

Abstract
Neural progenitor cell (NPC) migration is an essential process for brain development, adult neurogenesis, and neuroregeneration after brain injury. Stromal cell-derived factor-1 (SDF-1, CXCL12) and its traditional receptor CXCR4 are well known to regulate NPC migration. However, the discovery of CXCR7, a newly identified CXCL12 receptor, adds to the dynamics of the existing CXCL12/CXCR4 pair. Antagonists for either CXCR4 or CXCR7 blocked CXCL12-mediated NPC migration in a transwell chemotaxis assay, suggesting that both receptors are required for CXCL12 action. We derived NPC cultures from Cxcr4 knockout (KO) mice and used transwell and stripe assays to determine the cell migration. NPCs derived from Cxcr4 KO mice polarized and migrated in response to CXCL12 gradient, suggesting that CXCR7 could serve as an independent migration receptor. Furthermore, Cxcr4 KO NPCs transplanted into the adult mouse striatum migrated in response to the adjacent injection of CXCL12, an effect that was blocked by a CXCR7 antagonist, suggesting that CXCR7 also mediates NPC migration in vivo. Molecular mechanism studies revealed that CXCR7 interact with Rac1 in the leading edge of the polarized NPCs in the absence of CXCR4. Both CXCR7 and Rac1 are required for extracellular signal-regulated kinases (ERK) 1/2 activation and subsequent NPC migration, indicating that CXCR7 could serve as a functional receptor in CXCL12-mediated NPC migration independent of CXCR4. Together these results reveal an essential role of CXCR7 for CXCL12-mediated NPC migration that will be important to understand neurogenesis during development and in adulthood.
Keywords: Chemokine receptor, Cell migration, Neural stem cell, Cell signaling
Introduction
Neural stem cells (NSC), including early NSCs and late neural progenitor cells (NPCs), exist throughout the entire lifespan [1]. Neurogenesis, a process for NPCs to give rise to newly born neurons, is very active in the developing brain but continues to diminish in the adult and aging brain [2–6]. Multiple aspects of NPC functions, including NSC self-renewal, survival, migration, differentiation, and maturation, are crucial for proper neurogenesis during development and in adulthood (reviewed in ref. [2–6]). Among these NPC functions, migration is a pivotal process because it localizes NPCs to the neurogenic or gliogenic cues for differentiation or replacement. Deciphering the mechanism for the NPC migration will help develop future stem cell-based therapeutic approaches for neurodegenerative diseases [7–9].
Stromal cell-derived factor-1 (SDF-1, CXCL12) is one of the most important chemokines in the brain, participating in the morphogenesis in developing brains and homeostasis in adult brains [10, 11]. The axis of CXCL12/CXCR4 has been established as crucial signals for NPC functions, including the survival, proliferation, and migration [12–16]. CXCR4 was once considered the sole receptor for CXCL12, until CXCR7 was recently identified as the other receptor for CXCL12 [17, 18]. In different studies, CXCR7 has been demonstrated to regulate cell migration and survival through contradicting roles that include ligand scavenging [19–22], direct signal transduction [23, 24], and interaction with CXCR4 [25]. Genetic knockout (KO) of either CXCR7 or CXCR4 results in perinatal lethality, underscoring the importance of these receptors [12, 17, 26].
CXCR7 is a seven-transmembrane receptor but typically cannot mediate the classical Gi signaling pathway like CXCR4 and most other chemokine receptors [27]. This receptor was previously thought of as a “decoy” and atypical chemokine receptor for CXCL12 [22, 28]. However, CXCR7 is known to also signal through endocytosis pathways and could activate mitogen-activated protein kinases (MAPK) through interaction with β-arrestin 2 [17, 19, 23]. Our previous study demonstrated that upon engaging CXCL12, CXCR7 mediated endocytotic signaling and promoted NPC survival [24]. Furthermore, CXCR7 was found to activate MAPK in NPCs, but this signaling pathway was mainly investigated for NPC survival rather than migration.
While NPC migration plays a pivotal role in neurogenesis in both the developing and adult brains, the cell migration event could be seen as a fundamentally polarized process [29]. Activation of Rac1 in the leading edge of migrating cells regulates cytoskeleton reorganization [30] and the protrusion of the migration structure lamellipodia [31, 32]. Currently, there is no established molecular connection between CXCR7 and Rac1. We hypothesized that CXCR7 mediates NPC polarization and migration to CXCL12 in the absence of CXCR4 through extracellular signal-regulated kinases (ERK) 1/2 and Rac1 activation. We used Cxcr4 KO (Cxcr4−/−) mice, small molecule inhibitors, and in vitro and in vivo migration systems to illustrate the CXCR4-independent role of CXCR7 in CXCL12-mediated NPC migration and the underlying molecular mechanisms.
Materials and Methods
Reagents
Recombinant mouse CXCL12 was obtained from R&D Systems (Minneapolis, MN, www.rndsystems.com). AMD3100 was obtained from Sigma-Aldrich. CCX704 and CCX771 were obtained from ChemoCentryx (Mountain View, CA, www.che-mocentryx.com). Phospho-ERK1/2, total-ERK1/2, Rac1, and β-actin protein levels were detected using antiphospho-ERK1/2, anti-total-ERK1/2 antibodies (1:1000, Cell Signaling Technologies, Danvers, MA, www.cellsignal.com), anti-Rac1 (1:1000, Thermo Scientific, Waltham, MA, www.thermoscientific.com), and anti-β-actin antibody (1:5,000, Sigma Aldrich, St. Louis, MO, www.sigmaaldrich.com), respectively.
Animal
Cxcr4 KO mice and wild type C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, Maine, www.jax.org). Cxcr7-EGFP transcriptional reporter transgenic mice were kindly provided by Dr. Richard Miller from Northwest University. All mice were housed and bred in the Comparative Medicine facilities at the University of Nebraska Medical Center. All procedures were conducted according to protocols approved by the Institutional Animal Care and Use Committee at the University of Nebraska Medical Center.
NPC Culture
The forebrain of each embryo at gestational day E15 was dissected and dissociated into single cells by triturating the tissue with a 1-ml pipette. Cells from each forebrain were seeded into a 100-mm Petri dish separately at a density of 2 × 105 cells per milliliter in 10 ml of mouse NeuroCult NSC Proliferation Medium (StemCell Technologies, Vancouver, BC, Canada, www.stemcell.com) supplemented with epidermal growth factor (10 ng/ml, Sigma Aldrich) and basic fibroblast growth factor (20 ng/ml, Sigma Aldrich) for selective neurosphere cultures. After genotyping confirmation of Cxcr4 KO or wild type phenotypes by polymerase chain reactions, NPCs derived from wild type or Cxcr4 KO mice were selected and cultured in vitro. Neurospheres were passaged when they reached 100–150 μm in diameter.
Stripe Assay
Polydimethylsiloxane molds were casted on silicon base manufactured pattern of 100 μm wide by 100 μm deep micro channels, and then placed on fibronectin and poly-D-lysine precoated coverglasses to form a complete seal. Fluorescein conjugated albumin from bovine serum albumin (BSA, Life Technologies, Grand Island, NY, www.lifetechnologies.com), with or without CXCL12 (100 mg/ml), was added to one end of micro channels, and a vacuum was applied at the other end of micro channels to ensure BSA filled in all the micro channels. Polydimethylsiloxane molds were removed from coverglasses after 6-hour incubation, and coverglasses with micro patterns of BSA stripes were ready to use. NPCs were seeded on 12 mm diameter-coverglasses coated with micro stripes of BSA/BSA-CXCL12, and incubated at the indicated length of time to study either cell migration or polarization. Cells were then fixed on coverglasses and stained with nestin and 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). Images were taken by Zeiss 710 confocal microscope with a 20× objective and quantified by ImageProPlus. The ratio between the number of cells on stripes verses off stripes was calculated as migration index for the stripe assay to evaluate cell migration.
Transwell Chemotaxis Assay
NPC migration was evaluated using an 8-μm pore size trans-well system (Fisher Scientific, Pittsburgh, PA, www.fishersci.com) coated with fibronectin (Sigma-Aldirich) at 5 μg/ml in phosphate-buffered saline (PBS) for 2 hours at 37°C. Briefly, NPCs were dissociated into single cells and resuspended in mouse NSC NeuroCult Proliferation Medium at a density of 106 cells per milliliter. The top chamber of the transwell was loaded with 100 μl of cell suspension and cells were cultured for 12 hours to form an adherent monolayer culture. CXCL12 was added to the bottom chamber, and AMD3100, CCX704, and CCX771 were added to both chambers 2 hours before adding CXCL12 to the lower chamber. After 12 hours, the membrane of the transwell inserts was fixed with 4% paraformaldehyde (PFA) in PBS, and cells on top of the membrane were removed with a cotton swab. Cells that migrated to the bottom of the membrane were stained with DAPI in PBS at 10 ng/ml. For each insert, 10 fields were randomly selected for image-taking under microscope at 200× magnification, and cell numbers were quantified using Image-Pro Plus 7.0 (Media Cybernetics, Inc., Rockville, MD, www.mediacy.com). The ratio between cell number of each treated group and the cell number of the control group was calculated as the migration index.
Immunocytochemistry
NPCs cultured on coverglasses were fixed with 4% PFA and blocked by 2% BSA in PBS. After washing coverglasses, they were incubated overnight at 4°C with primary antibodies including monoclonal anti-CXCR4 (1:500; R&D Systems), anti CXCR7 (1:100; R&D Systems), and antinestin (1:5,000, Novartis, New York, NY, http://www.novartis.com/). Coverglasses were washed and incubated for 1 hour at room temperature with secondary antibodies including anti-mouse IgG (coupled with Alexa Fluor 488, Life Technologies), anti-rabbit IgG (coupled with Alexa Fluor 568, Life Technologies), and anti-chicken IgG (coupled with Alexa Fluor 633, Life Technologies). Nuclear DNA was stained with DAPI. F-actin was stained with phalloidin (coupled with Rhodamine, Life Technologies). Cover-glasses were mounted on glass slides with mounting buffer (Sigma-Aldrich). Immunostaining was examined by a Zeiss 710 confocal laser scanning microscope.
In Vivo NPC Migration
Four-week-old male C57BL/6 mice were housed in the animal facilities at the University of Nebraska Medical Center for 1 week before intracranial injection. All procedures were as described previously [33, 34] and conducted according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Nebraska Medical Center. Briefly, mice were anesthetized with Ketamine (120 mg/kg) and xylazine (16 mg/kg) intraperitoneally, placed in a stereotaxic apparatus (Stoelting, Wood Dale, IL, www.stoeltingco.com) for intracranial injection. A linear skin incision was made over the bregma and a 1-mm burr hole was drilled in the skull 0.2 mm posterior and 3.5 mm lateral to the bregma on the left side using a hand-held driller. NPCs at passage 3–5 were labeled with Qtracker565 Labeling Kit (Life Technologies) and injected into the left striatum. A medial parallel injection of CXCL12 (100 ng in 2.5 μl) was performed at 1 mm apart from the NPC injection track. Saline was used as a control for CXCL12 injection. Coordinates for inoculation were set as: 0.5–0.8 mm posterior to bregma, 4 mm (NPCs) and 3 mm (CXCL12 or PBS) lateral from the Sagittal midline, respectively, a depth and angle of 3.6 mm and 35°, respectively, from the vertical line. A Hamilton 10-μl syringe (Fisher) and 30-gauge needles were used for cell injection. Needles were used only for one injection and then disposed of to prevent contamination of Qtracker-labeled cells into the saline or CXCL12 tracks. Mice were killed 7 days post-injection after intracardiac perfusion and brains were quickly removed. Immunohistochemistry of the brain sections were performed in a free-floating method that we described previously [33, 34].
IP and Western Blotting
An active Rac1 pull-down and detection kit (Thermo Scientific) was used for the immunoprecipitation (IP) assay. The IP and detection was performed according to the manufacturer’s instruction. Briefly, cell lysates were collected using IP buffer and precleared using protein G (Thermo Scientific). Then, pre-cleared cell lysates were incubated with monoclonal antibody for CXCR7 (Abcam, Cambridge, MA, www.abcam.com) overnight on a rotator. Antibodies against CXCR7, β-arrestin 2 (Millipore), and Rac1 (Thermo Scientific) were used to detect the CXCR7-β-arrestin2 and CXCR7-Rac1 interaction by Western blotting.
Statistical Analysis
Data were analyzed as means ± SEM. The data were evaluated statistically by the analysis of variance followed by Tukey-test for paired observations. The two-tailed Student’s t test was used to compare two groups. Significance was considered when p < 0.05. All experiments were performed with at least three donors to account for any donor-specific differences. Assays were performed at least three times in triplicate or quadruplicate.
Results
CXCR7 is Expressed in Mouse NPCs
Our previous study has demonstrated the expression of CXCR4 and CXCR7 in human NPCs by flow cytometry [24]. Because cellular distribution of CXCR4 and CXCR7 is important to their functions, we further investigated their expression patterns in NPCs through immunostaining. Both CXCR4 and CXCR7 receptors were expressed in adherent cultured mouse NPCs in vitro (Supporting Information Fig. S1). CXCR4 was expressed both on the cell membrane and in the cytosol (Supporting Information Fig. S1A, S1F), whereas CXCR7 mainly localized within the cytosol (Supporting Information Fig. S1B, S1G). Notably, our in vitro mouse NPC cultures were enriched for nestin+ cells, with more than 99% of the cells positive for the NPC marker nestin (Supporting Information Fig. S1C, S1H). Next, Cxcr7-EGFP reporter transgenic mice were used to visualize the CXCR7 transcription in vivo. This transgenic mouse line has the GFP gene placed under CXCR7 promoter so that GFP was only transcribed in cells transcribing CXCR7. A strong GFP signal was observed in the dentate gyrus region of adult Cxcr7-EGFP mouse (Supporting Information Fig. S2). Interestingly, the expression of GFP and SOX2 showed similar patterns in this region, suggesting the presence of CXCR7 transcript in adult NPCs in vivo. Notably, most of the GFP-positive cells were observed to be SOX2-positive cells in the dentate gyrus region. However, GFP-positive/SOX2-negative cells also existed and those cells were likely endothelial cells that are known to express high levels of CXCR7 [35].
NPCs Migrate in Response to CXCL12 Gradient In Vitro
Stripe assays and transwell chemotaxis assays were used to measure CXCL12-mediated mouse NPC migration in a semi-quantitative manner. In stripe assays, mouse NPCs were seeded on coverglasses printed with micro stripes of fluorescein-conjugated BSA/CXCL12 and incubated for 48 hours. The BSA alone group served as the control. All of the cells on coverglasses remained nestin-positive (Fig. 1A, 1B). On coverglasses printed with BSA alone, NPCs distributed evenly across the stripes (vehicle control group, Fig. 1A). In contrast, NPCs congregated on the BSA/CXCL12 stripes and formed a cell pattern aligning to the BSA/CXCL12 stripes in the cultures (CXCL12 group, Fig. 1B). Quantification on the number of cells in the images revealed 52% of cells localized to the BSA stripes, whereas 86% cells localized to the BSA/CXCL12 stripes (Fig. 1C). Furthermore, we defined the migration index as the ratio between the number of cells on the stripes and the number of cells off the stripes. The migration index of BSA/CXCL12 group was sixfold higher than that of the BSA alone group (Fig. 1D), suggesting CXCL12 mediates NPC migration and the stripe assay could be used to quantify the levels of NPC migration. To validate the use of stripe assay for NPC migration, a commercially available transwell chemotaxis assay also was used to measure levels of CXCL12-mediated migration in NPCs. The migration index is defined as the fold changes in the number of migrated cells compared with that of the untreated control group. The migration index in 100 ng/ml CXCL12 group was 4.2-fold higher than that of the negative control (p <0.05), while migration index of the 1000 ng/ml CXCL12 group was 14.5-fold of the control (p <0.05), suggesting that CXCL12 induced mouse NPC migration in a dose-dependent manner (Fig. 1E). Because 100 ng/ml CXCL12 significantly induced NPC migration, we used this concentration for later transwell experiments.
Figure 1.

CXCL12 mediates mouse neural progenitor cell (NPC) migration and polarization. (A–D): Mouse NPCs were seeded on cover-glasses coated with bovine serum albumin (BSA) stripes (green) without CXCL12 (A, BSA stripes, vehicle group) or with CXCL12 (B, BSA/ CXCL12 stripes). Cells were stained with nestin (white) and 4′,6-diamidino-2-phenylindole (blue). The quantification of stripe assay were shown as (C) (percent of total cells on stripes to total cell number) and standardized to migration index as (D) (ratio of cell number on stripes to cell number off stripes). (E): NPCs on transwell insert were incubated in the presence of CXL12 as indicated concentration and quantified. *, p <0.05. (F–I): NPC polarity change was also determined through immunochemistry technique with cells seeded on BSA stripes (F, G) and BSA/CXCL12 stripes (H, I). NPCs were seeded for 10 minutes before the staining. Scale bar =60 μm (A, B), 20 μm (E, G), 10 μm (F, H). Abbreviations: BSA, bovine serum albumin; DAPI, 4′,6-diamidino-2-phenylindole.
Cellular migration starts as a change to the cell polarity and the formation of lamellipodia [36]. We further examined the lamellipodia formation early when NPCs were placed into the stripe assays. As early as 10 minutes after seeding on BSA/CXCL12 stripes, NPCs started to polarize towards the BSA/CXCL12 stripes forming lamellipodia at the leading edge of the migrating cell bodies (Fig. 1H, 1I). In the BSA alone (vehicle control) group, cells exhibited minimum polarization with random filopodia occurring either toward or away from BSA stripes, and there was no formation of lamellipodia (Fig. 1F, 1G). Together, these data suggest that CXCl12 induces NPC polarization and the formation of lamellipodia before cell migration.
CXCL12-Mediated NPC Migration Could be Blocked by CXCR7 Antagonist
To determine the chemokine receptors involved in CXCL12-mediated NPC migration, AMD3100, an antagonist for CXCR4, and CCX771, an antagonist for CXCR7, were used in the transwell system. We pretreated the wild type NPCs with doses of AMD3100 ranging from 0.01 nM to 1 nM, CXCL12-induced NPC migration, as determined by migration index in the transwell assay, decreased significantly in a dose-dependent manner (Fig. 2A). These data suggest that CXCR4 is required for CXCL12-mediated NPC migration. Similarly, pretreatment with doses of CCX771 (0.1 nM or 1 nM) decreased CXCL12-mediated NPC migration in a dose-dependent manner. In comparison, CCX704, a negative control for CCX771, did not alter the migration (Fig. 2B). These results indicate that both CXCR4 and CXCR7 are required for NPC migration towards CXCL12 gradient.
Figure 2.

CXCR4 and CXCR7 are required for CXCL12-mediated neural progenitor cell (NPC) migration. (A, B): NPCs on transwell insert were incubated under the indicated conditions in the presence of CXCL12 and other agents including CXCR4 inhibitor AMD3100 (A), CXCR7 inhibitor CCX771 (B), and its negative control CCX704. Migrated cell numbers of each condition were normalized to the negative control and quantified as migration index. *, p <0.05.
CXCR7/CXCL12 Mediates Migration in Cxcr4 KO NPCs
The requirement of CXCR4 for CXCL12-mediated cell migration is well known. Therefore, we focused on CXCR7 and used primary NPCs derived from Cxcr4 KO mice to keep the cells devoid of CXCR4 influence. Cxcr4 wild type NPCs displayed random distribution across the stripes in the BSA alone group (Fig. 3A, 3B). In contrast, 85% of cells aligned on the BSA/CXCL12 stripes (Fig. 3E, 3F). Similar to the wild type NPCs, Cxcr4 KO NPCs also displayed random distribution in the BSA stripes (Fig. 3C, 3D). We expected that Cxcr4 KO NPCs would distribute randomly in the BSA/CXCL12 stripes. Surprisingly, we found that 82% of Cxcr4 KO NPCs also aligned on the stripes of CXCL12/fluorescein (Fig. 3G, 3H). Quantification of the image revealed that both wild type and Cxcr4 KO NPCs migrated to the BSA/CXCL12 stripes, suggesting a likely role of CXCR7 in CXCR4-independent cell migration (Fig. 3I). Consistent with the stripe assay, the transwell assay revealed similar data on the migration of Cxcr4 KO NPCs. Importantly, the CXCL12-mediated migration in Cxcr4 KO NPCs was blocked by CCX771 but not by AMD3100 (Fig. 3J), suggesting that CXCR7 could serve as a functional receptor for CXCL112-mediated NPC migration in the absence of CXCR4.
Figure 3.

CXCR7 mediates neural progenitor cell (NPC) migration in response to CXCL12 in vitro. (A–I): Wild type and Cxcr4 knock out NPCs were seeded on bovine serum albumin (BSA) stripes (A–D) and BSA/CXCL12 stripes (E–H). Cells were stained with nestin (purple) and counted after 48 hours. Scale bar =60 μm (A–H). The quantification of Stripe Assay is shown in (I). (J): NPCs on transwell inserts were incubated under the indicated conditions in the presence of CXCL12 and other agents as indicated. *, p <0.05, **, p <0.01, ***, p <0.001, compared with the untreated control of the same cell type; #, p <0.001; NS, nonsignificant. Abbreviation: BSA, bovine serum albumin.
Cxcr4−/− NPCs Migrate to CXCL12 In Vivo
To determine whether CXCR7 mediates NPC migration in vivo, we used Qtracker, a nanocrystal with stable fluorescence, to label Cxcr4−/− NPCs and observe their movement in the brain with immunohistochemistry technique. Transplanted NPCs formed a track along the needle path cross striatum and cortex, which was labeled as a dash line (Fig. 4A, 4E, 4I). When saline was injected in a parallel track 1 mm away from the NPC track, minimum Qtracker-labeled cells were observed in the saline track (Fig. 4A–4D). In contrast, parallel injection of CXCL12 produced a glial fibrillary acidic protein-visible track and Qtracker-labeled cells were detected in the CXCL12 track (Fig. 4E–4H). Importantly, when CCX771, a CXCR7 antagonist, was used along the Cxcr4−/− NPC track, significantly fewer Qtracker-labeled cells were detected in the CXCL12 track (Fig. 4I–4M), suggesting that CXCR7 promotes the migration of NPC in vivo. Few migrating NPCs were observed in the brain parenchyma between the two tracks. Instead, migrating NPCs were detected along the copus colusm between the two tracks (Fig. 4G, 4H, arrows). Indeed, further examination of the copus colusm area between the two injection tracks revealed migrating Qtracker-labeled NPCs in the CXCL12/Cxcr4−/− NPC group (Fig. 4N, 4O). Together, these data suggest that CXCR7 is important for CXCL12-mediated NPC migration in vivo.
Figure 4.

CXCR7 mediates neural progenitor cell (NPC) migration in response to CXCL12 in vivo. (A–O): Qtracker-labeled Cxcr4−/− NPCs (A–H) or same number of NPCs with CCX771 (100 μM in cell suspensions, (I–L) were transplanted into the left striatum of C57BL/6 mice through intracranial injection. Immediately after NPC transplant, parallel injection of saline (A–D) and CXCL12 (E–L) was performed at 1 mm apart from the NPC injection track. Seven days after injection, mice were killed and migrated NPCs were determined through immunohistochemistry. Scale bar =250 μm (A–L). (M): Migrated NPCs were quantified as the percentage of Qtracker fluorescence in the saline or CXCL12 track to the total Qtracker fluorescence. **, p <0.01, compared with the saline group; #, p <0.05, compared with the CXCL12 group. N =5 for each group. (N–O): Migrating NPCs in the copus colusm were visualized in high magnification pictures taken from the indicated area (arrow) in (G) and (H). Scale bar =20 μm (N–O). Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; GFAP, glial fibrillary acidic protein; NPCs, neural progenitor cell.
CXCR7 Interacts With Rac1 to Induce Polarization and Migration of NPCs
The molecular mechanism of CXCR7-mediated NPC migration remains unclear. To further determine the mechanism of CXCR7-mediated migration, we investigated the polarization processes of the migrating cells. Polarization often precedes migration and is a fundamental process to initiate cell movement [29]. Activation of Rac1 was found to be highly associated with cytoskeleton reorganization [30] and the protrusion of lamellipodia [31, 32]. We performed fluorescence-conjugated phalloidin staining to label F-actin and found that Cxcr4 KO NPCs were polarized and had lamellipodia formation 1 hour after seeding on BSA/CXCL12 stripes (Fig. 5A, 5B). Both CXCR7 (Fig. 5C, 5D) and Rac1 (Fig. 5E, 5F) localized within the cytosol, concentrating at the leading edge of cells towards BSA/CXCL12 stripes. Image analysis revealed that CXCR7 colocalized with Rac1 at the leading edge of the polarized cells, particularly in the region of lamellipodia (Fig. 5G–5J).
Figure 5.
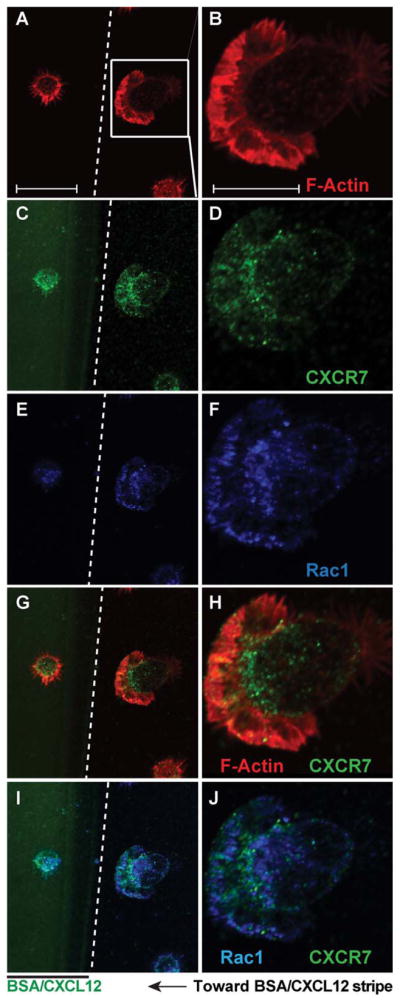
CXCR7 and Rac1 are colocalized in the polarized Cxcr4−/− neural progenitor cells (NPCs). (A–J): Cxcr4−/− NPCs were seeded on the bovine serum albumin (BSA)/CXCL12 stripes. Cells were fixed and stained with F-actin (A, B), CXCR7 (C, D) and Rac1 (E, F) 60 minutes later. (Images were acquired through a confocal microscope. Panels (G–J) were merged pictures of (A) and (C), (B) and (D), (C) and (E), (D) and (F), respectively. Scale bar =10 μm (A, C, E, G, I), 5 μm (B, D, F, H, J). Abbreviation: BSA, bovine serum albumin.
The concentration of Rac1 at the leading edge and its colocalization with CXCR7 indicates a possible involvement of Rac1 in CXCR7-mediated migration. To determine whether Rac1 is activated upon CXCR7 stimulation, cell lysates from Cxcr4 KO NPCs on BSA/CXCL12 stripes were collected at 0, 2, 5, and 10 minutes after seeding. Activated Rac1 was determined by Western blotting using an antibody specific to Rac1-GTP, the active form of Rac1. At as early as 5 minutes after seeding to the CXCL12 stripes, Rac1 in Cxcr4 KO NPCs were activated (Fig. 6A, 6B). Furthermore, IP pulled down by CXCR7 antibody followed by blotting for β-arrestin2 and Rac1 revealed that protein–protein interaction between Rac1 and CXCR7 increased at 2 hours after CXCL12 treatment, compared with 0 hour time point (Fig. 6C). Similarly, β-arrestin2 also increased interaction with CXCR7 upon CXCL12 treatment. These results suggest that upon CXCL12 treatment, CXCR7 interacts with Rac1 and β-arrestin2, which may initiate the intracellular signaling for NPC migration.
Figure 6.

CXCR7 interacts with Rac1 to induce neural progenitor cell (NPC) migration. (A): Cxcr4−/− NPCs were seeded on bovine serum albumin (BSA)/CXCL12 stripes and cell lysates were collected at the indicated time points and subjected to Western blotting for total Rac1 and Rac1-GTP detection. Actin was used as loading control. (B): Fold changes in Rac1GTP were normalized as a ratio of Rac1GTP to total Rac1 after densimetrical quantification and shown as fold changes relative to 0 minute time point. (C): Cxcr4−/− NPCs were treated with CXCL12 at the indicated time points. Cell lysates were treated with anti-CXCR7 antibody to immunoprecipitate CXCR7-Rac1 and CXCR7-β-arrestin2. Protein levels of β-arrestin2 and Rac1 were determined through immunoblotting. (D): NPCs on transwell insert were incubated under the indicated conditions in the presence of CXCL12 or NSC23766. Migrated cell numbers of each condition were normalized to the negative control and quantified as migration index. ***, p <0.001. Abbreviation: IP, immunoprecipitation.
To further investigate the role of Rac1 in CXCR7-mediated NPC migration, we pretreated Cxcr4+/+ and Cxcr4−/− NPCs with NSC23766, a Rac1 inhibitor, to block Rac1 activation. NSC23766-treated Cxcr4+/+ and Cxcr4−/− NPCs no longer responded to the CXCL12 in the transwell assay (Fig. 6D), suggesting that Rac1 is required for CXCR4-independent CXCR7-mediated NPC migration.
CXCR7/CXCL12 Mediates NPC Migration Through ERK1/2 Activation
The MAPK pathway is critical in regulating CXCL12-dependent cell migration [37]. Our previous study has demonstrated that CXCL12 mediated NPC survival through ERK1/2 activation [24]. To further investigate whether CXCR7 was able to activate ERK1/2, we used NPCs derived from Cxcr4 KO mice to rule out the effect of CXCR4 and its potential crosstalk with CXCR7. During a timecourse study, ERK1/2 was quickly phosphorylated upon CXCL12 treatment. After CXCL12 treatment, the dynamics of ERK1/2 phosphorylation in the Cxcr4 KO NPCs were similar to that of the wild type NPCs (Fig. 7A, 7B), suggesting CXCR7 can activate ERK1/2 in the absence of CXCR4 after CXCL12 treatment. However, the ERK1/2 phosphorylation in Cxcr4−/− NPCs was not as robust as that of the wild type NPCs, underscoring the potential effect of CXCR4 and its crosstalk with CXCR7 signaling.
Figure 7.

Extracellular signal-regulated kinases (ERK)1/2 is critical for CXCR7-mediated neural progenitor cell (NPC) migration. (A): Cxcr4−/− and wild type NPCs were treated with CXCL12 (100 ng/ml), and collected at the indicated time points. Total ERK1/2 and phospho-ERK1/2 were detected by Western blotting. Actin was used as loading control. (B): Fold changes of ERK1/2 activation were normalized as a ratio of phosphorylated ERK1/2 to total ERK1/2 after densimetrical quantification of panel (A) and shown as fold changes relative to 0 minute time point. (C): Wild type and Cxcr4−/− NPCs were pretreated with CCX771 or MSC23766 for 2 hours before the CXCL12 treatment. Fifteen minutes after CXCL12 treatment, cell lysates were collected and phosphorylated ERK1/2 and total ERK1/2 were determined through Western blotting. (D–H): Wild type and Cxcr4−/− NPCs were seeded on bovine serum albumin (BSA) stripes (12 panels to the left, E–H) or BSA/CXCL12 stripes (12 panels to the right (E–H) and with (F, H) or without (E, G) treatment of PD98059 for 48 hours. Scale bar =60 μm (E–H). (D): Cell migration was quantified as migration index. ***, p <0.001. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; DMSO, dimethyl sulfoxide; p-ERK, phospho-extracellular signal-regulated kinases.
Next, we investigated the mechanism of ERK1/2 activation in NPC migration. We used CCX771 and NSC23766 (Rac1 inhibitor), and determined the ERK1/2 phosphorylation through Western blotting. CCX771 and NSC23766 blocked CXCL12-mediated ERK1/2 phosphorylation in both Cxcr4+/+ and Cxcr4−/− NPCs, suggesting that both CXCR7 and Rac1 are required for the ERK1/2 activation in the migrating cells (Fig. 7C).
To further investigate whether ERK1/2 activation is essential for CXCR7-mediated NPC migration to CXCL12, ERK inhibitor PD98059 (50 μM) was used to pretreat cells before the stripe assays were performed. Treatment with PD98059 abolished the CXCL12-mediated migration in both wild type NPC and Cxcr4 KO NPCs (Fig. 5D–5H), suggesting that ERK1/2 activation is a critical signaling mechanism for CXCR7-mediated NPC migration. Together, these results further detemined that ERK1/2 activation contributes to CXCR7-mediated NPC migration to CXCL12 in the absence of CXCR4.
Discussion
The discovery of CXCR7 has added to the dynamics of the CXCL12/CXCR4 pair during development and in adulthood. Recent evidence has suggested that CXCR7 is able to induced endocytotic signaling. However, the significance of CXCR7-induced signaling remains to be fully defined. In this study, we demonstrated that CXCR7 is expressed in mouse NPCs, and plays a novel role in CXCR4-independent NPC migration in response to CXCL12 in both in vitro and in vivo NPC migration models. Furthermore, we identified Rac1 and ERK1/2 activation as a critical molecular mechanism for CXCR7-mediated migration. This essential role of CXCR7 in NPC migration is important to understand neurogenesis during development and in adulthood. Similar to CXCL12 and CXCR4, CXCR7 also displays marked expression in neuronal tissue. From E11.5 in mice, strong expression of CXCR7 mRNA is detected in the neural tube, the brain, and the septum transversum [17]. Previous work has demonstrated that CXCR7 regulates interneuron migration, either through activating MAPK or regulating chemokine responsiveness [20, 21, 38]. The exact role of CXCR7 in NPC migration has directed researchers’ attention to explore it in disease models [39]. However, whether this new receptor could independently mediate NPC migration remains unknown. Our study is the first line of evidence supporting the independent role of CXCR7 in mediating cell migration.
The stripe assay was originally established as an in vitro assay to study the response of axon growth cones and cell migration towards guidance cues [40–42]. The assay provided a microfluidic environment with patterns of stripes formed by BSA-fluorescein coating, which is an excellent carrier for small molecules including CXCL12. The assay is also a unique model to study cell migration since it provides a long-lasting concentration gradient of chemokines and the migrating or polarized cells can be visualized with ease. Our data with the stripe assay demonstrated that more than 85% NPCs migrated onto stripes coated with BSA-fluorescein conjugated with CXCL12. This result is consistent with the transwell chemotaxis assay, which showed increased migration index dependent on the concentration of CXCL12. AMD3100 and CCX771 were very specific for their blocking of individual CXCR4 and CXCR7. Interestingly, either CXCR4 or CXCR7 antagonist was able to block wild type NPC migration, suggesting that NPC migration to CXCL12 may depend on the interactions of CXCR4 and CXCR7 under the physiological condition. Cxcr4 KO mice are appropriate for the studying of the CXCR4-independent function. Due to the overwhelming evidence of CXCR4-mediated NPC migration [43, 44], we were surprised to find that Cxcr4 KO NPCs migrated to BSA/CXCL12 stripes and the effect could be blocked by the CCX771 but not by the AMD3100. These data further suggest that CXCR7 mediate NPC migration in the absence of CXCR4. Consistent with the migration data, Cxcr4 KO NPCs also were found to polarize with the formation of lamellipodia, which is a pivotal process for migration. These results suggest that CXCR7 is the essential receptor for CXCL12-mediated NPC migration in the absence of CXCR4.
CXCR7, a 7-Transmembrane receptor, was previously an orphan receptor until it was found as a receptor for CXCL12. Due to the mutation at “DRAILIV” motif [27], CXCR7 cannot activate G proteins that are associated with typical chemokine receptors. Therefore, CXCR7 has been proposed as a “decoy” receptor, mainly functioning to shape CXCL12 gradient for the guidance of cell migration in different models, including recent reports on self-guidance by shaping gradients of CXCL12 in the migration of posterior lateral line primordium in Zebrafish [45]. As a scavenger receptor for CXCL12, CXCR7 was also proposed to regulate the function of CXCR4 by modulating degradation of CXCR4 through excessive CXCL12 during the migration of interneurons during development [21]. On the other hand, in interneuron migration, it was proposed that CXCR7 functions through activation of MAPK with the interaction of β-arrestin2 cascade [38]. Although CXCR7 activation is proposed to mediate NPC migration [39], the detailed mechanism remains to be studied. MAPK is heavily involved in the functions of NPCs including survival, proliferation, and migration. Specifically, ERK1/2 is an important MAPK to modulate the cytoskeleton proteins, which are the effectors for cell migration. Our study demonstrated that the ERK1/2 inhibitor blocked migration towards CXCL12 of both wild type NPCs and Cxcr4 KO NPCs, underscoring the importance of ERK1/2 in independent CXCR7 function. Meanwhile, we have demonstrated the colocalization of CXCR7 and Rac1 in polarized NPCs towards CXCL12 by immunostaining, and IP data further confirmed the increased interaction between CXCR7 and Rac1 after CXCL12 treatment. Inhibiting Rac1 activation also blocked CXCR7-mediated NPC migration in both wild type and Cxcr4 KO, indicating that Rac1 is also required in CXCR7-mediated NPC migration to CXCL12. Rac1 is a member of the Rho family of small signaling G proteins [46]. It is well known that Rac1-GTPase cycles between GTP-active and GDP-inactive states [47]. CXCL12/CXCR4 could induce the polarization of a variety of cells by activating Rac1 [36, 48]. Rac1 is known to regulate cytoskeleton reorganization [30] and functions at the leading edge of cells. Activated Rac1 promotes lamellipodial protrusion [31, 32]. Our data suggest that Rac1 is activated at the leading edge of Cxcr4 KO NPCs towards CXCL12 and is essential for CXCR7-mediated NPC migration. Both immunostaining and IP results indicated the interaction between CXCR7 and Rac1 independent of CXCR4 in the Cxcr4 KO system, as evidence for CXCR7 functioning and signaling in NPC migration.
Cell migration is a process of integration of different signaling pathways. Polarization is a keystone of cell migration, and a cell must polarize first before migrating [36]. Specifically, Rac1 activation initiates and defines the protrusion/the leading edge of the migrating cell through formation of the lamellipodial protrusion [31, 32]. Our study in the Cxcr4 KO NPCs indicates that CXCR7 is specifically involved in NPC polarization. Treatment of CXCL12 for one hour on stripes was sufficient to initiate Cxcr4 KO NPC polarization. Our data revealed the interaction between CXCR7 and Rac1 during NPC polarization to CXCL12. Rac1 inhibitor NSC23766 blocked Cxcr4 KO NPC migration to CXCL12, further indicating that Rac1 is required in CXCR7-mediated NPC migration. Together, these results suggest ERK1/2 and Rac1 are required for CXCR7-mediated NPC migration in the absence of CXCR4.
Neurogenesis is a process that is reliant upon the proper proliferation, migration, survival, and differentiation of NPCs. NPC migration and survival are two of the pivotal functions strongly associated with in vivo CXCL12 gradient. Stem cell regenerative therapy is one of the promising therapeutic approaches for neurodegenerative diseases because stem cells have the potential to suppress brain inflammation, provide tropic support, and improve both the endogenous neurogenesis and exogenous neurogenesis for the brain [49–52]. CXCL12 has been proposed to be a chemokine to mediate transplanted stem cells migration [33, 53]. Our data provide the first evidence that CXCR7 mediates the migration of transplanted NPCs. However, the transplantation was performed in healthy mouse brains. It is unknown whether an inflammatory brain environment will affect NPC migration differently, which may account for the discrepancy between our data and the published literature [53].
Conclusion
Our study has shown the novel role of CXCR7 as a functional (migration and polarization) and signaling (ERK1/2 and Rac1) receptor for CXCL12-mediated NPC migration in the absence of CXCR4. These functional effects and molecular events of CXCR7 will provide important therapeutic targets in modulating NPC migration and neurogenesis.
Supplementary Material
Acknowledgments
We kindly acknowledge Beibei Jia, Kristin Leland Wavrin, Li Wu, Dr. Changhai Tian, and Hui Peng, who provided technical support for this work. We thank Janice A. Taylor and James R. Talaska of the Advanced Microscopy Core Facility at the University of Nebraska Medical Center for providing assistance with microscopy, the NIH S10-RR-027301, Nebraska Research Initiative and the Eppley Cancer Center (P30CA036727) for their support of the Core Facility. Julie Ditter, Lenal Bottoms, Jaclyn Ostronic, Myhanh Che, Johna Belling, and Robin Taylor provided outstanding administrative and secretarial support. This work was partly supported by National Basic Research Program of China (973 Program Grant 2014CB965000, project 1 Grant 2014CB965001, and project 3 Grant 2014CB965003), National Natural Science Foundation of China (Grants 81271419 and 81271420), and Joint Research Fund for Overseas Chinese, Hong Kong and Macao Young Scientists of the National Natural Science Foundation of China (Grant 81329002); NIH Grants R01 NS 41858-01, R01 NS 061642-01, the State of Nebraska, DHHS-LB606 Stem Cell 2009-10.
Footnotes
Author Contributions
J.C.Z., D.X., and Y.H.: conception and design, data analysis and interpretation, writing and final approval of the manuscript: Q.C., M.Z., Y.L., A.S., and B.Z.: participation in the design, data analysis and interpretation of the studies; Q.C., M.Z., Y.L., D.X., Y.W., A.S., B.Z., and Y.H.: data collection and provision of study materials; Q.C., M.Z.: manuscript writing. Q.C., M.Z., and Y.L. contributed equally to this article.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
See www.StemCells.com for supporting information available online.
References
- 1.Eriksson PS, Perfilieva E, Bjork-Eriksson T, et al. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- 2.Parent JM. Injury-induced neurogenesis in the adult mammalian brain. Neuroscientist. 2003;9:261–272. doi: 10.1177/1073858403252680. [DOI] [PubMed] [Google Scholar]
- 3.Gensert JM, Goldman JE. Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron. 1997;19:197–203. doi: 10.1016/s0896-6273(00)80359-1. [DOI] [PubMed] [Google Scholar]
- 4.Arvidsson A, Collin T, Kirik D, et al. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- 5.Alvarez-Buylla A, Seri B, Doetsch F. Identification of neural stem cells in the adult vertebrate brain. Brain Res Bull. 2002;57:751–758. doi: 10.1016/s0361-9230(01)00770-5. [DOI] [PubMed] [Google Scholar]
- 6.Kintner C. Neurogenesis in embryos and in adult neural stem cells. J Neurosci. 2002;22:639–643. doi: 10.1523/JNEUROSCI.22-03-00639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sukhikh GT, Malaitsev VV. Neural stem cell: Biology and prospects of neurotransplantation. Bulletin of Exp Biol Med. 2001;131:203–212. doi: 10.1023/a:1017503325833. [DOI] [PubMed] [Google Scholar]
- 8.Honmou O, Uede T, Hashi K. Neural stem cells derived from adult human brain: Implications for a cell therapy for CNS diseases [in Japanese] No shinkei geka (Neurol Surg) 2001;29:293–304. [PubMed] [Google Scholar]
- 9.Armstrong RJ, Rosser AE, Dunnett SB, et al. Neural stem cell technology as a novel treatment for Parkinson’s disease. Methods Mol Med. 2001;62:289–307. doi: 10.1385/1-59259-142-6:289. [DOI] [PubMed] [Google Scholar]
- 10.Banisadr G, Skrzydelski D, Kitabgi P, et al. Highly regionalized distribution of stromal cell-derived factor-1/CXCL12 in adult rat brain: Constitutive expression in cholinergic, dopaminergic and vasopressinergic neurons. Eur J Neurosci. 2003;18:1593–1606. doi: 10.1046/j.1460-9568.2003.02893.x. [DOI] [PubMed] [Google Scholar]
- 11.Li M, Ransohoff RM. Multiple roles of chemokine CXCL12 in the central nervous system: A migration from immunology to neurobiology. Prog Neurobiol. 2008;84:116–131. doi: 10.1016/j.pneurobio.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou YR, Kottmann AH, Kuroda M, et al. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 13.Lu M, Grove EA, Miller RJ. Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc Natl Acad Sci U S A. 2002;99:7090–7095. doi: 10.1073/pnas.092013799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006;7:243. doi: 10.1186/gb-2006-7-12-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein RS, Rubin JB, Gibson HD, et al. SDF-1 alpha induces chemotaxis and enhances Sonic hedgehog-induced proliferation of cerebellar granule cells. Development. 2001;128:1971–1981. doi: 10.1242/dev.128.11.1971. [DOI] [PubMed] [Google Scholar]
- 16.Imitola J, Raddassi K, Park KI, et al. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci U S A. 2004;101:18117–18122. doi: 10.1073/pnas.0408258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sierro F, Biben C, Martinez-Munoz L, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U S A. 2007;104:14759–14764. doi: 10.1073/pnas.0702229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balabanian K, Lagane B, Infantino S, et al. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem. 2005;280:35760–35766. doi: 10.1074/jbc.M508234200. [DOI] [PubMed] [Google Scholar]
- 19.Mahabaleshwar H, Tarbashevich K, Nowak M, et al. beta-arrestin control of late endosomal sorting facilitates decoy receptor function and chemokine gradient formation. Development. 2012;139:2897–2902. doi: 10.1242/dev.080408. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez-Alcaniz JA, Haege S, Mueller W, et al. Cxcr7 controls neuronal migration by regulating chemokine responsiveness. Neuron. 2011;69:77–90. doi: 10.1016/j.neuron.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 21.Abe P, Mueller W, Schutz D, et al. CXCR7 prevents excessive CXCL12-mediated downregulation of CXCR4 in migrating cortical inter-neurons. Development. 2014;141:1857–1863. doi: 10.1242/dev.104224. [DOI] [PubMed] [Google Scholar]
- 22.Naumann U, Cameroni E, Pruenster M, et al. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS One. 2010;5:e9175. doi: 10.1371/journal.pone.0009175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajagopal S, Kim J, Ahn S, et al. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A. 2010;107:628–632. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu B, Xu D, Deng X, et al. CXCL12 enhances human neural progenitor cell survival through a CXCR7- and CXCR4-mediated endocytotic signaling pathway. Stem Cells. 2012;30:2571–2583. doi: 10.1002/stem.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levoye A, Balabanian K, Baleux F, et al. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood. 2009;113:6085–6093. doi: 10.1182/blood-2008-12-196618. [DOI] [PubMed] [Google Scholar]
- 26.Gerrits H, van Ingen Schenau DS, Bakker NE, et al. Early postnatal lethality and cardiovascular defects in CXCR7-deficient mice. Genesis. 2008;46:235–245. doi: 10.1002/dvg.20387. [DOI] [PubMed] [Google Scholar]
- 27.Thelen M, Thelen S. CXCR7, CXCR4 and CXCL12: An eccentric trio? J Neuroimmunol. 2008;198:9–13. doi: 10.1016/j.jneuroim.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 28.Nibbs RJ, Graham GJ. Immune regulation by atypical chemokine receptors. Nat Rev Immunol. 2013;13:815–829. doi: 10.1038/nri3544. [DOI] [PubMed] [Google Scholar]
- 29.Etienne-Manneville S. Polarity proteins in migration and invasion. Oncogene. 2008;27:6970–6980. doi: 10.1038/onc.2008.347. [DOI] [PubMed] [Google Scholar]
- 30.Villalba M, Bi K, Rodriguez F, et al. Vav1/Rac-dependent actin cytoskeleton reorganization is required for lipid raft clustering in T cells. J Cell Biol. 2001;155:331–338. doi: 10.1083/jcb.200107080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ridley AJ, Paterson HF, Johnston CL, et al. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 32.Waterman-Storer CM, Worthylake RA, Liu BP, et al. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat Cell Biol. 1999;1:45–50. doi: 10.1038/9018. [DOI] [PubMed] [Google Scholar]
- 33.Wu Y, Chen Q, Peng H, et al. Directed migration of human neural progenitor cells to interleukin-1beta is promoted by chemokines stromal cell-derived factor-1 and monocyte chemotactic factor-1 in mouse brains. Transl Neurodegener. 2012;1:15. doi: 10.1186/2047-9158-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye L, Huang Y, Zhao L, et al. IL-1beta and TNF-alpha induce neurotoxicity through glutamate production: A potential role for neuronal glutaminase. J Neurochem. 2013;125:897–908. doi: 10.1111/jnc.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cruz-Orengo L, Holman DW, Dorsey D, et al. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J Exp Med. 2011;208:327–339. doi: 10.1084/jem.20102010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: Integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 37.Alsayed Y, Ngo H, Runnels J, et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood. 2007;109:2708–2717. doi: 10.1182/blood-2006-07-035857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Li G, Stanco A, et al. CXCR4 and CXCR7 have distinct functions in regulating interneuron migration. Neuron. 2011;69:61–76. doi: 10.1016/j.neuron.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Merino JJ, Oset-Gasque MJ. The CXCR7 activation by SDF1 induces Neural progenitor migration (NPC): A dual effect on CXCR4/CXCR7 axis within the vascular niche of ischemic rats. Int J Stroke. 2013;8:E56. doi: 10.1111/ijs.12174. [DOI] [PubMed] [Google Scholar]
- 40.Vielmetter J, Stolze B, Bonhoeffer F, et al. In vitro assay to test differential substrate affinities of growing axons and migratory cells. Exp Brain Res. 1990;81:283–287. doi: 10.1007/BF00228117. [DOI] [PubMed] [Google Scholar]
- 41.Wang HU, Anderson DJ. Eph family transmembrane ligands can mediate repulsive guidance of trunk neural crest migration and motor axon outgrowth. Neuron. 1997;18:383–396. doi: 10.1016/s0896-6273(00)81240-4. [DOI] [PubMed] [Google Scholar]
- 42.Barchi L, Bonnet J, Boudet C, et al. A high-resolution, intraspecific linkage map of pepper (Capsicum annuum L.) and selection of reduced recombinant inbred line subsets for fast mapping. Genome. 2007;50:51–60. doi: 10.1139/g06-140. [DOI] [PubMed] [Google Scholar]
- 43.Schultheiss C, Abe P, Hoffmann F, et al. CXCR4 prevents dispersion of granule neuron precursors in the adult dentate gyrus. Hippocampus. 2013;23:1345–1358. doi: 10.1002/hipo.22180. [DOI] [PubMed] [Google Scholar]
- 44.Stumm RK, Zhou C, Ara T, et al. CXCR4 regulates interneuron migration in the developing neocortex. J Neurosci. 2003;23:5123–5130. doi: 10.1523/JNEUROSCI.23-12-05123.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Venkiteswaran G, Lewellis SW, Wang J, et al. Generation and dynamics of an endogenous, self-generated signaling gradient across a migrating tissue. Cell. 2013;155:674–687. doi: 10.1016/j.cell.2013.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Didsbury J, Weber RF, Bokoch GM, et al. rac, a novel ras-related family of proteins that are botulinum toxin substrates. J Biol Chem. 1989;264:16378–16382. [PubMed] [Google Scholar]
- 47.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 48.del Pozo MA, Vicente-Manzanares M, Tejedor R, et al. Rho GTPases control migration and polarization of adhesion molecules and cytoskeletal ERM components in T lymphocytes. Eur J Immunol. 1999;29:3609–3620. doi: 10.1002/(SICI)1521-4141(199911)29:11<3609::AID-IMMU3609>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 49.Jeong SW, Chu K, Jung KH, et al. Human neural stem cell transplantation promotes functional recovery in rats with experimental intracerebral hemorrhage. Stroke. 2003;34:2258–2263. doi: 10.1161/01.STR.0000083698.20199.1F. [DOI] [PubMed] [Google Scholar]
- 50.Iguchi F, Nakagawa T, Tateya I, et al. Trophic support of mouse inner ear by neural stem cell transplantation. Neuroreport. 2003;14:77–80. doi: 10.1097/00001756-200301200-00015. [DOI] [PubMed] [Google Scholar]
- 51.Rosser AE, Zietlow R, Dunnett SB. Stem cell transplantation for neurodegenerative diseases. Current Opin Neurol. 2007;20:688–692. doi: 10.1097/WCO.0b013e3282f132fc. [DOI] [PubMed] [Google Scholar]
- 52.Shihabuddin LS, Aubert I. Stem cell transplantation for neurometabolic and neurodegenerative diseases. Neuropharmacology. 2010;58:845–854. doi: 10.1016/j.neuropharm.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 53.Carbajal KS, Schaumburg C, Strieter R, et al. Migration of engrafted neural stem cells is mediated by CXCL12 signaling through CXCR4 in a viral model of multiple sclerosis. Proc Natl Acad Sci U S A. 2010;107:11068–11073. doi: 10.1073/pnas.1006375107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.