

Abstract
Irreversible respiratory obstruction resulting from progressive airway damage, inflammation and fibrosis is a feature of several chronic respiratory diseases, including cystic fibrosis (CF), idiopathic pulmonary fibrosis (IPF) and chronic obstructive pulmonary disease (COPD). The cytokine transforming growth factor β (TGF-β) has a pivotal role in promoting lung fibrosis and is implicated in respiratory disease severity. In the present study, we show that a previously uncharacterized miRNA, miR-1343, reduces the expression of both TGF-β receptor 1 and 2 by directly targeting their 3′-UTRs. After TGF-β exposure, elevated intracellular miR-1343 significantly decreases levels of activated TGF-β effector molecules, pSMAD2 (phosphorylated SMAD2) and pSMAD3 (phosphorylated SMAD3), when compared with a non-targeting control miRNA. As a result, the abundance of fibrotic markers is reduced, cell migration into a scratch wound impaired and epithelial-to-mesenchymal transition (EMT) repressed. Mature miR-1343 is readily detected in human neutrophils and HL-60 cells and is activated in response to stress in A549 lung epithelial cells. miR-1343 may have direct therapeutic applications in fibrotic lung disease.
Keywords: fibrosis, lung, microRNA, transforming growth factor β
INTRODUCTION
The lung epithelium serves as a critical regulator of respiratory health. Not only does the epithelium perform a barrier function, protecting the pulmonary interstitium from harmful pathogens and environmental particles, but it also serves as an important site for host defence, mucociliary clearance and gas exchange [1]. Disruption of epithelial integrity underlies several chronic lung diseases, including cystic fibrosis (CF), idiopathic pulmonary fibrosis (IPF) and chronic obstructive pulmonary disease (COPD). In these disorders, a significant proportion of epithelial dysfunction results from lung fibrosis and architectural tissue remodelling. These processes gradually replace healthy elastic lung epithelium with fibrous connective tissue, composed mainly of collagens and extracellular matrix (ECM) components. The pathways of fibrosis impede normal epithelial function and lead directly to lung obstruction [2].
Transforming growth factor β (TGF-β) has a pivotal role in initiating mechanisms of tissue fibrosis. This cytokine is normally released in response to injury and stimulates cell differentiation and wound healing [3]. High levels of TGF-β are consistently observed in fibrotic lung diseases, in turn promoting excessive repair processes leading to organ dysfunction [4–7]. More specifically, TGF-β attracts and induces the differentiation of resident or circulating fibroblasts into contractile myofibroblasts in the lung, which migrate to sites of injury and produce ECM [3]. Furthermore, TGF-β promotes epithelial-to-mesenchymal transition (EMT), a process whereby alveolar epithelial cells in the lung can transdifferentiate into migratory fibroblastic cells [8].
The initiating events for each fibrotic lung disease are distinct; however, an absence of correlation between the primary insult and disease severity is a common feature. This implies possible genetic contributions that modify disease development and/or progression [9–11]. Universally, TGF-β is implicated as a major factor underlying fibrotic phenotypes, and polymorphisms promoting increased TGF-β expression were identified as genetic modifiers of COPD and CF lung disease severity [12–15]. However, in the absence of TGF-β-associated mutations, the molecular basis for dysregulated TGF-β signalling remains to be elucidated.
miRNAs, which are small 21–25-nt non-coding RNAs that repress genes post-transcriptionally, are compelling candidates for modulating fibrotic phenotypes and TGF-β signalling in the lung. Panels of misregulated miRNAs have been observed in a variety of human diseases, including pulmonary fibrosis, suggesting the importance of maintaining homoeostasis of miRNA expression [16–18]. More specifically, miR-155 exhibited pro-fibrotic and pro-inflammatory roles in models of both IPF and CF, in which it regulated expression of keratinocyte growth factor and interleukin-8 [19,20]. Furthermore, IPF and CF patient respiratory tissues showed up-regulation of miR-21 and miR-145 expression respectively and both miRNAs activated pulmonary fibroblasts and exacerbated experimental fibrosis in mice [21–23]. Conversely, overexpression of miR-29 and miR-31 inhibited markers of fibrosis in mouse models and normal lung fibroblasts, demonstrating protective roles [24–26].
In the present study, we describe the role of miR-1343 in attenuating TGF-β signalling and pathways of fibrosis in primary fibroblasts and lung epithelial cell lines. miR-1343 was identified in several small RNA-sequencing (RNA-seq) studies in humans [27,28], cows [29] and pigs [30,31], although it remains uncharacterized. It is not documented in rodents, suggesting low conservation through evolution. We found miR-1343 by using in silico tools to predict miRNAs targeting the 3′-UTR regions of both TGF-β receptor genes, which would in turn inhibit TGF-β signalling. The genomic location of miR-1343 adjacent to a modifier locus for CF lung disease severity [32] made it a compelling miRNA for further investigation. Our data show that miR-1343 represses TGF-β signalling as well as TGF-β-induced fibrotic markers and EMT. Hence, miR-1343 may have important roles in protecting against lung fibrosis and other TGF-β-induced processes.
MATERIALS AND METHODS
Cell culture
A549 lung adenocarcinoma cells [33], 16HBE14o- SV40 ori-transformed human bronchial epithelial (HBE) cells [34] and Caco2 colorectal adenocarcinoma cells [35] were cultured in Dulbecco’s modified Eagle’s medium (DMEM, low glucose) supplemented with 10% FBS. Primary lung fibroblasts were cultured in DMEM (high glucose) supplemented with 10% FBS and 1× penicillin/streptomycin for up to seven passages.
Cloning and mutagenesis
3′-UTRs were amplified from human genomic DNA according to TargetScan and UCSC gene annotations using Phusion Polymerase (New England Biolabs). Mutagenesis of seed sites was performed using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent). See Supplementary Table S3a for cloning primers and Supplementary Table S3b for mutagenesis primers.
Transient transfections and reporter assays
Transient transfections of hsa-miR-1343-3p Pre-miR miRNA precursor (PM20896, Life Technologies) and negative control (NC) miRNA precursor #2 (Life Technologies) were performed using Lipofectamine RNAiMax or Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocols to a final concentration of 20 nM.
Dual transient transfections of miRNA precursors plus plasmid DNA (pMIR-Report and pMIR-β-galactosidase, Promega) were performed as described previously [36] using Lipofectamine 2000 according to the manufacturer’s protocol. Cells were lysed 48 h post-transfection in 1× reporter lysis buffer (Promega). Luciferase and β-galactosidase assays were performed using the luciferase or β-galactosidase assay systems (Promega) respectively, according to standard protocols.
Transient transfections of p3TP-lux {Laboratories of Dr J. Wrana (Lunenfeld Tanenbaum Research Institute, Mount Sinai Hospital, Toronto, Ontario, Canada) and Dr J. Massagué (Memorial Sloan Kettering Cancer Center, New York City, NY, U.S.A.) [37]} and a modified pRL Renilla vector (Promega) were performed using Lipofectamine 2000 according to the manufacturer’s protocol. Cells were lysed in 1× passive lysis buffer (Promega) and luciferase assays were performed using the dual-luciferase reporter assay system (Promega).
RNA-sequencing
RNA-seq was carried out as described previously [38]. All data were deposited at GEO (http://www.ncbi.nlm.nih.gov/geo/GSE75591).
Cell adhesion assays
Cell adhesion assays were completed as described previously [39]. In the present study, 96-well plates were coated with 50 μl of 5 μg/ml collagen type 1 solution from rat tail (Sigma).
Quantitative PCR (qPCR)
For standard qPCR assays, RNA was isolated from cells using TRIzol (Life Technologies) by standard protocol 48 h following miRNA transfection. cDNA reactions were performed using the TaqMan Reverse Transcription Kit (Life Technologies) according to the manufacturer’s instructions. qPCRs were completed using Power SYBR Green (Life Technologies) using the following conditions: 50°C for 2 min, 95°C for 10 min, then cycling between 95°C for 15 s and 60°C for 1 min, 40 times. Ct values were normalized against β2-microglobulin (β2M). See Supplementary Table S3c for primers.
For miRNA qPCR assays, RNA was isolated from cells using the miRVana miRNA Isolation Kit (Life Technologies). miRNAs were reverse transcribed using the miRNA Reverse Transcription Kit (Life Technologies) and TaqMan Fast qPCR assays were performed using the hsa-miR-1343-3p TaqMan Assay (Life Technologies) using the following conditions: 50°C for 2 min, 95°C for 20 s, then cycling between 95°C for 3 s and 60°C for 30 s, 40 times. CT values were normalized to RNU6B (U6 small nuclear RNA; Life Technologies).
Western blot
Cell lysates were analysed by standard Western blot methods. Briefly, proteins were separated by SDS/PAGE and transferred on to membranes, which were blocked in 5% milk in TBS + 0.1% Tween 20 (TBST) for 1 h at 25°C. Primary antibodies were incubated overnight at 4°C either in 1% non-fat dried skimmed milk powder in TBST or 5% BSA in TBST. Secondary antibodies were incubated for 1 h at 25°C either in 1% or 5% non-fat dried skimmed milk powder in TBST. Antibodies were against TGFBR1 (TGF-β receptor 1; 1:500 dilution), pSMAD2/3 (phosphorylated SMAD2/3; 1:1000 dilution), pSMAD3 (1:500 dilution), SMAD2/3 (1:1000 dilution), GAPDH (glyceraldehyde-3-phosphate dehydrogenase; 1:5000 dilution) all from Cell Signaling Technology; TGFBR2 (TGF-β receptor 2; 1:500 dilution), E-cad (epithelial cadherin; 1:500 dilution), collagen 1A1 (1:500 dilution) all from Santa Cruz Biotechnology; ELMO2 (engulfment and cell motility 2; 1:500 dilution; Sigma–Aldrich); and αSMA (1:3000 dilution; Dako). Secondary antibodies were against mouse (P0447), rabbit (P0448) or goat (P0449) (1:5000 dilution;, all from Dako). Western blots were scanned by densitometry and quantified using ImageJ software (NIH).
TGF-β treatment
Cells were serum-starved in DMEM supplemented with 0.5% FBS for 6–16 h prior to TGF-β treatment. Human recombinant TGF-β1 (R&D Systems) was added to a final concentration of 5 ng/ml (unless otherwise noted) in serum-depleted medium for a period of 1–48 h.
Immunofluorescence
Immunofluorescence was completed under standard procedures using a primary antibody against αSMA (1:300 dilution; Dako) or SMAD2/3 (1:300 dilution; Cell Signaling Technology) and an Alexa Fluor 549-conjugated anti-mouse (1:300 dilution; Jackson ImmunoResearch) or Alexa Fluor 488-conjugated anti-mouse (1:300 dilution; Jackson ImmunoResearch) secondary antibody with DAPI counterstain. Microscopy utilized a Leica DMR-HC upright microscope and a QImaging Retiga 4000R camera.
Scratch wound healing assays
Wound healing assays were performed as previously described [39]. Cells were scratched with a p200 pipette tip and observed over a period of 24 h via microscopy. Scratch area was measured using ImageJ software (NIH).
Cell proliferation assays
The total number of viable cells was measured every 24 h using the CellTiter96 Aqueous Non-Radioactive Cell Proliferation Assay (MTS; Promega) according to the manufacturer’s instructions.
Northern blot
Northern blots to detect small RNAs were completed by standard protocol. Briefly, RNA was isolated from cells using TRIzol according to the manufacturer’s instructions. RNA (20 μg) was run on a 15% polyacrylamide/urea gel, which was then transferred on to Zeta Probe GT membranes (Bio-Rad Laboratories) by capillary action. Membranes were hybridized in ULTRAhyb-Oligo (LT, Life Technologies) overnight at 42°C with DNA probes complementary to miR-1343-3p (5′-GCGAGAGTGCGGGCCCCAGGAG-3′) or RNU6B (5′-CACGATTTGCGTGTCATCCTT-3′) that were γ32P-end-labelled with T4 polynucleotide kinase (New England Biolabs). Membranes were washed twice each in 2× SSC/0.1% SDS and 2× SSC/0.5% SDS before being exposed to a phosphoimager screen. Images were captured using a Typhoon FLA 7000 phosphoimager (GE Healthcare).
Statistical analysis and graphs
Results are expressed as means ± S.D. Statistics were performed using unpaired Student’s t tests on Prism software (GraphPad).
RESULTS
miR-1343 is predicted in silico to target both TGF-β receptors
Because TGF-β signalling is one of the most important promoters of lung fibrosis, we first sought to identify miRNAs that could directly repress expression of the two receptors that initiate the TGF-β signalling pathway: TGFBR1 and TGFBR2. The majority of miRNAs reduce gene expression post-transcriptionally by binding complementary seed sites located within the 3′-UTR of the transcript [40]. To identify miRNAs that target both TGFBR1 and TGFBR2 3′-UTRs, we employed TargetScan, a miRNA target prediction tool that is recognized as the most comprehensive and least error-prone in silico approach [40,41]. The TGFBR1 3′-UTR is 4886-bp-long within which TargetScan 6.2 predicts seed sites for 528 miRNAs. TGFBR2 has a 2543-bp 3′-UTR with 360 predicted miRNA-targeting sites. Intersection of these two data sets showed 175 miRNAs that were predicted to target both genes (Figure 1a). Fewer than 20 of these had context scores for both 3′-UTRs that were significant enough to warrant further study (<−0.15). Within this group, miR-1343 had top context scores for both TGFBR1 and TGFBR2 (−0.90 and −0.39 respectively; Figure 1b). miR-1343 is located within an intron of pyruvate dehydrogenase complex component X (PDHX), a gene mapping to chromosome 11p13. This genomic region was previously shown in a genome-wide association study (GWAS) to significantly associate with lung disease severity in CF patients carrying the F508del mutation [32] (Figure 1c). Because of the major impact of lung fibrosis on disease progression in CF, miR-1343 was a strong candidate for further analysis.
Figure 1. miR-1343 is the most significant miRNA predicted to target the TGF-β receptors.
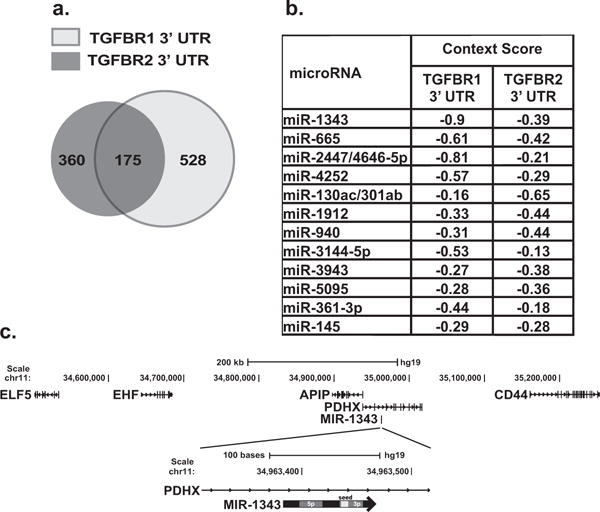
(a) Venn diagram illustrating the number of TargetScan-predicted miRNAs to target the TGFBR1 and TGFBR2 3′-UTRs. Overlapping region depicts miRNAs that are predicted to target both receptors. (b) Top miRNAs that are predicted to target both TGFBR1 and TGFBR2. Context scores are generated from TargetScan 6.2, with the more negative context scores being the most significant. (c) miR-1343 location at the chromosome 11p13 CF modifier locus and organization of its stem–loop structure.
miR-1343 targets the 3′-UTRs of TGFBR1 and TGFBR2
TGFBR1 was the most significant target gene predicted for miR-1343 according to TargetScan (Figures 1b and 2a). In addition to TGFBR2, SMAD-specific E3 ubiquitin protein ligase 1 (SMURF1), which acts as a negative regulator of TGF-β signalling, was also a predicted miR-1343 target (context score −0.28). Among other genes with high context scores that were relevant to lung biology and fibrosis were engulfment and cell motility (ELMO2, −0.89), collagen, type V, α1 (COL5A1, −0.70) and integrin α5 (ITGA5, −0.62; Figure 2a).
Figure 2. miR-1343 targets the 3′-UTRs of TGFBR1, TGFBR2 and ELMO2.
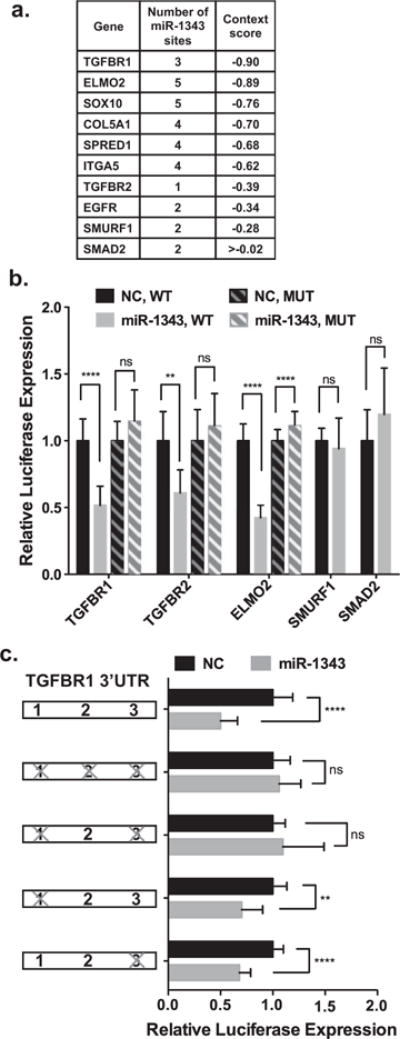
(a) TargetScan 6.2 analysis of miR-1343 predicted target genes. More negative context scores are most significant. (b) Luciferase assay of A549 cells transiently co-transfected with either wild-type (WT) or mutant (MUT) pMIR-REPORT-3′-UTR constructs and miR-1343 or NC miRNA. Luciferase values were normalized to pMIR-β-galactosidase levels and illustrated relative to the NC. n = 3. **P ≤ 0.01, ****P ≤ 0.0001; ns, not significant. (c) Luciferase assay of A549 cells transiently co-transfected with WT pMIR–REPORT–TGFBR1 3′-UTR or MUT constructs with seed sites mutations and miR-1343 or NC miRNA. Bars to the left illustrate the TGFBR1 3′-UTR with miR-1343 seed sites 1, 2 and 3. ‘X’ denotes miR-1343 seed sites that were mutated (Supplementary Figure S1). Luciferase values were normalized to pMIR-β-galactosidase levels and illustrated relative to the NC. n = 3; P-values same as for (b).
To establish whether miR-1343 targeted the 3′-UTRs of TGFBR1, TGFBR2, ELMO2 and SMURF1, these were cloned into the pMIR-Report vector downstream of a luciferase reporter gene. Luciferase expression from this vector decreases if the 3′-UTR is a target of the miRNA. Also included in these assays was the TGF-β effector SMAD2 (mothers against decapentaplegic homolog 2), which although a predicted target of miR-1343, has a very high context score (>0.02). The constructs were transiently transfected into A549 lung adenocarcinoma cells with precursor (pre-) miR-1343 or a non-targeting negative control (NC) miRNA. Luciferase activity was assayed 48 h post-transfection and normalized to a β-galactosidase transfection control (Figure 2b). miR-1343 significantly reduced luciferase expression from the TGFBR1, TGFBR2 and ELMO2 3′-UTR constructs in comparison with the NC miRNA. Mutation of the miR-1343 seed sites in TGFBR1, TGFBR2 and ELMO2 (Supplementary Figure S1) abolished this repression, demonstrating specific targeting by miR-1343. Additionally, miR-1343 had no impact on luciferase expression from the SMAD2 and SMURF1 3′-UTR constructs. Direct targeting of the 3′-UTR of TGFBR1, TGFBR2 and ELMO2 was confirmed in Caco2 intestinal epithelial cells (Supplementary Figure S2). These results indicate that miR-1343 reduces expression of both TGF-β receptors through direct targeting of their 3′-UTRs.
TGFBR1 contains three miR-1343 seed sites in its 3′-UTR (Figure 2a; Supplementary Figure S1), with sites 1 and 3 scored by TargetScan as well conserved, whereas site 2 is non-conserved. A combination of seed-site mutants were created within the pMIR-Report-TGFBR1 3′-UTR construct, with all three sites destroyed or sites 1 and 3 mutated individually or together (Figure 2c; Supplementary Figure S1). Using the same transfection protocol described above, mutation of all three miR-1343 sites abolished the impact of the miRNA on the TGFBR1 3′-UTR, as did combined destruction of sites 1 and 3. Single site mutations of either site 1 or site 3 alone still retained the effect of miR-1343. These data suggest that the two conserved sites are required to achieve full miR-1343 targeting potential.
miR-1343 controls the expression of genes important for lung health and disease
Since miRNAs often control biological processes at multiple levels, we next investigated the impact of miR-1343 genome-wide by RNA-seq. Pre-miR-1343 or NC miRNA were transiently overexpressed in A549 and 16HBE14o- lung epithelial cells. Total RNA was extracted 48 h post-transfection from four miR-1343 and four NC miRNA samples and RNA-seq was performed on libraries generated from them. Tophat and Cufflinks software were used with default parameters to generate fragments per kb per million mapped fragments (FPKM) values and CuffDiff was used to determine differentially expressed genes (DEGs) [42]. Transcripts of 4488 genes in A549 cells and 2393 genes in 16HBE14o- cells showed significant changes following miR-1343 overexpression compared with NC cells (Figure 3a) (GEO: GSE75591). Of these, 1225 were differentially expressed in both cell types (Figure 3b) with 713 decreasing and 391 increasing in the miR-1343-transfected cells. The remaining genes (121) showed opposite responses in the two cell types.
Figure 3. The impact of miR-1343 on gene expression in 16HBE14o- and A549 cells; RNA-seq identifies multiple pathways relevant to airway disease and fibrosis.
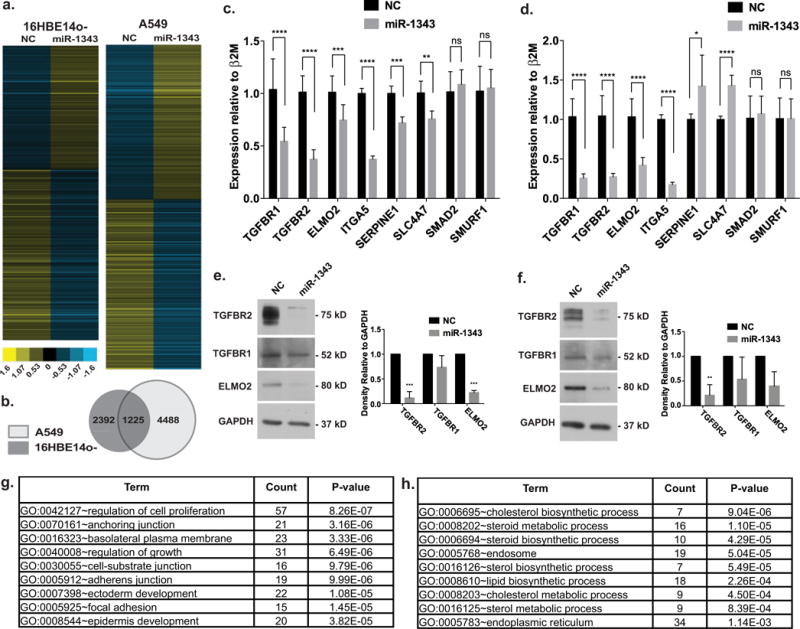
(a) Heat-maps show relative expression of DEGs following miR-1343 or NC miRNA transient expression in 16HBE14o- or A549 cells. Each line represents a different gene. (b) Venn diagram of DEGs identified by RNA-seq in A549 compared with 16HBE14o- cells. Overlapping region represents the number of DEGs identified in both cell types. (c) Gene expression levels measured by RT-qPCR 48 h after miR-1343 or NC miRNA transient transfection in A549 cells. Ct values normalized to β2M. n = 3. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001; ns, not significant. (d) Gene expression levels measured by RT–qPCR 48 h after miR-1343 or NC miRNA transient transfection in primary lung fibroblasts. CT values normalized to β2M. n = 3; P-values as for (c). (e) Western blots of A549 lysates transfected with miR-1343 or NC miRNA for 48 h and probed with antibodies specific to TGFBR1, TGFBR2 and ELMO2. Proteins were quantified relative to GAPDH loading control and are expressed as fold change compared with NC. n = 3; P-values as for (c). (f) Western blots of lysates from primary lung fibroblasts transfected with miR-1343 or NC miRNA for 48 h and probed and quantified as for (e). n = 3; P-values as for (c). (g and h) DAVID gene ontology analysis of down-regulated (g) or up-regulated (h) genes identified by RNA-seq in both A549and 16HBE14o- cells following miR-1343 or NC miRNA transient expression.
Overlapping the DEGs identified by RNA-seq with TargetScan-predicted miR-1343 targets yielded 96 genes in common, all of which probably represent true miR-1343 target genes. TGFBR1, TGFBR2, ELMO2 and ITGA5 were among those common genes and consistent with luciferase reporter gene assays (Figure 2b) SMAD2 and SMURF1 were not among the DEGs. Genes identified by both RNA-seq and TargetScan were further validated by reverse transcription (RT)-qPCR assays following NC or pre-miR-1343 transient expression in A549 cells (Figure 3c). Levels of TGFBR1, TGFBR2, ELMO2 and ITGA5 transcripts were significantly down-regulated in the presence of miR-1343 compared with the NC miRNA. Two additional genes that were differentially expressed by RNA-seq but were not predicted by TargetScan (suggesting that they are indirect targets) were also confirmed by RT–qPCR: serpin peptidase inhibitor, clade E member 1 (SERPINE1), a known TGF-β-responsive gene and solute carrier family 4, member 7 (SLC4A7), a sodium bicarbonate transporter. Also consistent with RNA-seq and luciferase assay results, levels of SMAD2 and SMURF1 transcripts were unaffected. Similar data for 16HBE14o- cells are shown in Supplementary Figure S3.
To confirm that the effects of miR-1343 were not limited to lung epithelial cells, miR-1343 and NC miRNA were transiently transfected into primary lung fibroblasts (Figure 3d). Again TGFBR1, TGFBR2, ELMO2 and ITGA5 expression levels were significantly decreased with pre-miR-1343 compared with the NC, whereas SMAD2 and SMURF1 levels were unaltered. However, in contrast with A549 cells, SERPINE1 and SLC4A7 expression increased in the primary lung fibroblasts. These observations, together with those from 16HBE14o- cells, suggest that genes indirectly targeted by miR-1343 can differ between cell types.
Since miRNAs can destabilize transcripts by several mechanisms, we next examined the effects of miR-1343 on the proteins encoded by its target genes. At 48 h after transient transfection of A549 cells with pre-miR-1343 or NC miRNA, cells were lysed and proteins were separated by SDS/PAGE followed by Western blotting. Blots were probed with antibodies specific for TGFBR1, TGFBR2 and ELMO2 and normalized to GAPDH as a loading control (Figure 3e). TGFBR2 and ELMO2 both showed statistically significant down-regulation of expression in miR-1343-treated cells compared with NC miRNA (between 88% and 98% and 73% and 82% reductions in expression respectively). The reduction in TGFBR1 with miR-1343 was less, at 12% to 60% reduction in three separate experiments (P = 0.17). Similar results showing significant down-regulation in TGFBR2 protein expression were seen after miR-1343 delivery in primary lung fibroblasts (84–92% reduction, Figure 3f) and 16HBE14o- cells (61–86% reduction; Supplementary Figure S4). TGFBR1 (17–97% reduction in lung fibroblasts; 20–30% in 16HBE14o-) and ELMO2 (27–81% reduction in lung fibroblasts; 11–77% in 16HBE14o-) were also reduced with miR-1343 treatment, although these values did not reach statistical significance (P = 0.21 P = 0.07 and P = 0.07, P = 0.07 respectively). These results indicate that miR-1343 is likely to regulate critical cellular processes.
To test this possibility, a gene ontology process enrichment analysis was performed on the DEGs that exhibited shared expression changes by RNA-seq in both airway cell lines using the Database for Annotation, Visualization and Integrated Discovery (DAVID) [43,44]. Among the DEGs down-regulated by miR-1343, pathways involved in lung epithelial function relating to cell growth, cell–substrate junction and cellular adhesion were significantly altered (Figure 3g; Supplementary Table S1). Consistent with these bioinformatic predictions, cell adhesion assays on collagen I-coated substrates showed that pre-miR-1343 significantly reduced adhesion of transfected A549 cells in comparison with NC miRNA at three time points (Supplementary Figure S5). Pathways involved in steroid/lipid biosynthesis and intracellular protein transport were over-represented among DEGs up-regulated by miR-1343 (Figure 3h; Supplementary Table S2). These results suggest that miR-1343 may regulate, both directly and indirectly, multiple cellular processes that are key to lung epithelial function.
miR-1343 perturbs the canonical TGF-β signalling pathway
Since miR-1343 targeted and repressed both of the TGF-β receptors in vitro, we next investigated whether this miRNA could influence the TGF-β signalling pathway. First, we used the p3TP-lux vector, which contains a portion of the SERPINE1 promoter and three TGF-β-activated TPA (tetradecanoylphorbol acetate) responsive elements driving luciferase expression [37]. Pre-miR-1343 or NC miRNA was transfected into A549 cells with p3TP-lux and a Renilla transfection control vector. After 48 h, cells were treated with TGF-β1 to induce the TGF-β signalling pathway (Figure 4a). TGF-β stimulation of luciferase expression in cells treated with miR-1343 was only 25% of the levels seen in NC miRNA-treated cells. This result suggests that miR-1343 reduces TGF-β signalling in these cells and that its activity diminishes TGF-β-responsive gene expression.
Figure 4. Overexpression of miR-1343 reduces canonical TGF-β signalling.
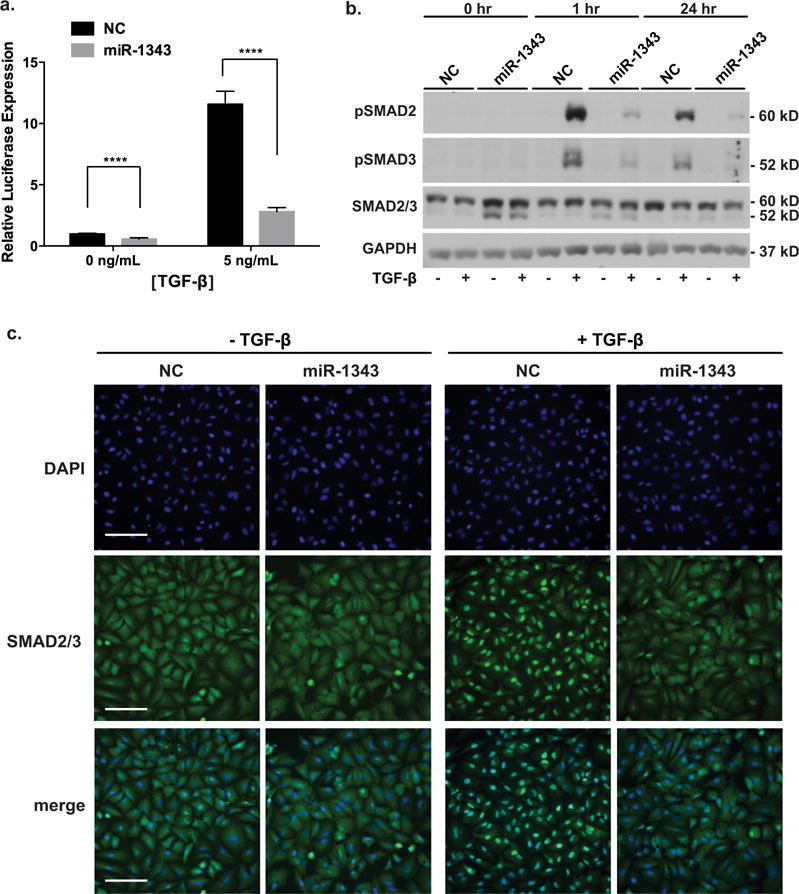
(a) Luciferase assay of A549 cells transiently transfected with the p3TP-lux vector and either miR-1343 or NC miRNA. Cells were treated with TGF-β1 (5 ng/ml) or vehicle control (0 ng/ml), 48 h after transfection, for 24 h. Luciferase values were normalized to pMIR-β-galactosidase levels and expressed relative to NC treated with vehicle. n = 3; ****P ≤ 0.0001. (b) miR-1343 represses phosphorylation of SMAD2/3. Western blot of lysates from A549 cells transiently transfected with miR-1343 or NC miRNA and treated with TGF-β1 (5 ng/ml, +) or vehicle control (−) for the indicated period of time. Blots were probed with antibodies specific for pSMAD2 [phosphorylated (active) SMAD2], pSMAD3 [phosphorylated (active) SMAD3] and total SMAD2/3; slower migrating species is SMAD2 and faster is SMAD3. GAPDH was the loading control. (c) miR-1343 inhibits nuclear localization of pSMAD2/3. Representative images of immunofluorescence in A549 cells transiently transfected with miR migrating species-1343 or NC miRNA and treated with TGF-β1 (50 ng/ml, +) or vehicle control (−) for 1 h. Green fluorescence shows total SMAD2/3 and blue is DAPI nuclear counterstain. Merge illustrates total SMAD2/3 plus DAPI. Scale bar = 100 μm.
In its canonical signalling pathway, TGF-β binds to TGFBR1 and TGFBR2, which then dimerize to activate receptor SMAD factors (R-SMADs), SMAD2 and SMAD3, by receptor serine kinase activity. Phosphorylated R-SMADs co-localize with the common SMAD (Co-SMAD) SMAD4 in the cytoplasm, which promotes R-SMAD translocation to the nucleus where it affects gene expression [45]. To determine whether miR-1343 reduces TGF-β signalling by this mechanism, we next investigated the endogenous cellular pathway by monitoring phosphorylation and hence activation of SMAD2 and SMAD3 (pSMAD2, pSMAD3). Pre-miR-1343 or NC miRNA was transiently transfected into A549 cells, which after 48 h were exposed to TGF-β1 for 1 or 24 h. Cell lysates were resolved by SDS/PAGE and Western blots were probed with antibodies specific for pSMAD2, pSMAD3 or total SMAD2/3 (Figure 4b). miR-1343 substantially reduced the ratio of pSMAD2 and pSMAD3 to total SMAD2/3 after 1 and 24 h of TGF-β treatment in comparison with NC miRNA. Total SMAD2/3 levels were unchanged. Consistent results were obtained when this experiment was repeated in primary lung fibroblasts (Supplementary Figure S6).
Because TGF-β1 activation/phosphorylation of SMAD2 and SMAD3 causes their nuclear translocation, we next investigated whether the reduced levels of pSMAD2/3 observed in miR-1343-transfected cells correlated with a lack of their translocation to the nucleus. A549 cells were transiently transfected with pre-miR-1343 or NC miRNA for 48 h. Cellular localization of total SMAD2/3 was examined by immunofluorescence after either TGF-β1 treatment (50 ng/ml) or serum-starvation conditions for 1 h (Figure 4c). In the absence of TGF-β, both NC and miR-1343-transfected cells showed diffuse SMAD2/3 localization throughout the cytoplasm. As expected, TGF-β1 treatment in NC cells induced the nuclear translocation of SMAD2/3; however, this movement was largely abolished in miR-1343-containing cells. These data demonstrate the potent effects of miR-1343 on the canonical TGF-β signalling pathway.
miR-1343 represses pathways of TGF-β-induced fibrosis
Next, to determine whether phenotypes associated with TGF-β-induced fibrosis are also influenced by miR-1343, we focused on collagen type I, α1 (COL1A1) and α-smooth muscle actin (αSMA). These are both structural protein markers of active myofibroblasts and are known to be induced by TGF-β [3]. Primary lung fibroblasts were transiently transfected with pre-miR-1343 or NC miRNA and treated with TGF-β1 for 48 h. Cell lysates were separated by SDS/PAGE and Western blots were probed with specific antibodies for each protein or GAPDH as a control (Figure 5a). TGF-β induced the expression of both COL1A1 and αSMA in NC miRNA-treated fibroblasts, demonstrating the transition of the lung fibroblasts to an active myofibroblast-like state. In contrast, induction of COL1A1 and αSMA were substantially attenuated in the miR-1343-treated cells. Expression of αSMA protein following TGF-β treatment was also investigated by immunofluorescence in primary lung fibroblasts (Figure 5b; Supplementary Figure S7). After TGF-β exposure, extensive fibres of αSMA were seen in NC miRNA-treated fibroblasts. However, in the presence of miR-1343, intracellular αSMA fibres were not evident and the small amounts of protein were diffuse. These results suggest that miR-1343 can repress phenotypes associated with TGF-β-induced transition of resident fibroblasts to disease-causing myofibroblasts in the lung.
Figure 5. Phenotypes associated with TGF-β-induced fibrosis are repressed in miR-1343-overexpressing cells.
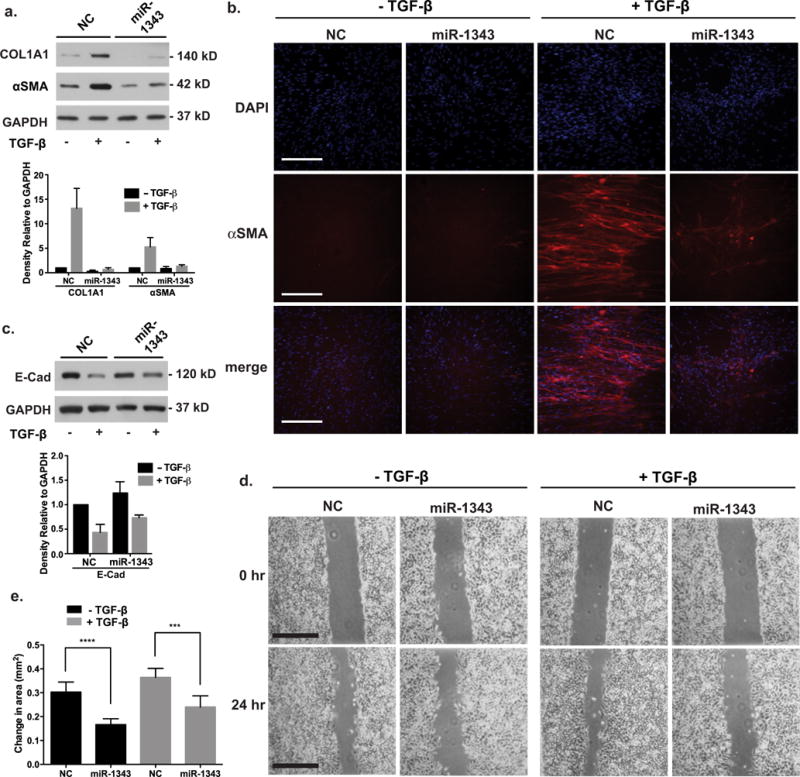
(a) Western blot of lysates from primary lung fibroblasts transiently transfected with miR-1343 or NC miRNA and treated with TGF-β1 (5 ng/ml, +) or vehicle control (−) for 48 h. Blots were probed with antibodies specific for COL1A1 and αSMA. GAPDH was the loading control. Proteins were quantified as in Figure 3(e). n = 3. (b) miR-1343 impairs synthesis and structural organization of αSMA after TGF-β exposure. Immunofluorescence in primary lung fibroblasts transiently transfected with miR-1343 or NC miRNA and treated with TGF-β1 (5 ng/ml, +) or vehicle control (−) for 48 h. Red fluorescence shows αSMA and blue fluorescence shows nuclei (DAPI). Merge illustrates αSMA staining plus DAPI. Scale bar = 250 μm. (c) Western blot of lysates from A549 cells transiently transfected with miR-1343 or NC miRNA and treated with TGF-β1 (5 ng/ml, +) or vehicle control (−) for 48 h. Blots were probed with antibodies specific for E-cad. GAPDH was the loading control. Proteins were quantified as in Figure 3(e). n = 3. (d) Wound scratch assay in A549 cells transiently transfected with miR-1343 or NC miRNA and treated with TGF-β1 (5 ng/ml, +) or vehicle control (−). TGF-β was added at the 0 h time point when the scratch was created and cells were imaged 24 h later. Scale bar = 0.5 mm. (e) Quantification of change in wound closure area between 0 and 24 h of A549 cells described in (d). ****P ≤ 0.0001, ***P ≤ 0.001. Results are from three biological replicates, with five technical replicates each.
Another cellular process that is directly induced by TGF-β is cancer-associated EMT. During EMT, E-cad relocates from epithelial intercellular junctions and becomes cytoplasmic, thus allowing cells to become more motile, a characteristic of mesenchymal cell types. This process was observed previously in A549 cells, where it is also associated with reduced levels of E-cad [46]. To test whether miR-1343 affects TGF-β-induced EMT, A549 cells were transiently transfected with pre-miR-1343 or NC miRNA and treated with TGF-β1 for 48 h. Cell lysates were resolved by SDS/PAGE and E-cad levels examined by Western blotting with a specific antibody (Figure 5c). NC miRNA-treated cells showed greatly reduced levels of E-cad when treated with TGF-β; however, this reduction was less evident in miR-1343-treated cells.
Next, since mesenchymal cells exhibit increased motility, we assessed the migration of A549 cells into an artificial scratch wound following miRNA transient transfection. Although the scratch-wounding assay functions only as an approximate model for an actual wound, it allows for the observation of cellular migration and proliferation over time. At 48 h after pre-miR-1343 or NC miRNA transfection, EMT was induced by TGF-β1 stimulation. Simultaneously, a scratch wound was made across the confluent A549 cell monolayer and movement into the scratch was monitored microscopically during the following 24 h (Figures 5d and 5e). NC-treated cells exhibited marked wound closure in the presence of TGF-β, with the wound area closing 60% from an average of 0.6 to 0.24 mm2. However, wound healing into the scratch was considerably reduced in miR-1343-treated cells, with wound area closing only 40% from 0.6 to 0.36 mm2. Pre-miR-1343 transfected into A549 cells was also shown to slightly impair cell proliferation compared with NC transfected cells (Supplementary Figure S8). These results suggest that miR-1343 reduces cell migration and proliferation into a wound.
miR-1343 is processed in haemopoietic cells and in airway epithelial cells under stress
Our observations on the effects of miR-1343 on the TGF-β signalling pathway in lung epithelial cells and fibroblasts primarily used exogenous pre-miRNA transfected into cells. Since it is not possible to calibrate the amount of processed miRNA in these experiments, we next investigated the abundance and maturation of endogenous miR-1343 in primary cells with an important role in lung biology and relevant derivative cell lines. First, we used a commercial RT–qPCR TaqMan assay (Life Technologies) which is designed to detect only mature miR-1343, with levels normalized to the small nuclear RNA RNU6B. miR-1343 levels were assayed in A549 and Calu3 lung adenocarcinoma cells, 16HBE14o-, primary HBE cells, human nasal epithelial cells (HNE, from nasal scrapes), primary lung fibroblasts, Caco2, HL-60 promyelocytic leukaemia cells, K562 erythroleukaemia cells, THP-1 acute monocytic leukaemia cells, primary human lymphocytes and primary human neutrophils (Figure 6a). (This neutrophil population may contain a minority of other myeloid cell types that were simultaneously extracted from blood by density gradient centrifugation). Expression levels of miR-1343 were similar in all cell types assayed with the exception of primary HBE cells, neutrophils and the HL-60 and K562 cell lines, which had ~20–40-fold more miRNA than the lowest-expressing A549 cells.
Figure 6. Mature miR-1343expression is most abundant in neutrophils and is induced by serum starvation in lung epithelial cells.
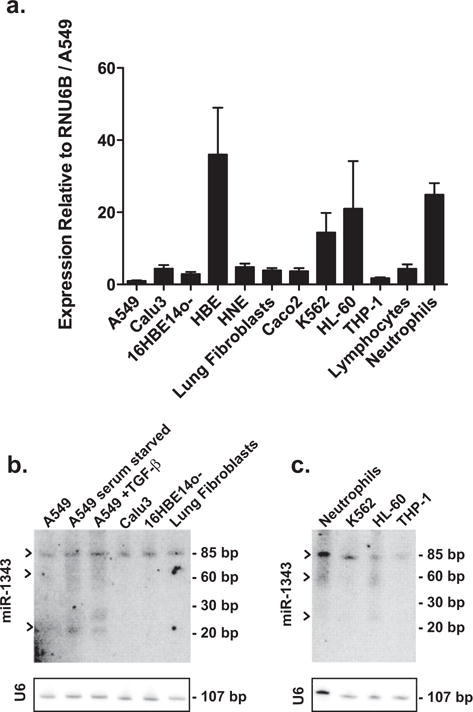
(a) miR-1343 expression levels measured in multiple cell types by TaqMan RT–qPCR assay. Values were normalized to RNU6B and are shown relative to A549 levels. For cell type description see the Results section. n = 3. (b) Northern blot showing levels of miR-1343in lung-derived cell types. Black arrows indicate unprocessed (~85 bp), precursor (~60 bp) and mature (~23 bp) miRNA forms. Blots were stripped and re-hybridized with a probe against RNU6B to confirm equal loading. For cell type description see the Results section. (c) Northern blot showing levels of miR-1343in various haematopoietic cell types. Black arrows as for (b). U6 probing to confirm equal loading as in (b).
To directly measure endogenous levels of miR-1343 and to distinguish between the precursor and mature forms of the miRNA, we also performed Northern blots of RNA extracted from the same cell types (Figures 6b and 6c). Northern blots were probed with γ-32P-labelled DNA probes complementary to human miR-1343-3p and RNU6B as a loading control. An unprocessed transcript (~85 bp) was equally abundant in all cell types. However, processed forms of miR-1343 (premature ~60 bp, mature ~23 bp), were only detected in primary neutrophils and HL-60 cells and in A549 cells that were serum-starved and/or treated with TGF-β (5 ng/ml) in serum-depleted medium for 48 h (Figures 6b and 6c). These results suggest that the main source of processed miR-1343 under normal conditions is within neutrophils and that it can be induced in lung epithelial cells upon stress.
DISCUSSION
In contrast with several previous reports that identified miRNAs involved in pathways of fibrosis through comparisons of normal and fibrotic tissue [18,20–22] or by differential gene expression in primary cells and cancer cell lines [16], we identified miR-1343 by searching for miRNAs that could directly target the TGF-β receptors.
By taking this approach we first demonstrated that the poorly characterized miR-1343 binds to seed sites in the 3′-UTR of TGFBR1 and TGFBR2 and represses endogenous levels of the receptors. In turn, miR-1343 represses canonical TGF-β signalling pathways in several cell types as shown by inhibition of SMAD2/3 phosphorylation and nuclear translocation. Subsequent to TGF-β exposure, miR-1343 also reduces expression of markers of fibrosis, such as αSMA and COL1A1.
Our luciferase assays and RT–qPCR data show miR-1343 to substantially repress both TGFBR1 and TGFBR2 expression by targeting their 3′-UTRs. However, the levels of modulation of mRNA are apparently not directly correlated with protein abundance. Western blots show that TGFBR2 protein is more sensitive to repression by miR-1343 than is TGFBR1. This observation may in part be due to poor specificity of the anti-TGFBR1 antibody in comparison with the reagent against TGFBR2. Lack of a direct correlation between mRNA and protein levels is not unexpected since there are many parameters of RNA and protein turnover that could affect the observed protein levels. Moreover, TGFBR1 and TGFBR2 may be subject to different mechanisms of miRNA-mediated repression and the subcellular localization of each mRNA may diverge [47]. Nevertheless, because TGFBR1 and TGFBR2 function as a heterodimer, dependent on one another for signalling [37,48,49], the biological activity of the receptor complex is likely to be determined by the expression levels of either subunit.
The genomic location of miR-1343 in a region marked by single nucleotide polymorphisms (SNPs) that associate with lung disease severity in F508del CF is noteworthy [32]. The GWAS implicated four genes that are close to the critical interval at chromosome 11p13, of which two [Ets homologous factor (EHF) and E74-like factor 5 (ELF5)] are epithelial-specific transcription factors and two [APAF interacting protein (APIP) and PDHX] are ubiquitously expressed genes involved in basic cellular processes [32]. We showed that EHF regulates genes important for maintenance of the lung epithelial barrier and its response to injury [38]. However, the mechanism(s) whereby this genomic region contributes to lung disease severity in CF is not understood. Since miR-1343 is located within an intron of the PDHX gene, which is involved in the conversion of pyruvate to acetyl -Co A in mitochondrial metabolism, it is likely that expression of these two genes may be co-regulated. This could have broad implications in lung health and disease. Also of relevance in the context of genetic modifiers of CF lung disease are correlations with TGF-β [14,50], although the molecular bases for these associations are not well established.
Our data showing the highest levels of processed miR-1343 in primary cells of myeloid lineage, specifically neutrophils and the HL-60 neutrophil-like cell line, suggest a predominant function in these cell types. Neutrophils, the most abundant myeloid cells in humans, are key regulators of respiratory function and are among the first cells recruited to sites of injury where they work to clear infection and amplify the inflammatory response [2]. Sustained neutrophil influx, survival and activation are all processes regulated by TGF-β signalling and are known to enhance lung fibrosis [51]. Based on our data showing miR-1343 to target TGFBR1 and TGFBR2 to reduce TGF-β signalling, it is likely that miR-1343 in neutrophils may interfere with the same pathways to block fibrosis. Another intriguing possibility is that miR-1343 may be released from neutrophils and affect TGF-β signalling pathways in nearby cells in the lung epithelium. Several studies showed that HL-60 cells and other myeloid cell types secrete exosomes containing a variety of RNA molecules, including miRNAs [52,53]. These exosomes can be taken up by other cells, for example in the bone marrow, where they alter gene expression [53]. By analogy, miR-1343-containing exosomes released by neutrophils in the lung could be delivered to epithelial cells or fibroblasts, thus altering their gene expression profiles and phenotype.
The observation that miR-1343 processing is induced by stress in A549 cells suggests that this miRNA may also regulate response to injury in lung epithelial cells. Although serum starvation is a known stress inducer in cultured cells [54], the impact of other forms of stress on miR-1343 is currently unknown. Moreover, it is not clear whether activation of TGF-β signalling has an additive effect to stress. miRNA expression is known to be induced by various forms of cell stress [55–57] and activity of miRNAs can also be modulated following amino acid starvation [47] or hippuristanol-induced stress [58].
Our work on miR-1343 is focused on human cells since it is not well conserved across species. Like many non-conserved miRNAs, miR-1343 may have evolved from a transposable element [59]. By using miRBase [60] and the Basic Local Alignment Search Tool (BLAST) [61], miR-1343 was not found to be conserved in rodents. However, it is conserved in chimpanzees, cows and pigs, where it is also located within an intron of the PDHX gene [29,31,61]. Furthermore, the miR-1343 seed sites in the TGFBR1 and TGFBR2 3′-UTRs are conserved across species that have miR-1343, but not in those lacking the miRNA [61]. This suggests co-evolution of miR-1343 and the 3′-UTRs that it regulates and also implies that miR-1343 cannot be accurately studied in species without the miRNA.
Despite the lack of a relevant animal model of lung fibrosis to investigate miR-1343, our data showing its role in regulating TGF-β signalling and fibrosis make it a compelling candidate for human-specific therapeutic approaches. Current treatments for fibrotic lung disease are largely based on targeting the immune system, which are ineffective at slowing or halting disease progression [62]. However, miRNAs are emerging as promising new targets for treatment of many human diseases [63]. Increasing miR-1343 levels in the airway epithelium could be a direct route to slow the progression of lung fibrosis. Although overexpression of miRNAs is a less well-characterized therapeutic approach than antisense inhibitor technology for blocking miRNAs, miRNA replacement therapy is a viable option [64]. Currently under development are therapies utilizing miR-29, which reduced the severity of bleomycin models of mouse pulmonary fibrosis [25], miR-16, which significantly slowed the growth of metastatic prostate cancer in mice [65], and miR-34, which inhibited models of non-small-cell lung cancers [66]. As drug formulation and delivery technology for miRNA mimics continue to be improved, miR-1343 stands out as a promising new candidate for therapy in diseases involving organ fibrosis.
Supplementary Material
Acknowledgments
We thank Dr Scott Randell for primary lung fibroblasts, HBE cells and nasal scrape RNA, Dr Lawrence Jennings, Dr Shih-Hsing Leir and Michael Mutolo for assistance, Lisa Jones for RNA-seq library preparation and Dr Michael Klüppel for helpful discussions.
FUNDING
This work was supported by the National Institutes of Health [grant numbers R01HL117843 (to A.H.) and F31HL126458 (to L.R.S.)].
Abbreviations
- CF
cystic fibrosis
- COL1A1
collagen type I α1
- COPD
chronic obstructive pulmonary disease
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- DEG
differentially expressed gene
- DMEM
Dulbecco’s modified Eagle’s medium
- E-cad
epithelial cadherin
- ECM
extracellular matrix
- EHF
Ets homologous factor
- ELMO2
engulfment and cell motility 2
- EMT
epithelial-to-mesenchymal transition
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GWAS
genome-wide association study
- HBE
human bronchial epithelial
- IPF
idiopathic pulmonary fibrosis
- ITGA5
integrin α5
- NC
negative control
- PDHX
pyruvate dehydrogenase complex component X
- pre-miR-1343
precursor miR-1343
- qPCR
quantitative PCR
- RNA-seq
RNA-sequencing
- RNU6B
U6 small nuclear RNA
- R-SMAD
receptor SMAD factor
- RT
reverse transcription
- SERPINE1
serpin peptidase inhibitor clade E member 1
- SLC4A7
solute carrier family 4 member 7
- SMAD2/3
mothers against decapentaplegic homolog 2/3
- SMURF1
SMAD-specific E3 ubiquitin protein ligase 1
- SSC
Saline Sodium Citrate
- TBST
TBS + 0.1 % Tween 20
- TGFBR1/2
TGF-β receptor1/2
- TGF-β
transforming growth factor β
- αSMA
α-smooth muscle actin
- β2M
β2-microglobulin
- TPA
tetradecanoylphorbol acetate
Footnotes
AUTHOR CONTRIBUTION
Lindsay Stolzenburg performed the majority of experiments, designed and optimized assays and analysed data. Sarah Wachtel assisted with experiments. Hong Dang analysed the RNA-seq data. Ann Harris participated in experimental planning, design and data interpretation. Lindsay Stolzenburg and Ann Harris wrote the manuscript. All authors contributed to manuscript edits before submission.
References
- 1.Rackley CR, Stripp BR. Building and maintaining the epithelium of the lung. J Clin Invest. 2012;122:2724–2730. doi: 10.1172/JCI60519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 4.Mak JCW, Chan-Yeung MMW, Ho SP, Chan KS, Choo K, Yee KS, Chau CH, Cheung AHK, Ip MSM. Elevated plasma TGF-beta1 levels in patients with chronic obstructive pulmonary disease. Respir Med. 2009;103:1083–1089. doi: 10.1016/j.rmed.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Harris WT, Muhlebach MS, Oster RA, Knowles MR, Noah TL. Transforming growth factor-beta(1) in bronchoalveolar lavage fluid from children with cystic fibrosis. Pediatr Pulmonol. 2009;44:1057–1064. doi: 10.1002/ppul.21079. [DOI] [PubMed] [Google Scholar]
- 6.Santana A, Saxena B, Noble NA, Gold LI, Marshall BC. Increased expression of transforming growth factor beta isoforms (beta 1, beta 2, beta 3) in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 1995;13:34–44. doi: 10.1165/ajrcmb.13.1.7541221. [DOI] [PubMed] [Google Scholar]
- 7.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci USA. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanscoy LL, Blackman SM, Collaco JM, Bowers A, Lai T, Naughton K, Algire M, McWilliams R, Beck S, Hoover-Fong J, et al. Heritability of lung disease severity in cystic fibrosis. Am J Respir Crit Care Med. 2007;175:1036–1043. doi: 10.1164/rccm.200608-1164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scholand MB, Coon H, Wolff R, Cannon-Albright L. Use of a genealogical database demonstrates heritability of pulmonary fibrosis. Lung. 2013;191:475–481. doi: 10.1007/s00408-013-9484-2. [DOI] [PubMed] [Google Scholar]
- 11.Silverman EK, Pierce JA, Province MA, Rao DC, Campbell EJ. Variability of pulmonary function in alpha-1-antitrypsin deficiency: clinical correlates. Ann Intern Med. 1989;111:982–991. doi: 10.7326/0003-4819-111-12-982. [DOI] [PubMed] [Google Scholar]
- 12.Celedón JC, Lange C, Raby BA, Litonjua AA, Palmer LJ, DeMeo DL, Reilly JJ, Kwiatkowski DJ, Chapman HA, Laird N, et al. The transforming growth factor-beta1 (TGFB1) gene is associated with chronic obstructive pulmonary disease (COPD) Hum Mol Genet. 2004;13:1649–1656. doi: 10.1093/hmg/ddh171. [DOI] [PubMed] [Google Scholar]
- 13.Wu L, Chau J, Young RP, Pokorny V, Mills GD, Hopkins R, McLean L, Black PN. Transforming growth factor-beta1 genotype and susceptibility to chronic obstructive pulmonary disease. Thorax. 2004;59:126–129. doi: 10.1136/thorax.2003.005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drumm ML, Konstan MW, Schluchter MD, Handler A, Pace R, Zou F, Zariwala M, Fargo D, Xu A, Dunn JM, et al. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443–1453. doi: 10.1056/NEJMoa051469. [DOI] [PubMed] [Google Scholar]
- 15.Arkwright PD, Laurie S, Super M, Pravica V, Schwarz MJ, Webb AK, Hutchinson IV. TGF-beta(1) genotype and accelerated decline in lung function of patients with cystic fibrosis. Thorax. 2000;55:459–462. doi: 10.1136/thorax.55.6.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 17.Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37:D98–D104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, Konishi K, Yousem SA, Singh M, Handley D, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:220–229. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pottier N, Maurin T, Chevalier B, Puisségur MP, Lebrigand K, Robbe-Sermesant K, Bertero T, Lino Cardenas CL, Courcot E, Rios G, et al. Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: implication in epithelial-mesenchymal interactions. PLoS One. 2009;4:e6718. doi: 10.1371/journal.pone.0006718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhattacharyya S, Balakathiresan NS, Dalgard C, Gutti U, Armistead D, Jozwik C, Srivastava M, Pollard HB, Biswas R. Elevated miR-155 promotes inflammation in cystic fibrosis by driving hyperexpression of interleukin-8. J Biol Chem. 2011;286:11604–11615. doi: 10.1074/jbc.M110.198390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang S, Cui H, Xie N, Icyuz M, Banerjee S, Antony VB, Abraham E, Thannickal VJ, Liu G. miR-145 regulates myofibroblast differentiation and lung fibrosis. FASEB J. 2013;27:2382–2391. doi: 10.1096/fj.12-219493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, Kaminski N, Abraham E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207:1589–1597. doi: 10.1084/jem.20100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Megiorni F, Cialfi S, Cimino G, De Biase RV, Dominici C, Quattrucci S, Pizzuti A. Elevated levels of miR-145 correlate with SMAD3 down-regulation in cystic fibrosis patients. J Cyst Fibros. 2013;12:797–802. doi: 10.1016/j.jcf.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Xiao J, Meng XM, Huang XR, Chung AC, Feng YL, Hui DS, Yu CM, Sung JJ, Lan HY. miR-29 inhibits bleomycin-induced pulmonary fibrosis in mice. Mol Ther. 2012;20:1251–1260. doi: 10.1038/mt.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montgomery RL, Yu G, Latimer PA, Stack C, Robinson K, Dalby CM, Kaminski N, van Rooij E. MicroRNA mimicry blocks pulmonary fibrosis. EMBO Mol Med. 2014;6:1347–1356. doi: 10.15252/emmm.201303604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang S, Xie N, Cui H, Banerjee S, Abraham E, Thannickal VJ, Liu G. miR-31 is a negative regulator of fibrogenesis and pulmonary fibrosis. FASEB J. 2012;26:3790–3799. doi: 10.1096/fj.11-202366. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Meunier J, Lemoine F, Soumillon M, Liechti A, Weier M, Guschanski K, Hu H, Khaitovich P, Kaessmann H. Birth and expression evolution of mammalian microRNA genes. Genome Res. 2013;23:34–45. doi: 10.1101/gr.140269.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Persson H, Kvist A, Rego N, Staaf J, Vallon-Christersson J, Luts L, Loman N, Jonsson G, Naya H, Hoglund M, et al. Identification of new microRNAs in paired normal and tumor breast tissue suggests a dual role for the ERBB2/Her2 gene. Cancer Res. 2011;71:78–86. doi: 10.1158/0008-5472.CAN-10-1869. [DOI] [PubMed] [Google Scholar]
- 29.Glazov EA, Kongsuwan K, Assavalapsakul W, Horwood PF, Mitter N, Mahony TJ. Repertoire of bovine miRNA and miRNA-like small regulatory RNAs expressed upon viral infection. PLoS One. 2009;4:e6349. doi: 10.1371/journal.pone.0006349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen T, Xi QY, Ye RS, Cheng X, Qi QE, Wang SB, Shu G, Wang LN, Zhu XT, Jiang QY, et al. Exploration of microRNAs in porcine milk exosomes. BMC Genomics. 2014;15:100. doi: 10.1186/1471-2164-15-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen C, Deng B, Qiao M, Zheng R, Chai J, Ding Y, Peng J, Jiang S. Solexa sequencing identification of conserved and novel microRNAs in backfat of Large White and Chinese Meishan pigs. PLoS One. 2012;7:e31426. doi: 10.1371/journal.pone.0031426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wright FA, Strug LJ, Doshi VK, Commander CW, Blackman SM, Sun L, Berthiaume Y, Cutler D, Cojocaru A, Collaco JM, et al. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet. 2011;43:539–546. doi: 10.1038/ng.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, Parks WP. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–1423. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 34.Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;10:38–47. doi: 10.1165/ajrcmb.10.1.7507342. [DOI] [PubMed] [Google Scholar]
- 35.Fogh J, Fogh JM, Orfeo T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst. 1977;59:221–226. doi: 10.1093/jnci/59.1.221. [DOI] [PubMed] [Google Scholar]
- 36.Gillen AE, Gosalia N, Leir SH, Harris A. MicroRNA regulation of expression of the cystic fibrosis transmembrane conductance regulator gene. Biochem J. 2011;438:25–32. doi: 10.1042/BJ20110672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wrana JL, Attisano L, Cárcamo J, Zentella A, Doody J, Laiho M, Wang XF, Massagué J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- 38.Fossum SL, Mutolo MJ, Yang R, Dang H, O’Neal WK, Knowles MR, Leir SH, Harris A. Ets homologous factor regulates pathways controlling response to injury in airway epithelial cells. Nucleic Acids Res. 2014;42:13588–13598. doi: 10.1093/nar/gku1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leir SH, Holgate ST, Lackie PM. Inflammatory cytokines can enhance CD44-mediated airway epithelial cell adhesion independently of CD44 expression. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1305–L1311. doi: 10.1152/ajplung.00255.2002. [DOI] [PubMed] [Google Scholar]
- 40.Alexiou P, Maragkakis M, Papadopoulos GL, Reczko M, Hatzigeorgiou AG. Lost in translation: an assessment and perspective for computational microRNA target identification. Bioinformatics. 2009;25:3049–3055. doi: 10.1093/bioinformatics/btp565. [DOI] [PubMed] [Google Scholar]
- 41.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 42.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 45.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 46.Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT) Respir Res. 2005;6:56. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 48.Anders RA, Leof EB. Chimeric granulocyte/macrophage colony-stimulating factor/transforming growth factor-beta (TGF-beta) receptors define a model system for investigating the role of homomeric and heteromeric receptors in TGF-beta signaling. J Biol Chem. 1996;271:21758–21766. doi: 10.1074/jbc.271.36.21758. [DOI] [PubMed] [Google Scholar]
- 49.Luo K, Lodish HF. Signaling by chimeric erythropoietin-TGF-beta receptors: homodimerization of the cytoplasmic domain of the type I TGF-beta receptor and heterodimerization with the type II receptor are both required for intracellular signal transduction. EMBO J. 1996;15:4485–4496. [PMC free article] [PubMed] [Google Scholar]
- 50.Dorfman R, Sandford A, Taylor C, Huang B, Frangolias D, Wang Y, Sang R, Pereira L, Sun L, Berthiaume Y, et al. Complex two-gene modulation of lung disease severity in children with cystic fibrosis. J Clin Invest. 2008;118:1040–1049. doi: 10.1172/JCI33754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lagraoui M, Gagnon L. Enhancement of human neutrophil survival and activation by TGF-beta 1. Cell Mol Biol. 1997;43:313–318. [PubMed] [Google Scholar]
- 52.Feng DQ, Huang B, Li J, Liu J, Chen XM, Xu YM, Chen X, Zhang HB, Hu LH, Wang XZ. Selective miRNA expression profile in chronic myeloid leukemia K562 cell-derived exosomes. Asian Pac J Cancer Prev. 2013;14:7501–7508. doi: 10.7314/apjcp.2013.14.12.7501. [DOI] [PubMed] [Google Scholar]
- 53.Huan J, Hornick NI, Shurtleff MJ, Skinner AM, Goloviznina NA, Roberts CT, Kurre P. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013;73:918–929. doi: 10.1158/0008-5472.CAN-12-2184. [DOI] [PubMed] [Google Scholar]
- 54.Pirkmajer S, Chibalin AV. Serum starvation: caveat emptor. Am J Physiol Cell Physiol. 2011;301:C272–C279. doi: 10.1152/ajpcell.00091.2011. [DOI] [PubMed] [Google Scholar]
- 55.Li G, Luna C, Qiu J, Epstein DL, Gonzalez P. Alterations in microRNA expression in stress-induced cellular senescence. Mech Ageing Dev. 130:731–741. doi: 10.1016/j.mad.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 57.Simone NL, Soule BP, Ly D, Saleh AD, Savage JE, Degraff W, Cook J, Harris CC, Gius D, Mitchell JB. Ionizing radiation-induced oxidative stress alters miRNA expression. PLoS One. 2009;4:e6377. doi: 10.1371/journal.pone.0006377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leung AKL, Calabrese JM, Sharp PA. Quantitative analysis of Argonaute protein reveals microRNA-dependent localization to stress granules. Proc Natl Acad Sci USA. 2006;103:18125–18130. doi: 10.1073/pnas.0608845103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Piriyapongsa J, Mariño-Ramírez L, Jordan IK. Origin and evolution of human microRNAs from transposable elements. Genetics. 2007;176:1323–1337. doi: 10.1534/genetics.107.072553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 62.Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5:167sr1. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]
- 63.Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov. 2014;13:622–638. doi: 10.1038/nrd4359. [DOI] [PubMed] [Google Scholar]
- 64.Bader AG, Brown D, Winkler M. The promise of microRNA replacement therapy. Cancer Res. 2010;70:7027–7030. doi: 10.1158/0008-5472.CAN-10-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takeshita F, Patrawala L, Osaki M, Takahashi R, Yamamoto Y, Kosaka N, Kawamata M, Kelnar K, Bader AG, Brown D, et al. Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol Ther. 2010;18:181–187. doi: 10.1038/mt.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wiggins JF, Ruffino L, Kelnar K, Omotola M, Patrawala L, Brown D, Bader AG. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010;70:5923–5930. doi: 10.1158/0008-5472.CAN-10-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.