

Abstract
Mutations in interleukin-1 receptor accessory protein like 1 (IL1RAPL1) gene have been associated with non-syndromic intellectual disability and autism spectrum disorder. This protein interacts with synaptic partners like PSD-95 and PTPδ, regulating the formation and function of excitatory synapses. The aim of this work is to characterize the synaptic consequences of three IL1RAPL1 mutations, two novel causing the deletion of exon 6 (Δex6) and one point mutation (C31R), identified in patients with intellectual disability. Using immunofluorescence and electrophysiological recordings we examined the effects of IL1RAPL1 mutants over-expression on synapse formation and function in cultured rodent hippocampal neurons. Δex6 but not C31R mutation leads to IL1RAPL1 protein instability and mislocalization within dendrites. Analysis of different markers of excitatory synapses and sEPSC recording revealed that both mutants fail to induce pre- and post-synaptic differentiation, contrary to WT IL1RAPL1 protein. Cell aggregation and immunoprecipitation assays in HEK293 cells showed a reduction of the interaction between IL1RAPL1 mutants and PTPδ that could explain the observed synaptogenic defect in neurons. However, these mutants do not affect all cellular signaling since their over-expression still activates JNK pathway. We conclude that both mutations described in this study lead to a partial loss of function of the IL1RAPL1 protein through different mechanisms. Our work highlights the important function of the trans-synaptic PTPδ/ IL1RAPL1 interaction in synaptogenesis and as such, in intellectual disability in the patients.
INTRODUCTION
Intellectual disability (ID) is defined as an overall intelligence quotient (IQ) lower than 70 and limitations in adaptive behavior, with an onset before the age of 18. ID affects around 3% of the population, and X-linked ID (XLID) is responsible for 10% of severe ID cases. To date, 116 of XLID genes have been identified. Mutations in one of these genes, interleukin-1 receptor accessory protein-like 1 (IL1RAPL1), are associated with cognitive impairment ranging from non-syndromic ID to autism spectrum disorder (ASD). Until now, described mutations include exon deletions and nonsense mutations that result in the absence of protein, in most of the cases (1–14).
IL1RAPL1 is a member of interleukin 1 receptor family, and shares 52% of homology with the IL-1 receptor accessory protein (IL1RAcP) (1). It contains 3 extracellular Immunoglobulin (Ig) –like domains, a single transmembrane domain, an intracellular Toll/IL-1R (TIR) domain and a C-terminal tail of 150 amino acids, that is not shared with other family members. IL1RAPL1 is expressed in the brain, and is located on excitatory synapses with an enrichment at the postsynaptic compartment (15).
The importance of IL1RAPL1 in brain function was demonstrated by studies of Il1rapl1 knockout mouse model (16). These mice show impaired associative learning and synaptic defects, including decrease in dendritic spines and synaptic plasticity in different brain regions (15, 17).
Growing body of evidence underlines the importance of IL1RAPL1 on synapse physiology. Several IL1RAPL1-interacting proteins necessary for IL1RAPL1-induced pre- and post-synaptic differentiation have been identified. IL1RAPL1 interacts through its C-terminal domain with the calcium sensor NCS-1, regulating the activity of N-type voltage-gated calcium channel (VGCC) in PC12 cells (18, 19). In neurons, IL1RAPL1 interacts with PSD-95, a major scaffolding protein of excitatory synapses, and modulates its synaptic localization by regulating JNK activity and PSD-95 phosphorylation (15). Interaction with RhoGAP2 and Mcf2l, two regulators of Rho GTPases activity, is required for IL1RAPL1 to induce dendritic spine formation and function (20, 21). Hayashi et al., identified other proteins interacting with the intracellular domain of IL1RAPL1, like PKCε, PLCβ1 and Rasal1 (21). Trans-synaptic interaction with the protein tyrosine phosphatase PTPδ through the extracellular domain of IL1RAPL1 was also shown to be essential for synaptogenesis (20, 22).
We identified two novel IL1RAPL1 mutations, an in frame deletion of exon 6 (Δex6), in two unrelated patients with ID. Unlike the majority of previously reported IL1RAPL1 mutations, which primarily lead to loss of IL1RAPL1 protein, this deletion and one point mutation in exon 3 (c.91T>C; p.(Cys31Arg)) (23), are compatible with IL1RAPL1 protein synthesis, but are predicted to affect the function of its extracellular domain. As part of this work we explore the impact of these mutations on synapse formation and function and how this can explain the intellectual disability of the patients.
RESULTS
Clinical characterization of patients and identification of two novel mutations on IL1RAPL1
P72 Family
The pedigree of family P72 is shown in Figure 1A. Patient II-2 (male, 30 years) presents moderate ID, autistic-like behaviour, is extraverted, aggressive, and has language and motor delay. He has large hands, big ears, long face and synophrys. Patient II-3 (male, 43 years) presents mild ID and has no major behavioural problems. He also has autistic-like behaviour and language and motor delay. He has facial dysmorphism, big ears and round face. Neurological examination was normal. The only clinical feature of III-2 (female, 10 years) is ID, needing special care.
Figure 1. Identification of two novel mutations on IL1RAPL1 associated with intellectual disability.
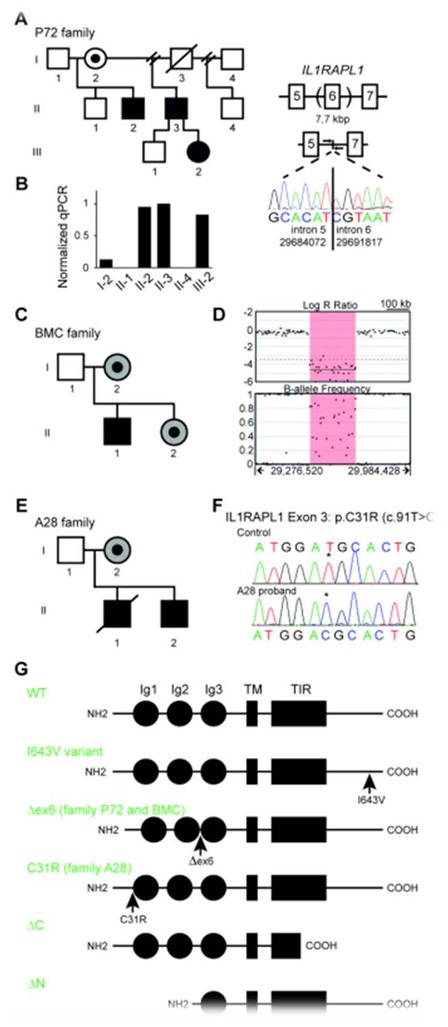
(A) Pedigree of family P72, were II-2, II-3 and III-2 present moderate to mild ID. In those individuals a ~ 7 kb deletion on IL1RAPL1 between intron 5 and 6 results in exon 6 deletion. This was confirmed by real-time PCR in fibroblast from the obligate carrier female I-2 and affected patients, using oligonucleotides flanking the deletion (B). This in frame deletion leads to an IL1RAPL1 protein lacking 25 amino acids in the extracellular domain (G). (C) Pedigree of family BMC, where II-1 has ID. I-2 and II-2 (shaded in gray) have learning problems but their developmental delay is less severe than that of the proband. (D) SNP array revealed a deletion of ~200 kb between intron 5 and 6 of IL1RAPL1 that results in the in frame exon 6 deletion, as in family P72.
(E) Pedigree of Family A28, were II-1 and II-2 present moderate to mild intellectual disability, and I-2 has learning problems (shaded in gray). (F) Both affected males inherited from I-2 a point mutation located in exon 3 of IL1RAPL1 (c.91T>C), which results in an amino acid change C31R. (c.91T>C mutation in II-2 was initially reported by Tarpey et al., (23)). This missense mutation is located before the first Ig-like domain (G).
Structure of I643V variant and mutants are shown in (G). IL1RAPL1 protein (696 aa) contains 3 extracellular Ig-like domains (Ig1–3), a single transmembrane domain (TM), an intracellular Toll/IL-1 Receptor (TIR) domain, and a 150 amino acid C-terminal tail. ΔC and ΔN mutants were used as controls in this study. The in frame deletion of IL1RAPL1 exon 6, found in P72 and BMC, is referred as Δex6.
During a search for mutations in IL1RAPL1 gene in male patients with XLID, we found a deletion of exon 6 in genomic DNA from patient II-2. This deletion was also found in the affected brother II-3. Physical mapping of the deletion by CGH array and long range PCR allowed us to characterize its size (7744 base pairs (bp)) and define the DNA breakpoints between intron 5 and 6 of IL1RAPL1 (g.29684073_29691812del; c.1212_1286del; hg19/LOVD3 IL1RAPL1_000009). Using oligonucleotides flanking the deletion breakpoints, we studied by real-time PCR the segregation of the deletion in P72 family. As shown in Figure 1B, the deletion is present in II-2, II-3 and III-2 but not in II-1 and II-4 DNA isolated from blood; the low level of amplification in obligate carrier I-2 suggests somatic mosaicism. The in frame deletion of IL1RAPL1 exon 6 is predicted to lead to a protein lacking 25 amino acids in the extracellular domain, between immunoglobulin domain (Ig) 2 and 3 (p.(Ala235_Leu259del); Fig. 1G).
In order to elucidate if III-2’s phenotype is due to a skewed X chromosome inactivation we evaluated her X-inactivation pattern using the AR, FMR1 or FMR2 loci in her fibroblasts. Unfortunately none of these markers was informative and given that IL1RAPL1 expression in fibroblasts and blood cells is very low, we assessed the X-inactivation skewing by testing the expression of one SNP (single-nucleotide polymorphism) in the 3′UTR of APOO, a gene located on the X chromosome at < 5 kb from IL1RAPL1, in fibroblasts from III-2. Using this SNP (rs8680) we were able to differentiate her parent’s contribution, and we found the expression of both alleles in III-2 cDNA suggesting random X-inactivation in her fibroblasts (Suppl. Fig. 1).
BMC Family
The proband II-1 (male, 27 months old) was born after an uneventful pregnancy as the second child of non-consanguineous parents (Fig. 1C). He had some delay of motor development, sitting at 9 months and walking at 25 months. At the age of 27 months he only speaks three words and formal developmental testing confirmed delay. He was advised to start in special education. Family history is significant for learning difficulties in the mother; she attended special education. The proband has one sister with learning difficulties. Physical examination reveals height of 84 cm (below third percentile), weight 12.7 kg (25th percentile) and head circumference 48.4 cm (25th percentile). He has mild facial dysmorphism with a prominent forehead. He has generalized joint hyperlaxity, normal male genitalia and skin significant eczema. Brain MRI was normal.
Microarray analysis revealed a deletion of ~200 kb with the proximal breakpoint in intron 5 and the distal breakpoint in intron 6 of IL1RAPL1 (g.29517322_29746541del; c.703+99897_778+59920del; hg19/ LOVD3 IL1RAPL1_000008) predicted to result in a deletion of the entire exon 6 (Fig. 1D). Additionally, a duplication on chromosome 19q13.41 of unknown clinical significance was observed (g.52860055_52996104dup; c.-41492_*76112dup; hg19, LOVD3 ZNF528_000001). The parents and sister of the proband were also tested and both the deletion and duplication are inherited from the mother, and present in the sister. Similarly to the above-described deletion of exon 6, this one is predicted to lead to the same IL1RAPL1 mutant protein lacking 25 amino acids in the extracellular domain, between immunoglobulin domain (Ig) 2 and 3 (p.(Ala235_Leu259del); Fig. 1G).
A28 Family
The pedigree of family A28 is shown in Figure 1E. II-1 (male, now deceased) had moderate ID (IQ assessed as 36–51), gynaecomastia, obesity, small testes, normal height (169 cm) and head circumference (54.5 cm), sexual deviant behavior (treated with an anti-androgen medication and necessitating living in care). II-2 (male, 57 years) presents mild ID, obesity, significant behavioral issues, normal head circumference, normal facial features, gynaecomastia, normal hands and feet. The female obligate carrier I-2 is phenotypically normal, with normal height (153 cm) and head circumference (54.2 cm). She appeared to have low average intelligence.
A missense substitution in IL1RAPL1 exon 3 (c.91T>C) (LOVD3 IL1RAPL1_000003) leading to an amino acid change p.(Cys31Arg) (C31R) was initially reported by Tarpey et al. in II-2 patient (23), but no clinical information about the family nor further characterisation of this variant (ie. if deleterious to IL1RAPL1 function or not) was studied. We first confirmed the segregation of this variant in the A28 family (Fig. 1F) and subsequently investigated its functional consequences. This point mutation is located in the extracellular domain of IL1RAPL1 protein before the first Ig domain (Fig. 1G). We assessed the pathogenicity of this variant by in silico analysis using the following software: Mutation taster (24), SIFT (25) and PolyPhen 2 (26). Mutation taster analysis predicted that this missense variant is a disease-causing mutation. PolyPhen analysis, which predicts possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations, of the missense mutation also considered it to be “probably damaging” (score 1.0). Finally, SIFT analysis predicted also the substitution at position 31 from Cys to Arg to affect protein function with a score of 0.00 (damaging). Alamut splicing predictions (Interactive Biosoftware) suggested no significant impact of this substitution on donor and acceptor splice sites.
Finally, a point mutation in IL1RAPL1 exon 11 (c.1927A>G) leading to an amino acid change p.(Ile643Val) (I643V), was found in a male with ID, but was not observed in his affected brother potentially ruling out this variant as the genetic cause of the disease in this XLID family. This IL1RAPL1 variant was reported before by Piton et al.,(5) and is unlikely to be pathogenic since in silico analysis considered it to be tolerated by the protein. In our study, we use this variant as a control, as this single amino acid change is located in the intracellular domain (Fig. 1G), contrary to Δex6 and C31R mutations.
In this and previous studies (15, 20) we used as controls two IL1RAPL1 mutant proteins, ΔC and ΔN, lacking a large part of intra or extra cellular domains, respectively (Fig. 1G and Table 1).
Table 1.
Reported mutations on IL1RAPL1 gene in ID patients, and their consequences for protein function.
| Reference | Mutation / exons | Protein | Functional consequences |
|---|---|---|---|
| (1) | deletion exon 3–5 | Probably not produced | |
| (1) | nonsense exon 11 | Y459X predicted to lead to a protein lacking part of the TIR domain and the entire C-ter domain | ΔC (15, 20)
|
| (2, 3) | nonsense exon 11 | W487X predicted to produce a protein lacking half of the TIR domain and the entire C-ter domain | |
| (4) | deletion exons 3–6* | Probably not produced | |
| (5) | nonsense exon 9 | I367SX6 predicted to produce a protein lacking part of the trans membrane domain as well as the entire C-ter domain | I367SX6 (5)
|
| (5) | deletion exon 3–7 | Frame shift A28EfxX15 predicted to produce a short protein containing only 8 amino acids in addition to the signal peptide | |
| (5) and current report | missense exon 11 | I643V variant produces a full length protein In silico analysis predicts it to be tolerated by the protein |
I643V (Current report)
|
| (6) | deletion exon 3–5 | The resulting protein should lack the first two Ig-like domains, but it is possible that synthesis stops after deletion | |
| (6) | deletion exon 2 | Probably not produced | |
| (7) | deletion exons 3–5 | Probably not produced | |
| (8) | deletion exons 1–5 | Probably not produced | |
| (8) | deletion exons 3–6* | In frame deletion (p.28_259del) predicted to produce a shorter protein devoid of the two first Ig-like domains | Δ N (20)
|
| (9) | deletion exons 2–6 | Probably not produced | |
| (10) | deletion exons 3–11 | Probably not produced | |
| (11, 13) | deletion exon 3 | Out of frame deletion leading to a premature stop codon A28EfxX7 Protein is probably not produced | |
| (12) | deletion exon 3–5 | Predicted to cause an in frame deletion of 207 amino acids (N29_A235del) | |
| Current report | deletion exon 6 | In frame deletion that results in a shorter extracellular domain Protein instability |
Δ ex6 (current report)
|
| (23) and current report | missense exon 3 | One amino acid change before the first Ig-like domain (C31R) In silico analysis predicts damage to the structure and function |
C31R (current report)
|
| (14) | deletion exon 7 | Predicted to produce a truncated protein, containing only the first two Ig-like domains |
Modified from original article, in accordance with hg38 assembly.
ΔC mutant corresponds to a nonsense IL1RAPL1 mutation in exon 11 (c.1377C>A) observed in a patient with non syndromic ID (1). The construct used in our study lacks the half of the TIR and the complete C-terminal domains (p.(Tyr459X)).
ΔN mutant protein lacks the first two Ig-like domains, and corresponds to the deletion of exons 1 to 6 of IL1RAPL1 (c.1_778del) (Fig. 1G and Table 1). Deletions of these exons were found in different patients with ID, but they probably lead to the absence of IL1RAPL1 expression (4, 6–9).
IL1RAPL1 protein expression and localization is affected by mutations in its extracellular domain
In order to evaluate the effect of IL1RAPL1 mutations on protein stability, HEK293 cells were co transfected with GFP and vectors bearing HA-tagged WT or mutant IL1RAPL1. Protein expression of mutants is significantly decreased (~75% for Δex6 (~108 KDa) and ~60% for C31R (~115 KDa), compared to the WT (~115 KDa) protein) 24 hours after transfection as revealed by immunoblot (Suppl. Fig. 2). The decrease in protein is not due to lower transfection efficiency (evaluated by GFP signal) suggesting that both mutations lead to decreased stability of the IL1RAPL1 protein in these cells. Protein expression of an IL1RAPL1 mutant lacking the half of the TIR and the complete C-terminal domains (ΔC, ~62 KDa) is decreased to similar levels than Δex6 and C31R. Next, we studied the stability of the mutants in mouse hippocampal neurons. Whereas IL1RAPL1 protein expression in Δex6 transfected neurons is severely abolished to the background level, C31R mutant protein has similar expression level to WT protein (Fig. 2A and 2B). I643V variant is more abundant compared to WT protein, but there are probably no consequences since this excess of protein does not affect other analyzed parameters (see below). All IL1RAPL1 variants including Δex6 are correctly targeted to the membrane of neurons as measured by the ratio of surface HA per total HA signal, compared to WT IL1RAPL1 protein (Fig. 2C and 2D). As WT IL1RAPL1 protein is present in the dendrites and enriched at the postsynaptic site in the dendritic spines (15), we analyzed the subcellular localization of the variants by measuring the coefficient of variation of HA-IL1RAPL1 signal along dendrites (CV, see material and methods) and also the distribution of IL1RAPL1 signal in spines versus dendrites. Both C31R and I643V mutant proteins are distributed in dendritic shafts and are also enriched in spines similarly to WT (Fig. 2E and 2F). In contrast and despite its low abundance, Δex6 mutant is predominantly observed forming discrete puncta within dendritic shafts. Coefficient of variation analysis of Δex6 IL1RAPL1 signal along the dendritic shaft clearly shows more variations than WT and other mutated proteins (*** p <0.001 compared to WT protein, n=14 neurons (not shown)). In addition, Δex6 mutant shows a decrease of IL1RAPL1 signal in dendritic spines of neurons compared to WT transfected neurons (Fig. 2F).
Figure 2. Protein expression and localization of Δex6 and C31R IL1RAPL1 mutants.
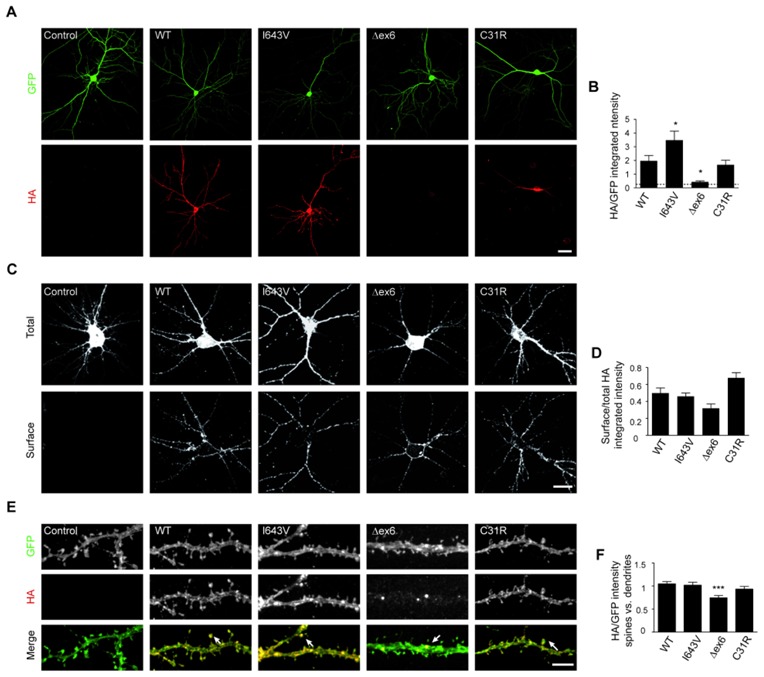
(A) Protein detection by immunofluorescence in mouse hippocampal neurons co-transfected with different HA-IL1RAPL1 constructs and GFP. IL1RAPL1 proteins were revealed by an anti-HA tag antibody, and signal was normalized to GFP expression (scale bar 20 μm). (B) Bar graphs show the mean + SEM of HA-IL1RAPL1 to GFP expression ratio (at least 35 neurons per each condition from 3 independent experiments, *p <0.01 compared to WT). (C) Total (top panel) and surface (bottom panel) staining of HA-IL1RAPL1 proteins in mature hippocampal neurons using an anti-HA tag antibody (scale bar 20 μm). The ratio of integrated intensity of surface HA signal per total HA signal was measured for each neuron, and the mean + SEM is shown in (D). (E) Localization of total IL1RAPL1 within dendrites at DIV 18 in hippocampal neurons co-transfected with GFP (green) and the different HA-IL1RAPL1 constructs (red). Arrows in merge images show IL1RAPL1 localization to spines, or forming puncta on dendritic shafts when Δex6 is expressed (scale bar 5 μm). (F) Bars show the mean + SEM of the ratio of HA/GFP integrated density in spines and the HA/GFP integrated density in dendritic shafts (*** p <0.001 compared to WT protein).
In conclusion, deletion of the region between Ig2 and Ig3 domains in Δex6 mutant is responsible for its instability and mislocalization in dendrite and spines. In contrast, these parameters are not altered by C31R mutation.
Impact of IL1RAPL1 mutations on excitatory synapse formation
Knocking-down or overexpressing IL1RAPL1 decreases or increases excitatory synapse formation, respectively (15, 20, 22, 27). In order to evaluate the impact of the three newly described mutants on this IL1RAPL1-dependent synaptogenic phenotype, we co-transfected cultured hippocampal neurons with GFP and HA-tagged IL1RAPL1 constructs and their effect on pre- and post-synapse formation was evaluated using specific markers.
WT and I643V IL1RAPL1 transfected neurons at DIV18 present a large increase of the pre-synaptic marker synaptophysin that is not observed in neurons overexpressing Δex6 or C31R mutant (Suppl. Fig. 3). As synaptophysin labels both excitatory and inhibitory pre-synapses, we stained transfected neurons with more specific markers using anti-VGLUT1 and anti-VGAT antibodies to label excitatory or inhibitory pre-synapses, respectively. We observed that IL1RAPL1 increases the excitatory pre-synaptic marker, an effect that is not observed after Δex6 or C31R mutant over-expression (Fig. 3A and 3B). Staining for the inhibitory pre-synaptic marker VGAT is not affected after WT or mutants expression confirming the specific function of IL1RAPL1 in excitatory synapses (Suppl. Fig. 4A).
Figure 3. Consequences of IL1RAPL1 mutations on excitatory synapse formation.
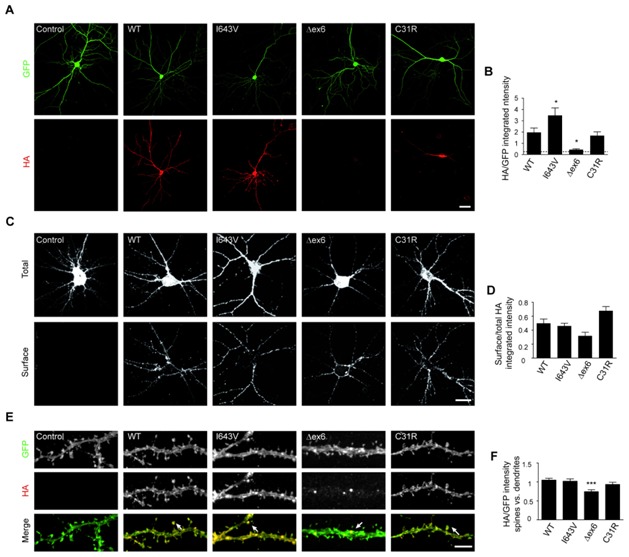
(A) Rat hippocampal neurons co-transfected with GFP and different HA-IL1RAPL1 constructs were stained with anti-VGLUT1 antibody to label excitatory pre-synapses. Each column of images shows double-labeling for GFP (top panel) and VGLUT1 (middle panel); the merged images are shown in the bottom panel (scale bar 20 μm). Quantification of VGLUT1 clusters intensity in neurons overexpressing IL1RAPL1 constructs is shown in (B). Bar graphs show the mean + SEM of VGLUT1 intensity (15 neurons from 3 independent experiments for each condition, ** p <0.005, *** p <0.001, compared to control neurons).
(C) Mouse hippocampal neurons were co-transfected with GFP and the different IL1RAPL1 constructs, and were stained at DIV18 with anti-PSD-95 antibody to label excitatory post-synapses. Each column of images shows double-labeling for GFP (top panel) and PSD-95 (middle panel); the merged images are shown in the bottom panel (scale bar 10 μm). Bar graphs in (D) show the mean + SEM of the PSD-95 clusters per micron in at least 26 neurons from 3 independent experiments (* p < 0.01 *** p < 0.001, compared to control neurons). The number of protrusions along dendrites was assessed from at least 25 neurons from each condition as showed in (E) (* p < 0.01, compared to control neurons).
(F) Typical recording of sEPSC from mouse hippocampal neurons at 18–21 DIV transfected with different IL1RAPL1 constructs. The average frequency and amplitude of these events is represented in (G) (6 to 10 transfected neurons per condition and 32 non transfected neurons (nt) * p< 0.01, compared to control neurons).
Over-expression of both WT and I643V IL1RAPL1 induces an increase of the excitatory post-synaptic marker PSD-95 (Fig. 3C and 3D, ~100 and ~150% respectively, no statistical differences between them) together with an increase in the number of dendritic protrusions (Fig. 3E, ~20% in both cases), compared to control neurons. In agreement with these data and as previously reported, WT and I643V IL1RAPL1 over-expression increase the frequency (~300% in both cases), but not the amplitude of spontaneous excitatory post-synaptic currents (sEPSC) (Fig. 3F and 3G). In contrast, none of the post-synaptic effects are observed in neurons overexpressing Δex6 or C31R mutants (Fig. 3C–E) suggesting that these mutants lose their synaptogenic properties. According to immunocytochemistry data, neither WT nor mutant protein altered the frequency and amplitude of spontaneous inhibitory post-synaptic currents (sIPSC, Suppl. Fig. 4B).
Worth noting, ΔC mutant lacking part of intracellular domain is able to increase synaptophysin (Suppl. Fig. 3) and VGLUT1 staining, but not to increase PSD-95 staining (n=16 neurons per group from 2 independent experiments) or the number of dendritic spines (20). On the other hand, a mutant lacking part of the extracellular domain (ΔN in Fig. 1G) is also unable to increase pre- and post-synaptic differentiation (Suppl. Fig. 3). These observations support the fact that pre-synaptic differentiation is dependent on IL1RAPL1 extracellular domain, while both extra and intracellular domains are important for post-synaptic differentiation (20, 22). This suggests that extracellular domain of IL1RAPL1 is damaged in C31R mutant, and that this could account for its synaptogenic deficit. In the case of Δex6 mutant, this deficit is probably due to the decrease in protein stability and mislocalization within dendrites as shown above.
Mechanism of synaptic deficits induced by IL1RAPL1 mutants
The synaptogenic activity of IL1RAPL1 is dependent on its interaction with a specific isoform of the tyrosine phosphatase PTPδ (20, 22, 28). This protein interacts with IL1RAPL1 extracellular domain, and was shown to be specific as other members of the protein family, like LAR and PTPσ, are able neither to interact with nor to induce IL1RAPL1-dependent synaptogenesis.
As C31R mutant lack synaptogenic activity, presumably because of changes in its extracellular domain structure, we hypothesized that this mutation perturbed the trans-synaptic interaction with PTPδ. In order to test this hypothesis, a group of HEK293 cells overexpressing either GFP or HA-IL1RAPL1 proteins and another group expressing Myc-PTPδ ectodomain were subjected to a cluster assay as previously described (20). After counting the number of red/green clusters (yellow in merge image in Fig. 4A), this assay revealed some but not significant interaction between C31R IL1RAPL1 mutant and PTPδ, compared to control cells and ΔN mutant that lacks the 2 first Ig-like domains (Fig. 4A). Similarly, the small amount of the Δex6 mutant expressed shows a severe reduction of clustering. However compared to WT IL1RAPL1, both mutants show significant decrease of clustering efficiency, suggesting that the mutants reduce somehow the interaction with PTPδ (~40% for both mutants). This deficit could contribute to the inability of C31R mutant to induce the formation of excitatory synapses.
Figure 4. Molecular mechanism accounting for synaptic deficits induced by IL1RAPL1 mutants.
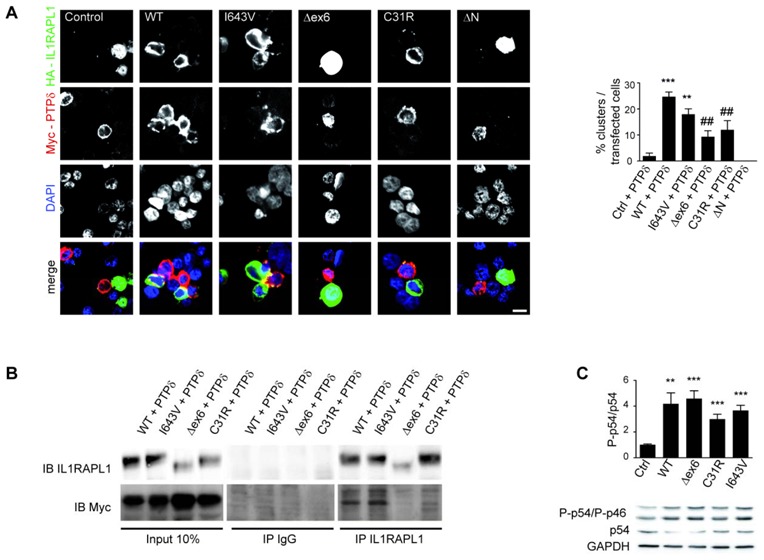
(A) HEK293 cells expressing either GFP or HA-IL1RAPL1 constructs (green), and HEK293 cells expressing Myc-PTPδ ectodomain (red) were subjected to a cluster assay. Nuclei (blue) were stained with DAPI (scale bar 10 μm). Clustering was assessed by counting the number of green/red clusters (yellow in merge images) and normalizing by the number of transfected cells (green + red) cells (** p< 0.005 *** p< 0.001 compared to Control + PTPδ; ## p< 0.005 compared to WT + PTPδ). ΔN mutant, which lacks the first 2 Ig-like domains, was used as negative control. (B) Lysates from HEK293 cells expressing the indicated HA-IL1RAPL1 constructs were mixed with lyzates from another group of cells expressing Myc-PTPδ ectodomain in a volume proportion of 1 (for IL1RAPL1) to 1.5 (for Myc-PTPδ) and subjected to an in vitro immunoprecipitation assay using IL1RAPL1 antibody. 10% of the mixed lyzates was loaded as control of IL1RAPL1 and Myc-PTPδ protein over-expression (left panel). IL1RAPL1 antibody immunoprecipitates were revealed after immunoblotting (IB) using IL1RAPL1 (K10) and Myc antibodies. Rabbit IgG antibody was used as a negative control (central panel).
(C) Lysates from HEK293 cells transfected with different IL1RAPL1 constructs were probed by immunoblot with antibodies against total p54 and phospho-specific (Thr183/Tyr185) p46 and p54 JNK isoforms. Protein loading was normalized by GAPDH expression. Bar graphs show the mean + SEM of phospho / total ratio of p54 JNK isoform (6 independent experiments, ** p <0.005 *** p <0.001 compared to control lysates).
To support this conclusion, we performed in vitro interaction tests by immunoprecipitating IL1RAPL1 from protein lysates containing both IL1RAPL1 and Myc-PTPδ proteins, and we evaluated by immunoblotting the presence of Myc- PTPδ ectodomain in the immunoprecipitate. Whereas WT or I643V efficiently interact with PTPδ, we observed a strong reduction of Myc staining after immunoprecipitation of both Δex6 and C31R mutants (Fig. 4B). However, decrease of Δex6 protein expression is likely to be also responsible for this observation (Suppl. Fig. 2 and Fig. 4B, input 10% and immunoprecipitated IL1RAPL proteins).
Taken together, cell aggregation and immunoprecipitation assays lead us to conclude that C31R mutation decreases the interaction of IL1RAPL1 with PTPδ.
Mutants regulate other IL1RAPL1-dependent signaling
Besides PTPδ, IL1RAPL1 interacts with NCS-1, PSD-95, RhoGAP2, Mcf2l, PKCε and PLCβ1 (15, 18, 20, 21). These proteins interact with the intracellular domain of IL1RAPL1, suggesting that signaling independent from the extracellular domain could still be induced in neurons expressing IL1RAPL1 mutants with intact intracellular domains.
Even if there is no evidence for direct interaction with c-jun N-terminal kinase (JNK), the role of IL1RAPL1 on the regulation of activity of this kinase has been reported (15, 29, 30). Over-expression of IL1RAcP and IL1RAPL1 was shown to increase JNK basal activity in HEK293 cells (29, 31). In order to evaluate the capacity of the mutants to activate JNK, we assessed by immunoblotting the basal activity of this kinase in HEK293 cells overexpressing different IL1RAPL1 constructs.
We show that over-expression of IL1RAPL1 mutants in HEK293 cells increases the basal JNK phosphorylation, to levels comparable to the WT protein (Fig. 4C). Even if only p56 JNK isoform was quantified, phosphorylation of p46 isoform appears also to be increased after IL1RAPL1 over-expression. This result suggests that Δex6 and C31R mutants do not lose all signaling capacity, independently from synaptogenesis, and that even low expression of the IL1RAPL1 Δex6 mutant is able to induce this signaling.
DISCUSSION
There are hundreds of genes in which mutations are known to cause intellectual disability or ASD or both. Since its discovery as a gene implicated in ID, several mutations of IL1RAPL1 were found in patients with different severity of ID. As shown in Table 1, the majority of the described mutations include large deletions. It is of particular interest that they mostly involve the first exons coding for extracellular domain of IL1RAPL1 protein. Some authors suggest that because of the incidence of genomic rearrangements, such as pericentromeric inversions, this region must be particularly prone to recombination (3, 32). Moreover, the majority of mutations likely results in the absence of the IL1RAPL1 protein, or is predicted to lead to truncated proteins. Until now only one frame shift mutation leading to a shorter IL1RAPL1 protein has been characterized functionally (5). The impact of mutations described so far on IL1RAPL1 protein production and function, when available, is summarized in Table 1.
Here we report two novel mutations of IL1RAPL1 related to non-syndromic ID and we characterize their functional consequences. Both mutations result in an in frame deletion of exon 6, leading to a 25 amino acids loss in the extracellular domain of IL1RAPL1. These mutations were identified in two unrelated families (P72 and BMC) and have different DNA breakpoints. In both cases, the deletion co-segregates with the ID phenotype in an X-linked recessive manner. Besides exon 6 deletion, patient II-1 in family BMC presents also a duplication on chromosome 19 that includes ZNF528 gene. Missense mutations of this gene were previously identified in two patients with mild ID (33). Due to the fact that Δex6 mutation was also found in members of the P72 family presenting ID, we propose the deletion in IL1RAPL1 as the major cause of ID in these 2 families, but we cannot rule out that the severity of cognitive impairment could be modulated by deletions or duplications in other genes, such as ZNF528.
We also characterized the functional consequences of a unique missense variant C31R previously reported, but not further investigated (23). This variant was predicted to be damaging to IL1RAPL1 protein, which can be due to the importance of this region for protein folding. To our knowledge, this is the only pathogenic IL1RAPL1 missense variant described so far.
Several studies on XLID genes, including IL1RAPL1, raise the question of the role of X chromosome inactivation on female phenotype (3, 4, 6, 8). In the present study, IL1RAPL1 mutations were found in healthy as well as in affected females from the three families (Fig. 1). Females I-2 and II-2 (BMC family), and I-2 (A28 family) have some learning problems or low average intelligence. But unlike III-2 from P72 family, they do not have ID. We speculate that, even if not observed in fibroblasts, the X chromosome inactivation pattern may be skewed in III-2’s brain or in particular subsets of neurons, resulting in a predominant expression of the mutant allele (34).
Together with Δex6, the C31R mutation allowed us to address the impact of relatively milder mutations, in comparison to large deletions or nonsense mutations, on IL1RAPL1 protein stability, localization and synaptic function. IL1RAPL1 is located at both pre- and post-synaptic compartments of excitatory synapses, but is enriched in the post-synaptic membrane (15), and it’s over-expression is known to increase the formation of this type of synapses on hippocampal neurons (15, 20, 21). We showed that Δex6 mutation lead to decreased protein stability in neurons, mislocalization within dendrites and decreased presence in spines, even if mutant protein is correctly targeted to the membrane. In the other hand, C31R mutation does not affect IL1RAPL1 stability in neurons nor localization on dendritic spines and shafts. Our experiments clearly show that Δex6 and C31R were not able to increase excitatory synapse number, after evaluation of either pre- or post-synaptic markers. In the case of Δex6 mutant, the lack of synaptogenic effect can be explained by the severe decrease in IL1RAPL1 protein expression and its miss localization, as shown in Figure 2A–B and 2E–F. However C31R mutant protein, whose expression is similar to WT, also fails to increase synaptic formation. This impairment was also observed in ΔN, which lack the majority of the IL1RAPL1 extracellular domain (Suppl. Fig. 3 and Table 1). As previously shown (20), ΔC mutant with intact extracellular domain is able to increase the pre-synaptic marker synaptophysin establishing that this domain is essential for pre-synaptic differentiation (Suppl. Fig. 3). We then hypothesized that C31R mutation affects this domain and the binding to interacting partners. PTPδ is the only partner known to interact with IL1RAPL1 extracellular domain and this interaction was shown to be essential for IL1RAPL1-mediated synaptogenesis (20, 22). In order to dissect the molecular mechanism underlying the synaptic deficits observed in neurons transfected with C31R, we evaluated mutant’s capacity to interact with PTPδ. The cell aggregation and immunoprecipitation assays shown in Figure 4 allowed us to conclude that the decrease of interaction with this tyrosine phosphatase participates to the inability of C31R IL1RAPL1 mutant to increase the number of excitatory synapses.
Despite reduced expression (Δex6) and perturbed synaptogenesis (C31R), we hypothesized that some of the signaling could be preserved in cells transfected with Δex6 and C31R mutants. Indeed, we observed that both mutant proteins were able to induce JNKs basal activation. The capacity of IL1RAPL1 to regulate JNK activity was previously shown (15, 29, 30), even if the mechanism is still unclear. PSD-95 phosphorylation by JNK has been shown to regulate PSD-95 at the excitatory synapses and we proposed that the reduction of excitatory synapses in Il1rapl1 knockout neurons was secondary to reduced JNK activity (15, 35). Our results suggest that Δex6 and C31R mutations decrease IL1RAPL1 synaptogenic activity while maintaining other signaling, like JNK activation, uncoupling the two events. Alternatively, JNKs belong to the MAPK family, and in neurons they are involved in diverse roles including cell death, radial migration, neurite formation, metabolism regulation, and behavioral control. JNK signaling has an impact on synaptic plasticity, as a regulator of AMPA receptors trafficking (36, 37). The functional role of JNK, in particular in response to IL1β stimulation, is still under investigation (30).
Finally, the I643V variant was reported in ID patients as well as in controls. This, together with in silico prediction suggests that this variant is not deleterious for IL1RAPL1 function. In this study we evaluated the potential functional consequences of this amino acid change within the intracellular domain of IL1RAPL1 with the aim to assess if it may act as a susceptibly variant to ID. We observed that I643V protein was significantly increased in transfected neurons, but the increase of excitatory synapse number was comparable to WT IL1RAPL1. These observations support the hypothesis that the functional interactions, but not the quantity of IL1RAPL1 protein is important for synapse formation. This functional characterization strongly suggests I643V to be a neutral IL1RAPL1 variant.
In conclusion, the cognitive deficits observed in patients carrying Δex6 mutations can be explained by the decrease of IL1RAPL1 protein stability in neurons, together with the fact that residual produced protein is mislocalized. In the other hand, deficits observed in patients with C31R mutation are caused by a decrease of the capacity to interact with PTPδ, and thus to increase synaptogenesis. In addition, these mutations allowed us to rule out the functional involvement of JNK in the PTPδ induced synaptogenic activity of IL1RAPL1.
MATERIALS AND METHODS
Genetic analysis
DNA was extracted from peripheral blood or skin fibroblasts using standard methods after parental and patients’ consent was obtained.
The following intronic primers were used to investigate the exon 6 deletion in P72 family: TGAAAGTGAAAAATATTTGGGAAA, and CACAATGTAACGAGAGCAGCA. Confirmation of the deletion was obtained by qPCR (LightCycler LC480, Roche) targeting exon 6 (CCCAAGCTTTTGTATCCTAT and ATGGATTTAGCTGCGAGTA) and exon 8 (ACATCAGATTCGGATTCATC and GCGTGTCGACGTCCATT) was used as a reference. CGH array (NimbleGen’s, Roche) and long range PCR (TGTGAGTGAGTGTGCATATGTGTGTATAGGTG and CGTGGGGACTAGACCAGGAGTTG) was used to map exon 6 deletion in P72 family members. Germinal mosaicism of the deletion was explored by qPCR (TGCTTGACAGAATTTTCCAAGGAGCA and GTTACCACTTTCATTTACCTTGGGATGA) were COL6A5 expression was used as a reference (ACCACTGGCAGCTTCTGGCAA and CGCCCCTGGACATCCTGCAA). The following primers were used to detect APOO polymorphism in patients’ fibroblasts: genomic DNA (TCCCAACTGTCTGGTTCTAGCTTGT andTGGTTTGACCCTGTCCCCCAT), and cDNA (TGGGATTAGCTGCCTCCCTCT and ACTGACTTCTATGCCATTTTTCTGT). X-chromosome inactivation studies were performed using the AR, FMR1 and FMR2–specific HpaII/PCR assay, to assess X-inactivation pattern.
SNP array analysis on BMC family members was performed using a HumanCytoSNP-12v2.1 beadchip following standard protocols as provided by the manufacturer on an iScan system (Illumina, San Diego, CA). CNV analysis was performed using CNV-WebStore (38). Familial relationships were validated by comparing the SNP patterns of the patient with those of the parents.
Identification of c.91T>C (C31R) mutation II-2 member of family A28 is described elsewhere (23). Segregation studies were performed by PCR and Sanger sequencing.
Newly identified variants were submitted to Leiden Open Variation Database 3.0 (LOVD 3.0)(39) (IL1RAPL1_000008 and IL1RAPL1_000009).
cDNA constructs
HA-tagged human IL1RAPL1 described before (15) was modified using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) to generate Δex6, C31R, I643V and ΔIg123 constructs. Myc-tagged PTPδ, ΔC, ΔN were described elsewhere (20).
Antibodies
The following primary antibodies were used: rabbit anti-IL1RAPL1 (K10,(15)), goat anti-IL1RAPL1 (R&D), mouse anti-GFP (Roche and Abcam), rabbit anti-VGlut1 (Synaptic Systems), rabbit anti-VGAT (Synaptic Systems), rabbit anti-HA-tag (Santa Cruz Biotechnology), mouse anti-HA-tag (Roche), mouse anti-c-Myc (Santa Cruz Biotechnology), mouse anti-PSD-95 (Affinity Bioreagents), rabbit anti-synaptophysin (Cell Signaling), rabbit anti-P- Thr183/Tyr185 JNK (Cell Signaling), mouse anti-JNK (Cell Signaling), mouse anti-GAPDH (Ambion). All fluorophore conjugated secondary antibodies were purchased from Jackson ImmunoResearch Labs.
HEK293 cells culture, transfection and immunoblotting
HEK293 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (all Invitrogen). Cells were seeded at 60–70% of confluence and transfected with the different constructs using Lipofectamine 2000 (Life Technologies). 24h after transfection, cells were lysed and an equal amount of protein was submitted to SDS-PAGE and transferred to nitrocellulose membrane. Membranes were incubated over night with HA tag, GFP, GAPDH or P-JNK antibodies. Total JNK was evaluated after stripping P-JNK signal. After incubation with HRP-conjugated secondary antibodies (Dako), Super Signal West Femto and ECL substrate (Pierce) were used for revelation. Acquisition was performed with LAS-4000 (General Electric) and quantification of band intensity was done with ImageJ software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2014.). IL1RAPL1 abundance was evaluated in lysates from cells co transfected with IL1RAPL1 constructs and GFP (control of transfection efficiency), by dividing HA intensity signal by GAPDH signal (protein loading control). JNK phosphorylation was measured by calculating the ratio between P-JNK (P-p54) and total JNK (p54). Statistical analysis was performed by one way analysis of variance (ANOVA) followed by Tukey’s post-hoc test for multiple comparisons.
Cell culture and transfection of primary rat and mouse hippocampal neurons
Low-density rat hippocampal neuronal cultures were prepared from embryonic day (E) 18–19 hippocampi as previously described with minor modifications (40, 41) and were grown in 12-well Petri dishes (Primo). Cultured mouse hippocampal neurons were prepared from E16.5 embryos, grown in 10 mm glass coverslips and maintained in Neurobasal B27-supplemented medium (Life Technologies). Neurons were transfected using Lipofectamine 2000 on Days In Vitro 11 (DIV11) and experiments were performed at DIV14–18. Experimental procedures on animals were approved by the local ethical committee.
Neuron surface staining
At DIV 14–15, live hippocampal neurons were labeled for 10 min at 37°C with anti-HA-tag rabbit antibody (10 μg/ml). After washing, neurons were fixed with paraformaldehyde (PFA) 4% plus 4% sucrose and incubated with anti-HA-tag mouse antibody in GDB (30 mM phosphate buffer, pH 7.4, 0.2% gelatin, 0.5% Triton X-100, 0.8 M NaCl (all Sigma-Aldrich)) for 3 h at room temperature. Cells were washed in 20 mM phosphate buffer containing 0.5 M NaCl and incubated with FITC- and Cy3- conjugated secondary antibodies.
Immunocytochemistry and image analysis
Cells were fixed in 4% PFA plus 4% sucrose at room temperature for 20 minutes, or 100% methanol at −20° for 10 min. Primary (1:100–1:800) and secondary (1:200) antibodies were applied in GDB buffer or in PBS (pH 7.4) containing 3% BSA and 0.2% Tween 20.
Confocal images were obtained using a Zeiss 510 confocal microscope (Carl Zeiss, a gift from Fondazione Monzino) or a Leica DMI6000 Spinning disk microscope. Quantification of synaptic protein staining was performed using MetaMorph (Molecular Devices, Downingtown, PA), and ImageJ software and NeuronJ plugin (42). Labeled, transfected cells were chosen randomly for quantification from six coverslips from 3 independent experiments for each condition and image analysis was performed under blind condition.
Coefficient of variation of IL1RAPL1 staining was calculated by dividing the standard deviation of HA signal by mean pixel intensity within dendrites (43). The dendritic spine number was measured as described previously (41, 44) with minor modifications. For each neuron we measured the number of protrusions present in all the dendrites along their entire length. Then we calculated mean and SEM (Standard Error of the Mean) for the neurons transfected with the same construct.
Quantification of protein surface staining was performed using MetaMorph (Molecular Devices, Downingtown, PA), and ImageJ software. The ratio of integrated intensity of surface rabbit anti-HA signal per total mouse anti-HA signal was measured for each neuron. Then we calculated the mean and SEM for the neurons transfected with the same construct.
HA-IL1RAPL1 in spines and dendritic shafts was quantified using IMARIS 7.2 software and Filament Tracer wizard (Bitplane). Integrated density of HA signal was normalized by GFP integrated density in each compartment. The ratio of HA/GFP in spines and in dendritic shafts was assessed for each neuron and mean + SEM was reported for neurons transfected with the same IL1RAPL1 construct.
Statistical analysis was performed by one way analysis of variance (ANOVA) followed by Tukey’s post-hoc test.
Electrophysiological recording on mouse hippocampal cultured neurons
Whole-cell patch-clamp recordings were made from GFP or IL1RAPL1 transfected mouse hippocampal neurons at 18–21 DIV. Non transfected cells from the same coverslip were also recorded as controls. Patch electrodes, fabricated from thick borosilicate glass, were pulled and fire polished to a final resistance of 2–4 MΩ and filled with internal solution containing (in mM): 125 CsMeSO3, 2 MgCl2, 1 CaCl2, 4 Na2ATP, 10 EGTA, 10 HEPES, 0.4 NaATP, and 5 QX314. Cultured neurons were superfused with an oxygenated external solution containing (in mM): 130 NaCl, 2.5 KCl, 2.2 CaCl2, 1.5 MgCl2, 10 HEPES, and 10 D-Glucose. Neurons were voltage clamped at −70 mV to record EPSCs and at 0V to record IPSCs. All of the experiments were performed at room temperature. Inward synaptic currents at −70 mV and outward currents at 0 mV were automatically detected by an automatic template based routine using pClamp 10.4 software (Molecular Devices). Recordings were performed under blind conditions. Typically, time periods of 120 seconds were used for analysis of synaptic events occurring at both membrane potentials.
Cell aggregation and immunoprecipitation assays
Two groups of HEK293 cells grown in 12-well plates were transfected, one with HA-IL1RAPL1 WT or mutants, and the other with Myc-PTPδ ectodomain. Cells transfected with GFP were used as negative control. After 12 h, cells were detached and counted for the cell aggregation assay, or lysated with 50 mM TRIS-HCl, 200 mM NaCl, 1 mM EDTA, 1% NP40, 1% Triton X-100 and protease inhibitors (RIPA buffer), for the immunoprecipitation assay.
For the cell aggregation assay, cell suspension was transferred to microtubes and gently centrifuged (800 g, 5 minutes, RT) to eliminate PBS-EDTA. The pellets were resuspended in aggregation medium (AM) containing HBSS 1x, MgCl2 1mM, and CaCl2 2mM. The two groups of transfected cells were mixed together and rotated at room temperature for 30 min to allow cells to aggregate. Cell mixtures (4 x 106 cells) were added to 1 ml AM on poly-L-Lys coated coverslips in multiwall (12 well) plate and let attach for some minutes at 37°C with 5% CO2. Once attached, cells were fixed and stained. Image analysis was performed under blind conditions, and aggregation coefficient was calculated by the number of green + red clusters (yellow in merge images) divided by the number of total transfected (green + red) cells and expressed as percent.
For the immunoprecipitation assay, protein A Sepharose beads (GE Healthcare) were washed in RIPA buffer. Anti-IL1RAPL1 antibody (K10, (15)) was added to the beads at 5μg/ml in RIPA buffer and incubated for 1 h. Lysates from the two groups of transfected cells in RIPA buffer were mixed in a in a volume proportion of 1 (for IL1RAPL1) to 1.5 (for Myc-PTPδ) and incubated overnight at 4° C with the beads/IL1RAPL1 antibody. The beads were washed 3 times with RIPA buffer and elution was performed in sample buffer for SDS-PAGE (5 min at 100°C), and loaded to 10% SDS-PAGE. Protein detection was performed as described in immunoblotting section.
Supplementary Material
Acknowledgments
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
This work was supported by the European Union’s FP7 large scale integrated network Gencodys (http://www.gencodys.eu/, HEALTH-241995); by the French Research Agency (ANR-2010-BLANC-1434-03); by the European Union’s EraNet program (ANR 2010-Neuro-001-01); by Institut national de la santé et de la recherche médicale (INSERM); by Ecole des Neurosciences de Paris (to M. R-B); by Comitato Telethon Fondazione Onlus (grant no. GGP13187 and GGP11095 (to C. S.)); Fondazione CARIPLO project number 2012-0593; Italian Institute of Technology; Seed Grant; Ministry of Health in the frame of ERA-NET NEURON; PNR-CNR Aging Program 2012–2014 and Foundation Jérôme Lejeune (to C. S.).
We thank Cherif Beldjord for X chromosome inactivation and segregation studies; Thierry Bienvenu for mutation analyzes and in silico prediction of genetic variant pathogenicity; Cellular imaging facility of Institut Cochin for image analysis advice.
ABREVIATIONS
- ID
Intellectual Disability
- XLID
X-linked Intellectual Disability
- IQ
intelligence quotient
- ASD
Autism Spectrum Disorder
Footnotes
CONFLICT OF INTEREST STATEMENT
None declared.
References
- 1.Carrié A, Jun L, Bienvenu T, Vinet MC, McDonell N, Couvert P, Zemni R, Cardona A, Van Buggenhout G, Frints S, et al. A new member of the IL-1 receptor family highly expressed in hippocampus and involved in X-linked mental retardation. Nat Genet. 1999;23:25–31. doi: 10.1038/12623. [DOI] [PubMed] [Google Scholar]
- 2.Kozak L, Chiurazzi P, Genuardi M, Pomponi MG, Zollino M, Neri G. Mapping of a gene for non-specific X linked mental retardation : evidence for linkage to chromosomal region Xp21.1–Xp22.3. J Med Genet. 1993;30:866–869. doi: 10.1136/jmg.30.10.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tabolacci E, Pomponi MG, Pietrobono R, Terracciano A, Chiurazzi P, Neri G. A Truncating Mutation in the IL1RAPL1 Gene Is Responsible for X-linked Mental Retardation in the MRX21 Family. Am J Genet. 2006;487:482–487. doi: 10.1002/ajmg.a.31107. [DOI] [PubMed] [Google Scholar]
- 4.Nawara M, Klapecki J, Borg K, Jurek M, Moreno S, Tryfon J, Bal J, Chelly J, Mazurczak T. Novel mutation of IL1RAPL1 gene in a nonspecific X-linked mental retardation (MRX) family. Am J Med Genet A. 2008;146A:3167–3172. doi: 10.1002/ajmg.a.32613. [DOI] [PubMed] [Google Scholar]
- 5.Piton A, Michaud JL, Peng H, Aradhya S, Gauthier J, Mottron L, Champagne N, Lafrenière RG, Hamdan FF, Joober R, et al. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum Mol Genet. 2008;17:3965–3974. doi: 10.1093/hmg/ddn300. [DOI] [PubMed] [Google Scholar]
- 6.Behnecke A, Hinderhofer K, Bartsch O, Nümann A, Ipach ML, Damatova N, Haaf T, Dufke A, Riess O, Moog U. Intragenic deletions of IL1RAPL1: Report of two cases and review of the literature. Am J Med Genet A. 2011;155A:372–379. doi: 10.1002/ajmg.a.33656. [DOI] [PubMed] [Google Scholar]
- 7.Whibley AC, Plagnol V, Tarpey PS, Abidi F, Fullston T, Choma MK, Boucher CA, Shepherd L, Willatt L, Parkin G, et al. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am J Hum Genet. 2010;87:173–188. doi: 10.1016/j.ajhg.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franek KJ, Butler J, Johnson J, Simensen R, Friez MJ, Bartel F, Moss T, DuPont B, Berry K, Bauman M, et al. Deletion of the immunoglobulin domain of IL1RAPL1 results in nonsyndromic X-linked intellectual disability associated with behavioral problems and mild dysmorphism. Am J Med Genet A. 2011;155A:1109–1114. doi: 10.1002/ajmg.a.33833. [DOI] [PubMed] [Google Scholar]
- 9.Mikhail FM, Lose EJ, Robin NH, Descartes MD, Rutledge KD, Rutledge SL, Korf BR, Carroll AJ. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am J Med Genet A. 2011;155A:2386–2396. doi: 10.1002/ajmg.a.34177. [DOI] [PubMed] [Google Scholar]
- 10.Youngs EL, Henkhaus R, Hellings JA, Butler MG. IL1RAPL1 gene deletion as a cause of X-linked intellectual disability and dysmorphic features. Eur J Med Genet. 2012;55:32–36. doi: 10.1016/j.ejmg.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barone C, Bianca S, Luciano D, Di Benedetto D, Vinci M, Fichera M. Intragenic ILRAPL1 deletion in a male patient with intellectual disability, mild dysmorphic signs, deafness, and behavioral problems. Am J Med Genet A. 2013;161A:1381–1385. doi: 10.1002/ajmg.a.35860. [DOI] [PubMed] [Google Scholar]
- 12.Mignon-Ravix C, Cacciagli P, Choucair N, Popovici C, Missirian C, Milh M, Mégarbané A, Busa T, Julia S, Girard N, et al. Intragenic rearrangements in X-linked intellectual deficiency: Results of a-CGH in a series of 54 patients and identification of TRPC5 and KLHL15 as potential XLID genes. Am J Med Genet A. 2014;9999:1–7. doi: 10.1002/ajmg.a.36602. [DOI] [PubMed] [Google Scholar]
- 13.Tucker T, Zahir FR, Griffith M, Delaney A, Chai D, Tsang E, Lemyre E, Dobrzeniecka S, Marra M, Eydoux P, et al. Single exon-resolution targeted chromosomal microarray analysis of known and candidate intellectual disability genes. Eur J Hum Genet. 2013;22:792–800. doi: 10.1038/ejhg.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Redin C, Gerard B, Lauer J, Herenger Y, Muller J, Quartier A, Masurel-Paulet A, Willems M, Lesca G, El-Chehadeh S, et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet. 2014 doi: 10.1136/jmedgenet-2014-102554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pavlowsky A, Gianfelice A, Pallotto M, Zanchi A, Vara H, Khelfaoui M, Valnegri P, Rezai X, Bassani S, Brambilla D, et al. A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation. Curr Biol. 2010;20:103–115. doi: 10.1016/j.cub.2009.12.030. [DOI] [PubMed] [Google Scholar]
- 16.Gambino F, Kneib M, Pavlowsky A, Skala H, Heitz S, Vitale N, Poulin B, Khelfaoui M, Chelly J, Billuart P, et al. IL1RAPL1 controls inhibitory networks during cerebellar development in mice. Eur J Neurosci. 2009;30:1476–1486. doi: 10.1111/j.1460-9568.2009.06975.x. [DOI] [PubMed] [Google Scholar]
- 17.Houbaert X, Zhang CL, Gambino F, Lepleux M, Deshors M, Normand E, Levet F, Ramos M, Billuart P, Chelly J, et al. Target-specific vulnerability of excitatory synapses leads to deficits in associative memory in a model of intellectual disorder. J Neurosci. 2013;33:13805–13819. doi: 10.1523/JNEUROSCI.1457-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bahi N, Friocourt G, Carrié A, Graham ME, Weiss JL, Chafey P, Fauchereau F, Burgoyne RD, Chelly J. IL1 receptor accessory protein like, a protein involved in X-linked mental retardation, interacts with Neuronal Calcium Sensor-1 and regulates exocytosis. Hum Mol Genet. 2003;12:1415–1425. doi: 10.1093/hmg/ddg147. [DOI] [PubMed] [Google Scholar]
- 19.Gambino F, Pavlowsky A, Béglé A, Dupont JL, Bahi N, Courjaret R, Gardette R, Hadjkacem H, Skala H, Poulain B, et al. IL1-receptor accessory protein-like 1 (IL1RAPL1), a protein involved in cognitive functions, regulates N-type Ca 2+ -channel and neurite elongation. Proc Natl Acad Sci U S A. 2007;104:2–7. doi: 10.1073/pnas.0701133104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valnegri P, Montrasio C, Brambilla D, Ko J, Passafaro M, Sala C. The X-linked intellectual disability protein IL1RAPL1 regulates excitatory synapse formation by binding PTPδ and RhoGAP2. Hum Mol Genet. 2011;20:4797–4809. doi: 10.1093/hmg/ddr418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi T, Yoshida T, Ra M, Taguchi R, Mishina M. IL1RAPL1 associated with mental retardation and autism regulates the formation and stabilization of glutamatergic synapses of cortical neurons through RhoA signaling pathway. PLoS One. 2013;8:e66254. doi: 10.1371/journal.pone.0066254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshida T, Yasumura M, Uemura T, Lee SJ, Ra M, Taguchi R, Iwakura Y, Mishina M. IL-1 receptor accessory protein-like 1 associated with mental retardation and autism mediates synapse formation by trans-synaptic interaction with protein tyrosine phosphatase δ. J Neurosci. 2011;31:13485–13499. doi: 10.1523/JNEUROSCI.2136-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, O’Meara S, Latimer C, Dicks E, Menzies A, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–43. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 25.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshida T, Mishina M. Zebrafish orthologue of mental retardation protein IL1RAPL1 regulates presynaptic differentiation. Mol Cell Neurosci. 2008;39:218–228. doi: 10.1016/j.mcn.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 28.Pulido R, Serra-Pagès C, Tang M, Streuli M. The LAR/PTP delta/PTP sigma subfamily of transmembrane protein-tyrosine-phosphatases: multiple human LAR, PTP delta, and PTP sigma isoforms are expressed in a tissue-specific manner and associate with the LAR-interacting protein LIP. 1. Proc Natl Acad Sci U S A. 1995;92:11686–11690. doi: 10.1073/pnas.92.25.11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan JA, Brint EK, O’Neill LAJ, Tong L. Crystal structure of the Toll/interleukin-1 receptor domain of human IL-1RAPL. J Biol Chem. 2004;279:31664–31670. doi: 10.1074/jbc.M403434200. [DOI] [PubMed] [Google Scholar]
- 30.Pavlowsky A, Zanchi A, Pallotto M, Giustetto M, Chelly J, Sala C, Billuart P. Neuronal JNK pathway activation by IL-1 is mediated through IL1RAPL1, a protein required for development of cognitive functions. Commun Integr Biol. 2010;10:245–247. doi: 10.4161/cib.3.3.11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brint EK, Fitzgerald KA, Smith P, Coyle AJ, Gutierrez-Ramos JC, Fallon PG, O’Neill LaJ. Characterization of signaling pathways activated by the interleukin 1 (IL-1) receptor homologue T1/ST2 A role for Jun N-terminal kinase in IL-4 induction. J Biol Chem. 2002;277:49205–49211. doi: 10.1074/jbc.M209685200. [DOI] [PubMed] [Google Scholar]
- 32.Leprêtre F, Delannoy V, Froguel P, Vasseur F, Montpellier C. Dissection of an inverted X(p21.3q27.1) chromosome associated with mental retardation. Cytogenet Genome Res. 2003;101:124–129. doi: 10.1159/000074167. [DOI] [PubMed] [Google Scholar]
- 33.Schuurs-Hoeijmakers JHM, Vulto-van Silfhout AT, Vissers LELM, van de Vondervoort IIGM, van Bon BWM, de Ligt J, Gilissen C, Hehir-Kwa JY, Neveling K, del Rosario M, et al. Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J Med Genet. 2013;50:802–811. doi: 10.1136/jmedgenet-2013-101644. [DOI] [PubMed] [Google Scholar]
- 34.Wu H, Luo J, Yu H, Rattner A, Mo A, Wang Y, Smallwood PM, Erlanger B, Wheelan SJ, Nathans J. Cellular resolution maps of X chromosome inactivation: implications for neural development, function, and disease. Neuron. 2014;81:103–119. doi: 10.1016/j.neuron.2013.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim MJ, Futai K, Jo J, Hayashi Y, Cho K, Sheng M. Synaptic Accumulation of PSD-95 and Synaptic Function Regulated by Phosphorylation of Serine-295 of PSD-95. Neuron. 2008;57:326–327. doi: 10.1016/j.neuron.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 36.Zhu Y, Pak D, Qin Y, McCormack SG, Kim MJ, Baumgart JP, Velamoor V, Auberson YP, Osten P, van Aelst L, et al. Rap2-JNK removes synaptic AMPA receptors during depotentiation. Neuron. 2005;46:905–916. doi: 10.1016/j.neuron.2005.04.037. [DOI] [PubMed] [Google Scholar]
- 37.Thomas GM, Lin DT, Nuriya M, Huganir RL. Rapid and bi-directional regulation of AMPA receptor phosphorylation and trafficking by JNK. EMBO J. 2008;27:361–372. doi: 10.1038/sj.emboj.7601969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vandeweyer G, Reyniers E, Wuyts W, Rooms L, Kooy RF. CNV-WebStore: online CNV analysis, storage and interpretation. BMC Bioinformatics. 2011;12:4. doi: 10.1186/1471-2105-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fokkema IFAC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, den Dunnen JT. LOVD v. 2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–63. doi: 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]
- 40.Sala C, Piëch V, Wilson NR, Passafaro M, Liu G, Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31:115–130. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- 41.Verpelli C, Piccoli G, Zibetti C, Zanchi A, Gardoni F, Huang K, Brambilla D, Di Luca M, Battaglioli E, Sala C. Synaptic activity controls dendritic spine morphology by modulating eEF2-dependent BDNF synthesis. J Neurosci. 2010;30:5830–5842. doi: 10.1523/JNEUROSCI.0119-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meijering E, Jacob M, Sarria JCF, Steiner P, Hirling H, Unser M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A. 2004;58:167–176. doi: 10.1002/cyto.a.20022. [DOI] [PubMed] [Google Scholar]
- 43.Lyles V, Zhao Y, Martin KC. Synapse formation and mRNA localization in cultured Aplysia neurons. Neuron. 2006;49:349–356. doi: 10.1016/j.neuron.2005.12.029. [DOI] [PubMed] [Google Scholar]
- 44.Piccoli G, Verpelli C, Tonna N, Romorini S, Alessio M, Nairn AC, Bachi A, Sala C. Proteomic Analysis of Activity-Dependent Synaptic Plasticity in Hippocampal Neurons research articles. J proteomic Res. 2007;6:3203–3215. doi: 10.1021/pr0701308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.