

Abstract
Low frequency coding variants in TREM2 are associated with increased Alzheimer disease (AD) risk, while loss of functions mutations in the gene lead to an autosomal recessive early-onset dementia, named Nasu-Hakola disease (NHD). TREM2 can be detected as a soluble protein in cerebrospinal fluid (CSF) and plasma, and its CSF levels are elevated in inflammatory CNS diseases. We measured solubleTREM2 (sTREM2) in the CSF of a large AD case-control dataset (n=180) and 40 TREM2 risk variant carriers to determine whether CSF sTREM2 levels are associated with AD status or mutation status. We also performed genetic studies to identify genetic variants associated with CSF sTREM2 levels. CSF, but not plasma, sTREM2 was highly correlated with CSF total tau and phosphorylated-tau levels (r=0.35, p<1×10-4; r=0.40, p<1×10-4 respectively), but not with CSF Aβ42. AD cases presented higher CSF sTREM2 levels than controls (P=0.01). Carriers of NHD-associated TREM2 variants presented significantly lower CSF sTREM2 levels, supporting the hypothesis that these mutations lead to reduced protein production/function (R136Q, D87N, Q33X or T66M; p=1×10-3). In contrast, CSF sTREM2 levels were significantly higher in R47H carriers compared to non-carriers (P=6×10-3), suggesting that this variant does not impact protein expression and increases AD risk through a different pathogenic mechanism than NHD variants. In GWAS analyses for CSF sTREM2 levels the most significant signal was located on the MS4A gene locus (P=5.45×10-07) corresponding to one of the SNPs reported to be associated with AD risk in this locus. Furthermore, SNPs involved in pathways related to virus cellular entry and vesicular trafficking were overrepresented, suggesting that CSF sTREM2 levels could be an informative phenotype for AD.
Keywords: soluble TREM2, cerebrospinal fluid, Alzheimer disease
Introduction
The gene encoding the triggering receptor expressed on myeloid cells 2 (TREM2, MIM 605086), a signaling receptor involved in microglial activation, is known to be involved in several neurodegenerative disorders. Homozygous loss-of-function mutations in TREM2 were initially associated with an autosomal recessive form of early-onset dementia, polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL; MIM 221770), also known as Nasu-Hakola disease (NHD) [20,28,34,36,37]. Subsequently, TREM2 mutations were also found in frontotemporal (FTD)-like dementia cases without any bone-associated symptoms [19]. In 2013 several studies reported the association of the R47H genetic variant in AD risk with Odds Ratios similar to those of APOE [3,4,6,18,22,23,31]. Subsequent studies replicated this association in several populations and reported additional coding variants in TREM2 that increase AD risk [14,22,23]. Jin et al. performed deep-resequencing in TREM2 in 3730 European Americans, replicating the association of R47H, and finding that the R62H variant is also associated with risk (P= 2.36 × 10-4, OR = 2.36)[22]. The R62H variant has been also associated with AD risk in Belgian population [14]. Additionally other three variants that are in perfect linkage disequilibrium (T96K, L211P, and W191X) have been reported to be associated with AD status in African-American, but not in Europeans [14,18,22,23,25]. A soluble form of TREM2 (sTREM2) is detected in the cerebrospinal fluid (CSF) and plasma or serum and very likely derives from proteolytic cleavage of the surface receptor [39]. Our previous studies have shown that sTREM2 levels in the CSF are elevated in multiple sclerosis (MS) [39] and other inflammatory central nervous system (CNS) diseases. A recent study reported that CSF sTREM2 levels were significantly lower in AD cases compared to controls [27]. Up to now, no reports have replicated these findings in an independent dataset. Additionally, the impact of the AD-associated variants on CSF sTREM2 has not yet been explored [27]. In this study we analyzed the correlation of CSF sTREM2 levels with AD status, and determined whether CSF sTREM2 is associated with other well-known biomarkers for AD (CSF tau and Aβ) or different risk-variants status for TREM2 (R47H, R62H and T96K/L211P/W191X). Additionally, we performed genetic studies using CSF sTREM2 levels as a phenotype to identify novel variants and genes regulating sTREM2 levels or implicated in AD.
Materials and Methods
Study population
CSF and plasma samples were collected at the Ospedale Maggiore Policlinico, University of Milan, Italy and at the Knight Alzheimer's Disease Research Center (ADRC) at Washington University in St. Louis. A total of 107 cognitive normal individuals, 73 AD cases and 40 TREM2 carriers (9 R47H, 10 R62H; 5 NHD-associated mutations and 16 T96K/L211P/W191X) were included in the study (8 non-demented individuals, 18 AD cases and 4 TREM2 carriers from the Ospedale Maggiore Policlinico; 99 non-demented individuals, 55 AD cases and 36 TREM2 carriers from the ADRC). Symptomatic cases received a diagnosis of dementia of the Alzheimer type using the National Institute of Neurological and Communication Disorders and Stroke-Alzheimer's Disease and Related Disorders Association criteria for probable AD [33]. Disease severity was measured using the clinical dementia rating (CDR) [35]. CDR 0 corresponds to cognitively normal, whereas CDR 0.5, 1, 2 and 3 correspond to very mild, mild, moderate and severe dementia, respectively. Most AD cases included in this study had very mild or mild dementia (CDR 0.5 or 1), but 5 cases with moderate dementia (CDR 2). Additionally CSF TREM2 levels were also measured in 10 Frontotemporal Dementia cases from the Ospedale Maggiore Policlinico as a comparison. CSF from both sites was collected by lumbar puncture (LP) after overnight fasting, centrifuged and frozen at -80°C as described previously [16]. Blood was collected at the time of LP and serum or plasma was obtained by centrifugation and stored at -80°C. Apolipoprotein E (APOE) genotypes were determined by the Knight ADRC Genetics Core and CSF samples were analysed by immunoassay for β-amyloid 1-42 (Aβ42), total tau, and tau phosphorylated at threonine 181 (p-tau) (INNOTEST, Fujirebio, Ghent, Belgium)[16]. CSF Aβ42, total tau, and p-tau were only available for the Knight-ADRC samples. Table 1 shows demographic characteristics of cases and controls. All individuals were of European descent. Study protocols were approved by local ethics committees at the two sites. Written informed consent was obtained for all study participants [11,21,22,41].
Table 1. Demographic characteristic.
| Controls | AD cases | |
|---|---|---|
| No. of patients | 107 | 73 |
| Gender (% female) | 53% | 49% |
| Mean age at LP (SD), years | 70.2 (8.5) | 76.6 (5.2) |
| APOE Genotype, % ε4+ | 48% | 64% |
Data are given as mean ± SD
ELISA for human sTREM2
Soluble TREM2 levels were measured by enzyme linked immunosorbant assay (ELISA) as described previously [39]. Briefly, an anti-human TREM2 monoclonal antibody (clone 20G2) was used as capture antibody and coated overnight at 4°C. For detection, biotinylated mouse anti-human TREM-2 mAb (clone 29E3) was incubated for 1h at room temperature. Recombinant soluble human TREM2 (Sino Biological Inc.) was used to generate the standard curve. One sample was included in all assays with an interassay coefficient of variation of 5%.
Genetic studies
For the analyses, we used the log-transformed values to approximate CSF sTREM2 levels to a normal distribution. Genome-wide genotype data was available for the Knight-ADRC samples only. We identified unanticipated duplicates and cryptic relatedness using pair-wise genome-wide estimates of proportion identity by descent (IBD) using PLINK1.9 [8]. When duplicate samples or a pair of samples with cryptic relatedness was identified, priority was given to samples with higher genotype call rates. Principal component analysis (PCA) was conducted using PLINK1.9 [8]. Samples were excluded if not within the CEU cluster. Samples were genotyped on either the Illumina 660 Chip, Omniexpress Chip, Biobank or HumanExomeCore. QC, and imputation was performed as described previously [13,26].
Gene ontology over-representation analyses
Before analysis, GWAS SNPs were pruned using the clump function in PLINK v1.9 [8]. Significance threshold for index and clumped SNPs was 1 to include all SNPs. SNPs were clumped if they were within 1Mb and in LD with the index (r2=0.8). Index SNPs were mapped to genes using a gene map created from the Table Browser tool on the UCSC genome browser using the Feb. 2009 (GRCh37/hg19) assembly. SNPs were mapped to a gene if they were located within 20kb of that gene; if SNPs were mapped to more than one gene, all genes were included in the analysis. Gene ontology analyses were performed using the Protein Analysis Through Evolutionary Relationships (PANTHER) statistical over-representation test v9.0 (http://www.go.pantherdb.org) which used data from the Gene Ontology Consortium (GOC) (http://www.geneontology.org). PANTHER utilizes a binomial distribution test to calculate over-representation of candidate genes, relative to background, for different gene ontology (GO) terms. Background gene sets came from each tool's default which included 20,814 genes for PANTHER (based on total number of genes in the human database from Ensembl, April 2014). PANTHER tested each GO term type separately; further separating genes that were manually assigned to that term based on experimental evidence from those assigned electronically based on bioinformatics algorithms.
Genetic hereditability for CSF TREM2
The algorithm GCTA (Genome-wide Complex Trait Analysis) was used to estimate the proportion of phenotypic variance explained by genome-wide SNPs [43]
Statistical Analyses
Data were analyzed using Mann-Whitney test when two groups were compared, Kruskal-Wallis test for multigroup comparison and Pearson correlation coefficient for correlation analyses (GraphPad Prism, GraphPad Software). CSF TREM2 protein levels were tested for association using an additive model in PLINK v1.9 [8]. Covariates were study, age, gender, and two principal component factors for population structure. Bonferroni corrected statistical significance was defined as p<5×10-8 and p<1×10-5 was considered suggestive association. The genomic inflation factor was 1.002, indicating no inflation due to population stratification. The analyses presented in the main text of the manuscript were performed excluding the outliers (4 subjects) for CSF TREM2 and tau levels. Analyses including all the samples can be found in the supplementary material (supplementary Fig.1).
Data availability
All the phenotypic and genetic data is available to qualified investigator by applying to the Knight-ADRC (http://www.adrc.wustl.edu/Research/ResourceRequest.htm).
Results
Correlation of CSF sTREM2 levels with demographic features and CSF biomarkers
Levels of sTREM2 in the CSF were evaluated for potential correlation with demographic variables including age at LP and gender (Fig. 1 and supplementary Fig. 1). Soluble TREM2 levels were correlated with age (Pearson r=0.28, Fig. 1a) and were slightly but significantly higher in males compared to females (mean ± SD were 1080 ± 507 and 903 ± 373 pg/mL, respectively; P=0.017 by Mann-Whitney test; Fig. 1b). Levels of sTREM2 in the CSF did not correlate with its levels in plasma (r=0.04, P=0.20; Fig. 1C) supporting the hypothesis that CSF sTREM2 is produced in situ. Next, to further explore the possible role of sTREM2 in AD pathology, we used samples from the Knight-ADRC (Washington University) to investigated the correlation of its CSF levels with other AD-related CSF biomarkers, including Aβ42, tau and p-tau. Soluble TREM2 levels did not correlate with CSF Aβ42 (r=-0.04, P=0.53; Fig. 1d), but did correlate with tau (r=0.35, P<0.0001; Fig. 1e) and p-tau. (r=0.40, P<0.0001; Fig. 1f). The analyses were done excluding outliers, but similar results are obtained when those were included. This suggests that sTREM2 levels in the CSF may reflect underlying pathological processes related to AD pathology.
Figure 1. Correlation between CSF sTREM2, and CSF Aβ, tau, p-tau181 and plasma TREM2.
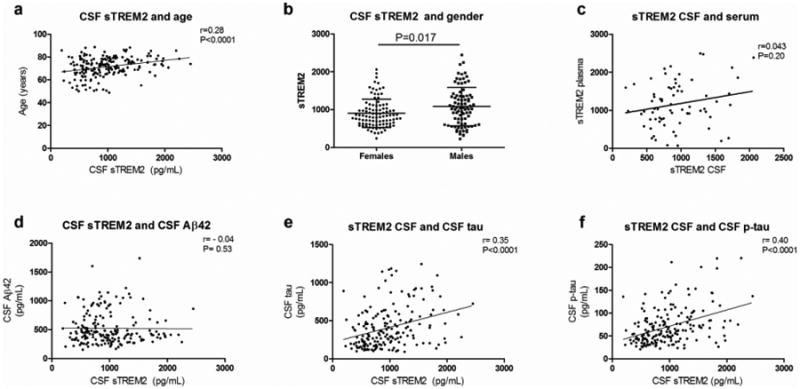
(a) CSF sTREM2 levels are correlated with age and (b) they are higher in males compared females. (c) CSF and plasma TREM2 levels are not correlated. (d) CSF sTREM2 levels are not correlated with CSF Aβ42, but they are significantly correlated with (e) CSF tau and (f) ptau. Pearson correlation was used for correlation analyses. Mann-Whitney test was used for two group comparison. Association of CSF sTREM2 levels with age and gender was performed using samples from Ospedale Maggiore Policlinico and the Knight-ADRC, while the association with CSF Aβ42, tau and p-tau only includes samples from the Knight ADRC.
sTREM2 levels in the CSF and peripheral blood of subjects with dementia
A recent study reported that in AD cases (n=56) CSF TREM2 levels were lower than controls (n=88) [27]. In this study we tried to replicate those findings but on a larger and very well characterized dataset. Soluble TREM2 levels were measured in the CSF in 107 non-demented CDR=0 participants and 73 AD cases (CDR>0) (Table 1; Fig. 2 and supplementary Fig. 2). All of the samples were screened for TREM2 variants, and only non-TREM2 variant carriers were included in these analyses. In our dataset, we saw that the CSF TREM2 levels were significant higher in AD cases compared to controls [median and range: 1028 (244-2570) and 832 (163-2196) pg/ml, respectively; P=0.015; Fig. 2a]. In this analysis 2 outliers were excluded (one in each group), but the results do not change when they are included (supplementary Fig 2a). In contrast, we did not see any significant differences in sTREM2 plasma levels between the 2 groups [1019 (190-2546) vs. 976 (65-2477) pg/ml in AD cases and controls, respectively; P=0.74; Fig. 2b]. Similarly, we show that FTD cases had significantly higher CSF sTREM2 levels [median and range: 1420 (492-2232) pg/ml] compared to controls (P=0.002; supplementary Fig. 3), but no differences were observed in plasma sTREM2 between the groups (data not shown).
Figure 2. CSF and blood sTREM2 levels in AD cases and controls.
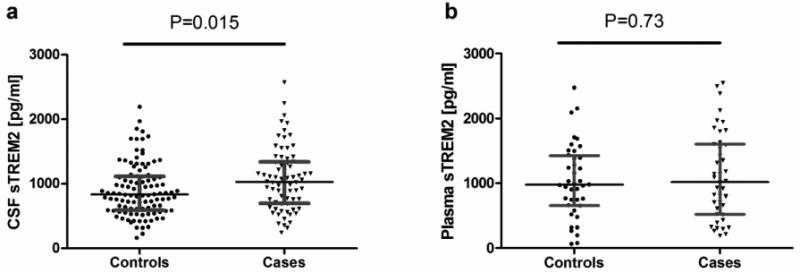
(a) sTREM2 levels were measured in CSF and (b) serum by ELISA in control and AD cases. Values are medians ± interquartile range. Mann-Whitney test was used for two group comparison.
Altered sTREM2 levels in the CSF of TREM2 genetic variant carriers
Next, we tested if there was an association between CSF sTREM2 levels and TREM2 variants. We measured sTREM2 in participants carrying different variants associated with increased AD risk (risk variants) or causative of NHD (pathogenic variants in homozygous state). CSF was available from participants carrying the R47H (n=8; 3 CDR 0, 3 CDR 0.5, 2 CDR≥1), R62H (n=10; 5 CDR 0, 5 CDR 0.5) and the T96K/L211P/W191X (n=15; 13 CDR 0, 2 CDR 0.5) risk variants associated with AD [22,23]. Levels of sTREM2 were also measured in the CSF of individuals heterozygous for the NHD-associated variants R136Q, D87N, Q33X, T66M (n=5, one for each genotype, but 2 carriers of Q33X), which in homozygosis cause NHD. Clinically, the carriers of NHD-associated pathogenic variants were diagnosed with AD, with exception of one D87N carrier with normal cognitive function. Notably, levels of sTREM2 in the CSF of individuals carrying the risk variant R47H were significantly higher compared to the non-carrier group (CDR 0; n=95) [median (range): 1732 (922-2452) vs. 856 (227-2196) pg/ml, respectively; P < 0.0001, Fig. 3 and supplementary Fig. 4]. In contrast, CSF sTREM2 levels in individuals carrying pathogenic TREM2 variants [379 (162-511) pg/ml] were significantly lower compared to the non-carrier groups (P<0.0001; Fig. 3). Similarly the T96K/L211P/W191X carriers [626 (373-1284) pg/ml] had also lower TREM2 levels compared to controls. We did not find a significant differences in CSF sTREM2 levels in R62H [818 (239-1830) pg/ml] compared to non-carrier controls. In this analysis 2 outliers were excluded (one R47H carrier and one in the T96K/L211P/W191X group), but the results do not change if included (supplementary Fig 4).
Figure 3. CSF sTREM2 levels in TREM2 variant carriers.
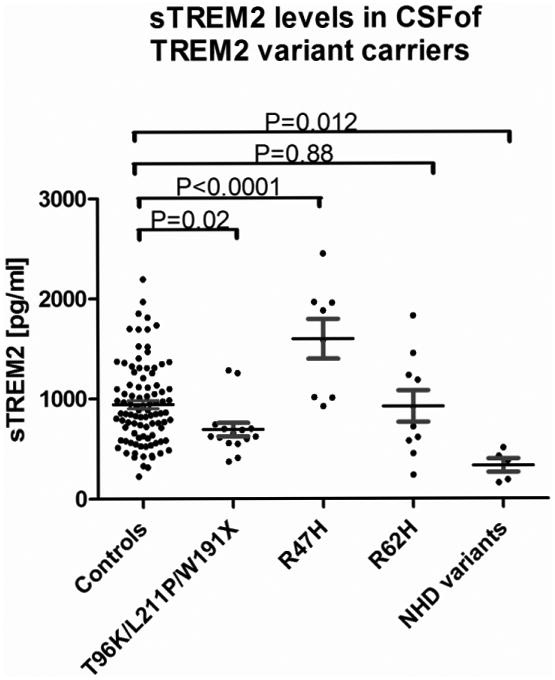
sTREM2 was measured in the CSF of cognitively normal (CDR 0) participants who were non-carriers for TREM2 genetic variants, carriers of the TREM2 AD-associated risk variants R47H (n=9), R62H (n=10), T96K/L211/W191X (n=16) and NHD pathogenic variants (R136Q, D87N, Q33X, T66M; n=5 heterozygous carriers), which in homozygosis cause NHD. Values are medians ± interquartile range. Multi-group statistical analysis was done by Kruskal-Wallis test.
Genetic architecture of CSF sTREM2 levels
Before performing any analysis, stringent quality control (QC) in both the genotype and the phenotype data was performed. For phenotype data we confirmed that the CSF sTREM2 level followed a normal distribution after log transformation. We also performed a stepwise regression analysis to include only the covariates that showed a significant association with CSF sTREM2 levels. We only included CSF sTREM2 levels for those individuals with no TREM2 coding variant. A total of 156 samples were included in the analyses. We used linear regression to test the additive genetic model of each SNP for association with CSF sTREM2 levels after adjustment for age, gender and the two principal component factors from population stratification analysis. A total of 7,566,684 imputed and genotyped SNPs were included. The inclusion of CDR or case/control status did not change the results significantly. No evidence of systematic inflation of p-values was found (λ = 1.0057). To estimate the proportion of variance in CSF sTREM2 levels explained by genetic variants we used a genome-partitioning analysis [43]. Approximately 37% (P=0.003) of the variability in the CSF levels are explained by common variants (minor allele frequency >2%), suggesting that CSF sTREM2 levels is highly regulated by common variation.
Despite the large proportion of the trait variability explained by common variants, we were not able to find any genome-wide significant signal, likely because of the limited sample size. However, the most significant signal was located on chromosome 11 in the membrane-spanning 4-domains subfamily (MS4A) gene locus (rs4939338, P-value: 5.45×10-07). Interestingly, this is the SNP associated with AD risk in the IGAP (International Genomics of Alzheimer's Project) GWAS (P-value for AD risk= 1.01×10-14) (Fig. 4). None of the SNPs in this locus were coding (supplementary Table 1), but some SNPs had a strong Regulon-DB Score (http://regulomedb.org/; rs61900467; score 3A and rs1582763 score 1f); suggesting that they could affect gene expression. Other loci identified by these analyses included PLD5 (rs74797562; P= 5.85×10-05) and PLA2G4E (rs7182355; P= 5.37×10-05) which has been recently reported to be associated with CSF APOE levels [12]. An annotated list including all the SNPs with a P <10-5 can be found in supplementary Table 1.
Figure 4. Manhattan and regional plots for CSF TREM2 levels.
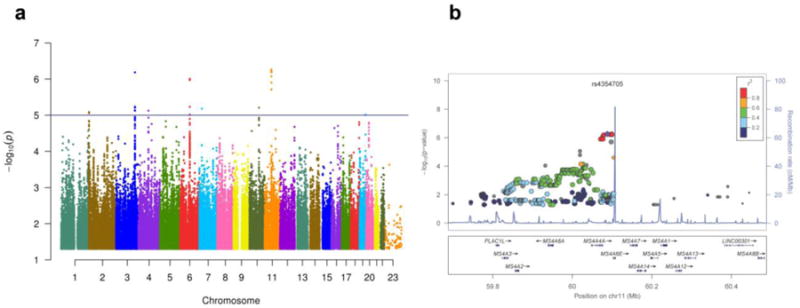
GWAS were performed on the Knight ADRC samples (n=156) using CSF TREM2 levels as endophenotype. (A) Manhattan plot of -log10 P-values for association with CSF TREM2 levels. Horizontal line indicates suggestive threshold of 1×10-5. There were no hits at genome-wide significance (5×10-8). (B) Regional plot for the most significant loci, which is located on the MS4A gene locus and overlaps with the same chromosomic region for risk for AD.
Pathway analyses
In the PANTHER analysis of the pruned CSF GWAS results there were 3 categories with P-values lower than 10-6 and five categories with lower than 10-5 (Table 2). Interestingly, the most significant categories are related to endocytosis and virus entry into host cells (receptor-mediated endocytosis of virus by host cell, P= 4.79×10-06). Other categories are also related to intracellular vesicular trafficking, including caveola assembly (P= 8.51×10-06), vesicle organization (P= 7.05×10-05) or multi-organism transport between others (P= 1.19×10-05).
Table 2. Top gene ontology categories from and PANTHER analyses of CSF TREM2 GWAS results.
| Category name | #Ref genes | #genes | expected | Fold enrichment | +/- | p |
|---|---|---|---|---|---|---|
| receptor-mediated endocytosis of virus by host cell | 3 | 2 | 0 | > 5 | + | 4.79E-06 |
| endocytosis involved in viral entry into host cell | 3 | 2 | 0 | > 5 | + | 4.79E-06 |
| caveola assembly | 4 | 2 | 0 | > 5 | + | 8.51E-06 |
| plasma membrane raft organization | 5 | 2 | 0.01 | > 5 | + | 1.33E-05 |
| plasma membrane raft assembly | 5 | 2 | 0.01 | > 5 | + | 1.33E-05 |
| membrane raft assembly | 10 | 2 | 0.01 | > 5 | + | 5.30E-05 |
| vesicle organization | 214 | 4 | 0.23 | > 5 | + | 7.05E-05 |
| membrane assembly | 13 | 2 | 0.01 | > 5 | + | 8.94E-05 |
| multi-organism localization | 15 | 2 | 0.02 | > 5 | + | 1.19E-04 |
| multi-organism transport | 15 | 2 | 0.02 | > 5 | + | 1.19E-04 |
| transport of virus | 15 | 2 | 0.02 | > 5 | + | 1.19E-04 |
| membrane biogenesis | 16 | 2 | 0.02 | > 5 | + | 1.35E-04 |
| membrane raft organization | 17 | 2 | 0.02 | > 5 | + | 1.52E-04 |
Discussion
Genetic studies in European-American (EA) and African-American (AA) populations demonstrate that there are multiple TREM2 genetic variants that increase risk for AD [18,22-24]. The R47H variant has shown the most significant association to AD risk in EA populations. Additional variants reported to be associated with AD risk are the R62H [14,22] and nine others (R52H, T66M, R136W, R136Q, H157Y, W191X, E202D, H215Q and T223I) which were exclusively found in AD cases [22]. However, the mechanisms by which they affect TREM2 function or increase AD risk are unknown. Furthermore, homozygous loss of function mutations in TREM2 are known to be the cause of NHD [28] and of some FTD-like cases [19]. In this study, we analyzed sTREM2 levels in CSF and plasma in AD cases compared to cognitively normal controls and in TREM2 mutation carriers. The goal was to evaluate a potential role of sTREM2 in AD pathogenesis as well as the impact of TREM2 genetic variants on sTREM2 levels.
TREM2 is expressed on microglia [38,39], with the release of sTREM2 in the extracellular space mainly due to proteolytic cleavage of the surface receptor [27]. We have shown that sTREM2 levels in the CSF are increased in individuals with inflammatory CNS diseases [39]. This supports the hypothesis that CSF sTREM2 levels could reflect microglia activation state as inflammation is associated with reactive microglia. To date only one report has investigated sTREM2 levels in individuals with dementia and reported lower CSF sTREM2 in AD and FTD cases compared to controls without apparent CNS disease [27]. In contrast, in our larger (n=180) dataset we found significantly higher CSF sTREM2 in AD and FTD cases compared to cognitively normal controls (CDR 0). The discrepancy between our results and what was previously reported may be due to differences in the cohorts that were analyzed, especially on the selection of controls and cases. Control subjects included in our analyses were cognitive normal individuals with CSF Aβ and tau biomarkers compatible with non-AD cases (high Aβ and low tau levels). In addition, most of AD cases included in our study had very mild or mild dementia (CDR 0.5 and 1), while disease severity of AD cases was not reported in the study published by Kleinberger et al. [27]. TREM2 expression by microglia could change when disease progress based on degree of cell activation, inflammation and tissue loss. Therefore the selection of AD cases can also explain the discrepancy between our results and what previously published. In any case, additional studies are needed to determine how CSF sTREM2 levels are correlated with diseases status and progression in AD. Notably, this study shows that CSF sTREM2 levels are increased also in FTD cases (with unknown underlying pathology) compared to healthy controls (supplementary Fig. 3). In a previous study, we found that CSF sTREM2 is increased also in MS patients [39]. All together these findings suggest that elevated CSF sTREM2 levels are not a specific maker of AD but a more general marker of microglia activation and neurodegeneration. Therefore, very likely CSF sTREM2 levels could be affected in other neurodegenerative disorders.
In our study, we specifically excluded from the analysis all TREM2 mutation carriers that can potentially skew the data. In fact, the amount of sTREM2 shed in the extracellular space is likely influenced by TREM2 genetic status as TREM2 mutations can affect protein production, maturation and cleavage. This was previously demonstrated in an in vitro system [27], but has not been investigated in vivo in TREM2 variant carriers. We have also measured soluble TREM2 in individuals carrying different TREM2 genetic variants, including those associated with increased AD risk and those that in homozygosis can cause NHD. We found that the NHD mutation carriers (heterozygous for the mutation) present significantly lower CSF sTREM2 levels compared to cognitively normal controls. This is consistent with what has been previously reported in one subject carrying a NHD mutation (T66M) in homozygosis [27] and could reflect defective protein production, maturation, surface expression or cleavage. On the other hand, we found significantly higher CSF sTREM2 levels in individuals with the AD-associated R47H variant. This variant was shown to minimally affect TREM2 protein expression on the cell surface and release in the extracellular space as a soluble form [27]. Overall, these findings indicate that AD and NHD-associated variants can predispose to CNS pathology by different functional mechanisms. Low sTREM2 levels in NHD-associated variants suggest that the pathology is due to loss of function, as reported by other studies [15,17,36]. More complex mechanisms could be implicated in AD-associated variants (e.g. R47H). Much of the uncertainty relates to the fact that TREM2 exact function and its ligand are still elusive though a recent report demonstrates that ApoE and specific classes of lipids bind and activate TREM2 [1,2,42].
Despite our relatively limited sample size, our genetic analyses suggest that a large proportion of the CSF sTREM2 variability can be explained by common genetic variants (MAF>2%) (h=0.37; P=0.003). As a comparison, a GWAS study of CSF levels of tau and p-tau estimated that the genetic effect of all the coding variants were around 6-9% and the genome-wide significant association located in APOE explained only 0.25-0.29% of the variability of CSF tau and ptau levels [13]. Our GWAS analyses suggest that CSF sTREM2 levels could be an informative endophenotype for genetic studies. First, the most significant signal is located on the MSA4 gene cluster, which was reported to be associated with AD risk [29]. Second, and more importantly, the top SNP for the CSF TREM2 (rs4939338, P= 5.45×10-07), is also one of the top SNPs for the GWAS for AD risk (P-value for AD risk= 1.01×10-14), indicating that the same signal is detected by the two phenotypes (sTREM2 levels in CSF and AD risk). The MS4A gene family encodes cell membrane proteins, the functions of which are still poorly understood but with a possible role in immune cells [32]. At present, it is unknown whether and how MSA4 genes interact with TREM2. Interestingly, among the closest gene to the GWAS top hits are MS4A6A, MS4A4A and MS4A7, and both MS4A6A, and MS4A7 are highly expressed in microglia, as is the case of TREM2 [44]. This suggests that either MS4A6A or MS4A7 could interact directly or indirectly with TREM2. Pathway analyses further support CSF sTREM2 as an informative endophenotype for AD. TREM2 is expressed in innate immune cells (e.g. dendritic cells and macrophages) implicated in host defense [9] while in the CNS TREM2 is involved in microglia activation and phagocytosis in response to injury [7,42]. Consistently with this, our genetic analyses found an enrichment of pathways involved in virus endocytosis, transport and intracellular vesicular trafficking.
In previous studies the TREM2 locus, and specifically the R47H variant has been shown to be associated with CSF p-tau levels [5,13,30]. In this study we found that CSF sTREM2 and p-tau levels are correlated (r=0.35, P<0.0001), suggesting a biological relationship between these two soluble proteins. Recent studies have also identified CSF YKL40 and VILIP1 levels as novel biomarker for AD [10,40], which are highly correlated with CSF p-tau levels (YKL40; r2=0.59; VILIP1; r2=0.62), and very likely expression of neuronal death or injury [10,40]. Along these lines sTREM2 levels in the CSF, which we found to be elevated in AD and FTD cases, could be a novel biomarker for neurodegeneration. On the other hand, the MS4A SNP (rs4939338) was not associated with CSF p-tau (p=0.54 [13]), suggesting that in this case the MS4A locus increases the risk for AD through a TREM2- and not tau-dependent mechanism.
TREM2 coding variants present very low frequency on the population (MAF<0.5%), therefore it is difficult to identify a large number of variant carriers. In this study we identified 40 carriers for TREM2 coding variants with CSF available that allowed us to perform variant-specific analyses.
To our knowledge this study has analyzed CSF TREM2 levels on the largest number of TREM2 variant carriers up to date. Additionally, our genetic studies provide new clues about TREM2 biology in microglia. However, given the limited sample size of variant-specific CSF and genetic analyses additional studies in larger datasets are needed to confirm these findings.
Supplementary Material
Supplementary figure 1. Correlation between CSF sTREM2, and CSF Aβ, tau, ptau181 and plasma TREM2. All the samples, including outliers were included in these analyses. (a) CSF sTREM2 levels are correlated with age and (b) they are higher in males compared females. (c) CSF and plasma TREM2 levels are not correlated. (d) CSF sTREM2 levels are not correlated with CSF Aβ42, but they are significantly correlated with (e) CSF tau and (f) p-tau. Pearson correlation was used for correlation analyses. Mann-Whitney test was used for two group comparison. Association of the CSF TREM2 levels with age and gender was performed using samples from Ospedale Maggiore Policlinico and the Knight-ADRC, and the association with CSF Aβ42, tau and p-tau only includes samples from the Knight ADRC
Supplementary figure 2. CSF and blood sTREM2 levels in AD cases and controls. All the samples, including outliers were included in these analyses. (a) sTREM2 levels were measured in CSF and (b) serum by ELISA in control and AD cases. Values are medians ± interquartile range. Mann-Whitney test was used for two group comparison.
Supplementary figure 3. CSF sTREM2 levels in AD, FTD cases and controls. sTREM2 levels were measured in CSF by ELISA in cognitively normal subjects, AD and FTD cases. Values are medians ± interquartile range. Multi-group statistical analysis was done by Kruskal-Wallis test.
Supplementary figure 4. CSF sTREM2 levels in TREM2 variant carriers. All the samples, including outliers were included in these analyses. sTREM2 was measured in the CSF of cognitively normal (CDR 0) participants who were non-carriers for TREM2 genetic variants, carriers of the TREM2 AD-associated risk variants R47H (n=9), R62H (n=10), T96K/L211/W191X (n=16) and NHD-pathogenic variants (R136Q, D87N, Q33X, T66M; n=5 heterozygous carriers) which in homozygosis cause NHD. Values are medians ± interquartile range. Multi-group statistical analysis was done by Kruskal-Wallis test.
Supplementary table 1: Summary statistics for all the SNPs with a p-value<10-5 for CSF TREM2
Acknowledgments
We thank Dr. Marco Colonna for providing the antibodies used in the sTREM2 assay. This work was supported by a Knight-ADRC pilot grant (to LP and CC) and grants from the National Institutes of Health (R01-AG044546, P01-AG003991, P50-AG005681), and Alzheimer Association (NIRG-11-200110). LP is a Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society (NMSS, JF 2144A2/1) and supported by Fondazione Italiana Sclerosi Multipla (2014/R/15). CC was a recipient of a New Investigator Award in Alzheimer's disease from the American Federation for Aging Research. CC is a recipient of a Bright Focus Foundation Alzheimer's Disease Research Grant (A2013359S). The recruitment and clinical characterization of research participants at Washington University were supported by NIH P50 AG05681, P01 AG03991, and P01 AG026276. This work was performed by accessing equipment available in the Hope Center for Neurological Disorders and the Departments of Neurology and Psychiatry at Washington University School of Medicine.
References
- 1.Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) J Biol Chem. 2015;290:26043–26050. doi: 10.1074/jbc.M115.679043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey CC, DeVaux LB, Farzan M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J Biol Chem. 2015;290:26033–26042. doi: 10.1074/jbc.M115.677286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benitez BA, Cooper B, Pastor P, Jin SC, Lorenzo E, Cervantes S, et al. TREM2 is associated with the risk of Alzheimer's disease in Spanish population. Neurobiol Aging. 2013;34:1711 e1715–1717. doi: 10.1016/j.neurobiolaging.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benitez BA, Cruchaga C. TREM2 and neurodegenerative disease. N Engl J Med. 2013;369:1567–1568. doi: 10.1056/NEJMc1306509#SA4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benitez BA, Jin SC, Guerreiro R, Graham R, Lord J, Harold D, et al. Missense variant in TREML2 protects against Alzheimer's disease. Neurobiol Aging. 2014;35:1510 e1519–1526. doi: 10.1016/j.neurobiolaging.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, et al. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA neurology. 2014;71:449–453. doi: 10.1001/jamaneurol.2013.6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R, et al. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 2015;129:429–447. doi: 10.1007/s00401-015-1388-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol. 2003;3:445–453. doi: 10.1038/nri1106. [DOI] [PubMed] [Google Scholar]
- 10.Craig-Schapiro R, Perrin RJ, Roe CM, Xiong C, Carter D, Cairns NJ, et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer's disease. Biological psychiatry. 2010;68:903–912. doi: 10.1016/j.biopsych.2010.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cruchaga C, Haller G, Chakraverty S, Mayo K, Vallania FL, Mitra RD, et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer's disease families. PLoS One. 2012;7:e31039. doi: 10.1371/journal.pone.0031039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruchaga C, Kauwe JS, Nowotny P, Bales K, Pickering EH, Mayo K, et al. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer's disease. Hum Mol Genet. 2012 doi: 10.1093/hmg/dds296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM, et al. GWAS of Cerebrospinal Fluid Tau Levels Identifies Risk Variants for Alzheimer's Disease. Neuron. 2013 doi: 10.1016/j.neuron.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, Gijselinck I, van der Zee J, et al. Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol Aging. 2014;35:726 e711–729. doi: 10.1016/j.neurobiolaging.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 15.Doragna D, Tupler R, Ratti MT, Montalbetti L, Papi L, Sestim R. An Italian family affected by Nasu-Hakola disease with a novel genetic mutation in the TREM2 gene. J Neurol Neurosur Ps. 2003;74:825–826. doi: 10.1136/jnnp.74.6.825-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 17.Guerreiro R, Bilgic B, Guven G, Bras J, Rohrer J, Lohmann E, et al. Novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol Aging. 2013;34:2890 e2891–2895. doi: 10.1016/j.neurobiolaging.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerreiro RJ, Lohmann E, Bras JM, Gibbs JR, Rohrer JD, Gurunlian N, et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA neurology. 2013;70:78–84. doi: 10.1001/jamaneurol.2013.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hakola HP. Neuropsychiatric and genetic aspects of a new hereditary disease characterized by progressive dementia and lipomembranous polycystic osteodysplasia. Acta Psychiatr Scand Suppl. 1972;232:1–173. [PubMed] [Google Scholar]
- 21.Jin SC, Pastor P, Cooper B, Cervantes S, Benitez BA, Razquin C, et al. Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer's disease Ibero-American cohort. Alzheimers Res Ther. 2012;4:34. doi: 10.1186/alzrt137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D, et al. Coding variants in TREM2 increase risk for Alzheimer's disease. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin SC, Carrasquillo MM, Benitez BA, Skorupa T, Carrell D, Patel D, et al. TREM2 is associated with increased risk for Alzheimer's disease in African Americans. Mol Neurodegener. 2015;10:19. doi: 10.1186/s13024-015-0016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jonsson T, Stefansson H, Ph DS, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 Associated with the Risk of Alzheimer's Disease. N Engl J Med. 2012 doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jonsson T, Stefansson K. TREM2 and neurodegenerative disease. N Engl J Med. 2013;369:1568–1569. doi: 10.1056/NEJMc1306509. [DOI] [PubMed] [Google Scholar]
- 26.Kauwe JS, Bailey MH, Ridge PG, Perry R, Wadsworth ME, Hoyt KL, et al. Genome-wide association study of CSF levels of 59 alzheimer's disease candidate proteins: significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet. 2014;10:e1004758. doi: 10.1371/journal.pgen.1004758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6:243ra286. doi: 10.1126/scitranslmed.3009093. [DOI] [PubMed] [Google Scholar]
- 28.Klunemann HH, Ridha H, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, et al. The genetic causes of basal ganglia calcification, dementia, and bone cysts DAP12 and TREM2. Neurology. 2005;64:1502–1507. doi: 10.1212/01.Wnl.0000160304.00003.Ca. [DOI] [PubMed] [Google Scholar]
- 29.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013 doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lill CM, Rengmark A, Pihlstrom L, Fogh I, Shatunov A, Sleiman PM, et al. The role of TREM2 R47H as a risk factor for Alzheimer's disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson's disease. Alzheimers Dement. 2015 doi: 10.1016/j.jalz.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luis EO, Ortega-Cubero S, Lamet I, Razquin C, Cruchaga C, Benitez BA, et al. Frontobasal gray matter loss is associated with the TREM2 p.R47H variant. Neurobiol Aging. 2014 doi: 10.1016/j.neurobiolaging.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma J, Yu JT, Tan L. MS4A Cluster in Alzheimer's Disease. Mol Neurobiol. 2015;51:1240–1248. doi: 10.1007/s12035-014-8800-z. [DOI] [PubMed] [Google Scholar]
- 33.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 34.Montalbetti L, Ratti MT, Greco B, Aprile C, Moglia A, Soragna D. Neuropsychological tests and functional nuclear neuroimaging provide evidence of subclinical impairment in Nasu-Hakola disease heterozygotes. Funct Neurol. 2005;20:71–75. [PubMed] [Google Scholar]
- 35.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 36.Numasawa Y, Yamaura C, Ishihara S, Shintani S, Yamazaki M, Tabunoki H, et al. Nasu-Hakola disease with a splicing mutation of TREM2 in a Japanese family. Eur J Neurol. 2011;18:1179–1183. doi: 10.1111/j.1468-1331.2010.03311.x. [DOI] [PubMed] [Google Scholar]
- 37.Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71:656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, Cross AH, et al. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. European journal of immunology. 2007;37:1290–1301. doi: 10.1002/eji.200636837. [DOI] [PubMed] [Google Scholar]
- 39.Piccio L, Buonsanti C, Cella M, Tassi I, Schmidt RE, Fenoglio C, et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain. 2008;131:3081–3091. doi: 10.1093/brain/awn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tarawneh R, Lee JM, Ladenson JH, Morris JC, Holtzman DM. CSF VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology. 2012;78:709–719. doi: 10.1212/WNL.0b013e318248e568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vallania FL, Druley TE, Ramos E, Wang J, Borecki I, Province M, et al. High-throughput discovery of rare insertions and deletions in large cohorts. Genome Res. 2010;20:1711–1718. doi: 10.1101/gr.109157.110. doi:gr.109157.110 [pii] 10.1101/gr.109157.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang J, Manolio TA, Pasquale LR, Boerwinkle E, Caporaso N, Cunningham JM, et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat Genet. 2011;43:519–525. doi: 10.1038/ng.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1. Correlation between CSF sTREM2, and CSF Aβ, tau, ptau181 and plasma TREM2. All the samples, including outliers were included in these analyses. (a) CSF sTREM2 levels are correlated with age and (b) they are higher in males compared females. (c) CSF and plasma TREM2 levels are not correlated. (d) CSF sTREM2 levels are not correlated with CSF Aβ42, but they are significantly correlated with (e) CSF tau and (f) p-tau. Pearson correlation was used for correlation analyses. Mann-Whitney test was used for two group comparison. Association of the CSF TREM2 levels with age and gender was performed using samples from Ospedale Maggiore Policlinico and the Knight-ADRC, and the association with CSF Aβ42, tau and p-tau only includes samples from the Knight ADRC
Supplementary figure 2. CSF and blood sTREM2 levels in AD cases and controls. All the samples, including outliers were included in these analyses. (a) sTREM2 levels were measured in CSF and (b) serum by ELISA in control and AD cases. Values are medians ± interquartile range. Mann-Whitney test was used for two group comparison.
Supplementary figure 3. CSF sTREM2 levels in AD, FTD cases and controls. sTREM2 levels were measured in CSF by ELISA in cognitively normal subjects, AD and FTD cases. Values are medians ± interquartile range. Multi-group statistical analysis was done by Kruskal-Wallis test.
Supplementary figure 4. CSF sTREM2 levels in TREM2 variant carriers. All the samples, including outliers were included in these analyses. sTREM2 was measured in the CSF of cognitively normal (CDR 0) participants who were non-carriers for TREM2 genetic variants, carriers of the TREM2 AD-associated risk variants R47H (n=9), R62H (n=10), T96K/L211/W191X (n=16) and NHD-pathogenic variants (R136Q, D87N, Q33X, T66M; n=5 heterozygous carriers) which in homozygosis cause NHD. Values are medians ± interquartile range. Multi-group statistical analysis was done by Kruskal-Wallis test.
Supplementary table 1: Summary statistics for all the SNPs with a p-value<10-5 for CSF TREM2
Data Availability Statement
All the phenotypic and genetic data is available to qualified investigator by applying to the Knight-ADRC (http://www.adrc.wustl.edu/Research/ResourceRequest.htm).