

Abstract
BACKGROUND AND PURPOSE:
In patients with premanifest (nonsymptomatic) and advanced Huntington disease, changes in brain iron levels in the basal ganglia have been previously reported, especially in the striatum. Quantitative susceptibility mapping by using MR phase imaging allows in vivo measurements of tissue magnetic susceptibility, which has been shown to correlate well with iron levels in brain gray matter and is believed to be more specific than other imaging-based iron measures. The purpose of this study was to investigate the use of magnetic susceptibility as a biomarker of disease progression.
MATERIALS AND METHODS:
Fifteen subjects with premanifest Huntington disease and 16 age-matched healthy controls were scanned at 7T. Magnetic susceptibility, effective relaxation, and tissue volume in deep gray matter structures were quantified and compared with genetic and clinical measures.
RESULTS:
Subjects with premanifest Huntington disease showed significantly higher susceptibility values in the caudate nucleus, putamen, and globus pallidus, indicating increased iron levels in these structures. Significant decreases in magnetic susceptibility were found in the substantia nigra and hippocampus. In addition, significant volume loss (atrophy) and an increase effective relaxation were observed in the caudate nucleus and putamen. Susceptibility values in the caudate nucleus and putamen were found to be inversely correlated with structure volumes and directly correlated with the genetic burdens, represented by cytosine-adenine-guanine repeat age-product-scaled scores.
CONCLUSIONS:
The significant magnetic susceptibility differences between subjects with premanifest Huntington disease and controls and their correlation with genetic burden scores indicate the potential use of magnetic susceptibility as a biomarker of disease progression in premanifest Huntington disease.
Huntington disease (HD) is known to be caused by a cytosine-adenine-guanine (CAG)-repeat expansion in the HTT gene,1,2 resulting in a protein with a long polyglutamine tract that has a toxic effect on neuronal populations.3,4 Signs and symptoms include motor, cognitive, and emotional dysfunction.5,6 Genetic testing can identify subjects at risk of HD many years before the onset of the disease when subjects are still in the prodromal or premanifest phase. In addition, CAG expansion length is inversely related to the age of disease onset, thereby allowing approximate prediction of the time to onset of motor dysfunction. Sensitive and robust imaging-based surrogate biomarkers are needed to monitor disease progression and response to possible treatment.
A well-studied MR imaging–based biomarker for disease progression in advanced and premanifest HD is brain atrophy, mainly in the striatum, but other subcortical structures such as the globus pallidus (GP), thalamus, hippocampus, and amygdala were also found to be affected to a certain extent in HD.7,8 Volumetric changes have been systematically reported in all stages of the disease, with atrophy starting around 12 years before predicted onset of motor dysfunction.7–9 In addition to structural changes, iron level change has also been suggested to play an important role in HD pathophysiology, likely preceding structure changes, and may serve as a potential biomarker for early diagnosis and monitoring of a response to a therapeutic approach. Previous studies have indicated that the mutated huntingtin protein increases intracellular calcium levels, which activate the Rhes protein, leading to an influx of intercellular iron through the divalent metal transporter 1, which further leads to increased oxidative damage.10,11 Such huntingtin-induced increased uptake of iron into the cells damages the neurons, inducing inflammation and neuronal loss, leading to degradation of striatopallidal projections.12,13
Another relevant pathologic model proposes that increased vulnerability of GABAergic medium spiny neurons in the early stages of HD could lead to the striatal iron accumulation and progressive volume loss.14 In fact, animal studies of advanced HD have shown significantly higher levels of iron in the basal ganglia structures.15 In addition, a postmortem study of advanced HD showed an increase in crude iron concentration of almost 150% in the putamen (PT) and GP.16 Rosas et al17 measured increased iron of 25%–30% in the same structures. These studies in advanced HD have sparked interest in investigating potentially abnormal iron accumulation during the earlier stages of the disease. Several recent studies by using transverse relaxation rate (R2*) measurements18,19 or magnetic susceptibility–related measures, such as phase,20 magnetic field correlation,21 and field shift,17 have indeed suggested that brain iron levels are increased in premanifest or patients with early-stage HD.
Previous MR imaging techniques for measuring tissue iron have been based on either relaxation or other indirect measures of tissue magnetic susceptibility, which could be masked by multiple confounding effects. For example, a major confounding effect in relaxation-based measures, including R2 and R2*, is the change of water content. Contributions from the macroscopic background field, which have no relationship to local tissue iron, may also contaminate R2*-based iron measures. Previous magnetic susceptibility–based measures such as phase or field shift are known to be nonlocal (ie, affected by the susceptibility values of surrounding tissue and the position of the head)22; thus, this feature makes them hard to interpret and less specific for measuring tissue iron. Recent developments in quantitative susceptibility mapping (QSM) techniques23–27 have made it possible to directly map brain tissue magnetic susceptibility. Compared with previous techniques, QSM is believed to give a more accurate and specific measure of tissue magnetic susceptibility, which has been shown to correlate well with tissue iron concentration in most brain gray matter regions.23,24,28–30
In the present study, QSM was used to assess and expand on previous findings31 of differences in magnetic susceptibility between healthy controls and patients with premanifest HD in the basal ganglia, focusing on regions that have shown altered iron content in advanced HD (ie, the caudate nucleus [CN], PT, GP,17 and other deep gray matter structures that have shown certain atrophy and may possibly be affected in HD [ie, amygdala, hippocampus, and thalamus]).8 Some deep iron-rich nuclei related to movement and cognition, including the substantia nigra and red nucleus, were also included in our analysis. To compare with previous studies, we also calculated R2* as another susceptibility-related measure for the 2 groups. In addition, we tested possible correlations between magnetic susceptibility and structure volume and the genetic burden of the CAG repeats. It is hypothesized that brain iron concentration changes in subjects with premanifest HD will affect the local tissue magnetic susceptibility, which may, therefore, serve as a potential biomarker for HD progression.
Materials and Methods
Subjects
Fifteen subjects with premanifest HD (5 men, 10 women; mean age, 42.4 ± 8.7 years) were recruited through the Baltimore Huntington's Disease Center at the Johns Hopkins University School of Medicine. Inclusion criteria for the premanifest HD group were a CAG repeat length in the HTT gene higher than 40 and a Unified Huntington's Disease Rating Scale32 motor score below 15. Of the subjects with premanifest HD, 7 were scanned in 2010, and 8, in 2013. Additionally, 16 age-matched healthy controls (8 men, 8 women; mean age, 43.3 ± 11.7 years) were recruited through Johns Hopkins University. Consent was obtained according to the Declaration of Helsinki and approved by the Johns Hopkins University Institutional Review Board.
The age of onset of HD correlates strongly with CAG-repeat length. To quantify the genetic burden at the time of the scan, we calculated the CAG-age product scaled (CAPs) score33 as CAPs = Age × (CAG − 33.66) / 432.3326). This indicates a probability of disease onset within 5 years of <.5, 0, and >.5 for CAPs scores <1, 1, and >1, respectively.
Clinical personnel, trained in neuropsychological subject evaluation, performed the following interviews and tests on the day of scanning for all gene-positive subjects: the Unified Huntington's Disease Rating Scale test to determine the total motor score, the Hamilton Depression Rating Scale,34 the Montreal Cognitive Assessment35 to screen for mild cognitive dysfunction, and the National Adult Reading Test36 as an estimate of premorbid intelligence. Subjects with premanifest HD with a known acute neuropsychiatric disease, severe cognitive impairment, affective disorders, or other acute medical disorders were excluded from the study. Because the mean age of the subjects with premanifest HD is below prevalence rates of sporadic neurodegenerative disease and vascular dementia, we did not expect subjects with these disorders. Healthy controls were recruited from a different study in which their status as healthy was determined by interviews and scores on the Montreal Cognitive Assessment, Mini-Mental State Examination, and memory tests (immediate and delayed recall).
MR Imaging Protocol
Subjects were imaged by using a 7T Achieva scanner (Philips Healthcare, Best, the Netherlands) equipped with a 1TX-32RX quadrature transmit head coil (Nova Medical, Wilmington, Massachusetts) and a 32-channel phased array receive coil. A T1-weighted MPRAGE image (TR/TE = 4.8/2.1 ms, resolution = 0.6 × 0.6 × 0.6 mm3, scan time = 6 minutes 32 seconds) was acquired for anatomic referencing and automated image segmentation. MR phase measurements used for QSM calculation were acquired by using a multiecho 3D gradient recalled-echo (GRE) sequence. Two types of scan parameters were used. For 8 subjects with premanifest HD and all healthy controls, multiecho 3D GRE scans with 8 echoes (TR/TE/ΔTE = 68/4/2 ms, flip angle = 9°, voxel size = 1 × 1 × 1 mm3, FOV = 220 × 220 × 100 mm3, bandwidth = 1529 Hz/voxel, scan time = 7 minutes 13 seconds) were acquired. For the other 7 patients, multiecho 3D GRE scans with 22 echoes (TR/TE/ΔTE = 61/2/2 ms, flip angle = 19°, voxel size = 1 × 1 × 1 mm3, FOV = 212 × 212 × 80 mm3, bandwidth = 1213 Hz/voxel, scan time = 6 minutes 12 seconds) were acquired. Images were inspected (by P.G.U.) for any imaging artifacts or abnormalities, but no additional inspections were performed for small white matter hyperintensities or vascular disease.
Although MR phase measurements depend on the B0 field and echo time, the tissue magnetic susceptibility calculated by using QSM is expected to be affected by scanning parameters to a small extent as long as the same TE range is used. To evaluate possible differences and variations in the calculated susceptibility values caused by the 2 scanning parameters used in this study, we scanned 4 healthy subjects (2 men, 2 women; age range, 27–36 years) with both scan settings and compared the corresponding susceptibility quantities. For all the GRE scans, the 5 echoes with a TE in the range of 10–18 ms were used to calculate magnetic susceptibility to obtain a good signal-to-noise ratio and minimize the effect of nonlinear phase evolution at short TEs (ie, with TE < 10 ms).37
Quantitative Susceptibility and R2* Mapping
Multiple processing steps were performed to calculate the quantitative susceptibility maps from acquired MR phase images. First, phase unwrapping was performed by using Laplacian-based phase unwrapping.27 A brain mask was then obtained by skull-stripping the GRE magnitude image acquired at a TE of 10 ms. The unwrapped phase images were then divided by 2π*TE to obtain an image of the frequency shift in hertz for each echo. Subsequently, background fields were eliminated with the variable spheric kernel size sophisticated harmonic artifact reduction for the phase-data (V-SHARP)29 approach with a maximum radius of 4 mm and a regularization parameter of 0.05.38 After removal of background fields, we averaged the resulting images of all 5 echoes to obtain a higher signal-to-noise ratio compared with single-echo reconstruction.39 Inverse dipole calculations to obtain the susceptibility maps were performed by using a Least Squares with QR factorization-based minimization.27 The central CSF region in the lateral ventricles of each subject was selected as a reference region for the final susceptibility quantification. All susceptibility values have been reported relative to the mean susceptibility value of this reference region for each subject. R2* maps were calculated by nonlinear fitting of a monoexponential curve to the square of the magnitude data at all available echoes in each voxel by using the power method.23
Image Segmentation
The T1-weighted MPRAGE image was coregistered to the GRE magnitude image at a TE of 10 ms. The coregistered T1 image was then segmented by using a multiatlas matching approach developed as part of the Johns Hopkins University brain atlas.40 After this atlas-based segmentation, ROIs in the CN, PT, GP, amygdala, hippocampus, and thalamus were extracted. Manual corrections on ROIs in the substantia nigra and red nucleus were performed on the basis of magnetic susceptibility contrast, because they show little or no contrast on T1-weighted images and the ROIs generated from the automated segmentation process based on T1 contrast generally deviate from the true anatomic locations.24 From each ROI, the volume of the structure was determined in an automated way by multiplying the amount of voxels in that ROI and the known voxel size. To account for different brain sizes across subjects, we corrected individual structural volume with the following approach: Corrected Structure Volume = Original Structure Volume × (Group Mean Intracranial Volume/Subject Intracranial Volume). Each of the ROIs was eroded by 2 pixels in 3D to eliminate partial volume effects and then was used as a mask to calculate average susceptibility and R2* values per region.
Statistics
To examine the differences between the controls and the premanifest HD group, we performed 1-way MANCOVA with the mean magnetic susceptibility, R2*, or tissue volume of each brain structure as the outcome variable, while controlling for age and sex. The P value after post hoc Bonferroni correction was considered significant at P ≤ .05.
To test how magnetic susceptibility, R2*, and tissue volume in the selected gray matter structures were correlated with clinical measures in the premanifest HD group, we performed a Pearson partial correlation analysis, controlling for age and sex. The outcome variable was set as the susceptibility value, R2*, or volume of each brain structure, and the predictor was set as the CAPs, Unified Huntington's Disease Rating Scale motor score, or Montreal Cognitive Assessment score. The CAPs score is a function of age; therefore, the correlation analysis of CAPs with susceptibility, R2*, and ROI volume was not corrected for age.
Results
The demographic information for all the subjects and the clinical test results on all patients with premanifest HD are summarized in Table 1. Sample susceptibility maps of a healthy control and a subject with premanifest HD are shown in Fig 1. Compared with the healthy control (Fig 1A), susceptibility increases in the PT, part of the GP, and CN can be seen in the subject with premanifest HD (Fig 1B). The On-line Figure shows selected ROIs generated by using the atlas-based segmentation overlaid on the QSM images of a healthy control subject. The comparison between the 2 GRE scan settings showed no significant differences in the calculated mean susceptibility values and SDs in any selected ROI; thus, combining the 2 datasets for susceptibility analysis is valid and should not give significant bias to our conclusion.
Table 1:
Demographic data and clinical assessment scores for controls and subjects with premanifest HDa
| Healthy Controls | Premanifest HD | |
|---|---|---|
| No. | 16 | 15 |
| Sex (male/female) | 8:8 | 5:10 |
| Age (yr) | 43.3 ± 11.7 | 42.4 ± 8.7 |
| Education (yr) | 18.1 ± 2.7 | 15.5 ± 2.6 |
| CAG length | – | 43.5 ± 2.6 |
| CAPs score | – | 0.9 ± 0.2 |
| Unified Huntington's Disease Rating Scale motor score | – | 7.2 ± 5.4 |
| Hamilton Depression Rating Scale | – | 4.7 ± 3.9 |
| Montreal Cognitive Assessment | 29.1 ± 0.7 | 26.0 ± 4.1 |
| Full-scale intelligence quotient | – | 111.0 ± 8.2 |
| Verbal intelligence quotient | – | 109.8 ± 9.2 |
Note:—– indicates results were not available for all subjects.
Data are presented as means.
Fig 1.

Sample axial and coronal QSM sections of a 41-year-old healthy subject (A). Similar sections in a 41-year-old subject with premanifest HD (B). Increased susceptibility values are visible in iron-rich deep gray matter structures, such as the caudate nucleus, putamen, and globus pallidus.
Controlling for age, group comparison between controls, and premanifest HD shows a significant susceptibility increase (Fig 2A and Table 2) in the CN, PT (P < .001), and GP (P < .01). The substantia nigra (P < .05) and hippocampus (P < .01) show a significant susceptibility decrease, with a similar trend also observed in the red nucleus and amygdala. Splitting the analysis by sex did not alter the results, with both male and female groups showing statistically significant differences in similar regions (data not shown). R2* values were found to be increased in the CN and PT (P < .01) (Table 2). In addition to susceptibility changes, a significant volume decrease was also observed in the CN and PT (P < .001) (Fig 2B and Table 2).
Fig 2.

Average susceptibility values (A) and average volumes (B) in each ROI of the control and premanifest HD groups. Error bars represent standard error (SE). One asterisk indicates a significant difference with P < .05; double asterisks, a significant difference with P < .01; triple asterisks, a significant difference with P < .001; TH, thalamus; AMG, amygdala; HIPP, hippocampus; SN, substantia nigra; RN, red nucleus.
Table 2:
Summary of average susceptibility (χ), average R2* values, and average volume for each selected ROI
| χ (ppm) ± SE |
R2* (s−1) ± SE |
Corrected Volume (mL) ± SE |
||||
|---|---|---|---|---|---|---|
| Controls | Premanifest HD | Controls | Premanifest HD | Controls | Premanifest HD | |
| Caudate nucleus | 0.023 ± 0.002 | 0.048 ± 0.004a,b,c | 43.69 ± 0.88 | 49.35 ± 1.63c,d | 7.1 ± 0.8 | 5.4 ± 0.9a,b |
| Putamen | 0.029 ± 0.002 | 0.067 ± 0.007a,b,c | 52.59 ± 1.39 | 64.21 ± 2.55b,c,d | 8.6 ± 0.7 | 6.1 ± 1.3a,b |
| Globus pallidus | 0.091 ± 0.005 | 0.117 ± 0.007d | 84.48 ± 2.64 | 92.75 ± 3.35 | 2.8 ± 0.4 | 3.0 ± 0.8 |
| Thalamus | −0.024 ± 0.002 | −0.021 ± 0.002 | 40.22 ± 0.70 | 40.07 ± 0.79 | 13.8 ± 0.8 | 12.9 ± 1.5 |
| Amygdala | −0.018 ± 0.003 | −0.023 ± 0.002 | 29.82 ± 0.78 | 31.59 ± 1.64 | 1.9 ± 0.6 | 2.0 ± 0.7 |
| Hippocampus | −0.017 ± 0.001 | −0.021 ± 0.002d | 31.32 ± 0.54 | 30.47 ± 1.12 | 6.1 ± 0.9 | 5.7 ± 1.1 |
| Substantia nigra | 0.080 ± 0.004 | 0.057 ± 0.009e | 75.45 ± 2.68 | 70.62 ± 3.05 | 1.3 ± 0.4 | 1.3 ± 0.4 |
| Red nucleus | 0.076 ± 0.005 | 0.066 ± 0.009 | 65.30 ± 2.71 | 67.00 ± 2.47 | 0.5 ± 0.2 | 0.6 ± 0.1 |
Significant difference with P < .001.
Significant correlation with CAPs score in subjects with premanifest HD.
Significant correlation with ROI volume in subjects with premanifest HD.
Significant difference with P < .01.
Significant difference between controls and premanifest HD with P < .05.
Correlation analysis with the predictor as the Unified Huntington's Disease Rating Scale motor score or Montreal Cognitive Assessment score did not result in significant correlations between those metrics and magnetic susceptibility or R2* in any selected ROI. In the CN and PT, strong inverse correlations were found between susceptibility and ROI volume (Fig 3A). The CAPs directly correlated with the susceptibility and inversely correlated with ROI volume in the CN and PT (Fig 3B, -C and Table 2). Using R2*, we observed a correlation with CAPs in the PT (P < .05, r = 0.52).
Fig 3.
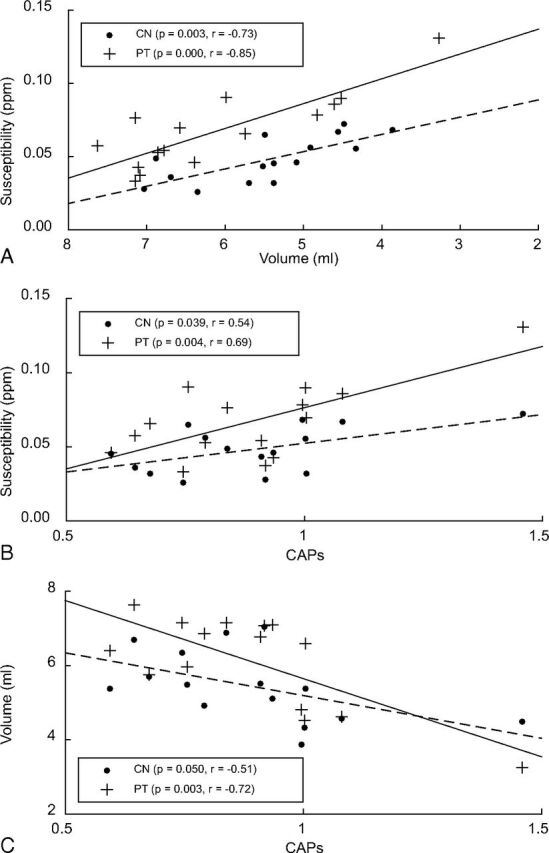
Correlation analysis of susceptibility and corrected structure volume while controlling for age (A), susceptibility with CAPs (B), and volume with CAPs (C).
Discussion
This cross-sectional study found significantly increased magnetic susceptibility values in the CN, PT, and GP of subjects with premanifest HD compared with control subjects. Due to the previously demonstrated strong direct correlation of susceptibility values with tissue iron levels in brain gray matter,24,30 such increase is attributed predominantly to an HD-related increase of tissue iron content in these regions. Such increased iron levels in the CN, PT, and GP in patients with premanifest HD are, in general, consistent with the findings of other in vivo MR imaging studies of these patients and those with symptomatic HD,13,17,18,20,41 indicating that altered iron homeostasis may be involved in HD pathophysiology and may happen at very early stages of the disease. Postmortem studies of the same regions in symptomatic HD also support these findings.16,17,42 In addition, the more significant changes in the CN and PT compared with the GP found in this study are consistent with the general understanding that HD-related neuronal damage begins in the striatum and then spreads to the GP and other regions.6 However, partly due to the use of different MR imaging–based iron measures, previous findings on iron level changes in HD have not always been consistent. For example, in comparison with the present study, Dumas et al,21 using magnetic field correlation, could detect only elevated iron levels in early symptomatic patients with HD but not in the premanifest HD group. Such discrepancy may come from the lower sensitivity of magnetic field correlation in detecting HD-induced tissue iron changes, lower spatial resolution, and more stringent patient inclusion criteria used in that study (ie, with a Unified Huntington's Disease Rating Scale score cutoff point of 5 for the premanifest HD group).
The increased iron level in the striatum in the premanifest HD group is further confirmed by the increased R2* in these regions. Given the strong correlation between R2* and tissue iron,30,43 the observation that susceptibility is more sensitive for detecting HD-related changes may hint at some other pathophysiologic features of the disease such as loss of diamagnetic myelin.44 Due to the microstructure of myelin and its diamagnetic nature, it is known that demyelination would decrease R2* and increase magnetic susceptibility. Similar higher sensitivity of susceptibility compared with R2* has been observed previously in detecting tissue changes in multiple sclerosis, in which demyelination is a known disease hallmark.45 In addition, some striatal demyelination and loss of tissue integrity in premanifest HD might also partly explain the increased mean diffusivity observed in another study.19 In most of the deep gray matter regions investigated in this study, myelin content was not expected to be very high; therefore, iron change is still likely to be the dominant source of the observed susceptibility and R2* increases in the premanifest HD group. Finally, R2* as a parameter is more difficult to quantify reproducibly due to its dependence on magnetic field inhomogeneity and the orientation of the brain. This explanation may be an alternative one for the somewhat lower significance.
This study also found decreased levels of iron in other brain regions related to movement and memory, such as the hippocampus, substantia nigra, and red nucleus, in subjects with premanifest HD. Decreased iron concentration has been reported in certain cortical areas18 and white matter regions44 during the progress of HD. Such iron decreases are generally explained by the redistribution of brain iron after the increase of oligodendrocyte density, thus, iron concentration is an attempt to restore initial neural or myelin loss caused by HD.18,44 The decrease in iron level in the hippocampus was previously observed as a nonsignificant trend by several in vivo studies21,44; however, to the best of our knowledge, there is no previous report on iron level changes in the substantia nigra in the premanifest HD group. The significant iron decrease in the substantia nigra and the decrease trend in the red nucleus may be caused by the iron redistribution processes or changes in myelin attenuation that are not yet fully understood. Increases in arterial blood volume could also be a cause of such a decrease because iron in deoxygenated venous blood has a strong effect on tissue susceptibility.30 Decreased cerebral blood flow in the basal ganglia and hippocampus of patients with premanifest HD has been reported,46–48 while increased arteriolar cerebral blood volume in the cortex has also been previously observed in patients49 and animal models.50
As expected, significant volume decreases (atrophy) in the CN and PT were observed in patients with premanifest HD in the present study, but not in the GP, where atrophy often occurs later in the disease.6 At this early stage, the significant iron-level difference observed in the GP may suggest that iron changes precede or have a greater magnitude than volume changes in the GP. In addition, iron levels were strongly inversely correlated with volumes in the CN and PT. Similar correlations were observed between R2* and volume in the CN and PT in this study. Such correlations were observed in the PT and GP in a very recent study by using R2*, but not in the CN.18 No correlations with volume were observed by using magnetic field correlation21 or phase imaging20 as iron measures. Both volume and susceptibility in the CN and PT showed strong correlations with CAPs, suggesting that both measures are closely related to the disease process. Correlation with CAPs was only observed in the PT by using R2*. Similar correlations in subjects with premanifest HD were observed previously only in the CN by using phase imaging20 and in the PT by using R2* measurements,18 but no correlations were observed by using the magnetic field correlation.21
Conclusions
Together with previous iron studies in premanifest and symptomatic HD, the present study suggests that disease-related iron increase in the striatum and GP happens in the early stage of HD and tissue magnetic susceptibility as obtained by QSM has a good potential to serve as a sensitive in vivo surrogate measure to track tissue iron changes in HD. However, due to the limited sample size used in the present study, the lack of a symptomatic HD group, and lack of controls for possible vascular diseases, quantitative susceptibility as biomarker of HD progression cannot be fully confirmed yet. Further investigation with a larger cohort and longitudinal follow-up is required in the future. In addition, tissue iron measures combined with other biomarkers sensitive to either macrostructure or microstructure impairment in a multimodal setup18,19 would further help understand the pathophysiology of HD.
Supplementary Material
Acknowledgments
The authors thank Dr James Pekar, Dr Andreia Faria, Mr Joseph Gillen, Ms Terri Brawner, Ms Kathleen Kahl, Ms Ivana Kusevic, and Dr Raj Stewart for their assistance with data acquisition.
ABBREVIATIONS:
- CAG
cytosine-adenine-guanine
- CAPs
CAG-age product scaled
- CN
caudate nucleus
- GP
globus pallidus
- GRE
gradient recalled-echo
- HD
Huntington disease
- QSM
quantitative susceptibility mapping
- PT
putamen
- R2*, R2
transverse relaxation rates
Footnotes
Disclosures: Issel Anne Lim—RELATED: Grant: National Institutes of Health Training Grant for Interdisciplinary Training in Psychiatry and Neuroscience (T32 MH015330).* Craig Jones—RELATED: Grant: Philips Healthcare,* Comments: My salary was paid, in part, by a grant to the Kennedy Krieger Institute from Philips Healthcare. Russell Margolis—RELATED: Grant: National Institutes of Health *; UNRELATED: Grants/Grants Pending: National Institutes of Health, Prana, Auspex, Teva, Child Health and Development Institute, Comments: treatment trials and pathogenic studies of HD. Christopher Ross—RELATED: Grant: Huntington's Disease Society of America–Centers of Excellence.* Peter van Zijl—RELATED: Grant: Philips Healthcare (technical development grant)*; Support for Travel to Meetings for the Study or Other Purposes: Philips Healthcare (supported travel to conferences); UNRELATED: Payment for Lectures (including service on Speakers Bureaus): Philips Healthcare, Comments: In the past, they have provided honoraria for lectures; Patents (planned, pending or issued): Philips Healthcare,* Comments: provided funds for patent licensing to the institution and also got income from such patent licenses; Travel/Accommodations/Meeting Expenses Unrelated to Activities Listed: Philips Healthcare, Comments: supported travel to conferences; OTHER: Dr Peter van Zijl is a paid lecturer for Philips Healthcare and is the inventor of technology that is licensed to Philips Healthcare. Xu Li—RELATED: Grant: Philips Healthcare, Comments: Dr Xu Li's salary is supported, in part, by a grant from Philips Healthcare. This arrangement has been approved by Johns Hopkins University in accordance with its conflict of interest policies. *Money paid to the institution.
This project was supported by the National Center for Research Resources and the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health through resource grant P41 EB015909, with educational funding from a National Institutes of Health Training Grant for Interdisciplinary Training in Psychiatry and Neuroscience T32 MH015330. In addition, a grant from the Huntington's Disease Society of America–Centers of Excellence and a grant from the David Mahoney Neuroimaging Program from the Dana Foundation supported this work. Dr Peter van Zijl is a paid lecturer for Philips Healthcare and is the inventor of technology that is licensed to Philips Healthcare. Dr Xu Li's salary is supported, in part, by a grant from Philips Healthcare. This arrangement has been approved by Johns Hopkins University in accordance with its conflict of interest policies.
References
- 1. Ross CA, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 2014;10:204–16 10.1038/nrneurol.2014.24 [DOI] [PubMed] [Google Scholar]
- 2. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes: the Huntington's Disease Collaborative Research Group. Cell 1993;72:971–83 10.1016/0092-8674(93)90585-E [DOI] [PubMed] [Google Scholar]
- 3. Gusella JF, Wexler NS, Conneally PM, et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature 1983;306:234–38 10.1038/306234a0 [DOI] [PubMed] [Google Scholar]
- 4. Tabrizi SJ, Scahill RI, Durr A, et al. ; TRACK-HD Investigators. Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol 2011;10:31–42 10.1016/S1474-4422(10)70276-3 [DOI] [PubMed] [Google Scholar]
- 5. Paulsen JS, Nopoulos PC, Aylward E, et al. ; PREDICT-HD Investigators and Coordinators of the Huntington's Study Group (HSG). Striatal and white matter predictors of estimated diagnosis for Huntington disease. Brain Res Bull 2010;82:201–07 10.1016/j.brainresbull.2010.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 2011;10:83–98 10.1016/S1474-4422(10)70245-3 [DOI] [PubMed] [Google Scholar]
- 7. Tabrizi SJ, Langbehn DR, Leavitt BR, et al. ; TRACK-HD investigators. Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol 2009;8:791–801 10.1016/S1474-4422(09)70170-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van den Bogaard SJ, Dumas EM, Acharya TP, et al. ; TRACK-HD Investigator Group. Early atrophy of pallidum and accumbens nucleus in Huntington's disease. J Neurol 2011;258:412–20 10.1007/s00415-010-5768-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aylward EH, Li Q, Stine OC, et al. Longitudinal change in basal ganglia volume in patients with Huntington's disease. Neurology 1997;48:394–99 10.1212/WNL.48.2.394 [DOI] [PubMed] [Google Scholar]
- 10. Browne SE, Beal MF. Oxidative damage in Huntington's disease pathogenesis. Antioxid Redox Signal 2006;8:2061–73 10.1089/ars.2006.8.2061 [DOI] [PubMed] [Google Scholar]
- 11. Muller M, Leavitt BR. Iron dysregulation in Huntington's disease. J Neurochem 2014;130:328–50 10.1111/jnc.12739 [DOI] [PubMed] [Google Scholar]
- 12. Douaud G, Behrens TE, Poupon C, et al. In vivo evidence for the selective subcortical degeneration in Huntington's disease. Neuroimage 2009;46:958–66 10.1016/j.neuroimage.2009.03.044 [DOI] [PubMed] [Google Scholar]
- 13. Dominguez DJ, Ng AC, Poudel G, et al. Iron accumulation in the basal ganglia in Huntington's disease: cross-sectional data from the IMAGE-HD study. J Neurol Neurosurg Psychiatry 2015. May 7. [Epub ahead of print] 10.1136/jnnp-2014-310183 [DOI] [PubMed] [Google Scholar]
- 14. Vonsattel JP. Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol 2008;115:55–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen J, Marks E, Lai B, et al. Iron accumulates in Huntington's disease neurons: protection by deferoxamine. PLoS One 2013;8:e77023 10.1371/journal.pone.0077023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen JC, Hardy PA, Kucharczyk W, et al. MR of human postmortem brain tissue: correlative study between T2 and assays of iron and ferritin in Parkinson and Huntington disease. AJNR Am J Neuroradiol 1993;14:275–81 [PMC free article] [PubMed] [Google Scholar]
- 17. Rosas HD, Chen YI, Doros G, et al. Alterations in brain transition metals in Huntington disease: an evolving and intricate story. Arch Neurol 2012;69:887–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sánchez-Castañeda C, Squitieri F, Di Paola M, et al. The role of iron in gray matter degeneration in Huntington's disease: a magnetic resonance imaging study. Hum Brain Mapp 2015;36:50–66 10.1002/hbm.22612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sánchez-Castañeda C, Cherubini A, Elifani F, et al. Seeking Huntington disease biomarkers by multimodal, cross-sectional basal ganglia imaging. Hum Brain Mapp 2013;34:1625–35 10.1002/hbm.22019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Apple AC, Possin KL, Satris G, et al. Quantitative 7T phase imaging in premanifest Huntington disease. AJNR Am J Neuroradiol 2014;35:1707–13 10.3174/ajnr.A3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dumas EM, Versluis MJ, van den Bogaard SJ, et al. ; TRACK-HD investigators. Elevated brain iron is independent from atrophy in Huntington's disease. Neuroimage 2012;61:558–64 10.1016/j.neuroimage.2012.03.056 [DOI] [PubMed] [Google Scholar]
- 22. Salomir R, De Senneville BD, Moonen CT. A fast calculation method for magnetic field inhomogeneity due to an arbitrary distribution of bulk susceptibility. Concept Magn Reson B 2003:19B:26–34 10.1002/cmr.b.10083 [DOI] [Google Scholar]
- 23. Deistung A, Schäfer A, Schweser F, et al. Toward in vivo histology: a comparison of quantitative susceptibility mapping (QSM) with magnitude-, phase-, and R2*-imaging at ultra-high magnetic field strength. Neuroimage 2013;65:299–314 10.1016/j.neuroimage.2012.09.055 [DOI] [PubMed] [Google Scholar]
- 24. Lim IA, Faria AV, Li X, et al. Human brain atlas for automated region of interest selection in quantitative susceptibility mapping: application to determine iron content in deep gray matter structures. Neuroimage 2013;82:449–69 10.1016/j.neuroimage.2013.05.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schweser F, Sommer K, Deistung A, et al. Quantitative susceptibility mapping for investigating subtle susceptibility variations in the human brain. Neuroimage 2012;62:2083–100 10.1016/j.neuroimage.2012.05.067 [DOI] [PubMed] [Google Scholar]
- 26. Wharton S, Schäfer A, Bowtell R. Susceptibility mapping in the human brain using threshold-based k-space division. Magn Reson Med 2010;63:1292–304 10.1002/mrm.22334 [DOI] [PubMed] [Google Scholar]
- 27. Li W, Wu B, Liu C. Quantitative susceptibility mapping of human brain reflects spatial variation in tissue composition. Neuroimage 2011;55:1645–56 10.1016/j.neuroimage.2010.11.088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schenck JF. The role of magnetic susceptibility in magnetic resonance imaging: MRI magnetic compatibility of the first and second kinds. Medi Phys 1996;23:815–50 10.1118/1.597854 [DOI] [PubMed] [Google Scholar]
- 29. Schweser F, Deistung A, Lehr BW, et al. Quantitative imaging of intrinsic magnetic tissue properties using MRI signal phase: an approach to in vivo brain iron metabolism? Neuroimage 2011;54:2789–807 10.1016/j.neuroimage.2010.10.070 [DOI] [PubMed] [Google Scholar]
- 30. Langkammer C, Schweser F, Krebs N, et al. Quantitative susceptibility mapping (QSM) as a means to measure brain iron? A post mortem validation study. Neuroimage 2012;62:1593–99 10.1016/j.neuroimage.2012.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Poynton CB, Lupo JM, Hess C, et al. Quantitative susceptibility mapping of Huntington's disease at 7 Tesla. In: Proceedings of the Joint Annual Meeting of the International Society for Magnetic Resonance in Medicine and the European Society for Magnetic Resonance in Medicine, Milan, Italy. May 10–16, 2014 [Google Scholar]
- 32. Unified Huntington's Disease Rating Scale: reliability and consistency—Huntington Study Group. Mov Disord 1996;11:136–42 10.1002/mds.870110204 [DOI] [PubMed] [Google Scholar]
- 33. Zhang Y, Long JD, Mills JA, et al. ; PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Indexing disease progression at study entry with individuals at-risk for Huntington disease. Am J Med Genet B Neuropsychiatr Genet 2011:156B:751–63 10.1002/ajmg.b.31232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960;23:56–62 10.1136/jnnp.23.1.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53:695–99 10.1111/j.1532-5415.2005.53221.x [DOI] [PubMed] [Google Scholar]
- 36. Bright P, Jaldow E, Kopelman MD. The National Adult Reading Test as a measure of premorbid intelligence: a comparison with estimates derived from demographic variables. J Int Neuropsychol Soc 2002;8:847–54 10.1017/S1355617702860131 [DOI] [PubMed] [Google Scholar]
- 37. Wharton S, Bowtell R. Fiber orientation-dependent white matter contrast in gradient echo MRI. Proc Natl Acad Sci U S A 2012;109:18559–64 10.1073/pnas.1211075109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu B, Li W, Guidon A, et al. Whole brain susceptibility mapping using compressed sensing. Magnetic Reson Med 2012;67:137–47 10.1002/mrm.23000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu B, Li W, Avram AV, et al. Fast and tissue-optimized mapping of magnetic susceptibility and T2* with multi-echo and multi-shot spirals. Neuroimage 2012;59:297–305 10.1016/j.neuroimage.2011.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Y, Zhang J, Hsu J, et al. Evaluation of group-specific, whole-brain atlas generation using Volume-based Template Estimation (VTE): application to normal and Alzheimer's populations. Neuroimage 2014;84:406–19 10.1016/j.neuroimage.2013.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bartzokis G, Cummings J, Perlman S, et al. Increased basal ganglia iron levels in Huntington disease. Arch Neurol 1999;56:569–74 10.1001/archneur.56.5.569 [DOI] [PubMed] [Google Scholar]
- 42. Dexter DT, Carayon A, Javoy-Agid F, et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson's disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991;114:1953–75 10.1093/brain/114.4.1953 [DOI] [PubMed] [Google Scholar]
- 43. Langkammer C, Krebs N, Goessler W, et al. Quantitative MR imaging of brain iron: a postmortem validation study. Radiology 2010;257:455–62 10.1148/radiol.10100495 [DOI] [PubMed] [Google Scholar]
- 44. Bartzokis G, Lu PH, Tishler TA, et al. Myelin breakdown and iron changes in Huntington's disease: pathogenesis and treatment implications. Neurochem Res 2007;32:1655–64 10.1007/s11064-007-9352-7 [DOI] [PubMed] [Google Scholar]
- 45. Langkammer C, Liu T, Khalil M, et al. Quantitative susceptibility mapping in multiple sclerosis. Radiology 2013;267:551–59 10.1148/radiol.12120707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harris GJ, Codori AM, Lewis RF, et al. Reduced basal ganglia blood flow and volume in pre-symptomatic, gene-tested persons at-risk for Huntington's disease. Brain 1999;122:1667–78 10.1093/brain/122.9.1667 [DOI] [PubMed] [Google Scholar]
- 47. Hasselbalch SG, Oberg G, Sorensen SA, et al. Reduced regional cerebral blood flow in Huntington's disease studied by SPECT. J Neurol Neurosurg Psychiatry 1992;55:1018–23 10.1136/jnnp.55.11.1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen JJ, Salat DH, Rosas HD. Complex relationships between cerebral blood flow and brain atrophy in early Huntington's disease. Neuroimage 2012;59:1043–51 10.1016/j.neuroimage.2011.08.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hua J, Unschuld PG, Margolis RL, et al. Elevated arteriolar cerebral blood volume in prodromal Huntington's disease. Mov Disord 2014;29:396–401 10.1002/mds.25591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lin C, Huang C, Lin M, et al. Magnetic resonance microscopic angiography visualization of abnormal microvasculature in a transgenic mouse model of Huntington's disease. In: Proceedings of the ISMRM 18th Scientific Meeting and Exhibition, Stockholm, Sweden. May 1–7, 2010 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.