

Abstract
Although chemotherapy-induced peripheral neuropathy (CIPN) affects approximately 5-60% of cancer patients, there are currently no treatments available in part due to the fact that the underlying causes of CIPN are not well understood. One contributing factor in CIPN may be persistence of DNA lesions resulting from treatment with platinum-based agents such as cisplatin. In support of this hypothesis, overexpression of the base excision repair (BER) enzyme, apurinic/apyrimidinic endonuclease 1 (APE1), reduces DNA damage and protects cultured sensory neurons treated with cisplatin. Here, we address stimulation of APE1's endonuclease activity through a small molecule, nicorandil, as a means of mimicking the beneficial effects observed for overexpression of APE1. Nicorandil was identified through high-throughput screening of small molecule libraries and found to stimulate APE1 endonuclease activity by increasing catalytic efficiency approximately 2-fold. This stimulation is primarily due to an increase in kcat. To prevent metabolism of nicorandil, an approved drug in Europe for the treatment of angina, cultured sensory neurons were pretreated with nicorandil and daidzin, an aldehyde dehydrogenase 2 inhibitor, resulting in decreased DNA damage but not altered transmitter release by cisplatin. This finding suggests that activation of APE1 by nicorandil in cisplatin-treated cultured sensory neurons does not imbalance the BER pathway in contrast to overexpression of the kinetically faster R177A APE1. Taken together, our results suggest that APE1 activators can be used to reduce DNA damage induced by cisplatin in cultured sensory neurons, although further studies will be required to fully assess their protective effects.
Keywords: chemotherapy-induced peripheral neuropathy, apurinic/aprymidinic endonuclease 1, small molecular activator, cisplatin, base excision repair
1. Introduction
Chemotherapy-induced peripheral neuropathy (CIPN) is one of the most prevalent dose-limiting toxicities of anticancer therapy. Of the patients receiving various anticancer drugs, 5-60% develop peripheral neuropathy depending on the drug used and the dosage administered [1], which often is severe enough to require alterations in therapy [1]. In addition, in a significant cohort of patients, the neuropathy persists even after therapy is discontinued [2]. To date, there are no therapies available to prevent or reverse CIPN [1], and the mechanisms that result in CIPN are unknown.
One potential mechanism that could account for neurotoxicity after chemotherapy is an increase in DNA damage, which in post-mitotic cells could result in alterations in protein expression and/or expression of mutated proteins and, in turn, could alter the function of peripheral neurons. In our previous studies, we established a causal relationship between CIPN and DNA damage and repair. Although cisplatin reacts with guanines to form intrastrand and interstrand DNA crosslinks, which are repaired by the nucleotide excision repair (NER) pathway, it also produces reactive oxygen species (ROS), which create oxidative DNA lesions that are repaired by the base excision repair pathway (BER) [3]. We demonstrated that compromising the BER pathway by reducing expression of APE1, a multi-functional DNA repair and redox signaling protein enhanced toxicity induced by cisplatin, whereas overexpression protected sensory neurons from DNA damage caused by cisplatin [4, 5]. Using APE1 mutants, we further demonstrated that the protective effects of APE1 required its endonuclease activity rather than its redox function [4-6]. We therefore hypothesized that if overexpression of APE1 increases DNA repair in cisplatin-treated sensory neurons, it might also be possible to increase DNA repair with a small molecule activator of APE1.
Activation of an enzyme requires acceleration of the slow step of the reaction, which for APE1 is proposed to be product release. Several laboratories have shown that under pre-steady state conditions, the chemistry for APE1's cleavage 5′ of an apurinic/apyrimidinic (AP) site is diffusion limited with turnover rates of 700-850 s-1 while steady state reaction rates are 2 s-1 [7, 8]. In crystal structures of APE1 bound to a substrate mimic, Arg 177 fills the void created by the AP site through intercalation [9, 10]. This finding led to a proposed role for R177 in slowing product release, an idea supported by the fact that R177A APE1 exhibits faster enzyme kinetics than the wild-type protein [9]. More recently, Arg 181 was also shown to contribute to slow product release through hydrogen bonding interactions with the phosphate backbone in the high resolution crystal structure of a product complex [10]. Slow product release by APE1 is proposed to allow recruitment of DNA polymerase β, XRCC1, and DNA ligase III, the BER enzymes required to complete repair and prevent release of the nicked product, which if left unrepaired could prove cytotoxic [9]. To date, this theory regarding slow product release has not been tested in a cellular assay.
The protective effects of APE1 overexpression in cisplatin-treated sensory neurons led us to explore the potential of augmenting APE1 DNA repair in sensory neurons using small molecule enhancers/stimulators since other modes of augmenting APE1 DNA repair function would be impractical; i.e. gene therapy, APE1 specific gene expression for just the repair function or epigenetic approaches. To accomplish this goal, we performed a high throughput screen (HTS) of chemical libraries for small molecules that enhanced APE1 DNA repair activity. Here, we characterize nicorandil as a small molecule activator of APE1 endonuclease activity, examine its effects on redox activity, and compare the effects of stimulating APE1 activity through use of nicorandil with overexpression of a catalytically faster R177A APE1 in sensory neurons treated with cisplatin.
2. Materials and Methods
2.1 Materials
Nicorandil used in initial screens was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY) as part of an FDA approved collection of compounds and was later obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX). The pharmacologically inactive denitrated metabolite of nicorandil, N-(2-hydroxyethyl)nicotinamide, was purchased from Enamine LLC (Monmouth Junction, NJ). Daidzin was purchased from TSZ Chemical/Biotang Inc (Lexington, MA). Tissue culture supplies were obtained from Invitrogen (Carlsbad, CA) and normocin from Invivogen (San Diego, CA). Capsaicin, 1-methyl-2-pyrrolidinone (MPL), poly-D-lysine, laminin, and routine chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific. MPL was the vehicle used to prepare water-insoluble drugs for use with cultured sensory neurons; the maximal concentration of MPL used was 0.01%. Nerve growth factor was purchased from Harlan Bioproducts for Science (Indianapolis, IN). Mouse monoclonal antihuman APE1 antibodies were raised in our laboratory and available from Novus Biologicals (Littleton, CO), whereas the mouse monoclonal anti-phospho-H2AX antibodies were from EMD Millipore (Billerica, MA) and the mouse monoclonal anti-Vinculin antibody from Sigma-Aldrich (St. Louis, MO). Goat anti-mouse HRP conjugated IgG secondary antibody was from Zymed Laboratories Inc. (San Francisco, CA); actin antibodies were purchased from Thermo (Fremont CA). Anti-HA-HRP was from Miltenyi Biotec Inc. (San Diego, CA). Chemiluminescence secondary antibodies were from Roche Diagnostics Corp. (Indianapolis, IN). Cisplatin was purchased from Sigma-Aldrich Inc. (St. Louis, MO), dissolved in MPL and stored at -20 °C as a 10 mM solution. Before drug treatment, the stock was diluted in F-12 growth medium and added to cultures and incubated for 8-72 hours as indicated. The CGRP antibody was a generous gift from Dr. M. Iadarola at the NIH. The Animal Care and Use Committee at Indiana University School of Medicine, Indianapolis, IN approved all procedures used in these studies.
2.2 Protein purification
Full length wild-type and R177A APE1 were purified as previously described [11]. In brief, the full-length protein was expressed as an N-terminal hexa-His-SUMO fusion protein in E. coli. Following lysis by using a French press, the crude extract was centrifuged at 35,000 rpm and the supernatant applied to a Ni-NTA column. The affinity tag was removed by on-column cleavage with the SUMO-specific Ulp1 protease, and the eluent was then applied to a Q-Sepharose column connected in series to an S-Sepharose column from which the protein was eluted using a NaCl gradient. The sample was then applied to a size exclusion column, concentrated, and stored at -80 °C.
2.3 Kinetic fluorescence-based APE1 endonuclease assays
The assay used to identify modulators of APE1 endonuclease activity and perform subsequent mechanistic studies provides a fluorescent signal following cleavage 5′ of tetrahydrofuran (THF) and release of a fluorescein-labeled product from the following substrate 5′-(6-FAM)-GAATCC-(THF)-CCATACGTATTATATCCAATTCC-3′ and 5′-GGAATTGGATATAATACGTATGGTGGATTC-(DABCYL)-3′ (Eurogentec, San Diego, CA) [12]. Prior to cleavage, the fluorescent signal is quenched due to its proximity to dabcyl in the complementary strand. The activator nicorandil was identified from a HTS of the FDA and clinically approved library from Enzo Life Sciences. Other libraries screened include 60,000 compounds from ChemDiv and selected compounds from ChemBridge, Vitas-M Laboratory Asinex, TimTec, Princeton BioMolecular Research, Life Chemicals, Enamine, and Maybridge. In initial screens, all compounds were assayed for inhibition or activation at concentrations of 10 μM. Enzyme concentrations, typically 0.15-0.35 nM were optimized to produce a linear signal at 37 °C over a 5 minute period using an Ultra Plate Reader (Tecan, Durham NC) in the kinetic mode with an excitation frequency of 495 nM and an emission frequency of 530 nM [12]. Assays were performed in 50 mM Tris, pH 7.5, 1 mM MgCl2 and 50 mM NaCl with varying concentrations of compounds and a final concentration of 1% DMSO in 50 μL total volumes in 384 well plates. The rate of reaction for APE1 without compound was considered as the 100% control, and subsequent stimulation of at least 50% by the compounds was considered relative to that of the control and further subjected to dose response analyses. A secondary gel-based assay was used to confirm the activation results obtained in the kinetic fluorescent based assay [13].
Crude extracts from dorsal root ganglion (DRG) cells were prepared as follows and assayed using the solution assay described above. The cell pellets were washed once and resuspended in 1× PBS with DTT (2 mM final concentration). The cell suspension was sonicated on ice 3× for 45 s with a 1 min rest between. The extract was then centrifuged at 13,000 rpm at 4 °C for 15 min and the supernatant removed from the remaining cell pellet. The protein concentration in the supernatant was measured using the Bradford Assay (Bio-Rad, Hercules, CA), then the extracts were aliquoted and stored at -80 °C until assayed. For APE1 repair activity assays of DRG cell extracts in the presence of nicorandil, each drug treatment was run in triplicate for each 96-well plate assay in a 100 μL volume. Three experiments were performed on cell extracts for a total of nine replicates of each treatment. All wells had a final DMSO concentration of 2%. DRG cell extract was added in a volume of 5 μL (3.3 μg final) to all wells and immediately assayed. The rate of the reaction for each dose was used to determine the change in APE1 repair activity as compared to the vehicle control.
For mechanistic studies, the enzyme concentrations of both wild-type APE1 (0.1 nM) and R177A APE1 (0.0125 nM) were optimized for 10 nM substrate using the same buffering conditions as in initial screens. Nicorandil concentrations were varied from 0.1 μM to 100 μM to determine a dose response curve. The substrate concentration was then varied from 0.1 μM to 100 μM in the presence of 12.5 μM nicorandil to determine its mechanism of action. Kinetic parameters in the presence and absence of nicorandil were calculated for wild-type and R177A APE1 enzymes using GraphPad Prism for data obtained from three independent experiments with triplicate measurements in each. In order to calculate kcat values, we used the total enzyme concentration as the concentration of active enzyme.
2.4 Differential Scanning Fluorimetry
The effects of small molecules on proteins can be assessed by differential scanning fluorimetry [13] with increases in melting temperature indicative of stabilizing interactions and decreases indicative of destabilization of the protein structure [11]. Purified proteins (final concentrations of 5 μM) were incubated with SYPRO orange (excitation at 483 nm and emission at 568 nm) and varying concentrations of nicorandil at 25 °C in 50 mM HEPES (pH 7.5), 50 mM KCl, 1 mM DTT. 1 mM MgCl2 was included where specified. The fluorescence was observed and processed with the LightCycle 480 Real-Time PCR System (Roche, Switzerland).
2.5 Cell culture
Sensory neuronal cultures were prepared as previously described [14]. Adult male Sprague-Dawley rats (150-175g; Harlan, Indianapolis, IN) were euthanized by CO2 asphyxiation and the dorsal root ganglia (DRG) were collected from all levels of the spinal column. After the nerve roots were trimmed, the DRGs were incubated in collagenase (1 mg/mL) for 1 hr at 37 °C and dissociated by mechanical agitation. For studies measuring cell viability, approximately 60,000 cells were plated into each well of 6-well culture plates precoated with poly-D-lysine and laminin. For release experiments and to assess DNA damage, approximately 30,000 cells were plated onto 12 well culture plates. Cultures were maintained in F-12 media (Invitrogen, Carlsbad, CA) supplemented with 10% horse serum, 2mM glutamine, 100 μg/mL normocin™, 50 μg/mL penicillin, 50 μg/mL streptomycin, 50μM 5-fluoro-2′-deoxyuridine (Invitrogen), 150 μM uridine, and 30 ng/mL of NGF (Harlan Bioproducts for Science, Inc. Indianapolis, IN) in 3% CO2 at 37 °C. Growth medium was changed every other day, and the cells were used after 11-13 days of culture.
2.6 Trypan blue exclusion
Trypan blue exclusion analysis was performed as previously described [15] to assess cell viability. After various treatments, cells in culture were suspended using a 0.05% trypsin–EDTA solution. Equal volumes of the cell suspension and 0.4% (w/v) trypan blue in phosphate-buffered saline (PBS) were mixed, and the cells scored under phase contrast microscope. Dead cells were those that took up the blue stain of trypan blue.
2.7 siRNA transfection
To decrease APE1 in sensory neuronal cultures, APE1siRNA and SCsiAPE1 (scrambled control) were transfected into cells as described previously [15, 16]. On day 4 in culture, the growth media was replaced with 0.5 mL of Opti-MEM® I medium containing 10 μL of the transfecting reagent Neuroporter in the absence or presence of the 21-mer oligonucleotide double-stranded siRNA to APE1 (APE1siRNA) or scrambled APE1siRNA (SCsiRNA). The APE1siRNA sequences are: 5′-GUCUGGUAAGACUGGAGUACC-3′ for the top strand and 5′-UACUCCAGUCUUACCAGACCU-3′ for the bottom strand. The SCsiRNA sequences are 5′-CCAUGAGGUCAGCAUGGUCUG-3′ for the top strand and 5′-AAGGUACUCCAGUCGUACCAG-3′ for the bottom strand. Fresh medium (0.5 mL) without antibiotics was added after 24 hrs of incubation and after an additional 24 hrs, the medium was replaced with normal medium containing antibiotics and cell growth maintained.
2.8 Viral Infection
The ViraPower™ Promoterless Lentiviral Gateway® Expression System protocol was performed per manufacturer's instructions for recombinational cloning to move R177A-APE1-IRES-EGFP from pENTR1A into the lentiviral transfer vector, pcsgw-DEST (Vector Production Facility, Indiana University School of Medicine). The recombination reaction is catalyzed by LR Clonase™ II enzyme mix resulting in the pLenti6-R4R2-V5 plasmid containing the CMVpromoter-R177AAPE1-IRES-EGFP. The construct was transformed into STBL3 E. coli (Invitrogen Life Technologies) and then selection was performed using ampicillin resistance and ccdBs sensitivity. On day one of viral production, T75 or T175 flasks were plated with 5×106 or 1.2×107 293FT cells (Invitrogen Life Technologies) in 12 or 25 ml of medium, respectively. Cells were grown at 37 °C in 5% CO2 for 24 hrs or until 80-85% confluent. On day two, calcium phosphate transfection was performed using the Profection Mammalian Transfection System (Promega) per manufacturer's instructions. On day 3, the media was changed to Opti-MEM® I Reduced Serum Media (Invitrogen Life Technologies) 15-17 hours post-transfection. On days 4-5, the medium is collected and the viral containing supernatant filtered using a 0.45 μM SFCA syringe or flask filter. The virus was concentrated using Centricon Plus-70 Centrifugal Filters (Millipore), collected, aliquoted, and stored at -80°C, and the viral titers were determined using QuickTiter™ Lentivirus Quantitation Kit (Cell Biolabs, Inc.). For lentiviral infections, DRG cells were cultured for 5 days and 150 pfu/cell of the lentivirus added to the media. Two days later the virus was removed and cells grown for an additional 5 days in regular media.
2.9 Western Blot Analysis
Cells in culture were scraped and lysed in 100 μL of lysis buffer (10 mM Tris, pH 7.5, 0.1 M NaCl, 1mM EDTA, 0.01% Triton X-100). Protein (40 μg) was quantified and electrophoresed in SDS gel-loading buffer on a 10% SDS–polyacrylamide gel. Protein concentration was quantified using the Lowry-based protein assay (Bio-Rad Laboratories, Hercules, CA) [17]. Protein (40 μg) was electrophoresed in SDS gel-loading buffer on a 10% SDS-polyacrylamide gel. After electrophoresis, the gel was transferred to a PVDF membrane. The membrane was blocked in blocking solution (TBST containing 5% nonfat dry milk) for 1 h at room temperature with agitation. The membrane was incubated with a mouse monoclonal antihuman APE1 (1:1000 for determination of immunoreactive-APE1 [4, 17]), antibodies to HA (1:1000; from Miltenyi Biotec Inc. (San Diego, CA) or mouse monoclonal anti-phospho-histone H2AX (1:500; EMD Millipore (Billerica, MA), for assessment of DNA double strand breaks, mouse monoclonal anti-vinculin (1:3000; Sigma-Aldrich (St. Louis, MO). The membrane was then washed three times (10 min each) with TBST. The washed membrane was incubated with HRP conjugated secondary antibody (1:3000) in blocking solution for 1 h at room temperature with agitation. The membrane was washed three times (10 min each) with TBST. The bands were visualized using autoradiographic film, density measured, and data expressed as density normalized to vinculin.
2.10 Release of Calcitonin gene-related peptide (CGRP)
Release studies were performed on the cells as previously described [4, 16]. On days 12-14 after harvest, the cultures were washed with HEPES buffer consisting of 25 mM HEPES, 135 mM NaCl, 3.5 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, 3.3 mM D-glucose, and 0.1% bovine serum albumin, pH 7.4 and maintained at 37 °C, then incubated for successive 10 min intervals with 0.4 mL of the same buffer. Basal or resting release was determined by exposing the cells to HEPES buffer alone, whereas stimulated release was determined by exposing the cultures to 30 nM capsaicin. During incubations, the cells were maintained in at 37 °C. After each incubation, the buffer was removed to measure the amount of CGRP using radioimmunoassay (RIA) as previously described [4, 16, 18]. At the end of each release experiment, the cells are scraped in 0.4 M HCl, sonicated, and an aliquot taken to measure total CGRP content in the cultures using RIA. Total content is determined by adding the total amount released in all incubations to the amount remaining in the cells.
2.11 Statistical analysis for experiments involving cultured sensory neurons
Data are expressed as the mean ± the standard error of the mean for at least three independent experiments from separate harvests. Data were subjected to parametric analysis by ANOVA followed by the Tukey test to determine statistically significant differences between treatment groups. Statistical significance was taken as p < 0.05 in all experiments.
2.12 Cell-based redox reporter assays
A cell-based transcription factor reporter assay was used as previously described [17, 19, 20] to determine the redox activity of nicorandil, daidzin, or nicorandil-daidzin combo along with the APE1 redox inhibitor, E3330 as a control. Briefly, the pRL Activator Protein 1 (AP-1) transcription factor firefly reporter vector (PathDetect cis-Reporting Systems, Stratagene) and the control pRL Renilla-TK (Promega Corp.) were cotransfected in patient-derived Pa02c cells at a 20:1 ratio (1.0 μg : 0.05 μg, respectively) using 3.0 μL/mL Lipofectamine®2000 (Invitrogen Life Technologies) in Opti-MEM® (ThermoFisher). After cells were incubated 24 hours (37°C, 5% CO2), media was exchanged, and following a 24-hour recovery, cells were washed in PBS and then treated with the various drug doses in Opti-MEM® of either E3330, nicorandil (Sigma-Aldrich), daidzin (Indofine Chemical), or nicorandil and 10 μM daidzin, along with vehicle (DMSO) and media controls. Following 24-hour exposure (37°C, 5% 2), firefly luciferase and Renilla activities were measured by the Promega Dual-Luciferase® system and quantified by the BioTek Synergy™ H4 fluorescent plate reader. For each dose, AP-1 luciferase RLU was normalized with Renilla-TK RLU and then further normalized to background media.
2.13 Redox electrophoretic mobility shift assay (EMSA)
Inhibition of APE1 redox activity was measured using an EMSA as routinely used in our laboratory and previously described [19, 21]. Briefly, purified APE1 protein (10 mg/mL) was reduced with DTT (1.0 mM) at 37 °C for 10 min and then diluted with PBS buffer to final concentrations of APE1 and DTT of 2 mg/mL and 0.2 mM, respectively. Reduced APE1 protein and 6 μg of oxidized nuclear extracts (Hey-C2 cells, treated with 0.01 mM diamide for 10 min) were added, incubated for 30 min, and one mL of poly(dI-dC) · poly(dI-dC) (1 mg/L, Amersham Biosciences, Piscataway, NJ) was added for 5 min followed by one mL of the 5′ hexachlorofluorescein phosphoramidite (HEX)-labeled double-stranded oligonucleotide DNA (0.1 pmol, The Midland Certified Reagent Company, Midland, TX) containing the AP-1 consensus sequence (5′-CGCTTGATGACTCAGCCGGAA-3′). The mixture was further incubated for 30 min at room temperature. The final concentration of DTT in the redox reactions was 0.02 mM. Samples were analyzed using nondenaturing polyacrylamide gel and detected using the Hitachi FMBio II Fluorescence Imaging System (Hitachi Genetic Systems, South San Francisco, CA).
3. Results
3.1 Activation of APE1 activity by nicorandil
Although a number of small molecule inhibitors of APE1 endonuclease activity have been identified through both experimental and in silico high-throughput screens [12, 22-31], to date no activators have been reported. Using a previously reported HTS [12], we identified and validated one compound, nicorandil (Figure 1A), which is approved for clinical use in Europe to treat angina and shown to be bioavailable in pharmacokinetic studies [32]. Nicorandil stimulates wild-type APE1 activity maximally by 40% as shown in a dose response curve (Figure 1B) with an EC50 of 0.82 μM. Similarly, nicorandil stimulates the kinetically faster R177A APE1 [9] with an EC50 of 2.2 μM (Figure 1C), although concentrations of nicorandil higher than 15 μM were found to inhibit R177A APE1. Nicorandil also stimulated APE1 repair activity in DRG cellular extracts at concentrations up to 0.5 mM (Figure 1D). Metabolism of nicorandil in cells releases nitric oxide, thus it was of interest to determine whether the nitro group of nicorandil was important for stimulating APE1 activity. However, the denitrated metabolite of nicorandil, N-(2-hydroxyethyl)nicotinamide [32] (Figure 1A) does not activate APE1 endonuclease but rather inhibits its activity with an IC50 of 65 μM (Figure 1E).
Figure 1.
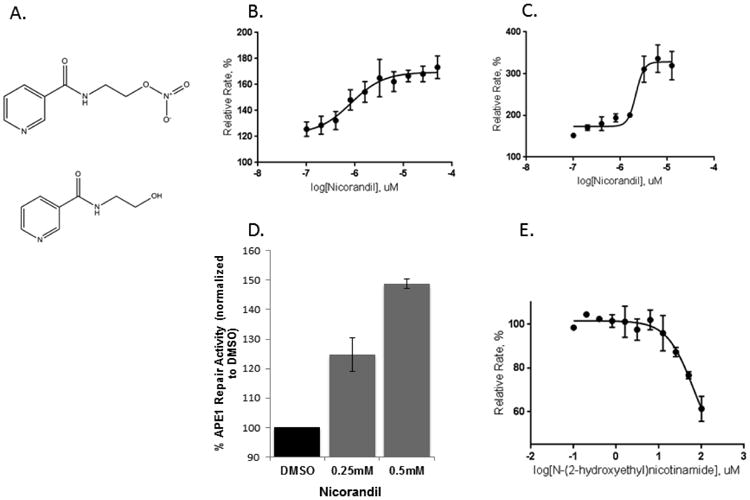
Effect of nicorandil on APE1 activity. (A) Chemical structures for nicorandil (top) and the denitrated metabolite, N-(2-hydroxyethyl)nicotinamide (bottom) are shown. Increasing concentrations of nicorandil stimulate wild-type (B) and R177A APE1 (C) endonuclease activity. A nicorandil titration assay was carried out at 0.1 nM wild-type APE1 or 0.0125 nM R177A APE1 with 10 nM substrate in the presence of 1% DMSO. The concentration range of nicorandil was varied from 0.1 to 50 μM for wild-type APE1 and 0.1 to 12.5 μM for R177A APE1 with triplicate measurements for each concentration. The rates in relative fluorescent units were calculated by normalizing to the rate of 1% DMSO control in (B, C, D, and E). The curves were fit to a four-parameter dose response equation in Graph Pad. The EC50 is 0.82 μM for wild-type APE1 and 2.2 μM for R177A APE1. (D) Effect of nicorandil on APE1 activity from DRG cell extracts. Nicorandil was added directly to untreated DRG cell extract at the concentrations shown and assayed for APE1 endonuclease activity as previously described. Three independent experiments were performed with triplicate measurements for reactions including a total volume of 100 μL with 50 nM substrate and 3.3 μg extract in 2 % DMSO. Reaction mixtures included the same buffering components as in (B). (E) The denitrated metabolite of nicorandil, N-(2-hydroxyethyl)nicotinamide, inhibits the activity of APE1. Using the same kinetic fluorescent assay as described in Figure 1B, the effects of increasing the concentration of N-(2-hydroxyethyl)nicotinamide on APE1 activity is shown. An IC50 of 65 μM was determined by using a four parameter logistics curve in non-linear regression as implemented in GraphPad Prism.
To determine the mechanism by which nicorandil activates APE1, kinetic studies were performed on both wild-type APE1 and R177A APE1 in the presence and absence of 12.5 μM nicorandil, the concentration found to maximally stimulate both wild-type and R177A APE1 (Figures 1B,C). In the absence of nicorandil, both enzymes exhibit a hyperbolic response in rate with increasing concentrations of substrate from 0.1 to 320 μM (Figure 2A and B). Kinetic parameters, kcat, KM, and kcat/KM, were calculated for both wild-type and R177A APE1. As shown in Table 1, the catalytic efficiency (kcat/KM) of R177A is approximately 4-fold higher than for the wild-type enzyme. The increase in catalytic efficiency arises primarily from a 7-fold increase in kcat for the R177A enzyme as compared to the wild-type enzyme as the KM values differ by less than a factor of 2.
Figure 2.
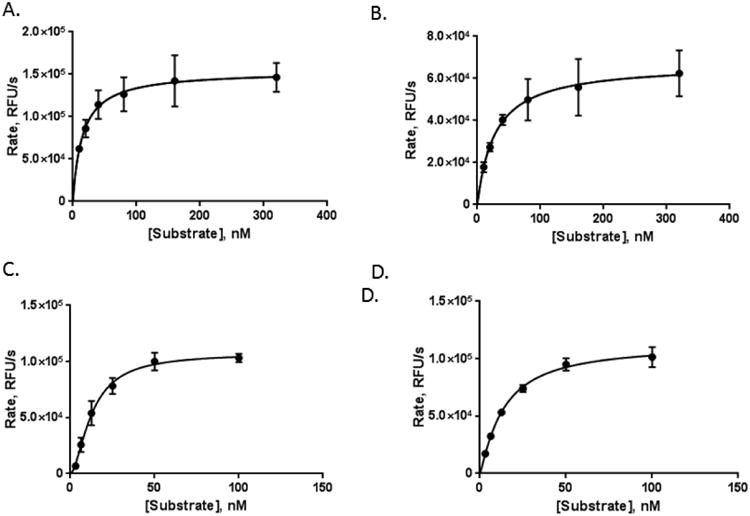
Nicorandil stimulates both wild-type and R177A APE1 endonuclease activity. APE1 endonuclease activity in the absence of nicorandil was measured for wild-type and R177A APE1. Enzyme concentrations of 0.2 nM and 0.0125 nM were used for wild-type and R177A APE1, respectively, for substrate concentrations of 10, 20, 40, 80, 160, and 320 nM. Similarly, the effect of 12.5 μM nicorandil on the kinetics of wild-type (C) and R177A (D) APE1 endonuclease activity was measured. Enzyme concentrations of 0.1 nM for wild-type and 0.0125 nM for R177A APE1 were used with substrate concentrations of 3.13, 6.25, 12.5, 25, 50, and 100 nM. The endonuclease activity was determined by monitoring the release of the fluorescein-labeled product in relative fluorescence units (RFUs) per second over the first five minutes of the reaction and plotted versus the substrate concentration. Curve fitting and kinetic parameters were calculated using GraphPad Prism. Data were obtained from three independent experiments with triplicate measurements for each substrate concentration.
Table 1. Effect of nicornadil on enzyme kinetic parameters.
| Enzyme | h | kcat * × 10-6 (s-1) | KM (nM) | kcat/KM (s-1) | Fold-increase# |
|---|---|---|---|---|---|
| Wild-type | 1.0 | 0.77 +/- 0.038 | 15 +/- 3 | 5.1 × 104 | |
| Wild-type + nicorandil | 1.7 | 1.1 +/- 0.046 | 13 +/- 1 | 9 × 104 | 1.7 |
| R177A | 1.0 | 5.4 +/- 0.034 | 28 +/- 6 | 19 × 104 | 3.8 |
| R177A + nicorandil | 1.2 | 9.1 +/- 0.40 | 14 +/- 2 | 67 × 104 | 3.4 |
h refers to the Hill slope calculated for an allosteric sigmoidal kinetics model in GraphPad.
kcat values are divided by 106 and were calculated as Vmax/[enzyme (nM)]. Vmax is in relative fluorescence units per second. Active enzyme concentration was estimated as the total enzyme concentration in this case.
# Fold increase is reported for wild-type stimulated by nicorandil vs. wild-type alone, R177A vs. wild-type, and R177A stimulated by nicroandil vs. R177A APE1 alone.
In the presence of nicorandil, the kcat/KM was increased 1.7-fold for wild-type and 3.4-fold for the R177A enzyme (Table 1). This increase in catalytic efficiency for the wild-type enzyme results from a 1.4-fold increase in kcat and a 1.2-fold decrease in KM. For the R177A enzyme, the kcat is increased by a factor 1.7 and KM is decreased by a factor of 2.1. However, in the presence of nicorandil, the wild-type enzyme exhibits a non-hyperbolic, sigmoidal response curve with increasing concentrations of substrate (Figure 2C and D) as indicated by a Hill slope of 1.7. R177A APE1 also exhibits a non-hyperbolic response with increasing substrate concentration but to a lesser extent with a Hill slope of 1.2. The non-hyperbolic kinetics confirms an apparent cooperative effect on the rates of reaction with increasing substrate concentrations.
As assessed by differential scanning fluorimetry, nicorandil does not bind directly to either wild-type or R177A enzymes in the presence or absence of Mg2+, which as previously reported stabilizes the wild-type enzyme [33]. R177A APE1 exhibited a Tm that was ∼4 °C lower than that of the wild-type APE1 but was stabilized by the addition of Mg2+ as shown by a ∼2 °C increase in melting temperature. The melting temperatures for both wild-type and R177A APE1 are unaffected by addition of nicorandil up to concentrations of 400 μM (Figure S1).
3.2 Pre-treatment of sensory neurons with nicorandil plus daidzin followed by cisplatin
Having established in vitro that nicorandil activates APE1 and that the nitro group is required for this activation, we sought to determine whether DNA repair could be enhanced in cisplatin treated sensory neurons by pre-treatment with nicorandil. Treatment with cisplatin is known to create oxidative damage to DNA, which is acted upon by APE1 [4, 5, 16]. In order for nicorandil to work in a cell, we concluded that it would be necessary to prevent or at least slow down denitration through metabolic processes. The primary enzyme responsible for metabolizing nitroglycerin and releasing nitric oxide is ALDH2 [34, 35]; this enzyme has also been shown to release nitric oxide from other organic nitrates, including nicorandil [36, 37]. ALDH2 can be inhibited by the drug daidzin [38, 39]; thus, we pre-treated sensory neurons with nicorandil alone (100 μM for 1 hour prior to and throughout cisplatin), daidzin alone (10 μM 3 days prior to and throughout cisplatin), and nicorandil plus daidzin and compared DNA damage following treatment with cisplatin as assessed by levels of phosphorylated γ-H2AX. Treatment with nicorandil or daidzin alone each decreased DNA damage less than two-fold; however, the combination of these two compounds resulted in an approximately 4-fold decrease in DNA damage 48 hours after treatment with cisplatin as shown in Figure 3A. Similarly, nicorandil plus daidzin treatment decreased DNA damage following treatment with menadione, a reagent known to cause oxidative DNA damage (Figure S2). Pretreatment with nicorandil and daidzin resulted in a small increase in cell survival as assessed by trypan blue exclusion for cultured sensory neurons treated with 30 μM cisplatin as compared to control samples (Figure 3B). This neuroprotective effect was not observed with nicorandil or daidzin when given alone and was overcome by exposing cells to 100 μM cisplatin for 24 hours (Figure 3B).
Figure 3.
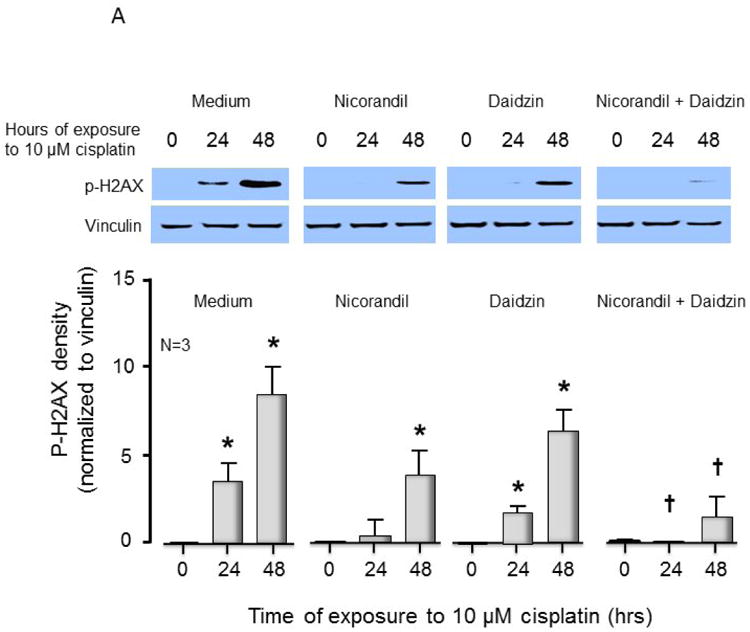
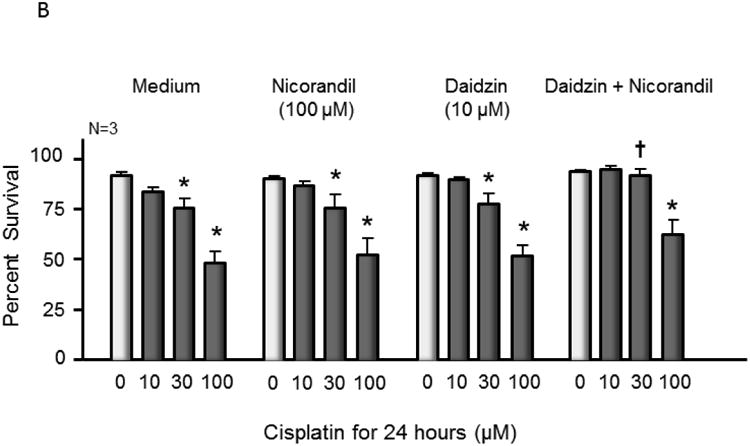
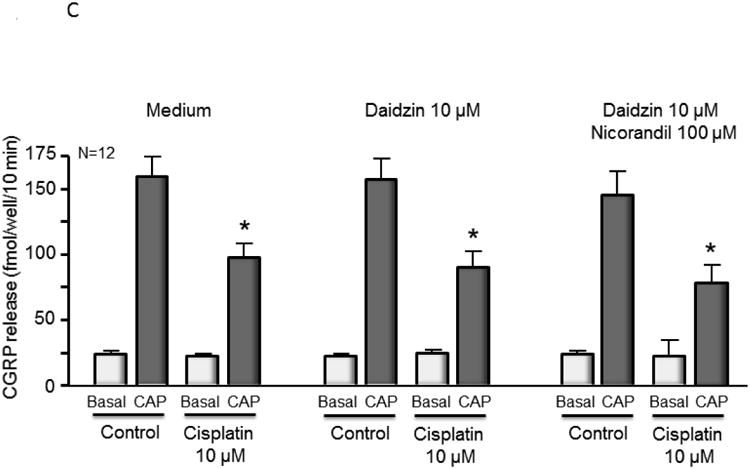
Nicorandil attenuates cisplatin-induced phosphor-H2AX and partially blocks cell death but does not alter cisplatin-induced changes in CGRP release. (A) The top panels show representative Western blots of phospho-H2AX (p-H2AX) and vinculin from cultures prior to and after 24 and 48 hours of exposure to 10 μM cisplatin after various drug treatments as indicated. The bottom panels represent the densitometry of p-H2AX expression normalized to vinculin from 3 independent experiments. The columns represent the mean ± SEM from cultures treated as indicated. An asterisk indicates a statistically significant increase in p-H2AX density compared to medium-treated controls, whereas a cross indicates a statistically significant reduction in p-H2AX compared to medium-treated controls. (B) Each column represents the mean ± SEM of percent survival of cells from control cultures (open columns) and those treated with various concentrations of cisplatin for 24 hours. Each panel represents cultures pretreated with medium, nicorandil, daidzin, or nicorandil and daidzin as indicated. An asterisk indicates a significant difference in survival after cisplatin compared to controls, whereas a cross indicates a significant difference in cultures treated with daidzin and nicorandil using ANOVA and Tukey's post hoc test. (C) Each column represents the mean ± SEM of basal CGRP release (open columns) or capsaicin-stimulated release (shaded columns) in fmol/well/min for untreated sensory neurons in culture (Medium) or cultures treated with daidzin or daidzin and nicorandil as indicated. Cultures were treated with medium (controls) or 10 μM cisplatin for 24 hours prior to release experiments. For release, wells of cells from 3 independent harvests were exposed for 10 min to HEPES alone (basal; open columns), or HEPES in the presence of 30 nM capsaicin (solid columns) as indicated. An asterisk indicates a significant difference in capsaicin-stimulated release compared to untreated cells.
We also examined the effectiveness of nicorandil plus daidzin to prevent damage to sensory neurons by cisplatin using a functional endpoint, release of the neurotransmitter CGRP. No difference was observed in resting or capsaicin-stimulated CGRP release for sensory neurons pretreated with nicorandil and/or daidzin prior to cisplatin treatment as compared to the control samples (Figure 3C). This result suggests that despite a reduction in DNA damage to the cells, pretreatment with nicorandil and daidzin did not attenuate the ability of cisplatin to alter the function of peptidergic sensory neurons.
3.3 Overexpression of R177A APE1 in cisplatin treated sensory neurons
Since R177A APE1 is catalytically more efficient than wild-type APE1, it was of interest to determine what effect overexpression of R177A APE1 would have in cisplatin-treated sensory neurons. The combination of increased catalytic activity and overexpression of R177A would be expected to provide an upper limit for the effects of activating APE1. At the same time, it provides a basis for evaluating the hypothesis that slow product release by APE1 is necessary for the base excision repair pathway to complete repair. Accordingly, endogenous rat APE1 was knocked down using rat APE1 siRNA, and human R177A APE1 was overexpressed using viral infection of sensory neurons that were treated with cisplatin. Protein levels of endogenous and exogenous APE1 after manipulation were measured using Western blotting (see Figure 4A and summary data in Figure 4B). Treating cultures with APE1siRNA reduced expression of the endonuclease approximately 80%, compared to cells treated with SCsiRNA, whereas, infection with lentivirvus containing the R177A APE1 construct restored APE1 levels in APE1siRNA treated cultures to the control level and resulted in a 70% increase in APE1 density in cells treated with SCsiRNA (Figure 4D). When neuronal cultures were exposed to cisplatin for 24 or 48 hours, there was a significant increase in the levels of DNA damage as assessed by levels of phosphorylated γ-H2AX (Figure 4A). For both vector and R177A APE1 overexpressing sensory neurons, a small increase in DNA damage is observed when endogenous APE1 is knocked down as compared to the SCsiRNA samples. Overexpression of R177A APE1 did not alter the DNA damaging effects of cisplatin (Figure 4A). In a similar manner, R177A APE1 overexpression was not effective in reducing cisplatin-induced neurotoxicity as measured by a loss in cell viability (Figure 4C) or by cisplatin-induced alterations in capsaicin-evoked CGRP release (Figure 4D).
Figure 4.
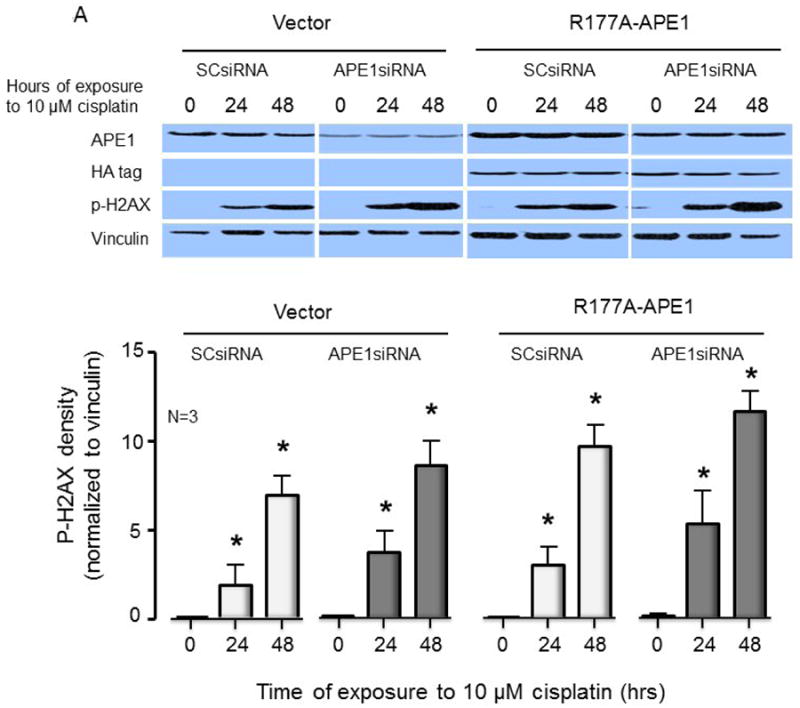
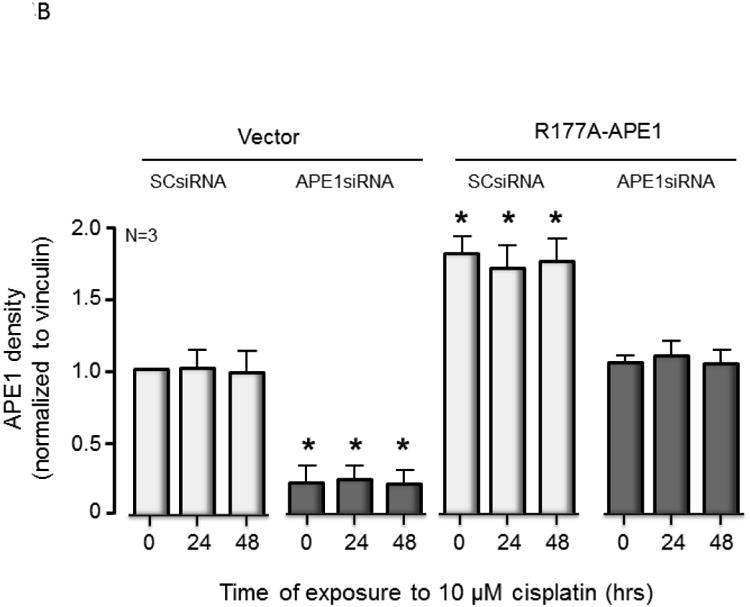
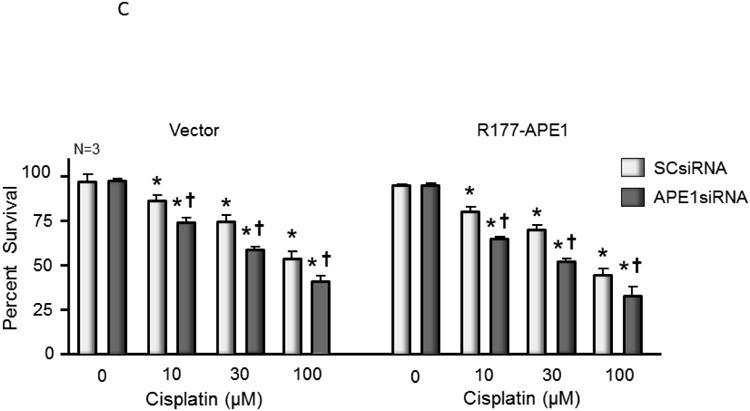
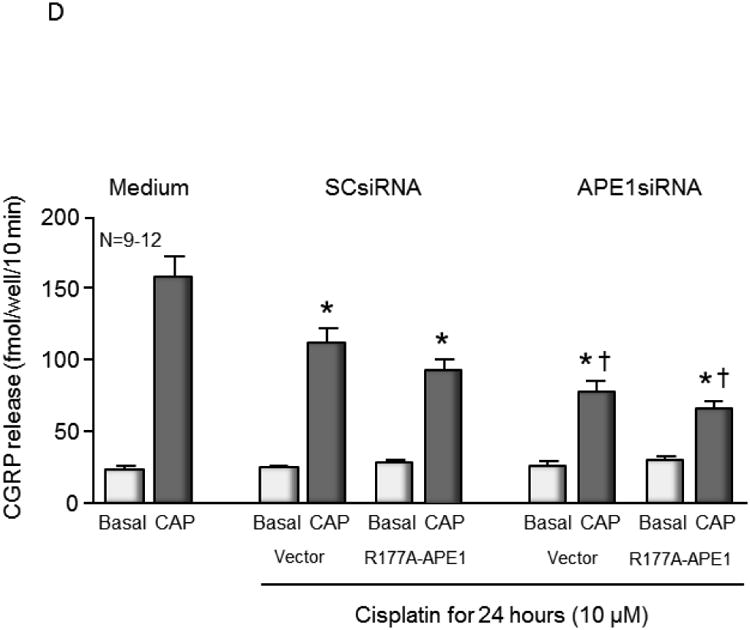
Overexpression of R177A APE1 in cisplatin-treated sensory neurons has no effect on DNA damage; levels of cell death or neurotransmitter release were comparable to control samples. (A) The top panels show representative Western blots of APE1, HA, phospho-H2AX (p-H2AX) and vinculin from cultures prior to and after 24 and 48 hours of exposure to 10 μM cisplatin. Cultures were exposed to 100 nM SCsiRNA or APE1siRNA and infected with lentiviral constructs containing EGFP (vector) or R177A-APE1 as indicated. The bottom panels represent the densitometry of p-H2AX expression normalized to vinculin from 3 independent experiments. The columns represent the mean ± SEM from cultures treated with SCsiRNA (lightly shaded columns) or APE1siRNA (heavy shaded columns) prior to or after exposure to 10 μM cisplatin. An asterisk indicates a statistically significant increase in P-H2AX density in each group compared to cells not exposed to cisplatin. (B) Each column represents the mean ± SEM densitometry of APE 1expression normalized to vinculin from 3 independent experiments from Figure 4A for cultures treated with SCsiRNA (lightly shaded columns) or APE1siRNA (heavy shaded columns) then viral infection prior to or after exposure to 10 μM cisplatin. An asterisk indicates a statistically significant increase in APE1 density cells treated with APE1siRNA compared to those treated with SCsiRNA. (C) Each column represents the mean ± SEM of percent survival of cells from cultures treated with SCsiRNA (open columns) or APE1siRNA (shaded columns) then treated with various concentrations of cisplatin for 24 hours. The panel on the left represents cells infected with control lentivirus, whereas the panel on the right represents cells infected with lentivirus containing the R177A APE1 construct. An asterisk indicates a significant difference in survival after cisplatin compared to controls, whereas a cross indicates a significant difference in cultures treated with APE1siRNA using ANOVA and Tukey's post hoc test. (D) Each column represents the mean ± SEM of basal CGRP release (open columns) or capsaicin-stimulated release (shaded columns) in fmol/well/min. Cultures are untreated (medium control from Figure 3C) or treated with SCsiRNA or APE1siRNA and infected with lentivirus containing EGFP (vector) or the R177A APE1construct as indicated prior to a 24 hour exposure to 10 μM cisplatin. An asterisk indicates a significant difference in capsaicin-stimulated release after cisplatin compared to medium controls, whereas a cross indicates a significant difference in APE1siRNA treated cultures compared to SCsiRNA treated cultures.
3.4 APE1 redox signaling activity is not affected by nicorandil or daidzin
Using our standard cell based AP-1 luciferase reporter assay [17, 19, 20] in human low passage Pa02c pancreatic tumor cells, we did not observe any inhibition of APE1 redox activity resulting from treatment with nicorandil or daidzin (Figure 5). E3330, a known selective APE1 redox inhibitor, was used as a positive control. Cells were treated with increasing amounts of either E3330, nicorandil, daidzin, or nicorandil with 10 μM daidzin for 24 hrs. Data shown (Figure 5) represent percent activity normalized to media with mean standard error and include background effects of the vehicle (DMSO). In a dose-dependent manner, the APE1 redox inhibitor E3330 reduced AP-1 activity as expected (50% response = 22.9 uM) and previously reported [19, 21] (Figure 5A), while no inhibition of AP-1 transactivation resulted from treatment with nicorandil (5B), daidzin (5C), and nicorandil with 10 uM daidzin/dose (5D).
Figure 5.
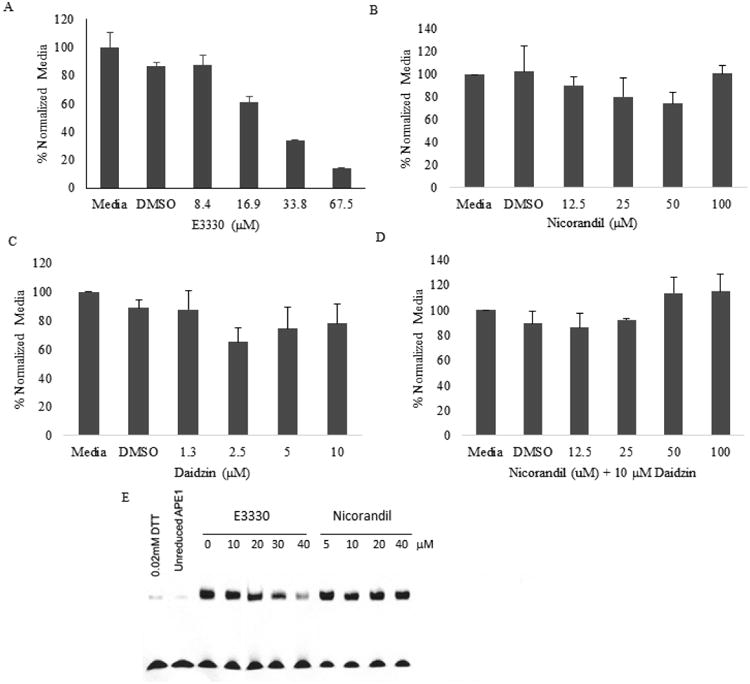
Nicorandil and/or daidzin do not inhibit APE1 redox signaling activity in a cell-based reporter transactivation or redox EMSA assays. Pa02c cells were cotransfected for 24 hours (37°C, 5% CO2) with AP-1 Luciferase Firefly vector and control pRL Renilla-TK vector in a 20:1 ratio, respectively, using 3.0 μL/mL of Lipofectamine®2000 (Invitrogen). Following media exchange and 24 hour recovery, cells were treated in doses for 24 hours of either E3330, Nicorandil (Sigma-Aldrich), daidzin (Indofine Chemical), or nicorandil with 10 μM daidzin. Firefly luciferase and Renilla activities were measured using the Promega Dual-Luciferase®system. For each dose, AP-1 luciferase RLU was normalized with Renilla-TK RLU, and then further normalized to media response. Data shown is percent normalized to media with mean standard error and includes vehicle (DMSO) response. Experiments were done in duplicate and repeated twice. In a dose-dependent manner, the APE1 redox inhibitor E3330 (A) reduced AP-1 activity as expected (50% response = 22.9 μM), while nicorandil (B), daidzin (C), and nicorandil with 10 μM daidzin/dose (D) did not elicit any APE1 redox inhibition. In a redox EMSA assay (E), E3330 (positive control) blocked APE1 redox signaling function while nicorandil had no effect.
To further validate the negative effect of nicorandil on APE1 redox activity, we performed a redox EMSA assay as previously described [19, 21]. In this assay, the positive control agent E3330 decreased APE1 redox activity as assessed by reduction of DNA-binding by c-Jun by 11, 28, 53 and 95% at 10, 20, 30 and 40 μM E3330 doses respectively, while nicorandil had no effect (Figure 5E).
4. Discussion
Overall, our studies are the first to investigate the use of a small molecule activator of APE1 endonuclease activity to protect cultured sensory neurons from DNA damage by cisplatin. Previous overexpression experiments using repair active, redox inactive or redox active, repair inactive APE1 suggest that the repair and not the redox activity of APE1 is necessary to confer protection to sensory neurons from damage by cisplatin, menadione, or ionizing radiation which produces oxidative DNA damage [4, 15, 16]. However, overexpression of a DNA repair active APE1 in neurons is not a clinically viable approach. Therefore, we hypothesized that use of small molecule activators as chemical probes would allow us to determine whether the protective effects are due only to APE1's endonuclease activity and have translational significance.
In these studies, we demonstrated that the small molecule nicorandil stimulates APE1 activity in vitro both for purified enzyme and for cellular extracts from sensory neurons. Nicorandil also stimulates activity of the catalytically faster R177A APE1 in vitro. Importantly, nicorandil does not affect APE1's redox activity in vitro or in pancreatic cancer cells as assessed by EMSA redox and redox transactivation assays, respectively. Mechanistically, nicorandil increases the kcat and lowers the KM thereby increasing the catalytic efficiency of both wild-type and R177A APE1. However, the resulting enzymatic rates plotted versus substrate concentration for wild-type APE1 are sigmoidal in the presence of nicorandil giving rise to an apparent cooperative effect with Hill slopes greater than one. Since there is a single active site in APE1, we speculate that the sigmoidal behavior arises from a more complex scenario in which two distinct species, enzyme-substrate and enzyme-substrate-activator, exist and give rise to product during the reaction at different rates. Substrate activation resulting in non-hyperbolic kinetics has been reported for ethanol and 1-butanol as substrates for human liver alcohol dehydrogenase γ2. In this case, an ordered mechanism was proposed to account for the apparent cooperativity [40].
Our in vitro studies suggested that the nitrate group of nicorandil is important for its stimulatory effect on APE1 endonuclease activity as a compound lacking this group actually inhibits the endonuclease activity. Thus, we concluded that nicorandil would be more effective in cell-based experiments if its metabolism by aldehyde dehydrogenase 2 was inhibited by daidzin [38, 39]. Individually, neither daidzin nor nicorandil had a significant effect on DNA damage as assessed by phosphorylation of γ-H2AX, but together DNA damage was significantly decreased in cisplatin-treated sensory neurons. Thus, the use of nicorandil in combination with daidzin is effective in ameliorating DNA damage induced by cisplatin, and we have established proof of concept in our studies with nicorandil.
It is well established that the chemistry of the endonuclease reaction catalyzed by APE1 is diffusion limited and that product release is the slow step of the reaction [7, 8]. Thus, accelerating APE1 activity necessarily involves increasing the rate of product release. The proposed role for slow product release is to allow transfer of the product which includes a single-strand break to downstream repair enzymes to ensure completion of DNA repair. Consequently, one possible outcome of activating APE1 with nicorandil might have been to imbalance BER resulting in more DNA damage from cisplatin rather than less damage as we observed. In fact, overexpression of a catalytically faster R177A APE1 did not decrease DNA damage consistent with imbalancing of the BER pathway nor did it confer any protective effects for any other functional endpoints in cisplatin-treated sensory neurons. In contrast, stimulation of wild-type APE1 activity by nicroandil does not imbalance the BER pathway. These results confirm that it is possible to activate APE1 without imbalancing BER in cisplatin treated sensory neurons.
Although pretreating the neuronal cultures with nicorandil and daidzin reduced cisplatin-induced DNA damage and partially attenuated the cisplatin-induced decrease in cell viability, it did not restore normal transmitter release from sensory neurons treated with cisplatin. In previous work, however, we showed a correlation between attenuation of cisplatin-induced DNA damage and attenuation of cisplatin-induced changes in viability and transmitter release when APE1 was overexpressed [3, 4]. The failure of nicorandil to affect CGRP release in cisplatin treated sensory neurons may result from the short duration of treatment, which while sufficient to effect increased DNA repair through stimulation of endogenous APE1 as measured by phospho-H2AX is not comparable to the overexpression of APE1, which remains in the system much longer. Alternatively, although the endonuclease function of APE1 is clearly important for protection in sensory neurons treated with cisplatin, other functional roles of APE1 may also play a role.
In conclusion, nicorandil in combination with daidzin is effective in decreasing overall DNA damage. Ideally a single agent that increases APE1 endonuclease activity with a better metabolic profile could prove even more useful in assessing the role of DNA repair in CIPN and ultimately as a therapeutic. Thus, identification of compounds related to nicorandil with different metabolic profiles is warranted. Yet to be explored is the effect of nicorandil and daidzin in cisplatin-treated cancer cells. Given that it is not currently possible to determine a priori which patients will suffer from long term CIPN following chemotherapy, we envision a post-chemotherapeutic application to remove persistent DNA damage in sensory neurons induced by chemotherapy. As proof of concept, the studies presented here examined pretreatment of cultured sensory neurons treated with cisplatin. Future studies will address post-treatment effects of APE1 activators in sensory neurons and effects in cancer cells.
Supplementary Material
Figure S1. Nicorandil does not bind directly to APE1 as assessed by differential scanning fluorimetry. The Tm is plotted for wild-type APE1 or R177A APE1 (final concentrations of 5 μM) versus the concentration of nicorandil in the presence and absence of 1 mM Mg2+, which was previously shown to stabilize APE1. Wild-type and R177A APE1 were incubated with SYPRO orange (excitation at 483 nm and emission at 568 nm) and nicorandil (1-400 μM) at 25 °C in 50 mM HEPES (pH 7.5), 50 mM KCl, 1 mM DTT. The fluorescence was processed with the LightCycle 480 Real-Time PCR System (Roche, Switzerland).
Figure S2. Nicorandil and daidzin attenuate menadione-induced phosphor-H2AX
The top panels show representative Western blots of phospho-H2AX (p-H2AX) and vinculin from cultures prior to and 2,4,6,16 and 24 of exposure to 10 μM cisplatin after various drug treatments as indicated. The bottom panels represent the densitometry of p-H2AX expression normalized to vinculin from 3 independent experiments. The columns represent the mean ± SEM from cultures treated as indicated. An asterisk indicates a statistically significant increase in p-H2AX density after menadione compared to controls, whereas a cross indicates a statistically significant reduction in p-H2AX compared to medium-treated controls.
Acknowledgments
This work was supported by grants from the National Institutes of Health NS091667 (to MRK and MRV), and CA167291 (to MRK), IU Simon Cancer Center Neurotoxicity Working Group Pilot Grant (to MMG and MRK) Hyundai Hope on Wheels, Jeff Gordon Children's Foundation and the Riley Children's Foundation (to MRK). We thank our colleague Thomas Hurley for helpful suggestions regarding inhibition of ALDH2 and providing daidzin for these studies.
Footnotes
Conflict of Interest: Mark R. Kelley has licensed E3330 though Indiana University Research & Technology Corporation to ApeX Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hershman DL, Lacchetti C, Dworkin RH, Lavoie Smith EM, Bleeker J, Cavaletti G, Chauhan C, Gavin P, Lavino A, Lustberg MB, Paice J, Schneider B, Smith ML, Smith T, Terstriep S, Wagner-Johnston N, Bak K, Loprinzi CL. Prevention and Management of Chemotherapy-Induced Peripheral Neuropathy in Survivors of Adult Cancers: American Society of Clinical Oncology Clinical Practice Guideline. Journal of Clinical Oncology. 2014;32:1941–1967. doi: 10.1200/JCO.2013.54.0914. [DOI] [PubMed] [Google Scholar]
- 2.Zafar SY, Marcello JE, Wheeler JL, Rowe KL, Morse MA, Herndon JE, 2nd, Abernethy AP. Treatment-related toxicity and supportive care in metastatic colorectal cancer. The journal of supportive oncology. 2010;8:15–20. [PubMed] [Google Scholar]
- 3.Jiang Y, Guo C, Vasko MR, Kelley MR. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer research. 2008;68:6425–6434. doi: 10.1158/0008-5472.CAN-08-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelley MR, Jiang Y, Guo C, Reed A, Meng H, Vasko MR. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PloS one. 2014;9:e106485. doi: 10.1371/journal.pone.0106485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang Y, Guo C, Vasko MR, Kelley MR. Implications of Apurinic/Apyrimidinic Endonuclease in Reactive Oxygen Signaling Response after Cisplatin Treatment of Dorsal Root Ganglion Neurons. Cancer research. 2008;68:6425–6434. doi: 10.1158/0008-5472.CAN-08-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HS, Guo C, Thompson EL, Jiang Y, Kelley MR, Vasko MR, Lee SH. APE1, the DNA base excision repair protein, regulates the removal of platinum adducts in sensory neuronal cultures by NER. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2015;779:96–104. doi: 10.1016/j.mrfmmm.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maher RL, Bloom LB. Pre-steady-state kinetic characterization of the AP endonuclease activity of human AP endonuclease 1. The Journal of biological chemistry. 2007;282:30577–30585. doi: 10.1074/jbc.M704341200. [DOI] [PubMed] [Google Scholar]
- 8.Schermerhorn KM, Delaney S. Transient-state kinetics of apurinic/apyrimidinic (AP) endonuclease 1 acting on an authentic AP site and commonly used substrate analogs: the effect of diverse metal ions and base mismatches. Biochemistry. 2013;52:7669–7677. doi: 10.1021/bi401218r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected] Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 10.Freudenthal BD, Beard WA, Cuneo MJ, Dyrkheeva NS, Wilson SH. Capturing snapshots of APE1 processing DNA damage. Nature structural & molecular biology. 2015;22:924–931. doi: 10.1038/nsmb.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Luo M, Marasco D, Logsdon D, LaFavers KA, Chen Q, Reed A, Kelley MR, Gross ML, Georgiadis MM. Inhibition of apurinic/apyrimidinic endonuclease I's redox activity revisited. Biochemistry. 2013;52:2955–2966. doi: 10.1021/bi400179m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bapat A, Glass LS, Luo M, Fishel ML, Long EC, Georgiadis MM, Kelley MR. Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells. The Journal of pharmacology and experimental therapeutics. 2010;334:988–998. doi: 10.1124/jpet.110.169128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nature protocols. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 14.Burkey TH, Hingtgen CM, Vasko MR. Isolation and culture of sensory neurons from the dorsal-root ganglia of embryonic or adult rats. Methods Mol Med. 2004;99:189–202. doi: 10.1385/1-59259-770-X:189. [DOI] [PubMed] [Google Scholar]
- 15.Vasko MR, Guo C, Kelley MR. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA repair. 2005;4:367–379. doi: 10.1016/j.dnarep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Vasko MR, Guo C, Thompson EL, Kelley MR. The repair function of the multifunctional DNA repair/redox protein APE1 is neuroprotective after ionizing radiation. DNA repair. 2011;10:942–952. doi: 10.1016/j.dnarep.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fishel ML, Wu X, Devlin CM, Logsdon DP, Jiang Y, Luo M, He Y, Yu Z, Tong Y, Lipking KP, Maitra A, Rajeshkumar NV, Scandura G, Kelley MR, Ivan M. Apurinic/Apyrimidinic Endonuclease/Redox Factor-1 (APE1/Ref-1) redox function negatively regulates NRF2. Journal of Biological Chemistry. 2014;290:3057–3068. doi: 10.1074/jbc.M114.621995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gracias N, Cummins T, Kelley M, Basile D, Iqbal T, Vasko MR. Vasodilatation in the rat dorsal hindpaw induced by activation of sensory neurons is reduced by Paclitaxel. NeuroToxicology. 2011;32:140–149. doi: 10.1016/j.neuro.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL, 2nd, Borch RF, Qiao X, Georgiadis MM, Kelley MR. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxidants & redox signaling. 2008;10:1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cardoso AA, Jiang Y, Luo M, Reed AM, Shahda S, He Y, Maitra A, Kelley MR, Fishel ML. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PloS one. 2012;7:e47462. doi: 10.1371/journal.pone.0047462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nyland RL, Luo M, Kelley MR, Borch RF. Design and synthesis of novel quinine inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/ref-1) Journal of medicinal chemistry. 2010;53:1200–1210. doi: 10.1021/jm9014857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaur G, Cholia RP, Mantha AK, Kumar R. DNA Repair and Redox Activities and Inhibitors of Apurinic/Apyrimidinic Endonuclease 1/Redox Effector Factor 1 (APE1/Ref-1): A Comparative Analysis and Their Scope and Limitations toward Anticancer Drug Development. Journal of medicinal chemistry. 2014 doi: 10.1021/jm500865u. [DOI] [PubMed] [Google Scholar]
- 23.Al-Safi RI, Odde S, Shabaik Y, Neamati N. Small-Molecule Inhibitors of APE1 DNA Repair Function: An Overview. Current Molecular Pharmacology. 2012;5:14–35. [PubMed] [Google Scholar]
- 24.Wilson DM, 3rd, Simeonov A. Small molecule inhibitors of DNA repair nuclease activities of APE1. Cellular and molecular life sciences : CMLS. 2010;67:3621–3631. doi: 10.1007/s00018-010-0488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian C, Li M, Sui J, Ren T, Li Z, Zhang L, Zhou L, Cheng Y, Wang D. Identification of a novel potential antitumor activity of gossypol as an APE1/Ref-1 inhibitor. Drug design, development and therapy. 2014;8:485–496. doi: 10.2147/DDDT.S62963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srinivasan A, Wang L, Cline CJ, Xie Z, Sobol RW, Xie XQ, Gold B. Identification and characterization of human apurinic/apyrimidinic endonuclease-1 inhibitors. Biochemistry. 2012;51:6246–6259. doi: 10.1021/bi300490r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruiz FM, Francis SM, Tintore M, Ferreira R, Gil-Redondo R, Morreale A, Ortiz AR, Eritja R, Fabrega C. Receptor-based virtual screening and biological characterization of human apurinic/apyrimidinic endonuclease (Ape1) inhibitors. ChemMedChem. 2012;7:2168–2178. doi: 10.1002/cmdc.201200372. [DOI] [PubMed] [Google Scholar]
- 28.Dorjsuren D, Kim D, Vyjayanti VN, Maloney DJ, Jadhav A, Wilson DM, 3rd, Simeonov A. Diverse small molecule inhibitors of human apurinic/apyrimidinic endonuclease APE1 identified from a screen of a large public collection. PloS one. 2012;7:e47974. doi: 10.1371/journal.pone.0047974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simeonov A, Kulkarni A, Dorjsuren D, Jadhav A, Shen M, McNeill DR, Austin CP, Wilson DM., 3rd Identification and characterization of inhibitors of human apurinic/apyrimidinic endonuclease APE1. PloS one. 2009;4:e5740. doi: 10.1371/journal.pone.0005740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seiple LA, Cardellina JH, 2nd, Akee R, Stivers JT. Potent inhibition of human apurinic/apyrimidinic endonuclease 1 by arylstibonic acids. Molecular pharmacology. 2008;73:669–677. doi: 10.1124/mol.107.042622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kotera N, Poyer F, Granzhan A, Teulade-Fichou MP. Efficient inhibition of human AP endonuclease 1 (APE1) via substrate masking by abasic site-binding macrocyclic ligands. Chemical communications. 2015 doi: 10.1039/c5cc06084b. [DOI] [PubMed] [Google Scholar]
- 32.Frydman A. Pharmacokinetic profile of nicorandil in humans: an overview. Journal of cardiovascular pharmacology. 1992;20(3):S34–44. doi: 10.1097/00005344-199206203-00008. [DOI] [PubMed] [Google Scholar]
- 33.He H, Chen Q, Georgiadis MM. High-resolution crystal structures reveal plasticity in the metal binding site of apurinic/apyrimidinic endonuclease I. Biochemistry. 2014;53:6520–6529. doi: 10.1021/bi500676p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lang BS, Gorren AC, Oberdorfer G, Wenzl MV, Furdui CM, Poole LB, Mayer B, Gruber K. Vascular bioactivation of nitroglycerin by aldehyde dehydrogenase-2: reaction intermediates revealed by crystallography and mass spectrometry. The Journal of biological chemistry. 2012;287:38124–38134. doi: 10.1074/jbc.M112.371716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Page NA, Fung HL. Organic nitrate metabolism and action: toward a unifying hypothesis and the future-a dedication to Professor Leslie Z. Benet. Journal of pharmaceutical sciences. 2013;102:3070–3081. doi: 10.1002/jps.23550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsou PS, Page NA, Lee SG, Fung SM, Keung WM, Fung HL. Differential metabolism of organic nitrates by aldehyde dehydrogenase 1a1 and 2: substrate selectivity, enzyme inactivation, and active cysteine sites. The AAPS journal. 2011;13:548–555. doi: 10.1208/s12248-011-9295-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao GY, Li DJ, Keung WM. Synthesis of daidzin analogues as potential agents for alcohol abuse. Bioorganic & medicinal chemistry. 2003;11:4069–4081. doi: 10.1016/s0968-0896(03)00397-3. [DOI] [PubMed] [Google Scholar]
- 39.Lowe ED, Gao GY, Johnson LN, Keung WM. Structure of daidzin, a naturally occurring anti-alcohol-addiction agent, in complex with human mitochondrial aldehyde dehydrogenase. Journal of medicinal chemistry. 2008;51:4482–4487. doi: 10.1021/jm800488j. [DOI] [PubMed] [Google Scholar]
- 40.Charlier HA, Jr, Plapp BV. Kinetic cooperativity of human liver alcohol dehydrogenase gamma(2) The Journal of biological chemistry. 2000;275:11569–11575. doi: 10.1074/jbc.275.16.11569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Nicorandil does not bind directly to APE1 as assessed by differential scanning fluorimetry. The Tm is plotted for wild-type APE1 or R177A APE1 (final concentrations of 5 μM) versus the concentration of nicorandil in the presence and absence of 1 mM Mg2+, which was previously shown to stabilize APE1. Wild-type and R177A APE1 were incubated with SYPRO orange (excitation at 483 nm and emission at 568 nm) and nicorandil (1-400 μM) at 25 °C in 50 mM HEPES (pH 7.5), 50 mM KCl, 1 mM DTT. The fluorescence was processed with the LightCycle 480 Real-Time PCR System (Roche, Switzerland).
Figure S2. Nicorandil and daidzin attenuate menadione-induced phosphor-H2AX
The top panels show representative Western blots of phospho-H2AX (p-H2AX) and vinculin from cultures prior to and 2,4,6,16 and 24 of exposure to 10 μM cisplatin after various drug treatments as indicated. The bottom panels represent the densitometry of p-H2AX expression normalized to vinculin from 3 independent experiments. The columns represent the mean ± SEM from cultures treated as indicated. An asterisk indicates a statistically significant increase in p-H2AX density after menadione compared to controls, whereas a cross indicates a statistically significant reduction in p-H2AX compared to medium-treated controls.