

Abstract
Aggregation of β-amyloid (Aβ) is widely believed to cause neuronal dysfunction in Alzheimer's disease. Transthyretin (TTR) binds to Aβ and inhibits its aggregation and neurotoxicity. TTR is a homotetrameric protein, with each monomer containing a short α-helix and two anti-parallel β-sheets. Dimers pack into tetramers to form a hydrophobic cavity. Here we report the discovery of a TTR mutant, N98A, that was more effective at inhibiting Aβ aggregation than wild-type (WT) TTR, although N98A and WT bound Aβ equally. The N98A mutation is located on a flexible loop distant from the putative Aβ-binding sites and does not alter secondary and tertiary structures nor prevent correct assembly into tetramers. Under non-physiological conditions, N98A tetramers were kinetically and thermodynamically less stable than WT, suggesting a difference in the tetramer folded structure. In vivo, the lone cysteine in TTR is frequently modified by S-cysteinylation or S-sulfonation. Like the N98A mutation, S-cysteinylation of TTR modestly decreased tetramer stability and increased TTR's effectiveness at inhibiting Aβ aggregation. Collectively, these data indicate that a subtle change in TTR tetramer structure measurably increases TTR's ability to inhibit Aβ aggregation.
Keywords: amyloid, protein aggregation, transthyretin
Introduction
Two pathological characteristics of Alzheimer's disease (AD) are intracellular neurofibrillary tangles, derived from phosphorylated tau protein, and extracellular plaques in the hippocampus and cerebral cortex, consisting of β-amyloid (Aβ) deposits (Wisniewski and Goñi, 2014). Aβ, a 4.3-kDa proteolytic cleavage product of amyloid precursor protein (APP), spontaneously self-assembles through a multi-step mechanism into soluble oligomers and insoluble fibrillar aggregates (Vivekanandan et al., 2011). While the exact mechanism of AD pathogenesis remains unknown, Aβ aggregates figure prominently in the dominant ‘amyloid cascade’ hypothesis of AD (Cleary et al., 2005; Overk and Masliah, 2014). Transgenic mouse models engineered to express human APP (APPsw) generate large amounts of Aβ and develop substantial amyloid deposits. However, these mice exhibit none of the signs of AD progression predicted by the amyloid cascade hypothesis, such as neurofibrillary tangles or widespread neuronal cell death (Stein and Johnson, 2002). The lack of AD-like pathology was explained when it was noted that APPsw mice up-regulated transthyretin (TTR) synthesis, as a response against Aβ deposition (Stein et al., 2004; Choi et al., 2007). This result suggested that TTR protects against Aβ toxicity, a conclusion supported by other in vitro and in vivo studies (Schwarzman et al., 1994; Giunta et al., 2005; Buxbaum et al., 2008; Costa et al., 2008; Li et al., 2011).
TTR, a 55-kDa homotetrameric transport protein, circulates in the blood (3–7 μM) and cerebrospinal fluid (CSF) (0.1–0.4 μM). Each 127-residue monomer contains two anti-parallel β-sheets and a short α-helix (Fig. 1) (Hamilton et al., 1993). The dimer forms via extensive hydrogen bonding between strands H and F of two monomers, while dimer–dimer association is mediated mainly via hydrophobic interactions between the AB loop and parts of strand H (Foss et al., 2005). Thyroxine binds to the hydrophobic pocket produced from tetramer assembly. TTR serves as the primary carrier for thyroxine in the CSF and a secondary carrier in the blood. TTR also functions as a carrier for retinol-binding protein (RBP). Both ligands reportedly stabilize the TTR tetramer (White and Kelly, 2001). TTR contains one free cysteine (Cys10) per monomer but no disulfide bonds. Post-translational oxidative modification at the Cys10 residue is common, with the most frequent reported to be S-cysteinylation, followed by S-sulfonation. Cys10 oxidation is generally more frequent in the blood than in the CSF; estimates vary, but typically it is reported that ∼80% of the plasma TTR, but <40% of the CSF TTR, is oxidized (Biroccio et al., 2006; Kingsbury et al., 2007; Trenchevska et al., 2011, 2014; Poulsen et al., 2012, 2014).
Fig. 1.
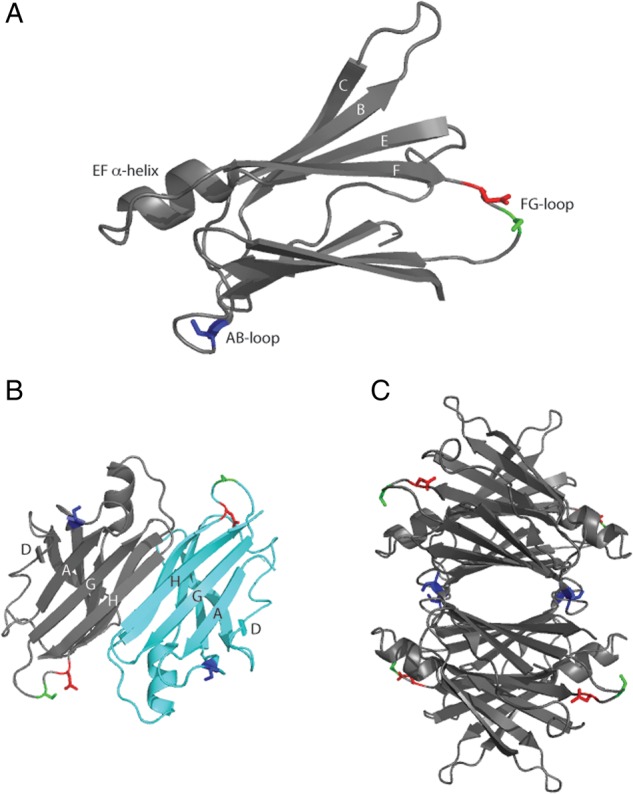
Ribbon structure of human TTR. (A) Monomer, (B) dimer and (C) tetramer with S23 (blue), S100 (green) and N98 (red) highlighted. Generated from PDB entry 1DVQ. Each monomer has an ‘inner sheet’ of strands D, A, G and H, and an ‘outer sheet’ of strands C, B, E and F as well as a lone α-helix between the E and F strands.
Wild-type (WT) TTR has a highly stable quaternary structure and is a robust protein as evidenced by the unusually large number of mutations that have been identified in the adult population: at least 124 TTR mutants are known, at 72 different positions (Rowczenio et al., 2014). The prevalence of variant TTR genes may be as high as 1 in 100 000 (Benson and Uemichi, 1996). Many of the TTR mutants are linked to late-onset inherited amyloid diseases such as familial amyloid polyneuropathy (FAP) (Planté-Bordeneuve and Said, 2011). Other mutations are benign or even protective (Hammarstrom et al., 2001). Invariably, the disease-associated mutations destabilize TTR's quaternary and/or tertiary structure (Hurshman Babbes et al., 2008). This and other observations have led to the hypothesis that TTR amyloid develops by tetramer dissociation to folded monomer, structural re-arrangement of the monomer and finally re-association of monomers into long fibrils (Lashuel et al., 1998; Quintas et al., 1999). WT TTR is the cause of senile systemic amyloidosis, a disease affecting up to 25% of those over 80 years of age (Westermark et al., 1990). WT TTR too is believed to initiate amyloidosis via tetramer destabilization, caused not by mutation but by destabilizing environmental or cellular factors (Foss et al., 2005; Palaninathan et al., 2008).
In previous work, we established that TTR binds directly to Aβ oligomers, slows its further aggregation and thereby reduces Aβ toxicity. We identified two specific residues, L82 on the EF helix and L110 on strand G, where mutation to alanine led to loss of binding, loss of inhibition of aggregation and loss of inhibition of toxicity (Du et al., 2012). The interior pocket (including strand G) was also identified as the Aβ binding site by another group, using different methodologies (Li et al., 2013). We proposed the hypothesis that Aβ oligomers bind to the EF helix on the exterior of the tetramer and that the binding induces a shift in tetramer structure and partial tetramer destabilization, which in turn facilitates stronger inhibition of aggregation (Yang et al., 2013). TTR mutants L55P (unstable tetramer), S112I (dimer) and F87M/L110M (monomer, mTTR) demonstrated enhanced binding to Aβ, and mTTR was a stronger inhibitor of Aβ aggregation than WT (Du et al., 2012). Those studies established that a partial (S112I, L55P) or complete (mTTR) loss of quaternary structure increases binding to Aβ and improves inhibition of Aβ aggregation.
In this work, we asked whether more subtle changes in TTR quaternary structure, without loss of tetramer formation, would also lead to greater Aβ binding and/or greater inhibition of Aβ aggregation. To examine this question, we mutated selected residues in flexible loop regions of TTR to alanine and characterized their interactions with Aβ. We extended these studies to also determine if oxidative modifications that occur naturally and affect TTR tetramer stability (Zhang and Kelly, 2003; Kingsbury et al., 2008) also change the nature of TTR's interaction with Aβ.
Materials and methods
TTR mutagenesis, production and purification
A recombinant plasmid of human WT TTR (pTWIN1-TTR) was assembled as previously described (Liu et al., 2009). The TTR mutant plasmids were created using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene, LaJolla, CA) using pTWIN1-TTR as the template. Sequencing of plasmids verified successful mutations. The mutant plasmids were transformed into BL21(DE3)pLysS cells (Promega, Madison, WI). WT and mutants S23A, S100A, N98A and F87M/L110M [mTTR, stable monomeric TTR (Jiang et al., 2001)] were produced and purified as previously described (Du et al., 2012). Size-exclusion chromatography (SEC) removed trace amounts of aggregates, and each protein stock was filtered through 0.22 µm filter. TTR tetramer concentration was determined by absorbance at 280 nm using an extinction coefficient of 77 600 M−1 cm−1 (Bateman et al., 2011). TTR was stored in PBSA-E (10 mM Na2HPO4/NaH2PO4, 150 mM NaCl, 0.02% w/v NaN3 and 1 mM EDTA at pH 7.4) unless otherwise indicated.
Size-exclusion chromatography
TTR (∼1.5 mg/ml) was injected into a BioAssist G3SWXL column (Tosoh Bioscience, King of Prussia, PA) on a high-performance liquid chromatography (HPLC) system using PBSA-E as the mobile phase. The elution peaks were detected by absorbance at 280 nm with the mobile phase flow rate at 1 ml/min.
TTR oxidation
To prepare S-cysteinylated protein (C-TTR), cystine (6 mg) was added to 20 ml of boiling PBSA (10 mM Na2HPO4/NaH2PO4, 150 mM NaCl, and 0.02% w/v NaN3 at pH 7.4), stirred until completely dissolved and then cooled to room temperature before use. Equal volumes of cystine (1.25 mM) and TTR (13 μM) were mixed and incubated overnight at room temperature with magnetic stirring. S-sulfonated TTR (S-TTR) was prepared by mixing sodium tetrathionate (40 mM) in PBSA with TTR (13 µM). Reactions were terminated by 3× dialysis against PBSA at room temperature. All samples were analyzed by a linear trap/FT-ICR MS (LTQ FT Ultra) hybrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Successful oxidation was confirmed: TTR, 13 761.6 Da (theoretical 13 761.04 Da); C-TTR, 13 880.78 Da (theoretical 13 880.05 Da, Δ = 119); and S-TTR, 13 841.71 Da (theoretical 13 841.89 Da, Δ = 80). From the mass spectra, we estimated that yield of oxidized species was close to 100% (not shown). All samples were filtered through 0.22 µm filters prior to experiments.
Aβ preparation
Aβ(1–40) (AnaSpec, Inc., San Jose, Ca) was dissolved in a filtered (0.22 μm) 50% acetonitrile solution to 1 mg/ml, frozen overnight at −80°C and re-lyophilized. To prepare the Aβ stock solution, lyophilized Aβ was dissolved in a filtered (0.22 μm) 8 M urea/100 mM glycine–NaOH buffer at pH 10 to a final concentration of 2.8 mM. Stock was divided into 4 or 8 μL aliquots, snap-frozen in an ethanol bath and stored at −80°C. Immediately before each experiment, aliquots were thawed and diluted into 0.22 μm filtered PBSA. Aβ monomer has an extinction coefficient of 1490 M−1 cm−1 at 280 nm and a molecular weight of 4330 Da (Christopeit et al., 2005).
Gel electrophoresis (SDS-PAGE)
TTR (3.6 μM) in PBSA-E was diluted into SDS to a final concentration of 2% (w/v) SDS. A duplicate of each sample was boiled for 10 min. Samples were loaded on a Precise 4–20% polyacrylamide gradient gel (Pierce, Rockford, IL) along with EZ-Run Protein Ladder (Fisher BioReagents, Fair Lawn, NJ). After electrophoresing with SDS-TRIS-HEPES buffer for 45 min at 125 V, the gel was stained with Coomassie blue.
For monitoring Aβ-induced destabilization, TTR (3.6 μM) with Aβ (60 or 80 μM) was incubated for 1–3 days at 37°C. Samples were loaded in a Precise 4–20% polyacrylamide gradient gel (Pierce, Rockford, IL), electrophoresed and stained.
ANS fluorescence
TTR and 1-anilinonaphthalene-8-sulfonic acid (ANS, AnaSpec, Fremont, CA, USA) were prepared to a final concentration of 1 and 29 μM, respectively, in PBSA-E. ANS fluorescence spectra were measured using a PTI QuantaMaster spectrofluorometer (Birmingham, NJ, USA) with excitation at 370 nm and emission recorded from 440 to 500 nm. For each sample, the average of triplicate spectra minus the background ANS signal was reported. ANS concentration was measured using an extinction coefficient of 4950 M−1 cm−1 at 350 nm (Hawe et al., 2008).
Circular dichroism
TTR was dialyzed overnight against phosphate–NaF buffer (10 mM Na2HPO4/NaH2PO4 and 150 mM NaF at pH 7.4). TTR was diluted to 2 μM in phosphate–NaF buffer, and the circular dichroism (CD) spectrum of each protein was measured on an Aviv 202SF CD spectrophotometer from Aviv Biomedical (Lakewood, NJ) at room temperature. Blank solvent spectra were measured and subtracted from the sample spectra.
Tryptophan (Trp) fluorescence
Fluorescence spectra of TTR (0.1 mg/mL) in PBSA were measured using a QuantaMaster spectrofluorometer with excitation at 290 nm and emission recorded from 300 to 420 nm. For each sample, the average of triplicate spectra minus the background was reported.
Acid-induced TTR aggregation
TTR in PBSA-E was diluted into acidic buffer (200 mM sodium acetate/acetic acid, 150 mM NaCl and 1 mM EDTA at pH 4.3) to a final tetramer concentration of 0.25 mg/mL and pH of 4.4 to initiate aggregation. Aggregate growth was monitored by dynamic light scattering (DLS).
Urea denaturation
Urea denaturation experiments were completed following the protocol of Hurshman Babbes et al. (Hurshman Babbes et al., 2008). Stock urea solutions were prepared in buffer, with the urea concentration determined by refractive index measurement. TTR at either 0.9 or 9 µM was prepared in buffers containing varying (0–7 M) urea and then incubated for 96 h at 4°C. Fluorescence measurements were taken in a PTI QuantaMaster spectrofluorometer. Samples were excited at 295 nm, and fluorescence emission spectra were recorded from 310 to 410 nm. Intensities at 335 and 355 nm were recorded, and solvent contribution was subtracted before calculating the ratio I355/I335 nm as a measure of the extent of unfolding.
TTR tetramer dissociation kinetics (S-TRAP)
TTR dissociation kinetics were measured using a method described in detail elsewhere (Xia et al., 2012). Briefly, TTR was prepared at 0.4 mg/mL in PBSA-E. SDS buffer [2 mM DTT, 25% (w/v) glycerol, 2% (w/v) SDS, 0.01% bromophenol blue and 62.5 mM Tris-HCl (pH 6.8)] (Bio-Rad Laboratories, Hercules, CA) was preheated at 90°C for 10 min. The preheated SDS buffer was added to an equal volume of TTR so that the final concentration of protein and SDS were 0.2 mg/mL (3.6 µM) and 1%, respectively. Upon mixing, samples were incubated at 80°C for various lengths of time. After incubation, samples were rapidly cooled on ice for 10 s and then centrifuged at 4000 rpm for 5 s at room temperature. Samples were loaded on a Precise 4–20% polyacrylamide gradient gel (Pierce, Rockford, IL) along with EZ-Run Protein Ladder (Fisher BioReagents, Fair Lawn, NJ). After electrophoresing with SDS-TRIS-HEPES buffer for 45 min at 125 V, the gel was stained with Coomassie blue. Destained gels were photocopied, and the densities of tetramer and monomer bands were quantified by ImageJ. The fraction of monomeric protein as a function of incubation time was fitted to a single exponential function to determine the tetramer dissociation constant (kd).
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) plates (Corning, Inc., Corning, NY) were coated with 5 μg/mL of TTR or mTTR (100 μl per well) in coating buffer (10 mM sodium carbonate, 30 mM sodium bicarbonate, 0.05% NaN3 at pH 9.6) overnight at room temperature. The plate was washed three times with wash buffer (PBS with 0.05% Tween 20) and incubated with protein-free blocking buffer (Pierce, Rockford, IL) for 1 h at room temperature. For a negative control, TTR was not coated, but wells were incubated with blocking buffer. Three to five replicate wells were prepared at each condition. Aβ (28 μM in PBSA) was incubated at room temperature for 2 days to allow the peptide to aggregate, then diluted to 0.5 μg/mL in PBS and immediately added to TTR-coated or negative control wells (50 μl/well). The plate was incubated at 37°C for 1 h. After washing the plate, anti-Aβ antibody 6E10 (Covance, Princeton, NJ) in wash buffer (1:3000) was added to each well (100 μl/well), and the plate was incubated at room temperature for 1 h with gentle shaking. After washing, anti-mouse HRP antibody (1:3000 dilution; Pierce, Rockford, IL) was added to each well (100 μl/well), and the plate was incubated for 1 h at room temperature with gentle shaking. The plate was washed three times with wash buffer, and then 100 μl of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate solution (Pierce, Rockford, IL) was added to each well. The plate was incubated at room temperature for 15–30 min; color development was stopped by adding 100 μl of 2 M sulfuric acid. Absorbance was measured at 450 nm with an EL800 Universal Microplate Reader (Bio-tek Instruments, Inc., Winooski, VT). Aβ binding was calculated as the mean of three or four replicate wells minus the mean of the negative control (Aβ alone) absorbance.
For experiments with oxidized TTR, Aβ was pre-aggregated at 0.8 mg/mL for 2 days at room temperature and then diluted to 5 μg/mL in PBS before adding to the plate. A 1:1500 dilution into wash buffer was used for both the anti-Aβ antibody (6E10) and anti-mouse HRP antibody.
Thioflavin T fluorescence
Thioflavin T (ThT) (Fisher Scientific, Fair Lawn, NJ) solution at 11 μM in PBSA was filtered through 0.22 μm; ThT concentration was measured using an extinction coefficient of 26 600 M−1 cm−1 at 416 nm in ethanol (Hawe et al., 2008). Aβ (28 μM) with or without TTR (3.6 μM) in PBSA was incubated at 37°C for 2–48 h. Immediately prior to measurement on a QuantaMaster spectrofluorometer, 10 μL of protein was added to 130 μL of 11 μM ThT (PTI, Birmingham, NJ) with an excitation at 440 nm and emission recorded from 450 to 550 nm. The average of triplicate signals at 480 nm minus the background signal of ThT in PBSA described the extent of Aβ fibril formation for each sample.
Dynamic light scattering
PBSA was filtered through 0.02 µm filter. Aβ alone (28 µM) or with TTR (3.6–4 µM) in PBSA was filtered through a 0.45 µm filter directly into a light-scattering cuvette and then placed into a bath of the index-matching solvent decahydronaphthalene with temperature controlled at 37°C. Light-scattering data at 90° scattering angle were measured using a Brookhaven BI-200SM system (Brookhaven Instruments Corp., Holtsville, NY) and an Innova 90C-5 argon laser (Coherent, Santa Clara, CA) operating at 488 nm and 150 mW. The z-averaged hydrodynamic diameter was determined from the autocorrelation function using the method of cumulants. The total scattered intensity was measured at 90° scattering angle and normalized by the total mass protein concentration. The normalized intensity is proportional to the weight-average molecular weight of the aggregates times a particle scattering factor that is a function of the shape and size of aggregates.
Nanoparticle tracking analysis
Nanoparticle tracking analysis (NTA) measurements were taken with a Nanosight LM10 (Nanosight, Amesbury, UK) equipped with a 405 nm laser. All buffers were filtered through 0.02 μm filters prior to use. Aβ (28 μM) mixed with TTR (3.6 μM) was filtered through 0.02 μm and injected into the sample chamber using a syringe. All measurements were collected at room temperature with the camera level set to the maximal value. One 90 s video was taken at various time points. The data were recorded and analyzed using NTA version 2.3.
Transmission electron microscopy
Aβ alone (28 μM) or with TTR (3.6 μM) in PBSA was incubated for 1–3 days at 37°C. A drop of sample was positioned on a pioloform-coated grid and stained with methylamine tungstate stain. Images were then taken with a Philips CM120 scanning transmission electron microscope (FEI Corp., Eindhoven, The Netherlands).
Results
Selection of mutants
Several groups have established that TTR inhibits Aβ aggregation and toxicity in vitro (Schwarzman et al., 1994; Giunta et al., 2005; Li et al., 2013). We previously showed that inhibition of aggregation is a direct result of TTR-Aβ binding, and we identified two critical residues for binding, on the solvent-exposed EF helix (L82) and in the interior of the TTR tetramer, on strand G (L110) (Du et al., 2012; Yang et al., 2013). We also identified other residues in or near strand G that reduced or eliminated Aβ binding (Cho et al., 2014). TTR mutants L55P (an unstable disease-associated mutant that is a mix of monomer and tetramer), S112I (dimer) and F87M/L110M (mTTR, an engineered monomer) all demonstrated enhanced binding to Aβ, and mTTR was a stronger inhibitor of Aβ aggregation than WT (Du and Murphy, 2010; Du et al., 2012).
In this study, we asked whether mutations in TTR, which did not interfere with tetramer formation, would influence its binding to Aβ and/or affect its inhibition of Aβ aggregation. We searched for polar non-charged residues in flexible loop regions, hypothesizing that these residues are unlikely to interfere with attainment of TTR's native secondary, tertiary or quaternary structure. We selected three positions: S23, N98 and S100 (Fig. 1). All three residues have neutral hydrophilic side chains. S23 is located on the AB loop near the dimer–dimer interface. N98 and S100 are located on the solvent-exposed FG loop and, based on analysis of the crystal structure, appear to hydrogen bond with the solvent. None were identified as critical residues for binding to Aβ (Du et al., 2012; Cho et al., 2014). We replaced each selected residue with alanine, the amino acid most commonly chosen in mutagenesis studies because of its relatively small size and neutral properties (Hecht et al., 2013). All mutants were readily expressed and purified. On denaturing SDS-PAGE, WT and all TTR mutants were monomers of the correct molecular weight (Supplementary Fig. S1). All eluted on a size-exclusion column as a single peak at the same elution volume as WT, demonstrating that mutations did not interfere with tetramer assembly (Supplementary Fig. S2).
TTR mutant interaction with β-amyloid
All three mutants bound Aβ, with slightly reduced binding to S23A and slightly enhanced to N98A relative to WT, but the differences were not statistically significant (Supplementary Fig. S3).
Thioflavin (ThT) fluorescence intensity is widely used as an indicator of the mass concentration of amyloid fibrils (Krebs et al., 2005). We used ThT to test whether the three mutants were able to inhibit Aβ aggregation. Aβ alone developed ThT fluorescence that increased over time, as expected (Fig. 2). WT, S23A and S100A all partially suppressed ThT fluorescence to similar extents. Unexpectedly, N98A was a significantly more effective inhibitor of amyloid fibril growth than WT (P < 0.01 at 24 and 48 h).
Fig. 2.

ThT analysis of TTR-mediated inhibition of Aβ aggregation. TTR (WT and mutants, 3.6 μM) was incubated with Aβ (28 μM) at 37°C for 2, 24 or 48 h. Fibrils were detected using ThT. Data shown are mean ± SD. TTR alone (WT or mutants) does not have any ThT fluorescence signal (not shown).
Measuring changes in ThT fluorescence intensity is not a foolproof means of detecting changes in amyloid content. For example, ThT intensity could be reduced upon addition of another compound due to competition for fibrillar binding sites between ThT and the added compound. Furthermore, some prefibrillar amyloid oligomers may be weakly ThT positive, and some amyloid fibrils are ThT negative (Cloe et al., 2011). To confirm the conclusion that N98A was a stronger inhibitor than WT, we measured the increase in the mean hydrodynamic diameter of Aβ aggregates in the presence of WT or N98A. Aβ aggregation (28 μM) with or without WT or N98A (3.6 μM) was monitored for 4 h at 37°C (Fig. 3). At these experimental conditions, WT and N98A are stable and do not aggregate (data not shown). Aβ alone exhibited the fastest increase in mean hydrodynamic diameter. Although WT partially slowed Aβ aggregate growth, N98A was measurably more effective than WT at reducing Aβ aggregate growth rate. We also examined Aβ aggregate size using NTA, a particle-by-particle scattering technique that yields both a particle size distribution and an absolute particle number concentration. Protein aggregates of 30 nm diameter or more are reliably detected (Filipe et al., 2010; Yang et al., 2014). Samples were analyzed at 20 min and 2 h after preparation (Fig. 4). Relative to WT, N98A reduced the number concentration of Aβ aggregates and shifted the particle size distribution toward smaller particles. Finally, we observed the effect of TTR (WT and mutants) on Aβ aggregate morphology using transmission electron microscopy (TEM) (Fig. 5). In the absence of TTR, the Aβ sample contained very dense packing of long, thin fibrils. Qualitatively, Aβ with WT contained fewer fibrils, and with N98A, fewer still, although there was no change in fibril morphology.
Fig. 3.

DLS analysis of TTR-mediated inhibition of Aβ aggregation. The increase in the mean hydrodynamic diameter of aggregates of Aβ (28 μM) alone (filled squares), with WT TTR (3.6 μM, empty circles) or with N98A (filled triangles) at 37°C was measured by DLS.
Fig. 4.

NTA of Aβ aggregate growth. Size distribution of Aβ (28 μM) incubated with WT (3.6 μM; black) or with N98A (3.6 μM; gray) for (A) 20 min and (B) 120 min.
Fig. 5.

TEM images of Aβ with TTR mutants. Aβ incubated alone (A) or with WT (B) or N98A (C) at 37°C for 24 h. All scale bars represent 500 nm in length.
Thus, N98A and WT bind similar amounts of Aβ, but N98A is a better inhibitor than WT, a conclusion consistent across several different measures of Aβ aggregation. Inhibition is achieved through both a reduction in the number of fibrillar aggregates and the mean size of aggregates. Those Aβ aggregates that remain are fibrillar; therefore, N98A inhibition of Aβ aggregation does not require a change from fibrillar to amorphous morphology.
N98a structure and stability
To try to determine the basis for N98A's enhanced activity against Aβ, we evaluated its structure and stability. CD spectra of N98A were identical to WT, demonstrating that the alanine mutation did not disrupt the secondary structure (Supplementary Fig. S4). Similarly, Trp fluorescence spectra were identical (Supplementary Fig. S5), indicating no change in tertiary fold (Lai et al., 1996; Hurshman Babbes et al., 2008).
TTR functions as a transport protein for thyroxine, which binds in a hydrophobic channel formed upon assembly of the tetramer (Hamilton and Benson, 2001). The fluorescent dye ANS can bind in this channel, upon which its fluorescence intensity is greatly enhanced and its emission maximum shifts from 515 to 465 nm (Cheng et al., 1977). Therefore, ANS fluorescence is used as an indicator of tetramer assembly and provides a measure of the compactness and stability of the core (Yang et al., 2013). ANS spectra were virtually identical for WT and S100A (Fig. 6), and fluorescence intensity was slightly higher for S23A. In contrast, ANS fluorescence intensity was lower for N98A compared with WT. This decrease could be attributed to a change in the structure or integrity of the thyroxine-binding pocket, an unexpected observation since N98 resides far away from the pocket.
Fig. 6.

ANS fluorescence of TTR alanine mutants. WT (filled circles), S23A (diamonds), S100A (plus symbols) and N98A (filled triangles). TTR concentration was 1 μM. ANS (29 μM) was excited at 370 nm, and emission spectra were collected.
TTR thermodynamic stability can be measured by the shift in Trp fluorescence spectra due to unfolding in the presence of chemical denaturants (Hurshman Babbes et al., 2008). Unfolding curves were identical for S23A and WT, while N98A unfolded at a lower urea concentration (Fig. 7A). TTR unfolding is a two-step process involving coupled steps of tetramer dissociation and monomer unfolding; the unfolding curves are therefore dependent on the concentration of the protein (Hurshman Babbes et al., 2008). While WT showed the expected increase in stability (shift in the unfolding curve toward higher urea) as its concentration was raised from 0.9 to 9 µM, there was much less shift in the unfolding curve with an increase in N98A concentration (Fig. 7B). This result indicates that the decrease in thermodynamic stability of N98A is due primarily to a reduction in the stability of the tetramer against dissociation, rather than in the stability of the monomer fold (Supplementary Table SI and accompanying supplemental text).
Fig. 7.

Thermodynamic stability of TTR mutants measured via urea denaturation. The ratio of Trp fluorescence intensities, a measure of the degree of unfolding, for (A) WT (filled circles), S23A (diamonds) and N98A (filled triangles) at 0.9 μM. (B) Concentration dependence of urea denaturation for WT at 0.9 µM (filled circles), WT at 9 µM (empty circles), N98A at 0.9 µM (filled triangles) and N98A at 9 µM (empty triangles). Curves are fits of the data to an apparent two-state model (Hurshman Babbes et al., 2008).
WT TTR is stable at neutral pH but will aggregate into amyloid fibrils at moderately acidic pH. We induced aggregation (WT or N98A) at pH 4.4 and monitored the growth of aggregates by DLS (Supplementary Fig. S6). The rate of growth of N98A aggregates was slightly faster than for WT, implying the mutant is a less stable tetramer than WT (Zhao et al., 2013). Finally, we measured the thermal stability of TTR tetramers at 80°C using the S-TRAP method (Xia et al., 2012). There was no statistically significant change in the tetramer dissociation rate constant kd for WT, S23A and S100A (Table I). For N98A, kd was 30% greater than WT (P = 0.02).
Table I.
TTR tetramer dissociation rate constants kd at 80°C
| kd (min−1) | |
|---|---|
| WT | 0.30 ± 0.05 |
| S23A | 0.34 ± 0.06 |
| S100A | 0.34 ± 0.06 |
| N98A | 0.40 ± 0.06 |
In summary, N98A exhibited no differences in secondary or tertiary structure compared with WT, and the mutant correctly and fully assembled into tetramers. However, the hydrophobic thyroxine-binding pocket, formed by tetramer assembly, is less compact than WT, and N98A tetramers are less resistant to dissociation, in the presence of chemical denaturants, low pH or high temperature. A subtle modification in tetramer structure and/or stability may explain both N98A's decreased resistance to dissociation and N98A's greater ability to inhibit Aβ aggregation.
Cys10 oxidation, stability and inhibition of Aβ aggregation
The single free cysteine in TTR, Cys10, is frequently oxidized in vivo, with S-cysteinylation and S-sulfonation the most common modifications (Biroccio et al., 2006; Kingsbury et al., 2007; Trenchevska et al., 2011; Poulsen et al., 2012). Oxidation affects TTR stability, with S-cysteinylation destabilizing and S-sulfonation stabilizing, relative to unoxidized WT (Zhang and Kelly, 2003; Kingsbury et al., 2008; Zhao et al., 2013). Given our results with N98A, we asked whether Cys10 oxidation would also affect TTR's ability to inhibit Aβ aggregation.
We oxidized TTR in a controllable manner to afford the S-cysteinylated (C-TTR) and S-TTR isoforms. CD spectra for S-TTR and C-TTR were identical to WT (not shown), indicating that the modifications had no effect on the secondary structure of the protein. Consistent with retention of native quaternary structure, all samples migrated as tetramers on SDS-PAGE in the absence of boiling (not shown). ANS fluorescence spectra for WT TTR and S-TTR were identical (Fig. 8), but for C-TTR, fluorescence intensity was reduced relative to the other proteins. These data confirm that S-sulfonation does not alter the thyroxine-binding cavity, while demonstrating the partial loss of the pocket with S-cysteinylation.
Fig. 8.

ANS fluorescence of oxidized TTR. WT (filled squares), S-TTR (empty circles) and C-TTR (empty triangles). TTR concentration was 1 μM. ANS (29 μM) was excited at 370 nm.
We next tested whether oxidation had any effect on the thermal stability of tetramers using the S-TRAP method (Xia et al., 2012). The dissociation rate constant kD at 80°C was 0.24 ± 0.05 min−1 for S-TTR and 0.52 ± 0.07 min−1 for C-TTR, demonstrating that S-TTR tetramers are kinetically more stable, and C-TTR is less stable, than unoxidized WT (kD = 0.30 ± 0.05 min−1). TTR aggregation was induced under mildly acidic (pH 4.4) conditions, and the kinetics were followed by DLS. The fastest growth rate was observed with C-TTR and the slowest with S-TTR (Supplementary Fig. S7). These results are consistent with the order of kinetic stability determined from S-TRAP and literature data (Zhang and Kelly, 2003; Kingsbury et al., 2008; Zhao et al., 2013).
We compared Aβ binding to TTR, S-TTR and C-TTR and observed no statistically significant differences in Aβ binding to unmodified TTR versus either of the oxidized forms (Supplementary Fig. S8). Finally, we tested whether oxidation altered the ability of TTR to inhibit Aβ aggregation. By ThT fluorescence, S-TTR was less effective, and C-TTR was more effective, than unoxidized WT at reducing Aβ aggregation (Fig. 9). By light scattering, we saw no significant difference between unmodified WT and S-TTR, but C-TTR inhibited Aβ aggregation more effectively than WT (Supplementary Fig. S9). After 3 days of incubation of Aβ at 37°C, we observed by TEM a large number of long (200 nm to >1 micron) unbranched fibrils (Fig. 10A). In samples of Aβ with unoxidized WT or S-TTR (seven-fold excess Aβ), fibrils were similar in morphology (Fig. 10B and C). Aβ fibrils formed in the presence of C-TTR were noticeably fewer in number and shorter, with typical lengths of ∼100 nm, and only a few fibrils >500 nm (Fig. 10D).
Fig. 9.

Effect of oxidation of TTR on inhibition of Aβ aggregation. TTR (3.6 μM) was incubated with Aβ (28 μM) at 37°C for 24 h. ThT was used to assess the extent of fibril formation.
Fig. 10.

TEM images of Aβ with oxidized TTR. Aβ incubated alone (A) or with TTR (B), S-TTR (C) or C-TTR (D) at 37°C for 3 days. All scale bars represent 500 nm in length.
Taken together, the data demonstrate that C-TTR tetramers are less resistant to dissociation, but C-TTR is a better inhibitor of Aβ aggregation. Functionally, S-cysteinylation has very similar effects as the N98A mutation on both TTR structure and its interaction with Aβ.
Aβ-induced destabilization
In SDS-PAGE, WT TTR migrates as tetramers if the samples are not boiled. N98A, S100A and S23A all formed tetramers similar to WT, as expected (Supplementary Fig. S1). N98A displayed an additional weak monomer band at ∼14 kDa. The appearance of monomers on gels is likely an effect of SDS and another indication that N98A tetramers are less resistant to dissociation compared with WT, because by SEC there was no evidence for N98A monomers in PBS (Supplementary Fig. S2).
Previously, we used non-boiling SDS-PAGE to detect that Aβ binding destabilizes TTR tetramers (Yang et al., 2013). We hypothesized that destabilization occurs upon Aβ binding to L82 on the EF helix. To determine if mutation to N98A, or Cys10 oxidation, affected the extent of Aβ-mediated dissociation, we incubated WT, N98A and C-TTR alone or with Aβ for 1–3 days at 37°C and then analyzed the samples by gel electrophoresis. In the absence of Aβ, WT and C-TTR remained fully tetrameric (Supplementary Fig. S10B). When Aβ was incubated with WT or C-TTR, 5–10% of TTR was now in monomer form, consistent with previous results. N98A alone was primarily tetrameric but with a minor monomer band as previously mentioned, the density of which increased after co-incubation with Aβ (Supplementary Fig. S10A). The net increase in TTR monomer due to Aβ was the same for N98A as for WT or C-TTR, suggesting that the destabilization of TTR tetramers caused by the N98A mutation or by S-cysteinylation is independent of the destabilization caused by Aβ binding.
Discussion
There are an unusually large number of naturally occurring TTR mutants (Rowczenio et al., 2014). These mutations are not intrinsically lethal, but some lead to TTR amyloidoses late in life. The disease-associated mutants invariably exhibit reduced tetramer stability, which correlates strongly with an enhanced tendency to self-associate into amyloid fibrils (Benson and Uemichi, 1996; Lashuel et al., 1998; Quintas et al., 1999). Even WT TTR, although generally considered to be a very stable tetrameric protein, will sometimes aggregate into amyloid fibrils, causing senile systemic amyloidosis, a disease that affects the elderly. Given the late-onset nature of these disorders, there has been some speculation that age-related changes in folding fidelity reduce TTR tetramer stability, thereby triggering susceptibility to aggregation.
In this study, we report the discovery of a mutation, N98A, which increases the effectiveness of TTR at inhibiting Aβ aggregation, as measured by several different methods (Figs. 2–5). The asparagine to alanine substitution is in a flexible loop region, distant from the putative Aβ-binding sites, and does not affect secondary or tertiary structure, prevent tetramer assembly, or change the amount of Aβ bound (Supplementary Fig. S3). Alanine substitution in a flexible loop, by itself, is insufficient to improve TTR's effectiveness, as two other mutants (S23A and S100A) were identical to WT in their ability to inhibit Aβ aggregation. Several lines of evidence indicate that the N98A mutation increases TTR susceptibility to tetramer dissociation. Specifically, we observed a shift in ANS binding and a decrease in the stability of tetramers in urea, at low pH, or at high temperature (Figs. 6, 7 and Supplementary Fig. S6; Table I). These changes were not observed for S23A or S100A. We attribute N98A's greater inhibition of Aβ aggregation to its reduced resistance to tetramer dissociation.
Besides mutation, post-translational modification can influence protein folding and stability. In the blood and CSF, a substantial fraction of TTR's lone cysteine (Cys10) is oxidized (Biroccio et al., 2006; Kingsbury et al., 2007; Trenchevska et al., 2011, 2014; Poulsen et al., 2012, 2014). We confirmed that mild oxidation did not affect TTR's secondary structure nor prevent assembly into tetramers and that S-cysteinylation, but not S-sulfonation, modestly reduced the kinetic stability of TTR. Again, we observed a clear connection between reduced tetramer stability and enhanced Aβ inhibition; S-cysteinylated TTR bound the same amount of Aβ as unoxidized WT but was more effective at slowing Aβ aggregation.
These data are useful in considering the mechanism by which TTR binds to Aβ and inhibits its aggregation. There are several possibilities, including: (1) TTR tetramers bind to Aβ monomers, reducing free Aβ concentration and thereby reducing both oligomer formation and further growth of oligomers by monomer addition; (2) TTR tetramers first dissociate into TTR monomers, which bind to Aβ monomers; (3) TTR tetramers bind to Aβ oligomers, sequestering them and preventing growth; and (4) TTR tetramers first dissociate into TTR monomers, which bind to Aβ oligomers.
In order to evaluate the feasibility of each alternative, we estimated the concentration of TTR tetramers and monomers at the experimental conditions used in most of our aggregation assays (3.6 µM TTR as tetramers, 28 µM Aβ as monomers). To obtain this, we used urea denaturation data to estimate equilibrium constants for tetramer dissociation to monomer, and monomer unfolding. (See Supplemental Information.) From this, we estimated that at 3.6 µM, WT contains ∼0.1 µM monomers (∼0.7% dissociated by mass), but N98A contains ∼1 µM monomers (∼7% dissociated by mass).
We consider each alternative in turn. First, TTR tetramers could bind Aβ monomers, thus decreasing the effective Aβ concentration available for aggregation. Li et al. estimated Kd ∼24 µM for this interaction (Li et al., 2013). Using this value and assuming a 1:1 stoichiometry, under our experimental conditions only 1.8 µM Aβ monomer is bound, so the free Aβ concentration is reduced by only 7% (from 28 to 26.2 µM). It is highly unlikely that this minor change in Aβ concentration can account for the significant inhibition of aggregation achieved by WT. N98A tetramer concentration is slightly lower (∼3.3 µM) due to greater dissociation, so the amount of Aβ monomer bound to N98A tetramers would be even less. One would therefore expect N98A to be a poorer inhibitor of Aβ than WT, which is opposite to our observations. Thus, we think TTR tetramer binding to Aβ monomer is the least likely mechanism of interaction leading to inhibition of aggregation.
A second alternative is that TTR monomers bind to Aβ monomers. We are not aware of experimental measurement of Kd for the TTR monomer–Aβ monomer interaction. Nonetheless, if we assume a strong affinity and a 1:1 stoichiometry, we can readily calculate that at most this mechanism could remove 0.1 µM Aβ (0.3% of the pool) with WT and 1 µM Aβ (3.5% of the pool) with N98A. Directionally, this is consistent with our observation that N98A is more effective than WT, but it seems highly unlikely in either case that this very minor reduction in concentration would have any measurable impact on Aβ aggregation rates.
Alternatively, the mode of inhibition could require TTR interaction with Aβ oligomers. We and others previously demonstrated that the amount of Aβ bound to adsorbed TTR was much higher if the Aβ preparation was pre-aggregated (Li et al., 2013; Yang et al., 2013). We do not have a direct measure of Kd for TTR-oligomer binding. Indeed, this is difficult to assay since neither the oligomer size nor the oligomer molar concentration is readily known. Li et al. reported an IC50 of 140 nM for adsorption of TTR, and 80 nM for adsorption of mTTR, to immobilized oligomeric Aβ (Li et al., 2013). If this IC50 is used to estimate Kd, then Aβ oligomer binding to TTR is roughly 150- to 300-fold higher affinity than Aβ monomer binding to TTR. In order to proceed with this analysis, we rather arbitrarily assumed that Aβ is fully associated into hexameric oligomers (28 µM Aβ monomers = 4.6 µM oligomers), with Kd = 140 nM, and we assumed that TTR tetramers bind only Aβ oligomers. With 3.6 µM TTR, ∼70% of the oligomers would be bound to WT, or ∼66% bound to N98A tetramers. It is easy to imagine that this level of sequestration would lead to significant inhibition of Aβ aggregation, thus supporting the hypothesis that TTR must interact with Aβ oligomers to inhibit its aggregation. But this model does not explain why N98A is more effective than WT. If on the other hand only TTR monomers bind Aβ oligomers, and we estimate Kd ∼80 nM, then the WT would bind ∼2% of the Aβ oligomers and N98A would bind ∼20%. This result explains why N98A would be more effective than WT. (If only a fraction of the Aβ is associated into oligomers and the rest remains as monomers, or if the oligomers were larger, then a higher fraction of the Aβ oligomers could be sequestered by WT, and an even higher fraction by N98A).
These analyses lead us to conclude that the most likely mechanism of inhibition of Aβ aggregation involves sequestration of Aβ oligomers, by destabilized TTR tetramers and/or by TTR monomers. Complete dissociation of TTR into monomers may not be required for strong binding to Aβ oligomers. For both N98A and C-TTR, there is reduced ANS binding, and a decrease in tetramer resistance to dissociation at low pH or high temperature, or in urea. These data are indicative of a change in the structure or packing of the tetramer (specifically a less compact hydrophobic cavity) that could allow access of relatively large Aβ oligomers to the inner binding pocket. The inner hydrophobic pocket of the tetramer forms a β-cylinder that is ∼0.8 nm in diameter and 5 nm in length (Blake et al., 1978), for a volume of ∼2.5 nm3. The entrance of the ‘cylinder’ can be seen in Fig. 1C. We calculated, using a density of 1.4 g/cm3, that an Aβ monomer occupies ∼5 nm3 and an Aβ hexamer ∼30 nm3. Thus, full accommodation of an Aβ monomer or oligomer in the inner pocket of the native WT TTR tetramer is problematic. Subunits in TTR undergo exchange, wherein monomers dissociate and then recombine into different tetramers, and the rate of subunit exchange is much faster for less stable mutants (Keetch et al., 2005; Wiseman et al., 2005). Aβ oligomer access to the interior site could be achieved during this exchange. We showed previously that Aβ partially competes with re-assembly of WT TTR monomers into tetramers (Du and Murphy, 2010). Incorporation of Aβ oligomers into TTR during exchange should be more efficient for N98A or C-TTR, with their faster dissociation kinetics, than for WT. In any case, our data support the hypothesis that TTR inhibition of Aβ aggregation is mediated by the interaction of aggregation-prone Aβ oligomers with the interior core of TTR.
Additional mechanisms may be in play. We have previously proposed that Aβ binding to TTR tetramers at the exterior EF helix destabilizes TTR quaternary structure (Yang et al., 2013), leading to either dissociation and/or greater access to the interior hydrophobic pocket. Data presented here (Supplementary Fig. S10) repeat this observation with WT and show that N98A and C-TTR also undergo further destabilization upon Aβ binding. This mechanism provides a means by which the effective TTR monomer concentration is increased in the presence of Aβ. Finally, we note that binding of Aβ to TTR was assayed using immobilized TTR. Inhibition of Aβ aggregation, on the other hand, was measured in solution. Immobilization typically stabilizes proteins, which could explain how N98A and C-TTR could bind similar quantities of Aβ as WT, but inhibit aggregation more effectively.
Why does the N98A mutation destabilize the tetramer? A preliminary molecular dynamic simulation of the dimer did not reveal any signs of perturbation of the folded structure due to the N98A mutation (not shown). To gain insight, we applied an alanine mutagenesis bioinformatics algorithm to TTR, which predicts ‘hot spots’ where mutations to alanine would significantly disrupt protein–protein interaction (Zhu and Mitchell, 2011). A strong hot spot was identified involving residues 92–95 in strand F; mutations in this hot spot are predicted to significantly disrupt the extensive hydrogen bonding between two monomers. N98 is at the transition from strand F to the FG loop. Since asparagine is a weak beta-breaker but alanine is a weak beta-former, it is possible that the N98A mutation extends strand F, distorts the FG loop and strains monomer–monomer interactions. Interestingly, S100 is only two residues C-terminal to N98 in the same FG loop, but mutation here had no net effect on TTR stability or inhibition of Aβ aggregation.
Cys10 is on the flexible N-terminus, with the side chain oriented toward H56 and G57 on the DE loop. S-sulfonation is believed to stabilize TTR via hydrogen bond interactions of the sulfite oxygens with H56 and G57, while S-cysteinylation decreases stability relative to WT because the bulky Cys group causes steric interference (Gales et al., 2007).
It is interesting to note that the modifications that make TTR less effective at one of its natural functions (thyroxine binding, as measured by ANS fluorescence), and slightly more amyloid-prone (as measured by acid-induced aggregation), are also the changes that make it more effective at inhibiting Aβ aggregation. Whether via oxidation, ligand binding (White and Kelly, 2001), or mutation, several natural avenues can subtly modify TTR tetramer stability, without wholesale disruption of protein structure. Our results suggest that these modest changes could significantly enhance or inhibit TTR's ability to slow Aβ aggregation and, presumably, to inhibit Aβ toxicity. The outcome is a delicate balance between small losses in TTR stability that might be advantageous for clearing Aβ, without hindering TTR's normal function as a transport protein or increasing the likelihood of developing TTR amyloidoses.
Supplementary data
Funding
This work was supported by National Institutes of Health Grant R01AG033493.
Supplementary Material
Acknowledgements
The authors thank Dr Julie Mitchell for the alanine mutagenesis analysis and the molecular dynamic simulation, Xiaomeng Lu for nanoparticle tracking data collection, and Claire Brickson for urea denaturation data collection.
References
- Bateman D., Tycko R., Wickner R. (2011) Biophys. J., 101, 2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson M.D., Uemichi T. (1996) Amyloid, 3, 44–56. [Google Scholar]
- Biroccio A., del Boccio P., Panella M. et al. (2006) Proteomics, 6, 2305–2313. [DOI] [PubMed] [Google Scholar]
- Blake C.C.F., Burridge J.M., Oatley S.J. (1978) Biochem. Soc. Trans., 6, 1114–1118. [DOI] [PubMed] [Google Scholar]
- Buxbaum J.N., Ye Z., Reixach N. et al. (2008) Proc. Natl. Acad. Sci. USA, 105, 2681–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S.Y., Pages R.A., Saroff H.A., Edelhoch H., Robbins J. (1977) Biochemistry, 16, 3707–3713. [DOI] [PubMed] [Google Scholar]
- Cho P., Joshi G., Johnson J., Murphy R.M. (2014) ACS Chem. Neurosci., 5, 542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.H., Leight S.N., Lee V.M.Y., Li T., Wong P.C., Johnson J.A., Saraiva M.J., Sisodia S.S. (2007) J. Neurosci., 27, 7006–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopeit T., Hortschansky P., Schroeckh V., Gührs K., Zandomeneghi G., Fändrich M. (2005) Protein Sci., 14, 2125–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary J.P., Walsh D.M., Hofmeister J., Shankar G., Kuskowski M.A., Selkoe D., Ashe K. (2005) Nat. Neurosci., 8, 79–84. [DOI] [PubMed] [Google Scholar]
- Cloe A.L., Orgel J., Sachleben J., Tycko R., Meredith S.C. (2011) Biochemistry, 50, 2026–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa R., Goncalves A., Saralva M., Cardoso I. (2008) FEBS Lett., 582, 936–942. [DOI] [PubMed] [Google Scholar]
- Du J., Murphy R.M. (2010) Biochemistry, 49, 8276–8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J., Cho P.Y., Yang D.T., Murphy R.M. (2012) Protein Eng. Des. Sel., 25, 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipe V., Hawe A., Jiskoot W. (2010) Pharm. Res., 27, 796–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss T., Wiseman R., Kelly J.W. (2005) Biochemistry, 44, 15525–15533. [DOI] [PubMed] [Google Scholar]
- Gales L., Saraiva M.J., Damas A.M. (2007) Biochem. Biophys. Acta, 1774, 59–64. [DOI] [PubMed] [Google Scholar]
- Giunta S., Valli M.B., Galeazzi R., Fattoretti P., Corder E.H., Galeazzi L. (2005) Clin. Biochem., 38, 1112–1119. [DOI] [PubMed] [Google Scholar]
- Hamilton J.A., Benson M.D. (2001) Cell. Mol. Life Sci., 58, 1491–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J., Steinrauf L., Braden B., Liepnieks J., Benson M.D., Holmgren G., Sandgren O., Steen L. (1993) J. Biol. Chem., 268, 2416–2424. [PubMed] [Google Scholar]
- Hammarstrom P., Schneider F., Kelly J.W. (2001) Science, 293, 2459–2462. [DOI] [PubMed] [Google Scholar]
- Hawe A., Sutter M., Jiskoot W. (2008) Pharm. Res., 25, 1487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht M., Bromberg Y., Rost B. (2013) J. Mol. Biol., 425, 3937–3948. [DOI] [PubMed] [Google Scholar]
- Hurshman Babbes A.R., Powers E.T., Kelly J.W. (2008) Biochemistry, 47, 6969–6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Smith C.S., Petrassi H.M., Hammarstrom P., White J., Sacchettini J., Kelly J.W. (2001) Biochemistry, 40, 11442–11452. [DOI] [PubMed] [Google Scholar]
- Keetch C., Bromley E., McCammon M.G., Wang N., Christodoulou J., Robinson C. (2005) J. Biol. Chem., 280, 41667–41674. [DOI] [PubMed] [Google Scholar]
- Kingsbury J.S., Théberge R., Karbassi J.A., Lim A., Costello C.E., Connors L.H. (2007) Anal. Chem., 79, 1990–1998. [DOI] [PubMed] [Google Scholar]
- Kingsbury J.S., Laue T.M., Klimtchuk E.S., Théberge R., Costello C.E., Connors L.H. (2008) J. Biol. Chem., 283, 11887–11896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs M., Bromley E., Donald A. (2005) J. Struct. Biol., 149, 30–37. [DOI] [PubMed] [Google Scholar]
- Lai Z., Colon W., Kelly J.W. (1996) Biochemistry, 35, 6470. [DOI] [PubMed] [Google Scholar]
- Lashuel H.A., Lai Z., Kelly J.W. (1998) Biochemistry, 37, 17851. [DOI] [PubMed] [Google Scholar]
- Li X., Masliah E., Reixach N., Buxbaum J. (2011) J. Neurosci., 31, 12483–12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Zhang X., Ladiwala A., Du D.G., Yadav J., Tessier P., Wright P., Kelly J.W., Buxbaum J. (2013) J. Neurosci., 33, 19423–19433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Hou J., Du J., Chumanov R.S., Xu Q., Ge Y., Johnson J.A., Murphy R.M. (2009) Protein Eng. Des. Sel., 22, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overk C.R., Masliah E. (2014) Biochem. Pharmacol., 88, 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaninathan S.K., Mohamedmohaideen N.N., Snee W.C., Kelly J.W., Sacchettini J.C. (2008) J. Mol. Biol., 382, 1157–1167. [DOI] [PubMed] [Google Scholar]
- Planté-Bordeneuve V., Said G. (2011) Lancet Neurol., 10, 1086–1097. [DOI] [PubMed] [Google Scholar]
- Poulsen K., Bahl J.M.C., Tanassi J.T., Simonsen A.H., Heegaard N.H.H. (2012) Methods, 56, 284–292. [DOI] [PubMed] [Google Scholar]
- Poulsen K., Bahl J.M.C., Simonsen A.H., Hasselbalch S.G., Heegaard N.H.H. (2014) Clin. Proteomics, 11, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas A., Saraiva M., Brito R. (1999) J. Biol. Chem., 274, 32943–32949. [DOI] [PubMed] [Google Scholar]
- Rowczenio D.M., Noor I., Gilmore J.D. et al. (2014) Hum. Mutat., 35, E2403–E2412. [DOI] [PubMed] [Google Scholar]
- Schwarzman A.L., Gregori L., Vitek M.P. et al. (1994) Proc. Natl. Acad. Sci. USA, 91, 8368–8372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein T., Johnson J. (2002) J. Neurosci., 22, 7380–7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein T., Anders N., Decarli C., Chan S., Mattson M.P., Johnson J. (2004) J. Neurosci., 24, 7707–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenchevska O., Kamcheva E., Nedelkov D. (2011) Proteomics, 11, 3633–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenchevska O., Phillips D., Nelson R., Nedelkov D. (2014) PLoS One, 9, e100713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivekanandan S., Brender J., Lee S., Ramamoorthy A. (2011) Biochem. Biophys. Res. Commun., 411, 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark P., Sletten K., Johansson B., Cornwell G.G. (1990) Proc. Natl. Acad. Sci. USA, 87, 2843–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J., Kelly J.W. (2001) Proc. Natl. Acad. Sci. USA, 98, 13019–13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman R.L., Green N.S., Kelly J.W. (2005) Biochemistry, 44, 9265–9274. [DOI] [PubMed] [Google Scholar]
- Wisniewski T., Goñi F. (2014) Biochem. Pharmacol., 88, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia K., Zhang S., Bathrick B., Liu S., Garcia Y., Colon W. (2012) Biochemistry, 51, 100–107. [DOI] [PubMed] [Google Scholar]
- Yang D., Joshi G., Cho P., Johnson J., Murphy R.M. (2013) Biochemistry, 52, 2849–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Lu X., Fan Y., Murphy R.M. (2014) AICHE J., 60, 1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Kelly J.W. (2003) Biochemistry, 42, 8756. [DOI] [PubMed] [Google Scholar]
- Zhao L., Buxbaum J., Reixach N. (2013) Biochemistry, 52, 1913–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., Mitchell J.C. (2011) Proteins Struct. Funct. Bioinforma., 79, 2671–2683. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.