

Summary
Covalent addition of a methyl group to the adenosine N6 (m6A) is an evolutionarily conserved and common RNA modification that is thought to modulate several aspects of RNA metabolism. While the presence of multiple m6A editing sites on diverse viral RNAs was reported starting almost 40 years ago, how m6A editing affects virus replication has remained unclear. Here, we used photo-crosslinking-assisted m6A sequencing techniques to precisely map several m6A editing sites on the HIV-1 genome and report that they cluster in the HIV-1 3’ untranslated region (3'UTR). Viral 3'UTR m6A sites or analogous cellular m6A sites strongly enhanced mRNA expression in cis by recruiting the cellular YTHDF m6A “reader” proteins. Reducing YTHDF expression inhibited, while YTHDF overexpression enhanced, HIV-1 protein and RNA expression, and virus replication in CD4+ T cells. These data identify m6A editing, and the resultant recruitment of YTHDF proteins, as major positive regulators of HIV-1 mRNA expression.
Introduction
Like proteins and DNA, RNA is subject to a number of covalent modifications that can impact its function and post-transcriptionally modified nucleotides have indeed been detected on eukaryotic mRNAs (Carlile et al., 2014; Dominissini et al., 2012; Dominissini et al., 2016; Meyer et al., 2012; Schwartz et al., 2014; Squires et al., 2012). Of these, the N6-methyladenosine (m6A) modification is the most common, with an average of ~3 m6A addition sites per mRNA and with ~25% of all cellular mRNAs containing generally multiple m6A residues (Desrosiers et al., 1975; Dominissini et al., 2012; Meyer et al., 2012). The importance of m6A is underlined by the fact that this modification is evolutionarily conserved from fungi to plants and animals, and that global inhibition of m6A addition is embryonic lethal in plants, insects and mammals (Meyer and Jaffrey, 2014; Yue et al., 2015).
The post-transcriptional addition of m6A to mRNAs occurs predominantly in the nucleus and is mediated by a heterotrimeric protein complex consisting of the two methyltransferase-like (METTL) enzymes METTL3 and METTL14 and their co-factor Wilms tumor 1-associated protein (WTAP) (Liu et al., 2014; Meyer and Jaffrey, 2014; Yue et al., 2015). This complex specifically methylates A residues in the consensus sequence (G/A/U)(G>A) m6AC (U/C/A), although only ~15% of sites that have this consensus are actually modified and the level of modification at any given site can vary significantly. In addition to these m6A “writers”, mammals also encode two RNA demethylases or “erasers” called ALKBH5 (α-ketoglutamarate-dependent dioxygenase homologue 5) and FTO (fat mass and obesity associated), which are found predominantly in the nucleus or cytoplasm, respectively (Jia et al., 2011; Zheng et al., 2013). Finally, the function of m6A residues on mRNAs is thought to be primarily mediated by three related cytoplasmic “reader” proteins called YTH-domain containing family 1 (YTHDF1), YTHDF2 and YTHDF3 (Meyer and Jaffrey, 2014; Wang et al., 2014; Wang et al., 2015; Yue et al., 2015). The three YTHDF proteins all contain a conserved carboxy-terminal YTH domain that binds m6A and a more variable amino-terminal effector domain of unclear function.
While the m6A modification of mRNAs is therefore well established and has been suggested to modulate several aspects of RNA metabolism (Meyer and Jaffrey, 2014; Yue et al., 2015), exactly how m6A editing regulates mRNA function remains largely unclear. Importantly, m6A modifications appear to be ubiquitous on mRNAs expressed by viruses that replicate in the nucleus, including SV40, the related retroviruses avian sarcoma virus and Rous sarcoma virus (RSV), adenovirus and influenza A virus (IAV) (Dimock and Stoltzfus, 1977; Kane and Beemon, 1985; Krug et al., 1976; Lavi and Shatkin, 1975; Sommer et al., 1976). As viruses invariably rapidly evolve to maximize their replication potential, and given that it would be simple to select for viral mutants that lack consensus m6A modification sites, this implies that the m6A modification of viral mRNAs enhances viral replication by enhancing some aspect(s) of mRNA function.
Despite the fact that the identification of m6A on viral mRNAs dates back over 40 years, no report has shown that m6A affects any aspect of viral mRNA function. Here, we first precisely map m6A modification sites on the RNA genome of human immunodeficiency virus 1 (HIV-1) and show that different HIV-1 isolates contain from four to six m6A clusters at the extreme 3’ end of the viral genome, i.e., primarily in the 3’ untranslated regions (3'UTRs) of the various HIV-1 mRNAs. We further present evidence that these 3'UTR m6A residues enhance HIV-1 gene expression and replication by increasing the steady state level of viral mRNA expression. Finally, we show that HIV-1 is sensitive to the level of YTHDF2 expression in infected T cells, demonstrating enhanced replication when YTHDF2 was overexpressed and strongly reduced replication when the YTHDF2 gene was knocked out by DNA editing. These data demonstrate that the m6A modification of HIV-1 plays a key role in promoting its replication and identifies this RNA modification as a potential target for antiviral drug development.
Results
Mapping m6A sites on the HIV-1 genome
Modification of adenosines to m6A on viral mRNAs has been reported for a range of viruses that replicate in the nucleus; however, with the exception of RSV, where seven m6A addition sites were mapped using biochemical approaches (Kane and Beemon, 1985), the location of individual m6A residues has remained unknown. To map m6A modifications in HIV-1, we used the previously described photo-crosslinking-assisted m6A sequencing (PA-m6A-seq) technique (Chen et al., 2015) to identify m6A residues on the HIV-1 genome in infected human CD4+ CEM-SS T cells. For this experiment, we pulsed HIV-1 infected T-cells with the nucleoside 4-thiouridine (4SU), isolated total poly(A)+ RNA (Fig. 1A), bound this RNA with an m6A-specific antibody and crosslinked the antibody to the RNA (Fig. 1B). RNA fragments bound to the m6A antibody were then reverse transcribed and sequenced. We identified several m6A sites that were all located in the 3’ most ~1.4kb of the ~9.2kb HIV-1 RNA genome (Fig. 1C). Expansion of this region of the HIV-1 genome (Fig.1D) reveals three major m6A peaks located in the overlap region between the env gene and the second coding exon of rev, in the “U3” region of the LTR, particularly in the conserved NF-κB binding sites, and finally in the “R” region of the LTR coincident with the TAR (trans-activation response) RNA hairpin, though several other minor m6A peaks were also visible.
Figure 1. m6A site discovery in HIV-1 isolate NL4-3.

(A) Overview of the general PAR-CLIP experimental design. (B) A schematic of the PA-m6A-seq and PAR-CLIP site discovery strategy is depicted. A typical transcript containing an m6A editing site is shown with an incorporated adjacent 4SU molecule (orange star). Upon binding, the m6A specific antibody or a host YTHDF reader protein is crosslinked to the 4SU. T>C transitions are generated from crosslinked 4SU during reverse transcription/cDNA synthesis. (C) PA-m6A-seq and PAR-CLIP were performed 64 h after infection with VSV-G pseudotyped HIV-1 strain NL4-3. Shown are the entire genome coverage tracks for PA-m6A-seq in CEM-SS cells, and then the FLAG-GFP control and YTHDF1, 2 and 3 tracks in 293T cells. (D) Expanded view of the 3'UTR region of HIV-1 containing the detected m6A editing sites. This ~1.4 kb region extends from the second coding exon of Rev to the end of the R region. Red/Blue bars indicate sites of T>C conversions. Reads are aligned to an HIV-1 genome that begins with the U5 region and ends with U3-R to avoid repeat alignments. The PA-m6A-seq has a Y axis of 0-200 reads, and all others are depicted with Y axes of 0-900 reads. For related data, see Fig. S1.
The function of m6A sites is primarily mediated by the cytoplasmic YTHDF proteins, though other potential nuclear or cytoplasmic m6A binding proteins have been reported (Meyer and Jaffrey, 2014; Meyer et al., 2015). To determine whether any of the m6A sites on the HIV-1 genome mapped using PA-m6A-seq are bound by one or more of the three YTHDF proteins in living cells, and hence likely to be functionally relevant, we generated clones of the human cell line 293T engineered to express FLAG-tagged versions of green fluorescent protein (GFP), YTHDF1, YTHDF2 or YTHDF3 (Fig. S1A). These cells were infected with a pseudotyped stock of the HIV-1 laboratory isolate NL4-3 (Adachi et al., 1986), cultured for 48 h and then incubated with 4SU for a further 16 h (Fig.1A). At this point, the cells were subjected to photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP), using a monoclonal anti-FLAG antibody, followed by deep sequencing (Fig. S1B) (Hafner et al., 2010).
Analysis of recovered reads detected T to C mutations, which are characteristic of crosslinked 4SU residues that have been subjected to reverse transcription, in 45-60% of all viral reads obtained from the three FLAG-YTHDF expressing clones but in <5% of the reads obtained from the clone expressing FLAG-GFP (Fig. S1C), consistent with known ability of all three YTHDF proteins, but not GFP, to bind RNA. Alignment of reads bearing T to C mutations to the human and HIV-1 genome (reads lacking T to C mutations were discarded) revealed that the HIV-1-specific reads showed a mean length of ~25 bp (Fig. S1D), which permitted their unequivocal assignment to the viral genome. Further analysis revealed that the HIV-1-specific reads for all three FLAG-tagged YTHDF proteins mapped to four binding clusters located in the 3’ ~1.4 kb of the HIV-1 RNA genome, three of which coincided with the three major sites of m6A addition mapped by in vitro crosslinking to an m6A specific antibody, as described above (Figs. 1C and D). Specifically, we noted binding clusters occupied by all three YTHDF proteins in the HIV-1 env/rev overlap; in the LTR NF-κB repeats and finally in the LTR R region. Of interest, all three YTHDF proteins also bound a site in the HIV-1 nef gene that was only detected as a minor binding site by PA-m6A-seq (Fig. 1D). None of the other, minor binding sites detected by PA-m6A-seq were bound at significant levels by any of the three YTHDF proteins. Therefore we can conclude that HIV-1 transcripts are modified by m6A editing at several sites located in the 3’ ~1.4kb of the viral RNA genome and that these m6A sites are bound in living cells by all three YTHDF effector proteins.
To determine if the m6A modification sites mapped on the NL4-3 laboratory isolate were conserved in primary HIV-1 isolates BaL and JR-CSF (Cann et al., 1990; Hwang et al., 1991), we repeated the PAR-CLIP analysis in the 293T clones expressing either FLAG-YTHDF1 or FLAG-YTHDF2 using pseudotyped stocks of BaL or JR-CSF. Analysis of the BaL isolate showed that all four clusters identified in NL4-3, in the env/rev overlap, in nef, in U3 and in TAR (Fig. 1D), were conserved in BaL and again bound by both YTHDF1 and YTHDF2 (Fig. 2A). However, we also noted a novel, intense binding site for both YTHDF proteins in the 3’ segment of nef that overlaps with the LTR U3 region (Fig. 2A, labeled BaL1). Sequence analysis revealed that this new site coincides with a consensus m6A modification site present in BaL (5-GGA*CC-3’) that is lacking in NL4-3 and JR-CSF (Fig. 2B).
Figure 2. m6A site discovery using primary HIV-1 isolates BaL and JR-CSF.

(A) YTHDF1 or YTHDF2 PAR-CLIP binding clusters were mapped for HIV-1 isolates NL4-3, BaL and JR-CSF for the 3’ region of the HIV-1 genome from the second exon of Rev to the end of the R region, as indicated. The three novel YTHDF protein binding clusters discovered for these two viruses are annotated below the relevant track. The Y axes for these alignments are NL4-3: 0-900 reads, BaL: 0-2000 reads, and JR-CSF: 0-1500 reads. (B) Alignment of two segments from the NL4-3 and BaL genome, with a putative novel methyl receptor adenosine present in BaL shown in red. (C) Similar to panel B, except aligning two regions of NL4-3 and JR-CSF, with the two novel methyl acceptor adenosines present in JR-CSF indicated. For related data, see Figs. S1 and S4.
The analysis of m6A editing sites in JR-CSF produced a similar result. Specifically, both YTHDF1 and YTHDF2 bound to the four m6A clusters previously identified in NL4-3 (Fig. 2A), while the novel m6A site identified in BaL was, as expected, lacking. However, in JR-CSF we identified two additional, novel m6A modification sites in the rev/env overlap region (JR-CSF1) and in the nef/U3 overlap region (JR-CSF2). The novel site in the rev/env overlap region again coincided with a novel “A” residue, present in JR-CSF but not NL4-3 or BaL, that forms part of an m6A consensus editing site (5’-GGA*CA-3’) while, in the case of the LTR U3 region target, the novel m6A site appeared to arise due to a change from a weaker m6A consensus sequence (5’-GAA*CU-3’) to a stronger consensus (5’-GGA*CU-3’) (Fig. 2C). In fact, we did detect a low level of YTHDF binding to this site in both NL4-3 and BaL, suggesting that this sequence may be subject to a low level of m6A modification in these viral strains (Fig. 2A). In conclusion, these data demonstrate that all four m6A clusters identified in the NL4-3 strain of HIV-1 are conserved in the primary isolates BaL and JR-CSF while BaL has also acquired one, and JR-CSF two, novel m6A sites, each lacking in the other two virus isolates. Of particular note is the fact that all YTHDF protein binding sites, including the novel ones present in BaL and JR-CSF, were located in the 3’ ~1.4 kb of all three virus strains, with no m6A sites being detected in the first ~7.8 kb of the genome (Figs. 1D and 1E, Fig. 2A). It has recently been proposed that m6A sites on cellular mRNAs are concentrated in the 3'UTR (Ke et al., 2015) and indeed the m6A sites identified in HIV-1 would be present in the 3’ UTR of all viral mRNAs (the U3/NF-kB and TAR clusters), in the 3’ UTR of all viral mRNAs except nef mRNAs (the nef and U3/nef overlap clusters) or in the 3'UTR of the viral Gag, Gag/Pol, Vif, Vpr, Rev and Tat mRNAs (the env/rev overlap clusters).
The introduction of m6A into 3'UTRs enhances mRNA function
While the YTHDF and m6A antibody-specific binding clusters detected in the NL4-3 strain of HIV-1 are <40 nt each (Fig. 1D), they nevertheless each contain two or three potential m6A sites that match the minimum m6A consensus sequence 5’-RA*C-3’ (Fig. 3A to D). To test the effect of these clusters on mRNA function in cis, we constructed two Renilla luciferase (RLuc)-based indicator plasmids containing either the entire ~1.4 kb region from the 3’ end of NL4-3, that encompasses all four NL4-3 m6A clusters, or containing just the most 3’ sequence, encompassing the U3/NF-kB and TAR clusters (Fig. 1D), in either a wildtype form or with all of the potential m6A modification sites listed in Fig. 3 mutated to G. These vectors contain all viral sequences required for poly(A) addition at the R/U5 junction and therefore are predicted to contain the same 3'UTRs as HIV-1 mRNAs. As shown in Fig. 4A, both indicator plasmids expressed significantly (p<0.01) higher levels of RLuc protein in transfected 293T cells when the wildtype HIV-1 sequence was utilized, compared to a similar m6A-deficient viral sequence. This effect was particularly notable for the indicator plasmid bearing the shorter viral 3'UTR characteristic of the viral early gene products Tat, Rev and Nef. Importantly, qRT-PCR analysis of the steady state level of the RLuc mRNA transcribed from these indicator plasmids (Fig. 4B) revealed that the viral 3'UTR m6A sites also exerted a very similar positive effect on the level of RLuc mRNA expression, thus suggesting that the increase in RLuc protein expression (Fig. 4A) is due to an equivalent change in the steady state level of RLuc mRNA (Fig. 4B). A closely similar positive effect of the HIV-1 3'UTR m6A editing sites on mRNA expression and function was also observed in the CD4+ human T-cell line CEM-SS (Figs. 4C-D).
Figure 3. Consensus m6A editing sites mapped to the NL4-3 genome.
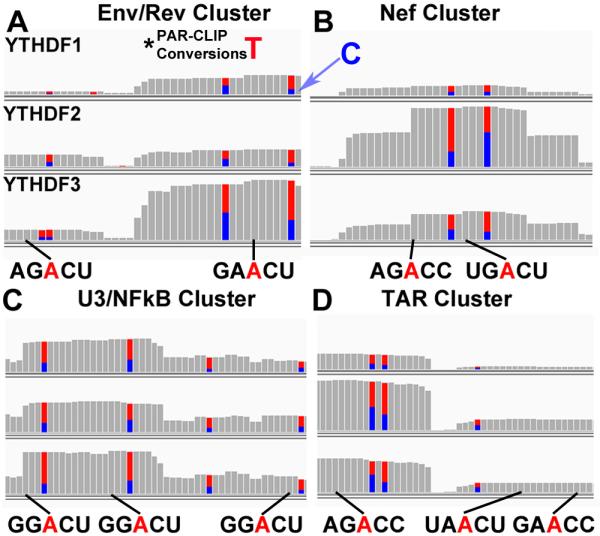
Shown in (A-D) are the 4 mapped YTHDF PAR-CLIP clusters present in NL4-3 with consensus m6A sites indicated. Adjacent T to C conversions, that result from 4SU photo-crosslinking (T=blue, C=red), are indicated. Below are the potential viral m6A editing sites shown in red, with a black line indicating the nucleotide position in the YTHDF binding cluster relative to the mutated T residue. This figure identifies all sites with a minimal (5’-RAC-3’) m6A consensus but this does not demonstrate that all of these A residues are actually modified. For related data, see Fig. S4.
Figure 4. 3'UTR m6A sites boost mRNA abundance and protein expression.

Dual luciferase indicators were constructed in which the 3'UTR of RLuc in psiCheck2 was replaced by HIV-1 3'UTR sequences in either a wildtype form or with the m6A sites listed in Fig. 3 replaced by G residues. The “HIV 3’ UTR” construct contains the entire ~1.4 kb 3'UTR region of HIV-1, encompassing all four m6A clusters, extending from the second coding exon of Rev through the viral poly(A) addition site. The U3/NF-kB/TAR indicator, which contains the viral 3’ UTR from 5’ of the LTR NF-kB repeats again through the viral poly(A) addition site, retains only the U3/NF-kB and TAR m6A sites. (A) The indicators were transfected into 293T cells and RLuc and internal control FLuc levels assayed at 48 h post-transfection. (B) This transfection was performed in 293T cells, as described in (A). Steady state transcript abundance was measured by qRT-PCR for both the internal control FLuc and the m6A cluster-containing RLuc mRNAs. RLuc mRNA abundance is shown normalized first to endogenous GAPDH mRNA and then to the control FLuc mRNA. (C) Similar to (A), except these luciferase assays were performed in transfected CEM-SS T-cells. (D) Similar to (B) except that this qRT-PCR analysis of FLuc and RLuc mRNA expression levels was performed in transfected CEM-SS T cells. (E) Cellular YTHDF PAR-CLIP clusters with 1, 2, 5, or 6 predicted m6A editing sites were compared using the same RLuc indicator assay as described in (A) and (C). These clusters were cloned into the 3'UTR of RLuc in a wildtype or mutant form, lacking m6A editing sites and RLuc activity determined. (F) YTHDF fusion proteins were constructed where the carboxy-terminal m6A binding domain was replaced with the MS2 coat protein, and these were compared to a negative control GFP-MS2 fusion after co-transfection into 293T cells along with a psiCHECK2 dual luciferase vector with and without MS2 binding sites inserted into the RLuc 3'UTR. (A through F). Average of from three to six independent experiments with SD indicated. For related data, see Fig. S2.
While we have mapped the m6A editing sites on the NL4-3 genome in the context of a virus infection (Fig.1), it could be argued that these same m6A sites might be inactive in the context of an indicator plasmid. To test this possibility, we performed PAR-CLIP on 293T cells transfected with wildtype or m6A-deficient versions of the U3/NF-κB/TAR indicator plasmid. As shown in Fig.S2A, we observed a high level of m6A editing of the LTR “R” region in the wildtype context and no m6A editing anywhere in either the HIV-1-derived 3'UTR sequence, or the RLuc open reading frame, in the mutated indicator plasmid lacking the viral m6A sites. Therefore, we can conclude that the differences in gene expression observed in Fig. 4 indeed reflect the presence or absence of 3’ UTR m6A modifications in cis.
Previously, m6A editing has been proposed to either enhance or decrease mRNA stability (Dominissini et al., 2012; Wang et al., 2014) raising the possibility that m6A editing might exert different effects dependent on, for example, RNA sequence context. To address the generalizability of the data shown in Figs. 4A through D, which show a clear enhancing effect of 3'UTR m6A editing sites of viral origin, we extended our analysis to cellular m6A editing sites that we had identified in human mRNAs in the course of our PAR-CLIP analyses (Figs. 1 and 2). Specifically, we identified m6A editing sites bound by the three YTHDF proteins in TBP (1 cluster), RHOB (2 clusters), GPBP1 (2 clusters), ASH1L (2 clusters), UBE2L3 (2 clusters), c-jun (5 clusters) and BTBD7 (6 clusters). These cellular m6A editing sites were cloned into the 3'UTR of the RLuc gene, either individually or together depending on their separation in their normal sequence context, as either the wildtype sequence or with the edited A residues mutated to G. As may be observed (Fig. 4E), in all nine vectors analyzed we observed significantly lower RLuc activity when the mutant form of these human RNA sequences, lacking a consensus m6A addition site(s), was tested, when compared to the wildtype sequence. The observed effect was generally greater the more m6A residues were present (Fig. 4E).
While the data shown in Fig. 4 are presented to show a loss of RLuc activity when comparing the mutant form, lacking m6A sites, to the wildtype form of the inserted 3'UTR, this does not, in fact, indicate inhibition of RLuc expression by 3'UTRs lacking m6A sites. Rather, these data reflect the activation of RLuc expression by m6A containing 3'UTRs. This is more obvious when these data are normalized to a control psiCheck2 plasmid lacking any inserted 3'UTR sequences. Thus, 3'UTR sequences that retain m6A editing sites substantially enhance RLuc expression in cis while the mutated 3'UTRs lacking m6A have no significant positive or negative effect (Figs. S2B and S2C).
As noted above, m6A sites are thought to function by recruiting one or more of the YTHDF proteins to the mRNA. Currently, it remains unclear whether these three proteins are functionally distinct, though our data indicate that all three YTHDFs are recruited to each of the m6A editing sites identified on the HIV-1 genome (Figs. 1 and 2) and we also observed this for the cellular m6A editing sites listed in Fig. 4C (data not shown). If this hypothesis is correct, then tethering of the effector domain of YTHDF1, 2 and/or 3 should reproduce the enhancing effect of 3'UTR m6A editing sites on RLuc expression seen in Figs. 4A and C. To test this hypothesis, we expressed fusion proteins consisting of the amino-terminal effector domains of YTHDF1, 2 and 3 linked to the bacteriophage MS2 coat protein. All three FLAG-tagged fusion proteins were expressed at levels comparable to the FLAG-tagged parental YTHDF proteins and showed the same cytoplasmic localization (Fig. S2D). When tested in combination with an RLuc indicator plasmid containing MS2 coat protein binding sites inserted into the 3'UTR, we saw a 3-4-fold enhancement in RLuc expression with all three YTHDF-MS2 fusions when compared to a control GFP-MS2 fusion (Fig. 4F), thus arguing that recruitment of YTHDF proteins to m6A editing sites mediates the positive effect of 3'UTRs containing m6A sites.
YTHDF protein overexpression enhances HIV-1 replication
While the experiments presented in Fig. 4 argue that the recruitment of cellular YTHDF proteins to m6A residues present in the 3'UTR of mRNAs can substantially enhance mRNA expression and protein production, these data do not address whether addition of m6A also enhances HIV-1 replication. Because the HIV-1 m6A editing sites listed in Fig. 3 are located in regions of the viral genome that play key roles in viral replication, e.g., the env/rev gene overlap, the NF-κB repeats and TAR, it is technically difficult to mutate these m6A sites without affecting other cis-acting RNA elements and any reduction in viral replication would therefore be very difficult to interpret.
As an alternative approach, we therefore asked whether overexpression of YTHDF1, YTHDF2 or YTHDF3 might enhance HIV-1 gene expression, presumably by facilitating the recruitment of these proteins to viral m6A editing sites. As shown in Figs. 5A and B, overexpression of the YTHDF proteins substantially enhanced the expression of the HIV-1 Nef, Tat and Rev mRNAs as well as the full-length viral genomic RNA (gRNA) at both 24 h and 48 h post-infection (hpi) in human 293T cells. This effect was especially marked when analyzing gRNA expression at 48 hpi, with an ~6-fold enhancement seen with both YTHDF2 and YTHDF3 overexpression. Analysis of viral protein expression at these same time points (Figs. 5C, D, E and F) revealed a very similar effect. For example, we observed an ~6-fold positive effect of YTHDF2 overexpression on p55 Gag expression at 24 hpi and an ~5-fold effect of YTHDF3 overexpression (Fig. 5E). Similarly, we observed an ~4-fold increase in HIV-1 p24 capsid expression at 48 hpi for both YTHDF2 and YTHDF3 overexpression. YTHDF1 overexpression exerted a more minor positive effect on viral gene expression (Figs. 5E and F), though the level of ectopic expression of these three proteins was comparable (Fig. S2D).
Figure 5. Overexpression of YTHDF m6A reader proteins boosts HIV-1 protein and RNA expression.

(A and B) qRT-PCR was used to quantify the expression level of the dominant spliced HIV-1 mRNA isoforms encoding Rev, Tat or Nef as well as the unspliced genomic RNA (gRNA). Assays were performed at 24 h (A) or 48 h (B) post-infection (hpi) using 293T cells stably overexpressing GFP (Neg) or one of the three YTHDF proteins (Y1 is YTHDF1 etc). Data were normalized to endogenous GAPDH mRNA. (C and D) Shown are representative Western blots from HIV-1 infection experiments similar to those described in (A and B). Infected 293T cells over-expressing GFP (Neg) or one of the YTHDF proteins were lysed at 24 hpi or 48 hpi then probed with an antibody specific for the HIV-1 p24 capsid protein, Nef, the FLAG tag on the overexpressed YTHDF protein or endogenous β-actin. Shown below the respective bands are actin-normalized quantifications. p55 represents uncleaved HIV-1 Gag polyprotein while p24 is the mature viral capsid (E and F). Shown are quantifications of band intensities from three independent Western experiments, similar to those shown in (C and D), performed at 24 hpi (E) or 48 hpi (F), with SD indicated.
We next tested whether overexpression or reduced expression of the YTHDF proteins would affect HIV-1 replication in CD4+ T cells. Western analysis of the expression of these three proteins showed a readily detectable level of expression of YTHDF2, low expression of YTHDF1 and no detectable expression of YTHDF3 in the CD4+ T-cell line CEM-SS (data not shown) and we therefore focused our attention on YTHDF2.
To examine how YTHDF2 affects HIV-1 replication in culture, we generated two subclones of CEM-SS, one in which the endogenous YTHDF2 gene was mutationally inactivated using CRISPR/Cas (Shalem et al., 2014) (Y2-KO) and a second cell line that overexpresses YTHDF2 by ~2-fold after transduction with a lentiviral YTHDF2 expression vector (Y2-OE). Analysis of these two cell lines, and a control CEM-SS cell line transduced with a GFP-expressing lentivirus, revealed comparable levels of CD4 and CXCR4 expression on their cell surface (Fig. S3). Nevertheless, analysis revealed a significant decline (p<0.006) in viral replication in the Y2-KO CEM-SS cells lacking YTHDF2, and a significant enhancement (p<0.009) in the replication of HIV-1 in the CEM-SS Y2-OE subclone that overexpresses YTHDF2 (Figs. 6A and C). Of note, this increase in viral replication occurred despite the observation of an enhanced viral cytopathic effect in the Y2-OE culture, which reduced the number of T-cells when compared to the Y2-KO culture (Fig.6B). Western analysis of viral protein expression at 72 hpi with HIV-1 confirmed a substantially higher level of HIV-1 Gag and Nef expression in the Y2-OE subclone, and a substantially lower level of HIV-1 Gag and Nef expression in the Y2-KO subclone, when compared to the control CEM-SS cells (Fig.6D). These data therefore further confirm the findings presented in Fig. 4 arguing that the m6A-mediated recruitment of YTHDF proteins to viral mRNAs enhances their expression and function and demonstrate that YTHDF protein expression is limiting in both 293T and CEM-SS cells. More importantly, these data argue that m6A editing, by recruiting YTHDF proteins to HIV-1 transcripts, significantly enhances the replication potential of HIV-1.
Figure 6. Recruitment of YTHDF2 to viral m6A editing sites boosts viral replication in CD4+ T cells.
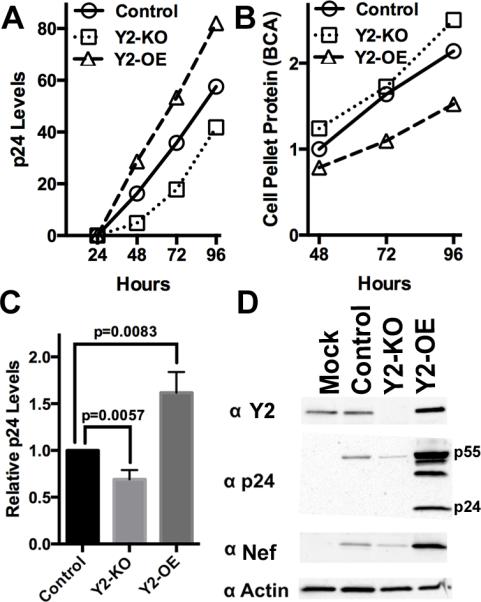
(A) A representative growth curve for HIV-1 NL4-3 in control CEM-SS cells, in a CEM-SS sub-clone lacking a functional YTHDF2 gene (Y2-KO) or in a CEM-SS sub-clone overexpressing YTHDF2 (Y2-OE). HIV-1 replication was monitored by p24 ELISA. (B) This graph shows the total level of protein recovered from the cell pellets harvested at the indicated time points from the cultures analyzed in (A). (C) This bar graph shows the average of 3 independent replicate p24 ELISA growth curve experiments at 96 hpi, with significance of differences indicated. (D) A representative Western blot of samples treated as in (A) at 72 hpi. This Western analyzes the level of intracellular expression of HIV-1 p24, Nef and YTHDF2, with endogenous β-actin used as a loading control. Equal quantities of protein, as determined by BCA analysis, were loaded in each lane. Mock: mock infected culture. For related data, see Fig. S3.
Discussion
Although m6A editing of viral mRNAs was first reported 40 years ago (Krug et al., 1976), the ability to precisely map these editing sites has only recently been achieved. Here, we have focused on the pathogenic human lentivirus HIV-1. We first used an in vitro technique, PA-m6A-seq (Chen et al., 2015), to map m6A editing sites on the genome of the HIV-1 laboratory strain NL4-3 using an m6A-specific antibody (Fig. 1). We then extended these data by using PAR-CLIP (Hafner et al., 2010) to map binding sites for the three human YTHDF reader proteins on the HIV-1 genome in infected cells (Fig. 1). These experiments identified four clusters on the HIV-1 RNA genome that not only bound all three cellular YTHDF proteins independently but also bound the m6A-specific antibody (Fig. 1). Analysis of m6A editing sites on two primary HIV-1 isolates, BaL and JR-CSF, demonstrated conservation of all four m6A binding clusters seen on NL4-3, though interestingly we also detected one (BaL) or two (JR-CSF) novel m6A editing sites on these primary isolates (Fig. 2).
Because all the m6A editing sites identified on the HIV-1 genome were located proximal to the viral polyadenylation site, in the 3'UTR region of many or all viral mRNAs, we next asked whether substitution of wildtype or m6A-deficient forms of the HIV-1 3’ UTR downstream of an indicator gene would affect its expression. As shown in Figs. 4A to D, we in fact observed a strong positive effect of the HIV-1 3'UTR on indicator gene expression that was entirely lost when the viral m6A editing sites were mutated to G. This effect, which was observed in both lymphoid and non-lymphoid cells, was equivalent at both the protein and RNA level, thus suggesting that m6A sites stabilize edited mRNAs. Moreover, this effect was not specific for HIV-1 as m6A sites derived from human mRNAs exerted a similar positive effect (Fig. 4E). Importantly, we were able to phenocopy the observed enhancement in mRNA function induced by 3'UTR m6A sites by recruiting any one of the three human YTHDF proteins to the 3'UTR of an indicator gene by fusion to an RNA binding site derived from the MS2 bacteriophage coat protein (Fig. 4F), thus arguing that m6A sites exert their effect by recruiting YTHDF proteins.
Several of the m6A editing sites mapped to the HIV-1 RNA genome were localized to sequences that are required for HIV-1 replication for other, unrelated reasons, including the overlap between the env gene and the second coding exon of rev, the LTR NF-κB binding sites and TAR, and mutational perturbation of any one of these would therefore be likely to reduce viral replication, thus making interpretation of any loss of viral fitness upon mutation of the viral m6A sites difficult. To circumvent this problem, and given our evidence that m6A sites primarily act to recruit YTHDF proteins (Figs. 1 and 4), we therefore instead asked if overexpression or knockdown of YTHDF proteins, especially YTHDF2, would induce the predicted up or down regulation of HIV-1 replication and gene expression, respectively. As shown most clearly in the CD4+ human T-cell line CEM-SS, we indeed observed a striking increase in HIV-1 replication when YTHDF2 was overexpressed (Y2-OE, Fig. 6) and a marked decline in HIV-1 replication in T cells in which the YTHDF2 gene had been inactivated by DNA editing (Y2-KO, Fig. 6). Together, these data therefore map several m6A editing sites to the 3’ UTR region of the HIV-1 genome (Figs. 1 and 2), reveal that 3'UTR m6A editing sites of either viral or cellular origin enhance gene expression in cis (Fig. 4) and finally demonstrate that the level of expression of cellular YTHDF proteins, especially YTHDF2, impacts the level of HIV-1 gene expression and replication in both non-lymphoid (Fig. 5) and lymphoid (Fig. 6) cells in culture, as indeed predicted if the role of m6A editing sites is to recruit YTHDF proteins to the mRNA (Fig. 4F).
If the m6A editing sites in the HIV-1 genome are important for maximizing virus replication, then one would predict that these would be conserved. The four m6A editing sites identified in the NL4-3 laboratory strain of HIV-1 were indeed found to be conserved in the primary isolates BaL and JR-CSF, though interestingly these also contained one or two additional m6A editing sites not seen in NL4-3 (Fig. 2). As regards the four m6A binding clusters mapped in all three HIV-1 variants, these each contain either two or three sites that bear the minimal m6A editing consensus 5’-RA*C-3’ (Fig. 3). Analysis of the conservation of these 10 possible m6A addition sites across the A, B, C and D clades of HIV-1 (Fig. S4A) shows that 7 out of 10 sites are highly conserved. Two of the three sites in the LTR U3 region are partly conserved, being found in three out of four of these HIV-1 clades, while one site, the second site in TAR, is only weakly conserved. As this potential TAR m6A site is a weak consensus editing site, we actually believe that editing at this site is unlikely. Indeed, the large majority of the crosslinking of all three YTHDF proteins to TAR occurs to the 5’ arm of TAR (Fig. S1E), arguing that the optimal m6A editing site flanking the bulged A residue at position 17 in the 5’ arm of TAR is the main target for m6A editing (Fig.S4B).
The TAR RNA hairpin forms part of the HIV-1 “R” region and is therefore present at both ends of the viral RNA genome (Hauber and Cullen, 1988). Many of the reads obtained during the YTHDF protein PAR-CLIP experiments extend past the R region into U3, thus demonstrating that the 3’ TAR is m6A edited (Fig. S1E). While no reads extending past R into U5 were recovered, it remains possible that the TAR m6A editing site(s) (Fig. S4) are also utilized at the 5’ end of the HIV-1 genome. This would be of interest given recent data arguing that m6A editing sites present in the 5'UTR can induce cap-independent translation, including under conditions of cell stress (Meyer et al., 2015). It has been proposed that HIV-1 mRNAs can also undergo cap independent translation initiation, despite the existence of a cap at the 5’ end of all viral mRNAs, yet no HIV-1 internal ribosome entry site has been identified (Monette et al., 2013). m6A editing of the 5’ TAR element might explain this apparent contradiction.
Our observation that m6A editing in 3’ UTRs, and the direct recruitment of the YTHDF proteins to 3'UTRs, can significantly enhance the level of mRNA expression and, hence, protein production contrasts with a previous paper arguing that YTHDF2 can destabilize bound mRNAs (Wang et al., 2015). We note, however, that earlier work had suggested that loss of m6A correlates with the reduced expression of edited transcripts (Dominissini et al., 2012), which is consistent with our data. While the location of m6A residues on a given mRNA, or perhaps their sequence context, could certainly regulate how m6A affects mRNA function, we do not believe that the positive effect of m6A residues present in the 3'UTR is a unique attribute of HIV-1, as several cellular m6A editing sites exerted a similar positive effect (Fig. 4E), as did RNA tethering of any of the three human YTHDF proteins (Fig. 4F). Moreover, it is well established that not only HIV-1, as demonstrated here, but also a wide range of other viruses contain multiple m6A editing sites (Dimock and Stoltzfus, 1977; Kane and Beemon, 1985; Krug et al., 1976; Lavi and Shatkin, 1975; Sommer et al., 1976). As viruses are under strong selective pressure to maximize their replication potential, and given that the random mutational inactivation of consensus m6A editing site would likely occur at high frequency, the observed conservation of the m6A editing sites in HIV-1 argues strongly for a positive role in the viral replication cycle.
If m6A is indeed important for viral replication, then the question arises whether a drug that inhibits m6A editing in HIV-1, or indeed other viruses, could act as an effective antiviral. Such a drug does in fact exist. Specifically, 3-deazaadenosine (DAA) has been shown to block m6A addition to mRNA substrates by blocking the hydrolysis of S-adenosylhomocysteine, a competitive inhibitor of S-adenosylmethionine, the methyl donor used by the METTL3/METTL14/WTAP complex (Chiang, 1998). Interestingly, DAA has also been reported to inhibit the replication of a range of viruses, including RSV, IAV and HIV-1, all of which display extensive m6A editing, though the mechanism of inhibition by DAA has remained uncertain (Bader et al., 1978; Fischer et al., 1990; Flexner et al., 1992). As shown in Fig. S5, we were also able to demonstrate the potent inhibition of HIV-1 replication by DAA. We observed a significant decline in m6A levels in total poly(A)+ RNA in cells treated with 50 μM DAA (Fig. S5A), a level that did not reduce cell growth or viability over the four day treatment period (Figs. S5C and S5D). Remarkably, this same 50 μM level of DAA effectively inhibited the replication of HIV-1 in CEM-SS cells (Fig. S5B), a result which copies the reduction in HIV-1 replication seen in CEM-SS cells lacking a functional YTHDF2 gene (Fig. 6). While these data do not prove that the sole inhibitory mechanism used by DAA to prevent HIV-1 replication in culture revolves around inhibition of m6A editing, they are intriguing in that they suggest that drugs that reduce m6A editing might have the potential to inhibit the replication of not only HIV-1 but also other viral pathogens, such as IAV.
While this manuscript was under review, Lichinchi et al. (2016) reported that HIV-1 contains 14 m6A editing site clusters, 11 of which were reported to be 5’ to all of the m6A sites reported here (Figs. 1 and 2). These authors focused particularly on two m6A sites they identified in the viral Rev response element (RRE) that they proposed regulated RRE function. We did not detect these proposed RRE m6A sites in any of our analyses, which used two distinct assays to map m6A editing sites on three HIV-1 isolates in two cell lines. While the reason for this discrepancy is currently unclear, it does not result from RRE sequence differences in the viruses analyzed.
Experimental Procedures
Western blots used the following primary antibodies: HIV-1 p24 (AIDS Reagent Program-3517), YTHDF2 (SC-162427, Santa Cruz), Actin (SC-4/778, Santa Cruz), FLAG (F1804, Sigma) and HIV-1 Nef (AIDS Reagent Program-2949). ELISAs utilized an HIV-1 p24 antigen capture kit (ABL Catalog #5421 and 5447). Total poly(A)+ RNA was purified using Ambion Poly(A)Purist MAG kits.
Molecular clones
YTHDF proteins were cloned into pEFTak expression vectors by PCR from MGC cDNA clones purchased from transOMIC Technologies (Huntsville, AL) (YTHDF1 – MGC cloneID: BC050284; YTHDF2 – MGC cloneID: BC002559; YTHDF3 – MGC cloneID: BC052970). These were then used to generate pLEX-based lentiviral vectors. For the YTHDF-MS2 coat protein fusions, pcDNA3 was modified to express pcGFP/MS2, pcYTHDF1/MS2, pcYTHDF2/MS2 and pcYTHDF3/MS2 chimeric proteins using the same YTHDF templates. The coordinates of the included N-terminal YTHDF segments are as follows: YTHDF1 (1-382), YTHDF2 (1-401), and YTHDF3 (1-409). The open reading frame for the MS2 bacteriophage coat protein was PCR amplified from pMS2-p65-HSF, (Addgene, #61426). Four copies of the MS2 RNA aptamer were inserted into psiCHECK2 (Promega) to generate the psiCHECK2-4XMS2 reporter plasmid. For the m6A site indicator plasmids, inserts were synthesized with predicted methyl receptor adenosines mutated to a guanosine. These m6A site mutant inserts, and the analogous WT inserts, were then cloned into psiCHECK2 (Promega) via the XhoI and NotI sites. The HIV-1 U3/NF-κB/TAR insert starts 34 bp 5’ of NF-κB site II in U3 and spans the entire R region, including TAR, before terminating 26 bp into the LTR U5 region. The 3'UTR construct has an identical 3’ terminus and initiates at the BamHI site in pNL4-3. All cellular m6A indicator constructs were constructed by insertion of oligonucleotides encompassing full length cellular m6A acceptor sites, in their wildtype or mutated form, into the 3'UTR of RLuc in psiCheck2.
Cell culture, HIV-1 production, and infections
293T cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) and antibiotics. CEM-SS cells were cultured in RPMI 1640 containing 10% FBS and antibiotics. HIV-1 was produced by transfection of 293T cells with the pNL4-3 molecular clone; at 72 h post-transfection, supernatant media were harvested, clarified by centrifugation and then filtered through a 0.45 μM filter (PALL). To prepare vesicular stomatitis virus glycoprotein (VSV-G) pseudotyped virus, pVSV-G was transfected at a 1:10 ratio relative to an HIV-1 proviral expression vector encoding NL4-3, BaL or JR-CSF. The supernatant media were harvested 72 h later, as described above. 293T cells were infected with the HIV-1 virus stock overnight and fresh media added the next morning. CEM-SS sub-clones were HIV-1 infected overnight, then washed with PBS and resuspended in fresh media next morning. Samples for p24 ELISA and Western analysis were collected over time from 6 ml infections per condition/biological replicate.
293T and CEM-SS clonal cell lines
Clonal YTHDF expressing 293T cell lines were produced by transduction with a constitutive lentiviral YTHDF expression vector followed by selection for the encoded puromycin resistance marker. Resistant cells were then sub-cloned by limiting dilution. CEM-SS (NIH AIDS Reagent Program catalog #776) overexpressing YTHDF2 were also obtained by lentiviral transduction, and puromycin resistant cells then sub-cloned by limiting dilution. YTHDF2 overexpression was confirmed by Western. YTHDF2 knockout CEM-SS cells were obtained by transduction with lentiCRISPRv2, with the sgRNA sequence 5’-GGAACCTTACTTGAGTCCAC-3’, obtained from a published library (Shalem et al., 2014), and were cloned by limiting dilution. The control for these cell lines was a puromycin selected GFP-expressing CEM-SS sub-clone.
PAR-CLIP and PA-m6A-Seq
PAR-CLIP was performed as described (Hafner et al., 2010). The three clonal 293T cell lines expressing FLAG-YTHDF proteins, or FLAG-GFP as a control, were infected with HIV-1 NL4-3 pseudotyped with VSV-G, incubated for 48 h and then pulsed with 100 μM 4SU in fresh media for 16 h. The cells were then harvested and the PAR-CLIP protocol performed. JR-CSF and BaL infections were conducted similarly. CEM-SS cells were infected with HIV-1, 4SU pulsed, total poly(A)+ RNA purified, and the rest of the PA-m6A-Seq protocol performed as described using an m6A specific polyclonal antibody (SySy). For the indicator plasmid PAR-CLIP experiment shown in Fig. S2A, 293T cells expressing FLAG-YTHDF2 were transfected with psiCHECK2-based constructs containing wildtype or mutant forms of the U3/NF-kB/TAR sequence. After 48 h, the cells were pulsed for 16 h with 4SU and harvested for PAR-CLIP.
PAR-CLIP libraries were sequenced on a HiSeq 2000, base calling was performed with CASAVA and processed with the fastx toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html). Reads >14 bp in length were used for bioinformatic analysis. All alignments were performed with Bowtie (Langmead et al., 2009). Reads were initially aligned to the human genome build hg19 allowing up to 1 mismatch, and unaligned reads were then aligned to the HIV-1 genome of interest, again with 1 mismatch. The HIV-1 aligned reads exhibited a substantial enrichment of reads containing T>C mutations when derived from cells expressing one of the YTHDF proteins (Fig. S1C), and these reads were of mean length >24 nt (Fig. S1D). For all visualizations, only reads containing T>C mutations were considered. Data was processed with in-house Perl scripts and Samtools, and visualized with IGV (Li et al., 2009; Robinson et al., 2011).
The raw sequencing data obtained from small RNA deep sequencing have been submitted to the NCBI Gene Expression Omnibus (GEO) and are available through accession number GSE77890.
Indicator assays and MS2-tethering
HIV-1 based indicators were transfected into 293T or CEM-SS cells utilizing the polyethylenimine (PEI) and Lipofectamine LTX (Invitrogen) transfection methods, respectively. Cells were harvested 48 h later and subjected to either cell lysis using Passive Lysis Buffer-PLB (Promega Dual Luciferase Kit), for protein extraction, or using TRIzol, for total RNA extraction. Protein lysates were analyzed for RLuc and FLuc levels using a Dual Luciferase Assay Kit (Promega). Total RNA was reverse-transcribed using a SuperScript III kit (Invitrogen) followed by SYBR green qPCR of cDNAs utilizing RLuc, FLuc, and GAPDH mRNA specific primers. RLuc mRNA abundance was determined by normalizing first to the endogenous GAPDH mRNA and then to the control FLuc mRNA. For the tethering assays, 293T cells were transfected with 50 ng psiCHECK2 or the psiCHECK2-4xMS2 reporter and 500 ng pcGFP/MS2, pcYTHDF1/MS2, pcYTHDF2/MS2 or pcYTHDF3/MS2 using PEI. Cells were harvested 72 h post-transfection and analyzed for RLuc (reporter) and FLuc (internal control) activity using the Dual-Luciferase Assay.
Supplementary Material
Acknowledgments
K.T. was supported by National Institutes of Health T32-CA009111.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
E.M.K and B.R.C. designed experiments, analyzed the data and wrote the manuscript. E.M.K, H.P.B., A.V.R.K., D.K., D.G., B.C.P., J.B.M. and K.T. performed the experiments, and N.G. and S.M.H. provided reagents.
Supplemental Information
Supplemental Information includes five figures and can be found with this article online.
References
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader JP, Brown NR, Chiang PK, Cantoni GL. 3-Deazaadenosine, an inhibitor of adenosylhomocysteine hydrolase, inhibits reproduction of Rous sarcoma virus and transformation of chick embryo cells. Virology. 1978;89:494–505. doi: 10.1016/0042-6822(78)90191-5. [DOI] [PubMed] [Google Scholar]
- Cann AJ, Zack JA, Go AS, Arrigo SJ, Koyanagi Y, Green PL, Koyanagi Y, Pang S, Chen IS. Human immunodeficiency virus type 1 T-cell tropism is determined by events prior to provirus formation. J Virol. 1990;64:4735–4742. doi: 10.1128/jvi.64.10.4735-4742.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515:143–146. doi: 10.1038/nature13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N, Han D, Dominissini D, Dai Q, Pan T, et al. High-resolution N(6) -methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew Chem Int Ed Engl. 2015;54:1587–1590. doi: 10.1002/anie.201410647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PK. Biological effects of inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol Ther. 1998;77:115–134. doi: 10.1016/s0163-7258(97)00089-2. [DOI] [PubMed] [Google Scholar]
- Desrosiers RC, Friderici KH, Rottman FM. Characterization of Novikoff hepatoma mRNA methylation and heterogeneity in the methylated 5′ terminus. Biochemistry. 1975;14:4367–4374. doi: 10.1021/bi00691a004. [DOI] [PubMed] [Google Scholar]
- Dimock K, Stoltzfus CM. Sequence specificity of internal methylation in B77 avian sarcoma virus RNA subunits. Biochemistry. 1977;16:471–478. doi: 10.1021/bi00622a021. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni A, Salmon-Divon M, Clark WC, et al. The dynamic N-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530:441–446. doi: 10.1038/nature16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AA, Muller K, Scholtissek C. Specific inhibition of the synthesis of influenza virus late proteins and stimulation of early, M2, and NS2 protein synthesis by 3-deazaadenosine. Virology. 1990;177:523–531. doi: 10.1016/0042-6822(90)90517-u. [DOI] [PubMed] [Google Scholar]
- Flexner CW, Hildreth JE, Kuncl RW, Drachman DB. 3-Deaza-adenosine and inhibition of HIV. Lancet. 1992;339:438. doi: 10.1016/0140-6736(92)90133-n. [DOI] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr., Jungkamp AC, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauber J, Cullen BR. Mutational analysis of the trans-activation-responsive region of the human immunodeficiency virus type I long terminal repeat. J Virol. 1988;62:673–679. doi: 10.1128/jvi.62.3.673-679.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SS, Boyle TJ, Lyerly HK, Cullen BR. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science. 1991;253:71–74. doi: 10.1126/science.1905842. [DOI] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane SE, Beemon K. Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol Cell Biol. 1985;5:2298–2306. doi: 10.1128/mcb.5.9.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, Haripal B, Zucker-Scharff I, Moore MJ, Park CY, et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015;29:2037–2053. doi: 10.1101/gad.269415.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug RM, Morgan MA, Shatkin AJ. Influenza viral mRNA contains internal N6-methyladenosine and 5′-terminal 7-methylguanosine in cap structures. J Virol. 1976;20:45–53. doi: 10.1128/jvi.20.1.45-53.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi S, Shatkin AJ. Methylated simian virus 40-specific RNA from nuclei and cytoplasm of infected BSC-1 cells. Proc Natl Acad Sci U S A. 1975;72:2012–2016. doi: 10.1073/pnas.72.6.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichinchi G, Gao S, Saletore Y, Gonzalez GM, Bansal V, Wang Y, Mason CE, Rana TM. Dynamics of the human and viral m6A RNA methylomes during HIV-1 infection of T cells. Nat Microbiol. 2016 doi: 10.1038/nmicrobiol.2016.11. doi:10.1038/nmicrobiol.2016.11, 16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monette A, Valiente-Echeverria F, Rivero M, Cohen EA, Lopez-Lastra M, Mouland AJ. Dual mechanisms of translation initiation of the full-length HIV-1 mRNA contribute to gag synthesis. PLoS One. 2013;8:e68108. doi: 10.1371/journal.pone.0068108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, Leon-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014;159:148–162. doi: 10.1016/j.cell.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer S, Salditt-Georgieff M, Bachenheimer S, Darnell JE, Furuichi Y, Morgan M, Shatkin AJ. The methylation of adenovirus-specific nuclear and cytoplasmic RNA. Nucleic Acids Res. 1976;3:749–765. doi: 10.1093/nar/3.3.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM, Preiss T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012;40:5023–5033. doi: 10.1093/nar/gks144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29:1343–1355. doi: 10.1101/gad.262766.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.