 242 are difficult to reconcile with the observations that 19 kDa‐GP1‐S‐S‐GP2‐mediated entry and infection are potently inhibited by both bafilomycin and E64d 77, 230, 238, and that only small structural changes were observed in the 19 kDa‐GP1‐S‐S‐GP2 ectodomain bound to the NPC1 C‐loop and crystallized at pH 5.0 243. Collectively, these findings suggest that in addition to NPC1 and low pH, another factor(s) is needed to mediate EBOV fusion in a manner that will lead to productive infection.
The sensitivity of 19 kDa‐GP1‐S‐S‐GP2‐mediated fusion and entry to E64d 77, 229, 230, 233, 238 suggests that further action of a cysteine protease may be required. However, neither the factor(s) responsible for the E64 sensitivity nor its target (19 kDa‐GP1‐S‐S‐GP2 or a host constituent) has yet been identified. The roles of other potential triggering factors also remain unclear. Interestingly, the fusion subunits of EBOV GP and ASLV Env share structural and functional similarities 152, 196. Both contain a CX6CC motif, but neither of their receptor‐binding subunits contains a thiol exchange motif (CXXC), as seen in the MLV and HTLV Envs, which use disulfide bond isomerization for fusion (see above). Hence, an analogous fusion‐triggering mechanism for EBOV GP would require exogenous reducing activity. While disulfide reducing agents (at low pH) can induce the 19 kDa‐GP1‐S‐S‐GP2 ectodomain to bind to target membranes 236, there is no evidence for a physiological role for disulfide bond reduction for EBOV entry. Even the role of NPC1 is still unclear. As for LAMP1 in LASV entry, does NPC1 play an active role in fusion triggering (i.e. induce significant conformational changes in 19 kDa‐GP1‐S‐S‐GP2 at physiological temperature) or is a main role, for example, to direct fusion to the limiting membrane of the endolysosome (i.e. away from the numerous small vesicles found within endolysosomes)? The role of TPC2 also remains to be elucidated 230, 232. Hence, triggering of primed 19 kDa‐GP1‐S‐S‐GP2 to form a productive fusion pore is complex, and it may represent a novel mode of fusion triggering.
Triggering HCV E1/E2 for fusion
HCV is a member of the Flaviviridae (Hepacivirus genus). Within the family, it groups most closely with pestiviruses (Pestivirus genus). Evidence suggests that the fusion mechanism of these viruses is novel. Both express two type I transmembrane glycoproteins (E1 and E2) required for fusion 244, 245, 246, 247, 248, 249, 250, but which is the bona fide fusion protein has been the subject of debate. While it was originally predicted that pestivirus and hepacivirus E2 proteins were class II fusion proteins, new evidence suggests this is not the case. First, the structures of the pestivirus 251, 252 and HCV 253, 254 E2 ectodomains do not conform to the class II fusion protein fold. Second, at low pH, the N‐terminal domain of pestivirus E2 is disordered 252, as seen for the E2 protein (the receptor binding, not fusion, protein) of Sindbis virus. Third, a consensus is emerging for a fusion loop (residues 264–293) in HCV E1 255, 256, 257. Fourth, a structure of the 79‐residue N‐terminal domain of HCV E1 (which does not include the predicted fusion loop) reveals a novel fold 258.
Additional findings point to a novel fusion mechanism for pestiviruses and hepaciviruses. Fusion for both viruses requires low pH 247, 259, 260, likely protonation of one or more histidines 251, 261, but simple low pH treatment of cells with prebound viruses does not lead to infection. While the combined action of low pH and a disulfide bond reducing agent induced a low level of pestivirus infection via fusion at the plasma membrane 262, similar treatments did not induce HCV infection 260. Instead, for HCV, a 60‐min incubation at neutral pH and 37°C followed by a low pH pulse induced some infection 259, 260. One study 263 suggested a role for binding of HCV E2 to CD81, one of the HCV receptors 264, 265, during the 60‐min preparatory period. However, details of the mechanism of HCV fusion remain to be resolved.
Concluding Remarks
The field of enveloped virus fusion in endosomes has come a long way since the inaugural paper by Helenius and coworkers 1. As elaborated above, detailed fusion mechanisms – encompassing key structural elements and key structural changes in the fusion protein, and key environmental triggering cues (low pH, receptors and proteases) – are now known for representatives of class I, II and III fusion proteins. Many questions remain. For examples: Are there additional classes of enveloped viral fusion proteins? Are there additional fusion‐triggering mechanisms? What are the common principles by which proteases trigger fusion proteins post receptor binding? How is fusion triggering orchestrated in multicomponent fusion machines? In another vein, recent work suggests that enveloped virus‐membrane fusion can spark signaling responses that trigger innate immune responses 266, 267, and that cells can use interferon‐inducible transmembrane proteins to block virus–cell fusion 268. For their part, some viruses, notably HIV, have evolved means to counter attempts by cells to thwart virus–cell fusion 269, 270. How exactly are these battles between enveloped viruses and cells enacted? And lastly, while there are drugs in use, in development or under consideration that block enveloped virus fusion 271, can we develop antifusion antiviral strategies that are broadly applicable, cost‐effective, non‐toxic and potent?
Acknowledgments
We thank numerous colleagues for helpful discussions, and we thank Kathryn Schornberg for preparing the figures. We apologize for citation omissions. National Institutes of Health Grants R21 AI103601 and RO1 AI114776 (to J. M. W.), and R21 AI111085, R21 AI117300 and National Science Foundation grant 1504846 (to G. R. W.) supported recent work in this area in our laboratories. G. R. W.'s research is funded in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under CEIRS Contract No. HHSN272201400005C. G. R. W. is also supported by grants from the Cornell Feline Health Center, the Winn Feline Health Foundation and the Morris Animal Foundation. The authors have no conflicts of interest to report.
References
- 1. Helenius A, Kartenbeck J, Simons K, Fries E. On the entry of Semliki forest virus into BHK‐21 cells. J Cell Biol 1980;84:404–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Matlin KS, Reggio H, Helenius A, Simons K. Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol 1981;91:601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marsh M, Helenius A. Adsorptive endocytosis of Semliki Forest virus. J Mol Biol 1980;142:439–454. [DOI] [PubMed] [Google Scholar]
- 4. Marsh M, Bolzau E, Helenius A. Penetration of Semliki Forest virus from acidic prelysosomal vacuoles. Cell 1983;32:931–940. [DOI] [PubMed] [Google Scholar]
- 5. Simons K, Garoff H, Helenius A. How an animal virus gets into and out of its host cell. Sci Am 1982;246:58–66. [DOI] [PubMed] [Google Scholar]
- 6. White J, Helenius A. pH‐dependent fusion between the Semliki Forest virus membrane and liposomes. Proc Natl Acad Sci USA 1980;77:3273–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. White J, Matlin K, Helenius A. Cell fusion by Semliki Forest, influenza, and vesicular stomatitis viruses. J Cell Biol 1981;89:674–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. White J, Kartenbeck J, Helenius A. Membrane fusion activity of influenza virus. EMBO J 1982;1:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. White J, Helenius A, Gething MJ. Haemagglutinin of influenza virus expressed from a cloned gene promotes membrane fusion. Nature 1982;300:658–659. [DOI] [PubMed] [Google Scholar]
- 10. Maeda T, Kawasaki K, Ohnishi S. Interaction of influenza virus hemagglutinin with target membrane lipids is a key step in virus‐induced hemolysis and fusion at pH 5.2. Proc Natl Acad Sci USA 1981;78:4133–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Quirin K, Eschli B, Scheu I, Poort L, Kartenbeck J, Helenius A. Lymphocytic choriomeningitis virus uses a novel endocytic pathway for infectious entry via late endosomes. Virology 2008;378:21–33. [DOI] [PubMed] [Google Scholar]
- 12. Pasqual G, Rojek JM, Masin M, Chatton J‐Y, Kunz S. Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog 2011;7:e1002232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lozach P‐Y, Huotari J, Helenius A. Late‐penetrating viruses. Curr Opin Virol 2011;1:35–43. [DOI] [PubMed] [Google Scholar]
- 14. Mercer J, Schelhaas M, Helenius A. Virus entry by endocytosis. Annu Rev Biochem 2010;79:803–833. [DOI] [PubMed] [Google Scholar]
- 15. Grove J, Marsh M. The cell biology of receptor‐mediated virus entry. J Cell Biol 2011;195:1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cossart P, Helenius A. Endocytosis of viruses and bacteria. Cold Spring Harb Perspect Biol 2014;6:a016972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Amara A, Mercer J. Viral apoptotic mimicry. Nat Rev Microbiol 2015;13:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mercer J, Helenius A. Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr Opin Microbiol 2012;15:1–10. [DOI] [PubMed] [Google Scholar]
- 19. Matlin KS, Reggio H, Helenius A, Simons K. Pathway of vesicular stomatitis virus entry leading to infection. J Mol Biol 1982;156:609–631. [DOI] [PubMed] [Google Scholar]
- 20. Mercer J, Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 2008;320:531–535. [DOI] [PubMed] [Google Scholar]
- 21. Rizopoulos Z, Balistreri G, Kilcher S, Martin CK, Syedbasha M, Helenius A, Mercer J. Vaccinia virus infection requires maturation of macropinosomes. Traffic 2015;16:814–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nanbo A, Imai M, Watanabe S, Noda T, Takahashi K, Neumann G, Halfmann P, Kawaoka Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein‐dependent manner. PLoS Pathog 2010;6:e1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. Cellular entry of Ebola virus involves uptake by a macropinocytosis‐like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog 2010;6:e1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mulherkar N, Raaben M, la Torre de JC, Whelan SP, Chandran K. The Ebola virus glycoprotein mediates entry via a non‐classical dynamin‐dependent macropinocytic pathway. Virology 2011;419:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krzyzaniak MA, Zumstein MT, Gerez JA, Picotti P, Helenius A. Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the f protein. PLoS Pathog 2013;9:e1003309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hetzenecker S, Helenius A, Krzyzaniak MA. HCMV induces macropinocytosis for host cell entry in fibroblasts. Traffic 2016;TRA.12355. [DOI] [PubMed] [Google Scholar]
- 27. Rossman JS, Leser GP, Lamb RA. Filamentous influenza virus enters cells via macropinocytosis. J Virol 2012; 86:10950–10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de Vries E, Tscherne D, Wienholts M. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog 2011;7:e1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sieczkarski SB, Whittaker GR. Influenza virus can enter and infect cells in the absence of clathrin‐mediated endocytosis. J Virol 2002;76:10455–10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen C, Zhuang X. Epsin 1 is a cargo‐specific adaptor for the clathrin‐mediated endocytosis of the influenza virus. Proc Natl Acad Sci USA 2008;105:11790–11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. HIV enters cells via endocytosis and dynamin‐dependent fusion with endosomes. Cell 2009;137:433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cox RG, Mainou BA, Johnson M, Hastings AK, Schuster JE, Dermody TS, Williams JV. Human metapneumovirus is capable of entering cells by fusion with endosomal membranes. PLoS Pathog 2015;11:e1005303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haid S, Grethe C, Bankwitz D, Grunwald T, Pietschmann T. Identification of a human respiratory syncytial virus (hRSV) cell entry inhibitor by using a novel lentiviral pseudotype (hRSVpp) system. J Virol 2015;90: 3065–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lennartz F, Bayer K, Czerwonka N, Lu Y, Kehr K, Hirz M, Steinmetzer T, Garten W, Herden C. Surface glycoprotein of Borna disease virus mediates virus spread from cell to cell. Cell Microbiol 2016;18:340–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clemente R, la Torre de JC. Cell entry of Borna disease virus follows a clathrin‐mediated endocytosis pathway that requires Rab5 and microtubules. J Virol 2009;83:10406–10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fritz R, Stiasny K, Heinz FX. Identification of specific histidines as pH sensors in flavivirus membrane fusion. J Cell Biol 2008;183:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Misinzo GM, Delputte PL, Nauwynck HJ. Involvement of proteases in porcine reproductive and respiratory syndrome virus uncoating upon internalization in primary macrophages. Vet Res 2008;39:55. [DOI] [PubMed] [Google Scholar]
- 38. Macovei A, Petrareanu C, Lazar C, Florian P, Branza‐Nichita N. Regulation of hepatitis B virus infection by Rab5, Rab7, and the endolysosomal compartment. J Virol 2013;87:6415–6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Gorp H, Van Breedam W, Delputte PL, Nauwynck HJ. The porcine reproductive and respiratory syndrome virus requires trafficking through CD163‐positive early endosomes, but not late endosomes, for productive infection. Arch Virol 2009;154:1939–1943. [DOI] [PubMed] [Google Scholar]
- 40. Hernáez B, Quetglas JI, Dalmau‐Mena I, Alonso C. PLOS ONE: Endosomal maturation, Rab7 GTPase and phosphoinositides in African swine fever virus entry. PLoS ONE 2012;7:e48853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kielian MC, Marsh M, Helenius A. Kinetics of endosome acidification detected by mutant and wild‐type Semliki Forest virus. EMBO J 1986;5:3103–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johannsdottir HK, Mancini R, Kartenbeck J, Amato L, Helenius A. Host cell factors and functions involved in vesicular stomatitis virus entry. J Virol 2009;83:440–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Galloway SE, Reed ML, Russell CJ, Steinhauer DA. Influenza HA subtypes demonstrate divergent phenotypes for cleavage activation and pH of fusion: implications for host range and adaptation. PLoS Pathog 2013;9:e1003151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zaraket H, Bridges OA, Duan S, Baranovich T, Yoon S‐W, Reed ML, Salomon R, Webby RJ, Webster RG, Russell CJ. Increased acid stability of the hemagglutinin protein enhances H5N1 influenza virus growth in the upper respiratory tract but is insufficient for transmission in ferrets. J Virol 2013;87:9911–9922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zaraket H, Bridges OA, Russell CJ. The pH of activation of the hemagglutinin protein regulates H5N1 influenza virus replication and pathogenesis in mice. J Virol 2013;87:4826–4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Imai M, Watanabe T, Hatta M, Das SC, Ozawa M, Shinya K, Zhong G, Hanson A, Katsura H, Watanabe S, Li C, Kawakami E, Yamada S, Kiso M, Suzuki Y, et al. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 2012;486:420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Herfst S, Schrauwen EJA, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Sorrell EM, Bestebroer TM, Burke DF, Smith DJ, Rimmelzwaan GF, Osterhaus ADME, Fouchier RAM. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 2012;336:1534–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Russier M, Yang G, Rehg JE, Wong S‐S, Mostafa HH, Fabrizio TP, Barman S, Krauss S, Webster RG, Webby RJ, Russell CJ. Molecular requirements for a pandemic influenza virus: an acid‐stable hemagglutinin protein. Proc Natl Acad Sci USA 2016;113:1636–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Costello DA, Whittaker GR, Daniel S. Variations in pH sensitivity, acid stability, and fusogenicity of three influenza virus H3 subtypes. J Virol 2015;89:350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kielian MC, Helenius A. Role of cholesterol in fusion of Semliki Forest virus with membranes. J Virol 1984;52:281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nieva JL, Bron R, Corver J, Wilschut J. Membrane fusion of Semliki Forest virus requires sphingolipids in the target membrane. EMBO J 1994;13:2797–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kleinfelter LM, Jangra RK, Jae LT, Herbert AS, Mittler E, Stiles KM, Wirchnianski AS, Kielian M, Brummelkamp TR, Dye JM, Chandran K. Haploid genetic screen reveals a profound and direct dependence on cholesterol for hantavirus membrane fusion. MBio 2015;6:e00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zaitseva E, Yang S‐T, Melikov K, Pourmal S, Chernomordik LV. Dengue virus ensures its fusion in late endosomes using compartment‐specific lipids. PLoS Pathog 2010;6:e1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Matos PM, Marin M, Ahn B, Lam W, Santos NC, Melikyan GB. Anionic lipids are required for vesicular stomatitis virus G protein‐mediated single particle fusion with supported lipid bilayers. J Biol Chem 2013;288:12416–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kadlec J, Loureiro S, Abrescia NGA, Stuart DI, Jones IM. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat Struct Mol Biol 2008;15:1024–1030. [DOI] [PubMed] [Google Scholar]
- 56. Li Z, Hung C, Paterson RG, Michel F, Fuentes S, Place R, Lin Y, Hogan RJ, Lamb RA, He B. Type II integral membrane protein, TM of J paramyxovirus promotes cell‐to‐cell fusion. Proc Natl Acad Sci USA 2015;112:12504–12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Read J, Clancy EK, Sarker M, de Antueno R, Langelaan DN, Parmar HB, Shin K, Rainey JK, Duncan R. Reovirus FAST proteins drive pore formation and syncytiogenesis using a novel helix‐loop‐helix fusion‐inducing lipid packing sensor. PLoS Pathog 2015;11:e1004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nunberg JH, York J. The curious case of arenavirus entry, and its inhibition. Viruses 2012;4:83–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moss B. Poxvirus cell entry: how many proteins does it take? Viruses 2012;4:688–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cooper RS, Heldwein EE. Herpesvirus gB: a finely tuned fusion machine. Viruses 2015;7:6552–6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Baquero E, Albertini AA, Gaudin Y. Recent mechanistic and structural insights on class III viral fusion glycoproteins. Curr Opin Struct Biol 2015;33:52–60. [DOI] [PubMed] [Google Scholar]
- 62. Sathiyamoorthy K, Jiang J, Hu YX, Rowe CL, Möhl BS, Chen J, Jiang W, Mellins ED, Longnecker R, Zhou ZH, Jardetzky TS. Assembly and architecture of the EBV B cell entry triggering complex. PLoS Pathog 2014;10:e1004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gallagher JR, Atanasiu D, Saw WT, Paradisgarten MJ, Whitbeck JC, Eisenberg RJ, Cohen GH. Functional fluorescent protein insertions in herpes simplex virus gB report on gB conformation before and after execution of membrane fusion. PLoS Pathog 2014;10:e1004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Delgado CL, Núñez E, Yélamos B, Gómez‐Gutiérrez J, Peterson DL, Gavilanes F. Spectroscopic characterization and fusogenic properties of preS domains of duck hepatitis B virus. Biochemistry 2012;51:8444–8454. [DOI] [PubMed] [Google Scholar]
- 65. White JM, Delos SE, Brecher M, Schornberg K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 2008;43:189–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Harrison SC. Viral membrane fusion. Virology 2015;479‐480C:498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bose S, Jardetzky TS, Lamb RA. Timing is everything: fine‐tuned molecular machines orchestrate paramyxovirus entry. Virology 2015;479–480:518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kielian M. Mechanisms of virus membrane fusion proteins. Annu Rev Virol 2014;1:171–189. [DOI] [PubMed] [Google Scholar]
- 69. Plemper RK. Cell entry of enveloped viruses. Curr Opin Virol 2011;1:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cogen GH. Herpes virus fusion and entry: a story with many characters. Viruses 2012;4:800–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schmidt FI, Bleck CKE, Mercer J. Poxvirus host cell entry. Curr Opin Virol 2012;2:20–27. [DOI] [PubMed] [Google Scholar]
- 72. Park HE, Gruenke JA, White JM. Leash in the groove mechanism of membrane fusion. Nat Struct Biol 2003;10:1048–1053. [DOI] [PubMed] [Google Scholar]
- 73. Böttcher‐Friebertshäuser E, Garten W, Matrosovich M, Klenk H‐D. The hemagglutinin: a determinant of pathogenicity. Curr Top Microbiol Immunol 2014;385:3–34. [DOI] [PubMed] [Google Scholar]
- 74. Zmora P, Blazejewska P, Moldenhauer A‐S, Welsch K, Nehlmeier I, Wu Q, Schneider H, Pöhlmann S, Bertram S. DESC1 and MSPL activate influenza A viruses and emerging coronaviruses for host cell entry. J Virol 2014;88:12087–12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Böttcher‐Friebertshäuser E, Klenk H‐D, Garten W. Activation of influenza viruses by proteases from host cells and bacteria in the human airway epithelium. Pathog Dis 2013;69:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005;308:1643–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J Virol 2006;80:4174–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pager CT, Dutch RE. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J Virol 2005;79:12714–12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pager CT, Craft WW, Patch J, Dutch RE. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology 2006;346:251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Diederich S, Sauerhering L, Weis M, Altmeppen H, Schaschke N, Reinheckel T, Erbar S, Maisner A. Activation of the Nipah virus fusion protein in MDCK cells is mediated by cathepsin B within the endosome‐recycling compartment. J Virol 2012;86:3736–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cao S, Zhang W. Characterization of an early‐stage fusion intermediate of Sindbis virus using cryoelectron microscopy. Proc Natl Acad Sci USA 2013;110:13362–13367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liu CY, Kielian M. E1 mutants identify a critical region in the trimer interface of the Semliki forest virus fusion protein. J Virol 2009;83:11298–11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Brandenberg OF, Magnus C, Rusert P, Regoes RR, Trkola A. Different infectivity of HIV‐1 strains is linked to number of envelope trimers required for entry. PLoS Pathog 2015;11:e1004595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Danieli T, Pelletier SL, Henis YI, White JM. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol 1996;133:559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Floyd DL, Ragains JR, Skehel JJ, Harrison SC, van Oijen AM. Single‐particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci USA 2008;105:15382–15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Xu K, Chan Y‐P, Bradel‐Tretheway B, Akyol‐Ataman Z, Zhu Y, Dutta S, Yan L, Feng Y, Wang L‐F, Skiniotis G, Lee B, Zhou ZH, Broder CC, Aguilar HC, Nikolov DB. Crystal structure of the pre‐fusion Nipah virus fusion glycoprotein reveals a novel hexamer‐of‐trimers assembly. PLoS Pathog 2015;11:e1005322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gibbons DL, Vaney M‐C, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA. Conformational change and protein‐protein interactions of the fusion protein of Semliki Forest virus. Nature 2004;427:320–325. [DOI] [PubMed] [Google Scholar]
- 88. Chao LH, Klein DE, Schmidt AG, Peña JM, Harrison SC. Sequential conformational rearrangements in flavivirus membrane fusion. Elife 2014;3:e04389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Markovic I, Pulyaeva H, Sokoloff A, Chernomordik LV. Membrane fusion mediated by baculovirus gp64 involves assembly of stable gp64 trimers into multiprotein aggregates. J Cell Biol 1998;143:1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Libersou S, Albertini AAV, Ouldali M, Maury V, Maheu C, Raux H, de Haas F, Roche S, Gaudin Y, Lepault J. Distinct structural rearrangements of the VSV glycoprotein drive membrane fusion. J Cell Biol 2010;191:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Maurer UE, Zeev‐Ben‐Mordehai T, Pandurangan AP, Cairns TM, Hannah BP, Whitbeck JC, Eisenberg RJ, Cohen GH, Topf M, Huiskonen JT, Grünewald K. The structure of herpesvirus fusion glycoprotein B‐bilayer complex reveals the protein‐membrane and lateral protein‐protein interaction. Structure 2013;21:1396–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Spruce AE, Iwata A, White JM, Almers W. Patch clamp studies of single cell‐fusion events mediated by a viral fusion protein. Nature 1989;342:555–558. [DOI] [PubMed] [Google Scholar]
- 93. Chernomordik LV, Kozlov MM. Mechanics of membrane fusion. Nat Struct Mol Biol 2008;15:675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cohen FS, Melikyan GB. The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J Membr Biol 2004;199:1–14. [DOI] [PubMed] [Google Scholar]
- 95. Lee KK. Architecture of a nascent viral fusion pore. EMBO J 2010;29:1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 1981;289:366–373. [DOI] [PubMed] [Google Scholar]
- 97. Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994;371:37–43. [DOI] [PubMed] [Google Scholar]
- 98. Tamm LK. Hypothesis: spring‐loaded boomerang mechanism of influenza hemagglutinin‐mediated membrane fusion. Biochim Biophys Acta 1614;2003:14–23. [DOI] [PubMed] [Google Scholar]
- 99. Lai AL, Freed JH. The interaction between influenza HA fusion peptide and transmembrane domain affects membrane structure. Biophys J 2015;109:2523–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chang D‐K, Cheng S‐F, Kantchev EAB, Lin C‐H, Liu Y‐T. Membrane interaction and structure of the transmembrane domain of influenza hemagglutinin and its fusion peptide complex. BMC Biol 2008;6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Reuven EM, Dadon Y, Viard M, Manukovsky N, Blumenthal R, Shai Y. HIV‐1 gp41 transmembrane domain interacts with the fusion peptide: implication in lipid mixing and inhibition of virus‐cell fusion. Biochemistry 2012;51:2867–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Vishwanathan SA, Hunter E. Importance of the membrane‐perturbing properties of the membrane‐proximal external region of human immunodeficiency virus type 1 gp41 to viral fusion. J Virol 2008;82:5118–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Howard MW, Travanty EA, Jeffers SA, Smith MK, Wennier ST, Thackray LB, Holmes KV. Aromatic amino acids in the juxtamembrane domain of severe acute respiratory syndrome coronavirus spike glycoprotein are important for receptor‐dependent virus entry and cell‐cell fusion. J Virol 2008;82:2883–2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tong S, Yi F, Martin A, Yao Q, Li M, Compans RW. Three membrane‐proximal amino acids in the human parainfluenza type 2 (HPIV 2) F protein are critical for fusogenic activity. Virology 2001;280:52–61. [DOI] [PubMed] [Google Scholar]
- 105. Jeetendra E, Ghosh K, Odell D, Li J, Ghosh HP, Whitt MA. The membrane‐proximal region of vesicular stomatitis virus glycoprotein G ectodomain is critical for fusion and virus infectivity. J Virol 2003;77:12807–12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Backovic M, Jardetzky TS, Longnecker R. Hydrophobic residues that form putative fusion loops of Epstein‐Barr virus glycoprotein B are critical for fusion activity. J Virol 2007;81:9596–9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Baquero E, Albertini AA, Raux H, Buonocore L, Rose JK, Bressanelli S, Gaudin Y. Structure of the low pH conformation of Chandipura virus G reveals important features in the evolution of the vesiculovirus glycoprotein. PLoS Pathog 2015;11:e1004756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Madu IG, Roth SL, Belouzard S, Whittaker GR. Characterization of a highly conserved domain within the severe acute respiratory syndrome coronavirus spike protein S2 domain with characteristics of a viral fusion peptide. J Virol 2009;83:7411–7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Walls AC, Tortorici MA, Bosch BJ, Frenz B, Rottier PJM, DiMaio F, Rey FA, Veesler D. Cryo‐electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 2016;531:114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kirchdoerfer RN, Cotrell CA, Wang N, Pallesen J, Yassine HM, Turner HL, Corbett KS, Graham BS, McLellan JS, Ward AB. Pre‐fusion structure of a human coronavirus spike protein. Nature 2016;531:118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lai AL, Park H, White JM, Tamm LK. Fusion peptide of influenza hemagglutinin requires a fixed angle boomerang structure for activity. J Biol Chem 2006;281:5760–5770. [DOI] [PubMed] [Google Scholar]
- 112. Lorieau JL, Louis JM, Schwieters CD, Bax A. pH‐triggered, activated‐state conformations of the influenza hemagglutinin fusion peptide revealed by NMR. Proc Natl Acad Sci USA 2012;109:19994–19999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lai AL, Moorthy AE, Li Y, Tamm LK. Fusion activity of HIV gp41 fusion domain is related to its secondary structure and depth of membrane insertion in a cholesterol‐dependent fashion. J Mol Biol 2012;418:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Qiang W, Sun Y, Weliky DP. A strong correlation between fusogenicity and membrane insertion depth of the HIV fusion peptide. Proc Natl Acad Sci 2009;106:15314–15319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Haque ME, Koppaka V, Axelsen PH, Lentz BR. Properties and structures of the influenza and HIV fusion peptides on lipid membranes: implications for a role in fusion. Biophys J 2005;89:3183–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008;454:177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gregory SM, Harada E, Liang B, Delos SE, White JM, Tamm LK. Structure and function of the complete internal fusion loop from Ebolavirus glycoprotein 2. Proc Natl Acad Sci USA 2011;108:11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Han X, Bushweller JH, Cafiso DS, Tamm LK. Membrane structure and fusion‐triggering conformational change of the fusion domain from influenza hemagglutinin. Nat Struct Biol 2001;8:715–720. [DOI] [PubMed] [Google Scholar]
- 119. Li Y, Han X, Lai AL, Bushweller JH, Cafiso DS, Tamm LK. Membrane structures of the hemifusion‐inducing fusion peptide mutant G1S and the fusion‐blocking mutant G1V of influenza virus hemagglutinin suggest a mechanism for pore opening in membrane fusion. J Virol 2005;79:12065–12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Qiao H, Armstrong RT, Melikyan GB, Cohen FS, White JM. A specific point mutant at position 1 of the influenza hemagglutinin fusion peptide displays a hemifusion phenotype. Mol Biol Cell 1999;10:2759–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Gregory SM, Larsson P, Nelson EA, Kasson PM, White JM, Tamm LK. Ebolavirus entry requires a compact hydrophobic fist at the tip of the fusion loop. J Virol 2014;88:6636–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Modis Y, Ogata S, Clements D, Harrison SC. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004;427:313–319. [DOI] [PubMed] [Google Scholar]
- 123. Kanai R, Kar K, Anthony K, Gould LH, Ledizet M, Fikrig E, Marasco WA, Koski RA, Modis Y. Crystal structure of west Nile virus envelope glycoprotein reveals viral surface epitopes. J Virol 2006;80:11000–11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Bressanelli S, Stiasny K, Allison SL, Stura EA, Duquerroy S, Lescar J, Heinz FX, Rey FA. Structure of a flavivirus envelope glycoprotein in its low‐pH‐induced membrane fusion conformation. EMBO J 2004;23:728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Cifuentes‐Muñoz N, Salazar‐Quiroz N, Tischler ND. Hantavirus Gn and Gc envelope glycoproteins: key structural units for virus cell entry and virus assembly. Viruses 2014;6:1801–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Roche S, Rey FA, Gaudin Y, Bressanelli S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 2007;315:843–848. [DOI] [PubMed] [Google Scholar]
- 127. Sun X, Belouzard S, Whittaker GR. Molecular architecture of the bipartite fusion loops of vesicular stomatitis virus glycoprotein G, a class III viral fusion protein. J Biol Chem 2008;283:6418–6427. [DOI] [PubMed] [Google Scholar]
- 128. Dubé M, Rey FA, Kielian M. Rubella virus: first calcium‐requiring viral fusion protein. PLoS Pathog 2014;10:e1004530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. DuBois RM, Vaney M‐C, Tortorici MA, Kurdi RA, Barba‐Spaeth G, Krey T, Rey FA. Functional and evolutionary insight from the crystal structure of rubella virus protein E1. Nature 2013;493:552–556. [DOI] [PubMed] [Google Scholar]
- 130. Hannah BP, Heldwein EE, Bender FC, Cohen GH, Eisenberg RJ. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J Virol 2007;81:4858–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006;313:217–220. [DOI] [PubMed] [Google Scholar]
- 132. Backovic M, Longnecker R, Jardetzky TS. Structure of a trimeric variant of the Epstein‐Barr virus glycoprotein B. Proc Natl Acad Sci USA 2009;106:2880–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Melikyan GB. HIV entry: a game of hide‐and‐fuse? Curr Opin Virol 2014;4:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hulme AE, Hope TJ. Live cell imaging of retroviral entry. Annu Rev Virol 2014;1:501–515. [DOI] [PubMed] [Google Scholar]
- 135. Chu VC, McElroy LJ, Chu V, Bauman BE, Whittaker GR. The avian coronavirus infectious bronchitis virus undergoes direct low‐pH‐dependent fusion activation during entry into host cells. J Virol 2006;80:3180–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Schowalter RM, Chang A, Robach JG, Buchholz UJ, Dutch RE. Low‐pH triggering of human metapneumovirus fusion: essential residues and importance in entry. J Virol 2009;83:1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Yun B, Guan X, Liu Y, Gao Y, Wang Y, Qi X, Cui H, Liu C, Zhang Y, Gao L, Li K, Gao H, Gao Y, Wang X. Trypsin‐ and low pH‐mediated fusogenicity of avian metapneumovirus fusion proteins is determined by residues at positions 100, 101 and 294. Sci Rep 2015;5:15584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wilen CB, Tilton JC, Doms RW. HIV: cell binding and entry. Cold Spring Harb Perspect Med 2012;2:a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Mothes W, Boerger AL, Narayan S, Cunningham JM, Young JA. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 2000;103:679–689. [DOI] [PubMed] [Google Scholar]
- 140. Matsuyama S, Delos SE, White JM. Sequential roles of receptor binding and low pH in forming prehairpin and hairpin conformations of a retroviral envelope glycoprotein. J Virol 2004;78:8201–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Melikyan GB, Barnard RJO, Markosyan RM, Young JAT, Cohen FS. Low pH is required for avian sarcoma and leukosis virus Env‐induced hemifusion and fusion pore formation but not for pore growth. J Virol 2004;78:3753–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Melikyan GB, Barnard RJO, Abrahamyan LG, Mothes W, Young JAT. Imaging individual retroviral fusion events: from hemifusion to pore formation and growth. Proc Natl Acad Sci USA 2005;102:8728–8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Dollery SJ, Wright CC, Johnson DC, Nicola AV. Low‐pH‐dependent changes in the conformation and oligomeric state of the prefusion form of herpes simplex virus glycoprotein B are separable from fusion activity. J Virol 2011;85:9964–9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Maeda T, Ohnishi S. Activation of influenza virus by acidic media causes hemolysis and fusion of erythrocytes. FEBS Lett 1980;122:283–287. [DOI] [PubMed] [Google Scholar]
- 145. Huang RT, Rott R, Klenk HD. Influenza viruses cause hemolysis and fusion of cells. Virology 1981;110:243–247. [DOI] [PubMed] [Google Scholar]
- 146. Skehel JJ, Bayley PM, Brown EB, Martin SR, Waterfield MD, White JM, Wilson IA, Wiley DC. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus‐mediated membrane fusion. Proc Natl Acad Sci USA 1982;79:968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 2000;69:531–569. [DOI] [PubMed] [Google Scholar]
- 148. White JM, Wilson IA. Anti‐peptide antibodies detect steps in a protein conformational change: low‐pH activation of the influenza virus hemagglutinin. J Cell Biol 1987;105:2887–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Daniels RS, Downie JC, Hay AJ, Knossow M, Skehel JJ, Wang ML, Wiley DC. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 1985;40:431–439. [DOI] [PubMed] [Google Scholar]
- 150. Thoennes S, Li Z‐N, Lee B‐J, Langley WA, Skehel JJ, Russell RJ, Steinhauer DA. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology 2008;370:403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Mair CM, Meyer T, Schneider K, Huang Q, Veit M, Herrmann A. A histidine residue of the influenza virus hemagglutinin controls the pH dependence of the conformational change mediating membrane fusion. J Virol 2014;88:13189–13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Delos SE, La B, Gilmartin A, White JM. Studies of the “chain reversal regions” of the avian sarcoma/leukosis virus (ASLV) and ebolavirus fusion proteins: analogous residues are important, and a His residue unique to EnvA affects the pH dependence of ASLV entry. J Virol 2010;84:5687–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Kalani MR, Moradi A, Moradi M, Tajkhorshid E. Characterizing a histidine switch controlling pH‐dependent conformational changes of the influenza virus hemagglutinin. Biophys J 2013;105:993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Zheng Y, Sánchez‐San Martín C, Qin Z‐L, Kielian M. The domain I‐domain III linker plays an important role in the fusogenic conformational change of the alphavirus membrane fusion protein. J Virol 2011;85:6334–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Nayak V, Dessau M, Kucera K, Anthony K, Ledizet M, Modis Y. Crystal structure of dengue virus type 1 envelope protein in the postfusion conformation and its implications for membrane fusion. J Virol 2009;83:4338–4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Nelson S, Poddar S, Lin T‐Y, Pierson TC. Protonation of individual histidine residues is not required for the pH‐dependent entry of west nile virus: evaluation of the “histidine switch” hypothesis. J Virol 2009;83:12631–12635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. de Boer SM, Kortekaas J, Spel L, Rottier PJM, Moormann RJM, Bosch BJ. Acid‐activated structural reorganization of the Rift Valley fever virus Gc fusion protein. J Virol 2012;86:13642–13652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Dessau M, Modis Y. Crystal structure of glycoprotein C from Rift Valley fever virus. Proc Natl Acad Sci USA 2013;110:1696–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Zheng A, Yuan F, Kleinfelter LM, Kielian M. A toggle switch controls the low pH‐triggered rearrangement and maturation of the dengue virus envelope proteins. Nat Commun 2014;5:3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Carneiro FA, Stauffer F, Lima CS, Juliano MA, Juliano L, Da Poian AT. Membrane fusion induced by vesicular stomatitis virus depends on histidine protonation. J Biol Chem 2003;278:13789–13794. [DOI] [PubMed] [Google Scholar]
- 161. Li Z, Blissard GW. Autographa californica multiple nucleopolyhedrovirus GP64 protein: roles of histidine residues in triggering membrane fusion and fusion pore expansion. J Virol 2011;85:12492–12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Rachakonda PS, Veit M, Korte T, Ludwig K, Böttcher C, Huang Q, Schmidt MFG, Herrmann A. The relevance of salt bridges for the stability of the influenza virus hemagglutinin. FASEB J 2007;21:995–1002. [DOI] [PubMed] [Google Scholar]
- 163. Garcia NK, Guttman M, Ebner JL, Lee KK. Dynamic changes during acid‐induced activation of influenza hemagglutinin. Structure 2015;23:665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Mueller DS, Kampmann T, Yennamalli R, Young PR, Kobe B, Mark AE. Histidine protonation and the activation of viral fusion proteins. Biochem Soc Trans 2008;36:43–45. [DOI] [PubMed] [Google Scholar]
- 165. Ivanovic T, Rozendaal R, Floyd DL, Popovic M, van Oijen AM, Harrison SC. Kinetics of proton transport into influenza virions by the viral M2 channel. PLoS One 2012;7:e31566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Wang J, Wu Y, Ma C, Fiorin G, Wang J, Pinto LH, Lamb RA, Klein ML, Degrado WF. Structure and inhibition of the drug‐resistant S31N mutant of the M2 ion channel of influenza A virus. Proc Natl Acad Sci USA 2013;110:1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Batishchev OV, Shilova LA, Kachala MV, Tashkin VY, Sokolov VS, Fedorova NV, Baratova LA, Knyazev DG, Zimmerberg J, Chizmadzhev YA. pH‐dependent formation and disintegration of the influenza A virus protein scaffold to provide tension for membrane fusion. J Virol 2015;90:575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Fontana J, Steven AC. At low pH, influenza virus matrix protein M1 undergoes a conformational change prior to dissociating from the membrane. J Virol 2013;87:5621–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Stauffer S, Feng Y, Nebioglu F, Heilig R, Picotti P, Helenius A. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration. J Virol 2014;88:13029–13046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Mire C, Dube D, Delos S, White J. Glycoprotein‐dependent acidification of vesicular stomatitis virus enhances release of matrix protein. J Virol 2009;83:12139–12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Hover S, King B, Hall B, Loundras E‐A, Taqi H, Daly J, Dallas M, Peers C, Schnettler E, McKimmie C, Kohl A, Barr JN, Mankouri J. Modulation of potassium channels inhibits bunyavirus infection. J Biol Chem 2016;291:3411–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Bron R, Wahlberg JM, Garoff H, Wilschut J. Membrane fusion of Semliki Forest virus in a model system: correlation between fusion kinetics and structural changes in the envelope glycoprotein. EMBO J 1993;12:693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Umashankar M, Sánchez‐San Martín C, Liao M, Reilly B, Guo A, Taylor G, Kielian M. Differential cholesterol binding by class II fusion proteins determines membrane fusion properties. J Virol 2008;82:9245–9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. van der Schaar HM, Rust MJ, Chen C, van der Ende‐Metselaar H, Wilschut J, Zhuang X, Smit JM. Dissecting the cell entry pathway of dengue virus by single‐particle tracking in living cells. PLoS Pathog 2008;4:e1000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Nour AM, Li Y, Wolenski J, Modis Y. Viral membrane fusion and nucleocapsid delivery into the cytoplasm are distinct events in some flaviviruses. PLoS Pathog 2013;9:e1003585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Esposito DLA, Nguyen JB, DeWitt DC, Rhoades E, Modis Y. Physico‐chemical requirements and kinetics of membrane fusion of flavivirus‐like particles. J Gen Virol 2015;96:1702–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Roth SL, Whittaker GR. Promotion of vesicular stomatitis virus fusion by the endosome‐specific phospholipid bis(monoacylglycero)phosphate (BMP). FEBS Lett 2011;585:865–869. [DOI] [PubMed] [Google Scholar]
- 178. Le Blanc I, Luyet P‐P, Pons V, Ferguson C, Emans N, Petiot A, Mayran N, Demaurex N, Fauré J, Sadoul R, Parton RG, Gruenberg J. Endosome‐to‐cytosol transport of viral nucleocapsids. Nat Cell Biol 2005;7:653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Bitto D, Halldorsson S, Caputo A, Huiskonen JT. Low pH and anionic lipid dependent fusion of Uukuniemi phlebovirus to liposomes. J Biol Chem 2016;M115.691113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Wallin M, Ekström M, Garoff H. Isomerization of the intersubunit disulphide‐bond in Env controls retrovirus fusion. EMBO J 2004;23:54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Li K, Zhang S, Kronqvist M, Wallin M, Ekström M, Derse D, Garoff H. Intersubunit disulfide isomerization controls membrane fusion of human T‐cell leukemia virus Env. J Virol 2008;82:7135–7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Tran EEH, Borgnia MJ, Kuybeda O, Schauder DM, Bartesaghi A, Frank GA, Sapiro G, Milne JLS, Subramaniam S. Structural mechanism of trimeric HIV‐1 envelope glycoprotein activation. PLoS Pathog 2012;8:e1002797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Munro JB, Gorman J, Ma X, Zhou Z, Arthos J, Burton DR, Koff WC, Courter JR, Smith AB, Kwong PD, Blanchard SC, Mothes W. Conformational dynamics of single HIV‐1 envelope trimers on the surface of native virions. Science 2014;346:759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 2005;433:834–841. [DOI] [PubMed] [Google Scholar]
- 185. Jardetzky TS, Lamb RA. Activation of paramyxovirus membrane fusion and virus entry. Curr Opin Virol 2014;5:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Wong JJW, Paterson RG, Lamb RA, Jardetzky TS. Structure and stabilization of the Hendra virus F glycoprotein in its prefusion form. Proc Natl Acad Sci USA 2016;113:1056–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Delos SE, Godby JA, White JM. Receptor‐induced conformational changes in the SU subunit of the avian sarcoma/leukosis virus A envelope protein: implications for fusion activation. J Virol 2005;79:3488–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Smith JG, Cunningham JM. Receptor‐induced thiolate couples Env activation to retrovirus fusion and infection. PLoS Pathog 2007;3:e198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Hernandez LD, Peters RJ, Delos SE, Young JA, Agard DA, White JM. Activation of a retroviral membrane fusion protein: soluble receptor‐induced liposome binding of the ALSV envelope glycoprotein. J Cell Biol 1997;139:1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Hernandez LD, White JM. Mutational analysis of the candidate internal fusion peptide of the avian leukosis and sarcoma virus subgroup A envelope glycoprotein. J Virol 1998;72:3259–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Damico RL, Crane J, Bates P. Receptor‐triggered membrane association of a model retroviral glycoprotein. Proc Natl Acad Sci USA 1998;95:2580–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Cardone G, Brecher M, Fontana J, Winkler DC, Butan C, White JM, Steven AC. Visualization of the two‐step fusion process of the retrovirus avian sarcoma/leukosis virus by cryo‐electron tomography. J Virol 2012;86:12129–12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Markosyan RM, Bates P, Cohen FS, Melikyan GB. A study of low pH‐induced refolding of Env of avian sarcoma and leukosis virus into a six‐helix bundle. Biophys J 2004;87:3291–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Netter RC, Amberg SM, Balliet JW, Biscone MJ, Vermeulen A, Earp LJ, White JM, Bates P. Heptad repeat 2‐based peptides inhibit avian sarcoma and leukosis virus subgroup a infection and identify a fusion intermediate. J Virol 2004;78:13430–13439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Padilla‐Parra S, Marin M, Kondo N, Melikyan GB. Synchronized retrovirus fusion in cells expressing alternative receptor isoforms releases the viral core into distinct sub‐cellular compartments. PLoS Pathog 2012;8:e1002694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Aydin H, Smrke BM, Lee JE. Structural characterization of a fusion glycoprotein from a retrovirus that undergoes a hybrid 2‐step entry mechanism. FASEB J 2013;27:5059–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Côté M, Zheng Y‐M, Liu S‐L. Receptor binding and low pH coactivate oncogenic retrovirus envelope‐mediated fusion. J Virol 2009;83:11447–11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. Ebola virus entry requires the cholesterol transporter Niemann‐Pick C1. Nature 2011;477:340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Jae LT, Raaben M, Herbert AS, Kuehne AI, Wirchnianski AS, Soh TK, Stubbs SH, Janssen H, Damme M, Saftig P, Whelan SP, Dye JM, Brummelkamp TR. Lassa virus entry requires a trigger‐induced receptor switch. Science 2014;344:1506–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Jae LT, Brummelkamp TR. Emerging intracellular receptors for hemorrhagic fever viruses. Trends Microbiol 2015;23:392–400. [DOI] [PubMed] [Google Scholar]
- 201. Li S, Sun Z, Pryce R, Parsy M‐L, Fehling SK, Schlie K, Siebert CA, Garten W, Bowden TA, Strecker T, Huiskonen JT. Acidic pH‐induced conformations and LAMP1 binding of the Lassa virus glycoprotein spike. PLoS Pathog 2016;12:e1005418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. Characterization of severe acute respiratory syndrome‐associated coronavirus (SARS‐CoV) spike glycoprotein‐mediated viral entry. Proc Natl Acad Sci USA 2004;101:4240–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Simmons G. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci USA 2005;102:11876–11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Bosch BJ, Bartelink W, Rottier PJM. Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J Virol 2008;82:8887–8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Matsuyama S, Ujike M, Morikawa S, Tashiro M, Taguchi F. Protease‐mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc Natl Acad Sci USA 2005;102:12543–12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Li F, Berardi M, Li W, Farzan M, Dormitzer PR, Harrison SC. Conformational states of the severe acute respiratory syndrome coronavirus spike protein ectodomain. J Virol 2006;80:6794–6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Bertram S, Glowacka I, Müller MA, Lavender H, Gnirss K, Nehlmeier I, Niemeyer D, He Y, Simmons G, Drosten C, Soilleux EJ, Jahn O, Steffen I, Pöhlmann S. Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin‐like protease. J Virol 2011;85:13363–13372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Belouzard S, Chu VC, Whittaker GR. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci USA 2009;106:5871–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Millet JK, Whittaker GR. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res 2015;202:120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Gierer S, Bertram S, Kaup F, Wrensch F, Heurich A, Krämer‐Kühl A, Welsch K, Winkler M, Meyer B, Drosten C, Dittmer U, Hahn von T, Simmons G, Hofmann H, Pöhlmann S. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J Virol 2013;87:5502–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Shirato K, Kawase M, Matsuyama S. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol 2013;87:12552–12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Qian Z, Dominguez SR, Holmes KV. Role of the spike glycoprotein of human Middle East respiratory syndrome coronavirus (MERS‐CoV) in virus entry and syncytia formation. PLoS One 2013;8:e76469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Millet JK, Whittaker GR. Host cell entry of Middle East respiratory syndrome coronavirus after two‐step, furin‐mediated activation of the spike protein. Proc Natl Acad Sci USA 2014;111:15214–15219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Burkard C, Verheije MH, Wicht O, van Kasteren SI, van Kuppeveld FJ, Haagmans BL, Pelkmans L, Rottier PJM, Bosch BJ, de Haan CAM. Coronavirus cell entry occurs through the endo‐/lysosomal pathway in a proteolysis‐dependent manner. PLoS Pathog 2014;10:e1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Sjöberg M, Lindqvist B, Garoff H. Activation of the alphavirus spike protein is suppressed by bound E3. J Virol 2011;85:5644–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. de Haan CAM, Stadler K, Godeke G‐J, Bosch BJ, Rottier PJM. Cleavage inhibition of the murine coronavirus spike protein by a furin‐like enzyme affects cell‐cell but not virus‐cell fusion. J Virol 2004;78:6048–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Licitra BN, Millet JK, Regan AD, Hamilton BS, Rinaldi VD, Duhamel GE, Whittaker GR. Mutation in spike protein cleavage site and pathogenesis of feline coronavirus. Emerg Infect Dis 2013;19:1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Le Coupanec A, Desforges M, Meessen‐Pinard M, Dubé M, Day R, Seidah NG, Talbot PJ. Cleavage of a neuroinvasive human respiratory virus spike glycoprotein by proprotein convertases modulates neurovirulence and virus spread within the central nervous system. PLoS Pathog 2015;11:e1005261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Licitra BN, Sams KL, Lee DW, Whittaker GR. Feline coronaviruses associated with feline infectious peritonitis have modifications to spike protein activation sites at two discrete positions. arXiv 2014:1412.4034.
- 220. Wicht O, Burkard C, de Haan CAM, van Kuppeveld FJM, Rottier PJM, Bosch BJ. Identification and characterization of a proteolytically primed form of the murine coronavirus spike proteins after fusion with the target cell. J Virol 2014;88:4943–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Wicht O, Li W, Willems L, Meuleman TJ, Wubbolts RW, van Kuppeveld FJM, Rottier PJM, Bosch BJ. Proteolytic activation of the porcine epidemic diarrhea coronavirus spike fusion protein by trypsin in cell culture. J Virol 2014;88:7952–7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Yamada Y, Liu DX. Proteolytic activation of the spike protein at a novel RRRR/S motif is implicated in furin‐dependent entry, syncytium formation, and infectivity of coronavirus infectious bronchitis virus in cultured cells. J Virol 2009;83:8744–8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Edinger TO, Pohl MO, Yángüez E, Stertz S. Cathepsin W is required for escape of influenza A virus from late endosomes. MBio 2015;6:e00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. White JM, Schornberg KL. A new player in the puzzle of filovirus entry. Nat Rev Microbiol 2012;10:317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Hofmann‐Winkler H, Kaup F, Pöhlmann S. Host cell factors in filovirus entry: novel players, new insights. Viruses 2012;4:3336–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Hunt CL, Lennemann NJ, Maury W. Filovirus entry: a novelty in the viral fusion world. Viruses 2012;4:258–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Miller EH, Chandran K. Filovirus entry into cells – new insights. Curr Opin Virol 2012;2:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, Ruthel G, Pfeffer SR, Dye JM, Whelan SP, Brummelkamp TR, et al. Ebola virus entry requires the host‐programmed recognition of an intracellular receptor. EMBO J 2012;31:1947–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Mingo RM, Simmons JA, Shoemaker CJ, Nelson EA, Schornberg KL, D'Souza RS, Casanova JE, White JM. Ebola virus and severe acute respiratory syndrome coronavirus display late cell entry kinetics: evidence that transport to NPC1+ endolysosomes is a rate‐defining step. J Virol 2015;89:2931–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Simmons JA, D'Souza RS, Ruas M, Galione A, Casanova JE, White JM. Ebolavirus glycoprotein directs fusion through NPC1+ endolysosomes. J Virol 2016;90:605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. Small molecule inhibitors reveal Niemann‐Pick C1 is essential for Ebola virus infection. Nature 2011;477:344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Sakurai Y, Kolokoltsov AA, Chen C‐C, Tidwell MW, Bauta WE, Klugbauer N, Grimm C, Wahl‐Schott C, Biel M, Davey RA. Ebola virus. Two‐pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015;347:995–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Spence JS, Krause TB, Mittler E, Jangra RK, Chandran K. Direct visualization of Ebola virus fusion triggering in the endocytic pathway. MBio 2016;7:e01857‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Tran EEH, Simmons JA, Bartesaghi A, Shoemaker CJ, Nelson E, White JM, Subramaniam S. Spatial localization of the Ebola virus glycoprotein mucin‐like domain determined by cryo‐electron tomography. J Virol 2014;88:10958–10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Dube D, Brecher MB, Delos SE, Rose SC, Park EW, Schornberg KL, Kuhn JH, White JM. The primed ebolavirus glycoprotein (19‐kilodalton GP1,2): sequence and residues critical for host cell binding. J Virol 2009;83:2883–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Brecher M, Schornberg KL, Delos SE, Fusco ML, Saphire EO, White JM. Cathepsin cleavage potentiates the Ebola virus glycoprotein to undergo a subsequent fusion‐relevant conformational change. J Virol 2012;86:364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Bale S, Liu T, Li S, Wang Y, Abelson D, Fusco M, Woods VL, Saphire EO. Ebola virus glycoprotein needs an additional trigger, beyond proteolytic priming for membrane fusion. PLoS Negl Trop Dis 2011;5:e1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Wong AC, Sandesara RG, Mulherkar N, Whelan SP, Chandran K. A forward genetic strategy reveals destabilizing mutations in the Ebolavirus glycoprotein that alter its protease dependence during cell entry. J Virol 2010;84:163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Harrison JS, Higgins CD, Chandran K, Lai JR. Designed protein mimics of the Ebola virus glycoprotein GP2 α‐helical bundle: stability and pH effects. Protein Sci 2011;20:1587–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Harrison JS, Koellhoffer JF, Chandran K, Lai JR. Marburg virus glycoprotein GP2: pH‐dependent stability of the ectodomain α‐helical bundle. Biochemistry 2012;51:2515–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Koellhoffer JF, Malashkevich VN, Harrison JS, Toro R, Bhosle RC, Chandran K, Almo SC, Lai JR. Crystal structure of the Marburg virus GP2 core domain in its postfusion conformation. Biochemistry 2012;51:7665–7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Markosyan RM, Miao C, Zheng Y‐M, Melikyan GB, Liu S‐L, Cohen FS. Induction of cell‐cell fusion by Ebola virus glycoprotein: low pH is not a trigger. PLoS Pathog 2016;12:e1005373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Wang H, Shi Y, Song J, Qi J, Lu G, Yan J, Gao GF. Ebola viral glycoprotein bound to its endosomal receptor Niemann‐Pick C1. Cell 2016;164:258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo‐particles containing functional E1‐E2 envelope protein complexes. J Exp Med 2003;197:633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Hsu M, Zhang J, Flint M, Logvinoff C, Cheng‐Mayer C, Rice CM, McKeating JA. Hepatitis C virus glycoproteins mediate pH‐dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci USA 2003;100:7271–7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Petracca R, Falugi F, Galli G, Norais N, Rosa D, Campagnoli S, Burgio V, Di Stasio E, Giardina B, Houghton M, Abrignani S, Grandi G. Structure‐function analysis of hepatitis C virus envelope‐CD81 binding. J Virol 2000;74:4824–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Wang Z, Nie Y, Wang P, Ding M, Deng H. Characterization of classical swine fever virus entry by using pseudotyped viruses: E1 and E2 are sufficient to mediate viral entry. Virology 2004;330:332–341. [DOI] [PubMed] [Google Scholar]
- 248. Douam F, Dao Thi VL, Maurin G, Fresquet J, Mompelat D, Zeisel MB, Baumert TF, Cosset F‐L, Lavillette D. Critical interaction between E1 and E2 glycoproteins determines binding and fusion properties of hepatitis C virus during cell entry. Hepatology 2014;59:776–788. [DOI] [PubMed] [Google Scholar]
- 249. Douam F, Lavillette D, Cosset F‐L. The mechanism of HCV entry into host cells. Prog Mol Biol Transl Sci 2015;129:63–107. [DOI] [PubMed] [Google Scholar]
- 250. Fénéant L, Potel J, François C, Sané F, Douam F, Belouzard S, Calland N, Vausselin T, Rouillé Y, Descamps V, Baumert TF, Duverlie G, Lavillette D, Hober D, Dubuisson J, et al. New insights into the understanding of hepatitis C virus entry and cell‐to‐cell transmission by using the ionophore monensin A. J Virol 2015;89:8346–8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Li Y, Wang J, Kanai R, Modis Y. Crystal structure of glycoprotein E2 from bovine viral diarrhea virus. Proc Natl Acad Sci USA 2013;110:6805–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Omari El K, Iourin O, Harlos K, Grimes JM, Stuart DI. Structure of a pestivirus envelope glycoprotein e2 clarifies its role in cell entry. Cell Rep 2013;3:30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, Dai X, Stanfield RL, Burton DR, Ward AB, Wilson IA, Law M. Hepatitis C virus E2 envelope glycoprotein core structure. Science 2013;342:1090–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Khan AG, Whidby J, Miller MT, Scarborough H, Zatorski AV, Cygan A, Price AA, Yost SA, Bohannon CD, Jacob J, Grakoui A, Marcotrigiano J. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 2014;509:381–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Drummer HE, Boo I, Poumbourios P. Mutagenesis of a conserved fusion peptide‐like motif and membrane‐proximal heptad‐repeat region of hepatitis C virus glycoprotein E1. J Gen Virol 2007;88:1144–1148. [DOI] [PubMed] [Google Scholar]
- 256. Li H‐F, Huang C‐H, Ai L‐S, Chuang C‐K, Chen SSL. Mutagenesis of the fusion peptide‐like domain of hepatitis C virus E1 glycoprotein: involvement in cell fusion and virus entry. J Biomed Sci 2009;16:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Perin PM, Haid S, Brown RJP, Doerrbecker J, Schulze K, Zeilinger C, Schaewen von M, Heller B, Vercauteren K, Luxenburger E, Baktash YM, Vondran FWR, Speerstra S, Awadh A, Mukhtarov F, et al. Flunarizine prevents hepatitis C virus membrane fusion in a genotype‐dependent manner by targeting the potential fusion peptide within E1. Hepatology 2016;63:49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Omari El K, Iourin O, Kadlec J, Sutton G, Harlos K, Grimes JM, Stuart DI. Unexpected structure for the N‐terminal domain of hepatitis C virus envelope glycoprotein E1. Nat Commun 2014;5:4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Meertens L, Bertaux C, Dragic T. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin‐coated vesicles. J Virol 2006;80:11571–11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, Rice CM. Time‐ and temperature‐dependent activation of hepatitis C virus for low‐pH‐triggered entry. J Virol 2006;80:1734–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Boo I, teWierik K, Douam F, Lavillette D, Poumbourios P, Drummer HE. Distinct roles in folding, CD81 receptor binding and viral entry for conserved histidine residues of hepatitis C virus glycoprotein E1 and E2. Biochem J 2012;443:85–94. [DOI] [PubMed] [Google Scholar]
- 262. Krey T, Thiel H‐J, Rümenapf T. Acid‐resistant bovine pestivirus requires activation for pH‐triggered fusion during entry. J Virol 2005;79:4191–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Sharma NR, Mateu G, Dreux M, Grakoui A, Cosset FL, Melikyan GB. Hepatitis C virus is primed by CD81 protein for low pH‐dependent fusion. J Biol Chem 2011;286:30361–30376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. Binding of hepatitis C virus to CD81. Science 1998;282:938–941. [DOI] [PubMed] [Google Scholar]
- 265. Fénéant L, Levy S, Cocquerel L. CD81 and hepatitis C virus (HCV) infection. Viruses 2014;6:535–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, Gonzalez‐Dosal R, Rasmussen SB, Christensen MH, Yarovinsky TO, Rixon FJ, Herold BC, Fitzgerald KA, Paludan SR. Virus‐cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol 2012;13:737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Hare DN, Collins SE, Mukherjee S, Loo Y‐M, Gale M, Janssen LJ, Mossman KL. Membrane perturbation‐associated Ca2+ signalling and incoming genome sensing are required for the host response to low‐level enveloped virus particle entry. J Virol 2015;90:3018–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Bailey CC, Zhong G, Huang I‐C, Farzan M. IFITM‐family proteins: the cell's first line of antiviral defense. Annu Rev Virol 2014;1:261–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, McCauley SM, Nowosielska A, Antonarakis SE, Luban J, Santoni FA, Pizzato M. HIV‐1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015;526:212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Usami Y, Wu Y, Göttlinger HG. SERINC3 and SERINC5 restrict HIV‐1 infectivity and are counteracted by Nef. Nature 2015;526:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Vigant F, Santos NC, Lee B. Broad‐spectrum antivirals against viral fusion. Nat Rev Microbiol 2015;13:426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]