

Abstract
Type II diabetes is the most prevalent form of diabetes. One of the primary complications of diabetes that significantly affects quality of life is bladder dysfunction. Many studies on diabetic bladder dysfunction have been performed in models of type I diabetes; however, few have been performed in animal models of type II diabetes. Using the Zucker Diabetic Fatty (ZDF) rat model of type II diabetes, we examined the contractility and sensitivity of bladder smooth muscle in response to mediators of depolarization-induced contraction, muscarinic receptor-mediated contraction, ATP-induced contraction, and neurogenic contraction. Studies were performed at 16 and 27 wk of age to monitor the progression of diabetic bladder dysfunction. Voiding behavior was also quantified. The entire bladder walls of diabetic rats were hypertrophied compared with that of control rats. Contractility and sensitivity to carbachol and ATP were increased at 27 wk in bladder smooth muscle strips from diabetic rats, suggesting a compensated state of diabetic bladder dysfunction. Purinergic signaling was increased in response to exogenous ATP in bladders from diabetic animals; however, the purinergic component of neurogenic contractions was decreased. The purinergic component of neurogenic contraction was reduced by P2X receptor desensitization, but was unchanged by P2X receptor inhibition in diabetic rats. Residual and tetrodotoxin-resistant components of neurogenic contraction were increased in bladder strips from diabetic animals. Overall, our results suggest that in the male ZDF rat model, the bladder reaches the compensated stage of function by 27 wk and has increased responsiveness to ATP.
Keywords: diabetic bladder dysfunction, Zucker diabetic fatty rat, muscarinic receptors, purinergic receptors, electrical field stimulation
diabetes mellitus (dm) is a disorder of carbohydrate metabolism typically characterized by defective insulin secretion or insulin resistance and subsequent hyperglycemia. In the past few decades, the prevalence of DM has been on the rise (1, 33), and lower urinary tract symptoms (LUTS) are among the most common complications of diabetes affecting up to 80% of patients (14). The most common and bothersome LUTS in diabetes is bladder dysfunction, with over 50% of diabetic patients experiencing some form of bladder dysfunction (48). The primary classic symptom diabetic patients exhibit is increased residual urine volume after voiding (23). More recent research suggests a combination of both storage and voiding problems (14). Experimental evidence now suggests a temporal profile of the development of diabetic bladder dysfunction (15). There is a progression from the compensated state, during which the bladder undergoes hypertrophy and the muscle becomes hypercontractile, to the decompensated state, when the muscle is hypocontractile and there is a decrease in voiding ability. In mouse and rat models of type I diabetes [high-dose streptozotocin (STZ)], the compensated/hypercontractile state was seen in vitro using strips of detrusor smooth muscle from animals 6–9 wk of age. In similar animals, the decompensated/hypocontractile state was noted later, between 12 and 20 wk of age (29). Similarly, cystometric studies demonstrated bladder function increased from 3 to 9 wk in a STZ type I diabetes rat model, and then function declined below controls between 12 and 20 wk (15). In the present study, we measured voiding behavior, morphometric analysis of the bladder, and contractility at 16 and 27 wk of age in a male rat model of type II diabetes.
Mechanistic studies on alterations in bladder smooth muscle regulation in animal models of type I diabetes are abundant in the literature (13). This is primarily due to the relative ease of inducing type I diabetes in animal models, typically using either alloxan or high doses of STZ, and the lower costs associated with developing and maintaining animals with type I diabetes. Unfortunately, the literature on the effects of type II diabetes on bladder function is sparse (21). Studies determining the time course of diabetic bladder dysfunction in type II diabetic animal models are severely lacking along with studies determining whether bladder dysfunction is similar in type I vs. type II diabetic animal models (13). Alterations in bladder contraction can be due to a variety of reasons. In addition to changes in the nerves innervating the bladder or changes in signaling between the urothelial layer and smooth muscle, there may also be changes within the smooth muscle itself. While many studies initially focused on neuropathic effects on diabetic bladder dysfunction, we are now focusing on changes within the bladder smooth muscle.
There are many animal models of type II diabetes such as the ob/ob mouse, the db/db mouse, the spontaneously diabetic Torii (SDT) rat, the Goto-Kakizaki (GK) rat, and the Zucker diabetic fatty (ZDF) rat (22). Our studies investigated bladder dysfunction in the male ZDF rat model. In these animals, hyperglycemia is initially observed at ∼7 wk of age, and almost all male ZDF (fa/fa) rats spontaneously develop diabetes by ∼8–12 wk of age on a normal chow diet (10, 19, 46). Bladder dysfunction has also been investigated in female ZDF rats; they require a high-fat diet to induce diabetes (25). Our studies focused on changes in the contractility and sensitivity of bladder smooth muscle tissue to exogenous agonists and release of endogenous transmitters.
In this study, we tested the hypothesis that during the compensated state of type II diabetic bladder dysfunction, the bladder wall would show a hypertrophic response, increased voiding behavior, and hypercontractile smooth muscle. This hypothesis was tested by investigating the changes in bladder morphometry, voiding behavior, and contractility in response to the addition of exogenous agonists, membrane depolarization, or to the release of endogenous neurotransmitters using electrical field stimulation. The purinergic response was specifically investigated as previous studies have suggested that nonadrenergic noncholinergic (NANC) mediators of contraction are more prominent in diseased states of the bladder and that ATP is one of the primary NANC mediators (9, 31, 43).
METHODS
Materials.
All reagents for solutions, unless otherwise specified, were purchased from Fisher Scientific (Pittsburgh, PA) and were of analytic grade or better. Atropine, bethanechol, carbachol, ATP, suramin, pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS), anti-P2X1 purinergic receptor antibody (P7857), and α,β-methylene ATP were purchased from Sigma-Aldrich (St. Louis, MO).
Animals.
Male ZDF rats [ZDF-Leprfa/Crl, fa/fa (diabetic) and fa/+ (control)] were obtained from Charles River at 12 wk of age and then housed on site in AALAC-accredited animal facilities at Drexel University College of Medicine. Rats were fed a normal chow diet throughout the study (Purina Formulab Diet 5008) and provided water ad libitum. Ten control and 10 diabetic rats were euthanized at 16 wk. Twenty-eight control and 18 diabetic rats were euthanized at 27 wk. Animal weights were recorded on a weekly basis, and blood glucose measurements were obtained biweekly using an Accu-Check Advantage blood glucometer (Roche Diagnostics, Indianapolis, IN). Blood was obtained by nicking the tail vein of the rat with a needle. The protocol was approved by the Drexel University College of Medicine's Institutional Animal Care and Use Committee.
Rat bladder strip preparation.
Rats were euthanized at 16 or 27 wk of age by CO2 inhalation followed by bilateral thoracotomy. After removal from the peritoneal cavity, the bladder was placed directly into 4°C physiological salt solution (PSS). PSS contained (in mM) 140 NaCl, 4.7 KCl, 1.2 MgSO4, 1.6 CaCl2, 1.2 Na2HPO4, 2 MOPS (pH 7.4), 5.5 d-glucose, and 0.02 EDTA. Adipose tissue was dissected away from the exterior of the bladder, and the bladder was opened longitudinally. Wet weight of the bladder was then determined, after draining of urine. The dome of the bladder (portion farthest from the urethra) was removed along with the trigone region (portion of the bladder below the level of the ureters), leaving the middle body of the bladder for experimentation. The urothelial/mucosal layer was removed, leaving a layer of predominantly bladder smooth muscle. The bladder smooth muscle layer was then cut into three to four longitudinal strips using both ventral and dorsal sides of the bladder body. The strips were stored in 4°C PSS for a maximum of 4 h until used. All strips were used for experimentation the same day the rat was euthanized.
Measurement of isometric contraction.
Bladder strips were mounted between a Grass FT.03 force transducer and a stationary clip in water-jacketed muscle organ baths containing PSS at 37°C and aerated with 100% O2 (MOPS-buffered PSS, pH 7.4 at 37°C in 100% O2). The strips were equilibrated for ∼ 45 min until a stable basal force recording was obtained. A passive force was applied to the mounted tissues by stretching, followed by stress-relaxation until the strips reached the optimal length for active stress development (Lo) approximated by 1 g of passive force. Partial length-tension curves were performed to determine that 1-g passive force approximated Lo in bladder strips from both control and diabetic rats. After equilibration and achieving Lo, strips were stimulated with 110 mM KCl (equimolar substitution for NaCl) and then relaxed with PSS. This contraction-relaxation cycle was repeated several times until the maximal active force in response to KCl stimulation did not change >10% between contractions (3–5 contractions on average). Maximal active force was defined as the difference between the maximal force in response to the stimulus and the basal force before the stimulus.
Concentration-response curves to KCl, carbachol, bethanechol, and ATP were performed on bladder strips from both control and diabetic rats. Both noncumulative and cumulative concentration-response curves were generated in response to KCl in PSS containing 4.7, 10, 20, 30, 50, 80, or 110 mM KCl (equimolar substitution for NaCl); carbachol using semi-log increases in concentration from 10 nM to 100 μM; bethanechol using semi-log increases in concentration from 100 nM to 3 mM; and ATP using semi-log increases in concentration from 100 nM to 10 mM. Although carbachol and bethanechol are both cholinergic agonists, bethanechol is specific for muscarinic receptors, whereas carbachol affects both muscarinic and nicotinic acetylcholine receptors. We used carbachol, as the majority of studies looking at activation of muscarinic receptors in bladders use carbachol, but we also used bethanechol to confirm that any changes in carbachol-mediated contraction were not due to its possible action at nicotinic acetylcholine receptors. All concentration-response curves were performed in the presence of 1 μM tetrodotoxin (TTX) to inhibit sodium channel activation and therefore prevent any activation of nerves within the preparation. Forces recorded were therefore only in response to agonist activation of receptors on or membrane depolarization of the smooth muscle cells. Recorded force was either normalized to the maximal force in response to 110 mM KCl or normalized by the tissue dry weight (mg).
Measurement of neurogenic contraction.
Neurogenic contractions were measured in bladders from control and diabetic rats by the use of electrical field stimulation (EFS). Bladder strips devoid of urothelium were mounted between a Grass FT.03 force transducer and a stationary glass hook in a water-jacketed muscle organ bath containing PSS at 37°C and aerated with 100% O2. The tissue on the glass hook was surrounded by 2 zig-zag platinum-wire electrodes ∼2 mm apart (Radnoti, Monrovia, CA). The platinum wire electrodes were connected to a Grass S88 stimulator through a Grass SIU-10 stimulus isolation unit. Output from the stimulator, stimulus isolation unit, and electrodes were all verified with the use of an oscilloscope to determine that the output settings on the stimulator were the same as the output of the electrodes within the organ bath. Equilibration, determination of Lo, and contraction-relaxation cycles to 110 mM KCl were performed as described above.
EFS experiments followed a modified version of the protocol described by Daneshgari's laboratory (28). After the multiple KCl contraction-relaxation cycles, frequency-response curves were performed with the following stimulation settings: 120 V, 8-ms pulse width, and a frequency range of 1–32 Hz. Each new stimulus was applied 3 min after the previous stimulation within the frequency-response curve to allow for tissue recovery from EFS. Initially, a control frequency-response curve was performed without any compounds present in the organ baths. This was followed by the addition of 1 μM atropine to the bathing solution for 30 min to inhibit muscarinic receptors before a second frequency-response curve was performed. Third, 10 μM α,β-methylene ATP was added, in the presence of 1 μM atropine, at least three times at intervals of 10 min to desensitize purinergic receptors (28). Desensitization was considered complete when the next application of 10 μM α,β-methylene ATP did not produce an increase in force. After a minimum of 30 min from the time of the initial α,β-methylene ATP addition, a third frequency-response curve was performed. At this point, the tissues were washed repeatedly to remove the inhibitors and 1 μM TTX was added to inhibit neurogenic contraction. Tissues were incubated in TTX for 30 min before a final frequency-response curve was performed. In separate experiments, the same protocol was followed except that in place of α,β-methylene ATP, 30 μM PPADS (51) was added to the bath to inhibit rather than desensitize purinergic receptors. After EFS experiments were completed, the length and dry weight of each tissue strip were recorded to calculate tension development. In select experiments, TTX-resistant contractions were inhibited by the addition of cumulative concentrations of nifedipine, a voltage-dependent calcium channel inhibitor, or removal of extracellular calcium.
Before data analysis, the TTX-resistant component of the contraction was subtracted from all EFS-induced force measurements, allowing only the neurogenic responses to be compared between conditions. The cholinergic portion of the contraction was quantified by subtracting the response to EFS after addition of atropine from the response to EFS before addition of atropine. The purinergic portion of the contraction was quantified by subtracting the response to EFS after addition of both atropine and α,β-methylene ATP (or PPADS) from the response to EFS after addition of atropine alone. The residual NANC portion of the contraction was quantified as the contraction remaining after addition of both atropine and α,β-methylene ATP (or PPADS). All of the components of contraction were then normalized to the maximal active force in response to 110 mM KCl, the maximal active force in response to control EFS at 32 Hz, or were converted to tension measurements.
Voiding behavior.
Selected rats were used for voiding behavior studies at 16 (8 lean, 8 diabetic) and 27 (10 lean, 10 diabetic) wk of age. Rats were removed from their housing unit and placed in metabolic cages for 6 h between the hours of 10 AM and 4 PM. Rats were provided with water ad libitum while in the metabolic cages. The number of voids and volume per void were measured for each rat during the 6-h study period. The number of voids and voiding volume were monitored by a single investigator with the voiding mass measured immediately at the end of the study period using an analytic balance and converted to volume using an approximate density of 1 g/ml. The same investigator measured both the number of voids and voiding volume, and all measurements were taken at the same time of the day for all animals.
Bladder histology.
In select experiments, whole bladders from 27-wk-old rats were equilibrated in 37°C PSS for 15 min after removal. The bladder dome and trigone were dissected away, and the detrusor body was cut open longitudinally. Then, longitudinal ventral strips of whole thickness bladder wall, which includes the urothelial layer, were excised and placed in 10% buffered formalin and shipped to AML Labs (Rosedale, MD) for paraffin embedding, sectioning, and staining. Cross sections of 5-μm thickness were taken from the paraffin-embedded bladder wall and stained with Masson's trichrome. The cross sections were transverse/orthogonal to the longitudinal strip to ensure the accuracy of the bladder wall thickness measurements. Masson's trichrome-stained tissue sections were then photographed under a bright light field on a Zeiss M2 imaging microscope and analyzed for bladder smooth muscle and urothelial/mucosal content. The thickness of the tissue section that was stained as smooth muscle or urothelium/mucosa was measured and compared with the entire thickness of the tissue section. The width of the bladder sections, as a measurement of wall thickness, was also compared between control and diabetic tissue sections (26). Images were analyzed for smooth muscle content and wall thickness using National Institutes of Health ImageJ software.
Purinergic receptor expression.
Bladder smooth muscle tissue strips (with urothelium removed by microdissection) were frozen in a dry ice/acetone slurry containing 6% wt/vol trichloroacetic acid and 10 mM dithiothreitol. Tissues were thawed and then homogenized in a buffer containing 10% vol/vol glycerol and 1% wt/vol SDS. Twenty micrograms of sample extract was subjected to SDS-PAGE (10% gel) and then transferred to nitrocellulose membranes. The membranes were then blocked with Odyssey blocking buffer (Li-Cor) for 1 h and incubated with a 1:10,000 dilution of rabbit anti -P2X1 antibody (P7857, Sigma-Aldrich) and a 1:200,000 dilution of anti-calponin antibody overnight. Calponin was used as a loading control. Membranes were then washed three times in PBS containing 0.1% vol/vol Tween 20 and incubated in both anti-mouse and anti-rabbit Li-Cor secondary antibodies (1:10,000) for 45 min. Membranes were again washed three times in PBS containing 0.1% vol/vol Tween 20 with a final rinse in PBS not containing Tween 20. The membranes were scanned using an Odyssey imaging system (Li-Cor) and were quantified using the software accompanying the instrument. The ratio of the intensity of the P2X1 band to the intensity of the loading control calponin band was used as a semiquantitative measurement of P2X1 protein expression.
Statistical analysis.
Statistical significance between two means was determined using Student's t-test with or without the Bonferroni correction. For determination of the EC50 of concentration-response curves and fitting of the curves, Graphpad Prism software was used. Curves were either three-parameter fits using the Hill slope or four-parameter fits with variable slope. All n values refer to the number of bladder smooth muscle strips used for measurement; each strip was from a different animal.
RESULTS
Rat characteristics.
All rats were weighed weekly throughout the experimental time period. Body weights were significantly increased in the diabetic rats compared with control rats at 16 and 27 wk of age (Fig. 1, A and B). Blood glucose levels were also measured in all rats biweekly. Blood glucose levels were significantly elevated in the diabetic rats compared with the control rats at 16 and 27 wk of age (Fig. 1, C and D).
Fig. 1.

Characteristics of male control and diabetic Zucker diabetic fatty (ZDF) rats. A: body weight of control and diabetic rats at 16 and 27 wk of age. Body weights of diabetic rats were significantly increased at both ages. Body weights are means ± SE of 40 animals at 16 wk of age and 28 animals at 27 wk of age. B: body weights of control and diabetic animals measured from 12 wk until 27 wk of age. C: blood glucose levels of control and diabetic rats at 16 and 27 wk of age. Blood glucose levels were significantly increased in diabetic rats at both ages. Glucose levels are means ± SE of 14 animals at 16 wk of age and 11 animals at 27 wk of age. D: blood glucose levels of control and diabetic rats measured from 12 wk through 27 wk of age. E: bladder wet weights of control and diabetic rats at 16 and 27 wk of age. Bladder weights were significantly increased in diabetic rats at both ages. Bladder weights are means ± SE of 10 animals at 16 wk of age and 17 animals at 27 wk of age. Statistical comparisons were made between control and diabetic rats using Student's t-test. *P < 0.05 compared with control rats at same age.
Bladder hypertrophy.
Wet weight of the urinary bladders, after draining of urine, was measured to determine whether bladder hypertrophy occurred in diabetic rats (Fig. 1E). The wet weights of bladders from diabetic rats were significantly increased at 16 and 27 wk of age compared with those of control rats. The width of the cross sections of the bladder wall, which were equal to the thickness of the bladder wall, was used as one measure of general hypertrophy of the bladder (Fig. 2, Table 1). The thickness of the entire bladder wall from diabetic rats was significantly larger than that from control animals. To determine whether the bladder smooth muscle layer was hypertrophied, we measured the area of smooth muscle relative to the area of the entire cross section of the bladder wall (Fig. 2, Table 1). Masson's trichrome-stained sections showed no difference in the area of smooth muscle within the cross section as a percentage of the area of the entire cross section of the bladder wall in bladders from control and diabetic animals. Similarly, the thicknesses of the smooth muscle and urothelial/mucosal layers of bladders from diabetic animals when expressed as a percentage of the entire bladder wall thickness were no different from the smooth muscle and urothelial/mucosal layers in bladders from control animals (Table 1). However, the absolute thicknesses of the urothelial/mucosal layer and the smooth muscle layer from diabetic rats were both significantly increased compared with the thicknesses of the respective layers of the bladder wall from control animals. This suggests a general hypertrophy of all the layers of the bladder wall as well as an increase in bladder wet weight.
Fig. 2.

Masson's trichrome staining of bladder cross sections of 27-wk-old male control (A) and diabetic (B) rats. The red staining to the right in each image is smooth muscle bundles (SM). The blue staining in each image is collagen fibers that are part of the submucosa and extracellular matrix between smooth muscle bundles. Cross sections of bladder wall from diabetic rats show a thickened urothelial layer (URO; stained red).
Table 1.
Bladder histology quantification
| Bladder Wall Thickness, μm | Smooth Muscle Thickness, μm | Smooth Muscle Area, % | |
|---|---|---|---|
| Control | 1,320 ± 119 | 837 ± 69 | 48.7 ± 9.0 |
| Diabetic | 2,055 ± 121* | 1,336 ± 222* | 46.5 ± 8.6 |
| Urothelial/Mucosal Thickness, μm | Urothelial/Mucosal Thickness, % | ||
| Control | 319 ± 49 | 24.1 ± 0.6 | |
| Diabetic | 521 ± 13* | 25.5 ± 1.2 |
Values are means ± SE of measurements of 3 separate bladders from either control or diabetic animals at 27 wk of age. Statistical comparisons were made between control and diabetic animals using Student's t-test.
P < 0.05 compared with control rats.
Voiding behavior.
Bladder voiding was measured at 16 and 27 wk of age (Table 2). The number of voids during a 6-h period and the volume of urine per void were measured. At 16 wk of age, the diabetic rats had significantly decreased void volume over the 6-h period but a significantly increased number of voids compared with control rats. At 27 wk of age, the diabetic animals had significantly increased void volume and number of voids compared with control rats. The increase in the number of voids, but not void volume, at 16 wk in diabetic rats suggests the bladders have not yet entered the compensated state. The increase in void volume in the diabetic rats at 27 wk of age suggests the bladders have entered the compensated state.
Table 2.
Voiding behavior
| Volume per Void, ml |
Number of Voids per 6 h |
|||
|---|---|---|---|---|
| Control | Diabetic | Control | Diabetic | |
| 16 wk old | 1.93 ± 0.22 | 0.53 ± 0.09* | 1.75 ± 0.16 | 2.75 ± 0.31* |
| 27 wk old | 0.95 ± 0.16 | 3.28 ± 0.35* | 1.67 ± 0.24 | 3.81 ± 0.64* |
Values are means ± SE of measurements from 8–11 animals. Statistical comparisons were made between control and diabetic rats using Student's t-test.
P < 0.05 compared with the control rats at same age.
Contractility of bladder smooth muscle from 16-wk-old rats.
Concentration-response curves were performed in response to KCl, carbachol, bethanechol, and ATP in bladder smooth muscle strips from both control and diabetic rats at 16 wk of age. The concentrations were administered both cumulatively and noncumulatively. Concentration-response curves were used to calculate both maximal tension (Fig. 3A) and EC50 values (Table 3) in response to each stimulus. At 16 wk of age, maximal tension in bladder smooth muscle was similar in response to both the cumulative and noncumulative addition of KCl-, bethanechol-, and ATP-induced stimulation between control and diabetic animals (Fig. 3A). Maximal tension in response to the cumulative addition of carbachol was significantly decreased in bladders from diabetic compared with control rats. As shown in Table 3, EC50 values were similar between control and diabetic rats for both the cumulative and noncumulative addition of carbachol, bethanechol, and ATP. However, the EC50 in response to the cumulative addition of KCl was significantly decreased in bladders from diabetic rats compared with control rats. This demonstrates that bladder smooth muscle from diabetic rats is more sensitive to depolarization-induced, non-receptor-mediated contraction compared with control rats at 16 wk of age. While maximal tension in response to the cumulative addition of ATP was not increased at 16 wk of age, the contractility of the smooth muscle to ATP, quantified as a percentage of the response to 110 mM KCl-induced contraction, was increased in diabetic animals throughout the concentrations tested (Fig. 3B). This is most likely explained by the decrease in tension to KCl, although not significant, coupled with the lack of change in maximal tension in response to ATP (see Fig. 3A).
Fig. 3.
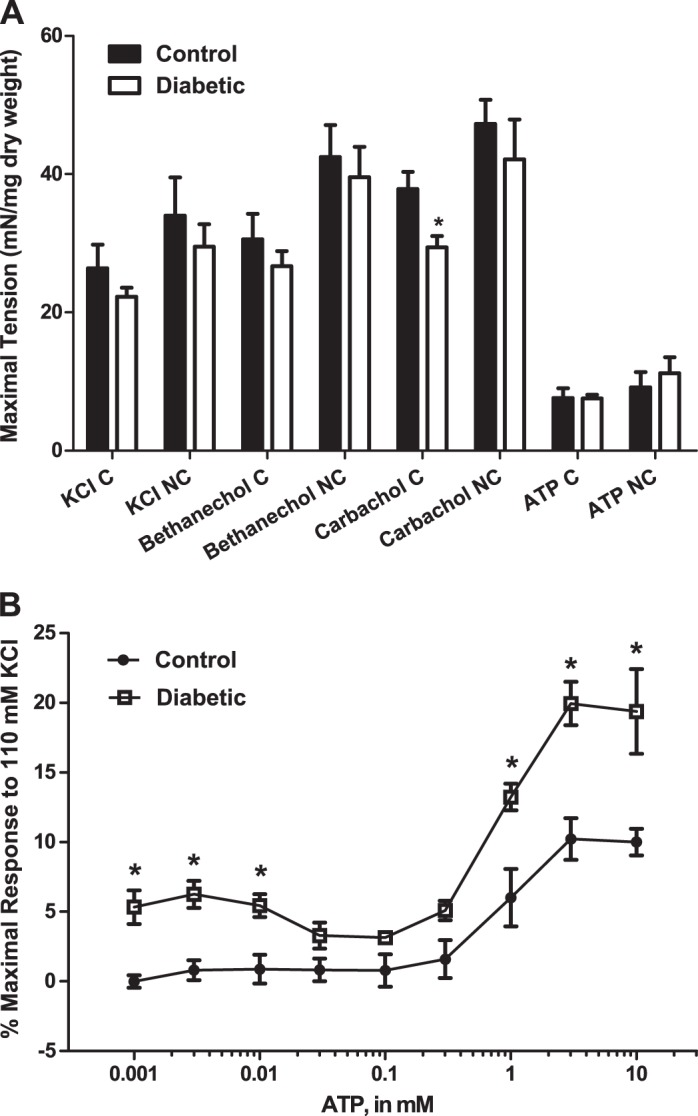
A: maximal tension in response to the cumulative (C) or noncumulative (NC) addition of KCl, bethanechol, carbachol, or ATP in 16-wk-old ZDF rats. Values are means ± SE of 4–6 determinations, each from a different animal. Statistical comparisons were made between control and diabetic animals using Student's t-test. Comparisons were only made within treatment groups (i.e., KCl C treatment group or ATP NC treatment group). *P < 0.05 compared with the control value in same treatment group. B: responses to the cumulative addition of ATP measured as the percentage of the maximal response to 110 mM KCl-physiological salt solution (PSS). Force in strips from diabetic animals was increased at almost all concentrations measured. Statistical comparisons were made between control and diabetic animals at each concentration using Student's t-test. *P < 0.05 compared with the control value.
Table 3.
EC50 values for 16-wk-old rats
| KCl, mM | Carbachol, μM | Bethanechol, μM | ATP, mM | |
|---|---|---|---|---|
| Cumulative | ||||
| Control | 36.4 ± 2.8 | 0.6 ± 0.2 | 16.0 ± 2.9 | 0.7 ± 0.2 |
| Diabetic | 27.2 ± 2.1* | 0.7 ± 0.2 | 11.4 ± 2.4 | 1.3 ± 0.4 |
| Noncumulative | ||||
| Control | 48.5 ± 2.4 | 0.4 ± 0.0 | 11.4 ± 1.7 | 1.1 ± 3.2 |
| Diabetic | 43.5 ± 1.6 | 0.4 ± 0.1 | 9.2 ± 0.9 | 0.7 ± 0.2 |
Values are means ± SE of 5–6 determinations, each from a different animal. Statistical comparisons were made between control and diabetic animals within the same treatment group using Student's t-test.
P < 0.05 compared with control animals.
Contractility of bladder smooth muscle from 27-wk-old rats.
At 27 wk of age, concentration-response curves for KCl, bethanechol, and carbachol were performed by the cumulative addition of stimulus. Since contractility changes were only seen following the cumulative addition of KCl, bethanechol, and carbachol at 16 wk, only cumulative response curves were performed at 27 wk. Concentration-response curves for ATP were performed by both the cumulative and noncumulative addition of ATP. There were no significant differences in maximal tension generated by KCl, bethanechol, or carbachol (Fig. 4A). However, there was a trend toward increased maximal tension in the bladders from diabetic rats in response to both carbachol and bethanechol, agonists that activate muscarinic receptors. This trend is opposite of the results at 16 wk, when the responses to carbachol and bethanechol tended to be decreased in diabetic animals (Figs. 4A vs. 3A). Maximal tension in response to the cumulative addition of ATP was significantly increased in the bladders from diabetic rats compared with control rats (Fig. 4A). EC50 values generated from cumulative concentration-response curves to KCl, carbachol, and ATP were not significantly different between control and diabetic animals (Table 4). The EC50 value for the cumulative addition of bethanechol was decreased in the bladders from diabetic animals, suggesting the smooth muscle of the bladder wall from diabetic animals is more sensitive to muscarinic stimulation. The EC50 value for the cumulative addition of ATP tended to be decreased in the bladders of diabetic animals (Table 4). The EC50 value was significantly decreased during the noncumulative addition of ATP in the bladders from diabetic rats (Table 4). This demonstrates that bladder smooth muscle from diabetic animals is more sensitive to purinergic stimulation at 27 wk of age. However, a true maximal effect in response to ATP could not be achieved due to ATP solubility (Fig. 4C). It should also be noted that the response to ATP was significantly greater in the noncumulative vs. the cumulative addition, most likely due to receptor desensitization during the cumulative addition of ATP (Fig. 4, C vs. B). The contractility of bladder smooth muscle from diabetic rats was increased in response to both the cumulative and noncumulative addition of ATP compared with bladders from control rats (Fig. 4, B and C). These results are similar to those at 16 wk (Fig. 3B), suggesting that the enhanced contractility at 16 wk remained at 27 wk.
Fig. 4.
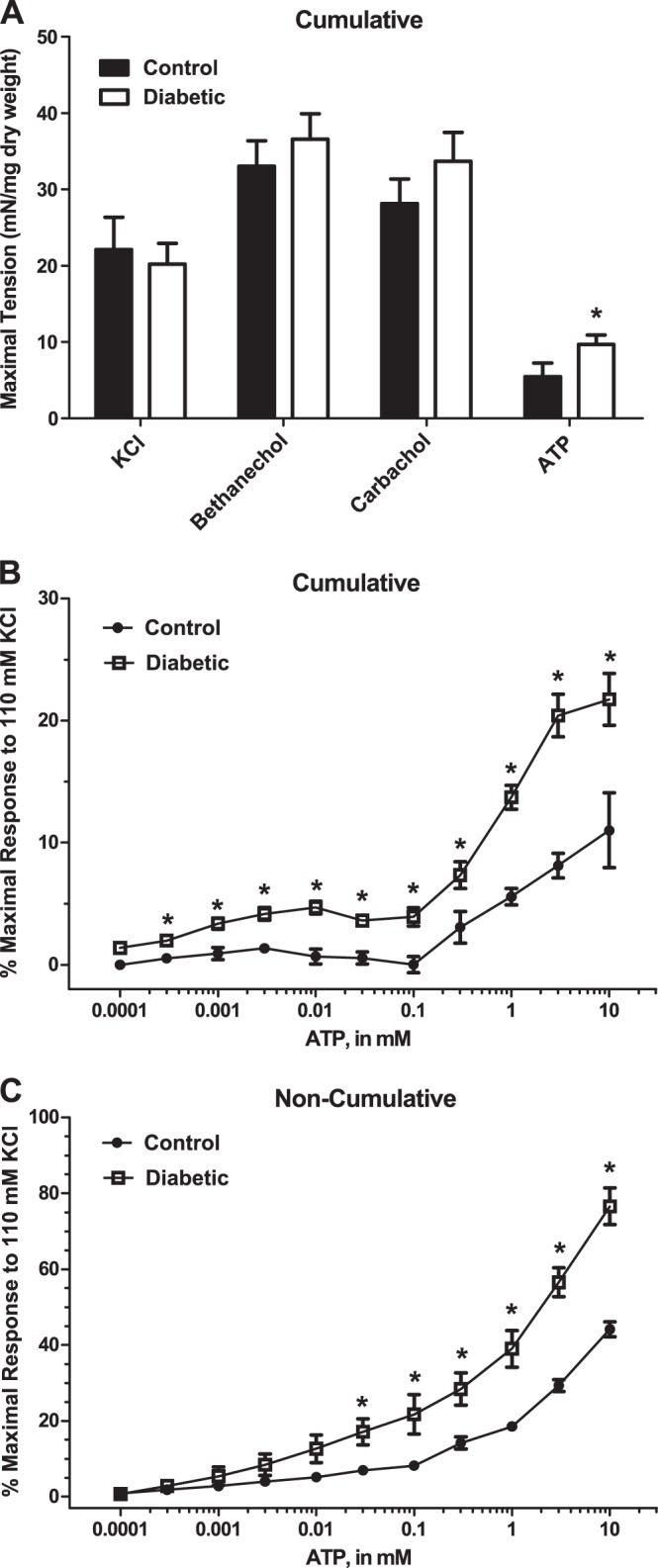
A: maximal tension in response to the cumulative addition of KCl, bethanechol, carbachol, or ATP in 27-wk-old control and diabetic rats. Values are means ± SE of 4–7 determinations, each from a different animal. Statistical comparisons were made between control and diabetic animals using Student's t-test. Statistical comparisons were only made between control and diabetic animals within each treatment group. *P < 0.05 compared with the control value in the same treatment group. B: responses to the cumulative addition of ATP measured as the percentage of the maximal response to 110 mM KCl-PSS. C: responses to the noncumulative addition of ATP measured as the percentage of the maximal response to 110 mM KCl-PSS. Force in strips from diabetic animals was increased at almost all concentrations measured (B and C). Statistical comparisons were made between control and diabetic animals at each concentration using Student's t-test. *P < 0.05 compared with the control value.
Table 4.
EC50 values for 27-wk-old rats
| KCl, mM | Carbachol, μM | Bethanechol, μM | ATP Cumulative, mM | ATP Noncumulative, mM | |
|---|---|---|---|---|---|
| Control | 24.8 ± 1.5 | 0.5 ± 0.0 | 7.7 ± 1.0 | 3.1 ± 1.6 | 2.2 ± 0.3 |
| Diabetic | 22.1 ± 1.2 | 0.6 ± 0.1 | 5.3 ± 0.4* | 1.2 ± 0.3 | 1.1 ± 0.3* |
Values are means ± SE of 4–8 determinations, each from a different animal. Statistical comparisons were made between control and diabetic animals within the same treatment group using Student's t-test.
P < 0.05 compared with control animals.
Contractility of bladder smooth muscle to EFS from 27-wk-old rats.
EFS was used as an initiator of contraction to test how the smooth muscle responded to the release of endogenous neurotransmitters from local nerve endings. A time control for EFS-induced stimulation was performed to ensure that any decrease in contraction was due to inhibition of receptors and not a decline of the contractility of the bladder smooth muscle to EFS with repeated stimulation (Fig. 5A). There was no run-down of the EFS-induced contractions during the time frame of our experiments. Representative experimental force tracings are shown in Fig. 5.
Fig. 5.

A: representative tracing of a time control performed using a bladder strip from a 27-wk-old diabetic rat. The tracing shows that there is no reduction in the electrical field stimulation (EFS)-induced response throughout the duration of our experimental protocol. B and C: representative tracings of EFS experiments performed using bladder strips from 27-wk-old control (B) or diabetic (C) rats. Experiments were performed as follows: an EFS frequency-response curve without inhibitors present, followed by a frequency-response curve in the presence of atropine, and then a frequency-response curve in the presence of atropine and α,β-methylene ATP or PPADS. Desensitization of P2X receptors by α,β-methylene ATP reduced the atropine-resistant portion of contractions in bladders strips from control animals, but not in bladder strips from diabetic animals (B vs. C).
Maximal tension in response to EFS was similar between bladder muscle strips from control and diabetic rats at 27 wk of age [40.0 ± 5.4 mN/mg (control); 33.7 ± 5.1 mN/mg (diabetic)]. Frequency-response curves performed without pharmacological inhibition tended to be similar between bladder muscle strips from control and diabetic rats at 27 wk of age, although the response at 16 Hz was significantly decreased in muscle strips from diabetic rats (Fig. 6A). Components of the EFS-induced contractions that were due to activation of receptors by acetylcholine or ATP were determined through the use of receptor antagonists. EFS was performed in the presence of 1 μM atropine to determine the cholinergic component of EFS-induced contractions. The cholinergic portion of contraction was similar between bladder muscle strips from control and diabetic rats at higher frequencies (8, 16, 32 Hz) but significantly decreased at lower frequencies (2, 4 Hz) (Fig. 6B).
Fig. 6.

EFS experiments performed in 27-wk-old rats. A: frequency-response curves in response to EFS from 1 to 32 Hz as a percentage of the maximal response to EFS at 32 Hz. Values are means ± SE of 7–9 determinations, each from a different animal. B: frequency-response curves for the atropine-sensitive/cholinergic component of EFS-induced contractions in bladder smooth muscle strips from 27-wk-old control and diabetic rats. Values are means ± SE of 9–12 determinations, each from a different animal. C: frequency-response curves for the α,β-methylene ATP-sensitive/purinergic component of EFS-induced contractions in bladder smooth muscle strips from 27-wk-old control and diabetic rats. D: frequency-response curves for the atropine and α,β-methylene ATP-resistant/residual component of EFS-induced contractions. Values are means ± SE of 8–9 determinations, each from a different animal. E: frequency-response curves for the PPADS-sensitive/purinergic component of EFS-induced contractions of 27-wk-old ZDF rats. Responses of bladder strips from diabetic animals were similar compared with the responses of bladder strips from control animals at all frequencies measured (1–32 Hz) in the presence of PPADS. Values are means ± SE of 8–9 determinations, each from a different animal. F: frequency-response curves for the atropine- and PPADS-resistant/residual component of EFS-induced contractions. Values are means ± SE of 5–6 determinations, each from a different animal. G: frequency-response curves for the tetrodotoxin (TTX)-resistant component of EFS-induced contractions. Values are means ± SE of 9–12 determinations, each from a different animal. All statistical comparisons were made between control and diabetic animals using Student's t-test. *P < 0.05 compared with the EFS response in control bladders at the same frequency.
The purinergic component of contraction was determined by two methods. P2X receptors were either desensitized by repeated exposure to α,β-methylene ATP or inhibited by PPADS (51). The α,β-methylene ATP-sensitive purinergic component of EFS-induced contraction was reduced in bladder strips from diabetic rats compared with responses in bladder strips from control rats at all frequencies measured except 4 Hz (Fig. 6C). Because the cholinergic component of EFS was unchanged and the purinergic component was decreased in bladders from diabetic animals, the residual (NANC) component of EFS-induced contraction was increased in bladder strips from diabetic rats compared with control rats across all frequencies measured except 4 Hz (Fig. 6D).
In contrast to the results using α,β-methylene ATP, the PPADS-sensitive purinergic component of EFS-induced contractions was similar between muscle strips from control and diabetic animals at all frequencies measured (Fig. 6E). The residual (NANC) component of EFS-induced contraction was increased at 1, 2, and 8 Hz in bladder strips from diabetic rats compared with control rats (Fig. 6F). Purinergic receptors were also blocked using the antagonist suramin. Preliminary results suggest that suramin, like PPADS, inhibited a portion of atropine-resistant contraction in bladder strips from both control and diabetic ZDF rats (data not shown).
EFS-induced contractions were exposed to TTX to determine whether they were fully neurogenic. Figure 6G shows that there was a TTX-resistant component of EFS-induced contractions, suggesting that at the stimulation settings used the EFS-induced contractions are not fully neurogenic. This non-neurogenic portion of the contraction was subtracted from all other measurements before comparisons were made to ensure that only the components of the neurogenic portion of the contraction were being compared. Interestingly, the TTX-resistant component of the contraction was increased at all frequencies measured in bladder strips from diabetic rats compared with the bladder strips from control rats at 27 wk of age (Fig. 6G). The TTX-resistant component of an EFS-induced contraction is typically the result of direct muscle stimulation. This non-neurogenic contraction in bladder muscle strips from both control and diabetic animals was sensitive to removal of extracellular calcium or the addition of nifedipine, suggesting it was the result of direct muscle stimulation. TTX-resistant contractions of bladder muscle strips were inhibited by similar concentrations of nifedipine in control and diabetic rats (data not shown).
Purinergic receptor expression.
Because the response to ATP was significantly altered in bladder tissues from diabetic animals, we measured the expression levels of the most common purinergic receptor, P2X1. Figure 7 shows a representative Western blot of P2X1 receptors in bladders from control and 27-wk-old diabetic animals. Figure 7 also shows the quantitative results of several such blots. There was no difference in total expression levels of P2X1 receptors between the two animal groups.
Fig. 7.
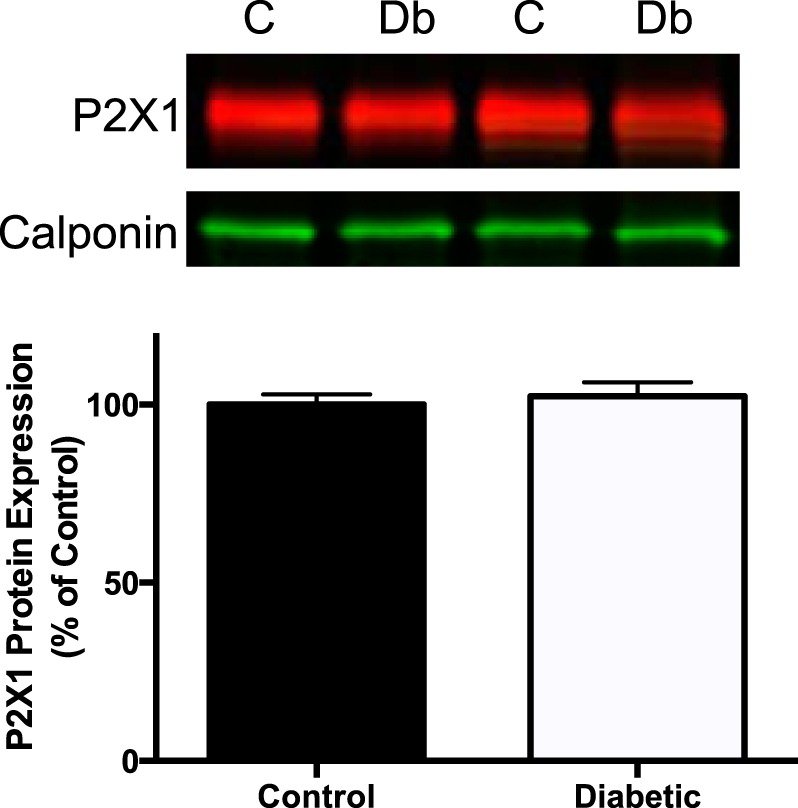
P2X1 receptor expression in bladder smooth muscle from 27-wk-old male control and diabetic animals. Top: representative Western blot of P2X1 receptor expression in bladders from control (C) and diabetic (Db) animals. Bottom: quantified data from 9 control and 6 diabetic animals There was no difference in total P2X1 receptor expression in bladders from the 2 animal groups.
Baseline spontaneous activity.
Treatment with α,β-methylene ATP or PPADS produced an increase in baseline spontaneous activity in bladder muscle strips from 27-wk-old diabetic rats (Fig. 8B) but not in strips from control rats (Fig. 8A). The α,β-methylene ATP-induced spontaneous activity was reversed with washout of the drug. PPADS-induced effects were only partially reversible by washout. The increase in baseline spontaneous activity was TTX resistant but nifedipine sensitive (data not shown). Spontaneous contractile activity was measured as an integral (g * s) and compared before and after the addition of α,β-methylene ATP. Quantified data from several such tracings are shown in Fig. 8C. Spontaneous activity was significantly increased after the addition of α,β-methylene ATP in strips from diabetic animals but was not affected by α,β-methylene ATP in strips from control animals.
Fig. 8.

A: Representative tracings of a muscle strip from the bladder of a 27-wk-old control rat before and after the addition of α,β-methylene ATP. Note the lack of change in baseline spontaneous activity after the addition of α,β-methylene ATP. B: representative tracings of a muscle strip from the bladder of a 27-wk-old diabetic rat before and after the addition of α,β-methylene ATP. Note the increase in baseline spontaneous activity after the addition of α,β-methylene ATP. C: quantified data analyzed as area under the curve of the spontaneous activity. Spontaneous activity was significantly greater in response to the addition of α,β-methylene ATP in bladders from diabetic animals compared with bladders from control animals; n = 8. *P < 0.05 by Student's t-test.
DISCUSSION
The majority of studies of diabetic bladder dysfunction have used animal models of type I diabetes. Our goal was to perform a longitudinal study of diabetic bladder dysfunction in an animal model of type II diabetes, the male ZDF rat, as this is the most prevalent form of diabetes in the world population (49). In this study we determined the diabetes-induced changes in the smooth muscle layer of the bladder wall from the type II diabetic ZDF rat. Specifically, we measured diabetes-dependent changes in 1) bladder morphology; 2) voiding behavior; 3) bladder smooth muscle contractility and sensitivity in response to the addition of exogenous agonists; and 4) bladder smooth muscle contractility in response to the release of endogenous neurotransmitters.
Both body weight and blood glucose concentration were increased in diabetic rats and similar to those in the original report on ZDF rats (38). When examining morphological changes in the bladder, we noted both increased bladder wet weight and increased wall thickness, which suggested diabetic rat bladder hypertrophy. Our analysis of the layers of the bladder wall showed that the increase in thickness of the wall was not specific to the smooth muscle layer but a generalized bladder wall hypertrophy. Both smooth muscle and urothelial/mucosal layers were hypertrophied in bladders from diabetic animals compared with bladders from control animals. However, the thicknesses of the smooth muscle and urothelial/mucosal layers when expressed as a percentage of total wall thickness were similar in bladders from the diabetic and control animals. This supports our conclusion that the hypertrophy was generalized over the entire bladder wall. Similar results were found in the STZ-induced type I diabetes rat model (11, 27). However, multiple studies suggest that this hypertrophy may be mostly due to diuresis as opposed to diabetes per se (17, 27).
Obesity is a well-accepted risk factor for the development of type II diabetes (41). The male ZDF rats used in this study weighed significantly more than age-matched control rats at 16 and 27 wk of age. However, the weights were somewhat variable over the 27-wk experimental time frame. In terms of potential effects on bladder structure, a PubMed search provided little literature to support or refute a relationship between obesity and bladder structure. A similar conclusion was reached by Christ et al. (12). Demaser's group (21) did find an increase in edema and vascular pathology in the bladder wall and an increase in fibrosis in the urethral sphincter in obese female ZDF rats regardless of the diabetic state. However, these changes were worsened in obese plus diabetic animals. Also, Demaser's group (21) found decreased voiding frequency and decreased contraction pressure in obese nondiabetic ZDF rats. These results may suggest that obesity itself could also modulate bladder function in animal models of type II diabetes.
In addition to the effects of obesity, the results of Demaser's study compared with the current study may represent a distinct difference in the response of the bladder to type II diabetes in female rats compared with that in male diabetic rats. In women, obesity has been shown to be associated with urinary incontinence in the absence of diabetes (16). However, Longhurst's group (18) suggested that there were only minor differences in bladder functionality from male or female rats with STZ-induced diabetes. It will continue to be important to consider gender of animal models in future studies of diabetic bladder dysfunction.
Mice fed a high-fat diet resulted in overactive bladder and enhanced contractility to both agonist- and membrane depolarization-induced bladder contractions; obesity and diabetes, however, were not individually studied (27). In contrast, Fan et al. (20) demonstrated that mice fed a high-fat diet exhibited reduced bladder smooth muscle contractile responses to agonist and membrane depolarization stimulation. Thus we suggest that, while obesity may affect bladder function, there is variable evidence suggesting that contraction and function may be increased or decreased. Furthermore, in our male ZDF rat model, as the induction of diabetes is spontaneous, it is not possible to completely separate the effects of diabetes and obesity. Since obesity and diabetes are common comorbidities in the human population, we feel it is best they are studied together.
Our voiding behavior measurements showed that diabetic rats had an increased average void volume and an increased number of voids over a 6-h measurement period at 27 wk. Similar findings have been made in other male type II diabetic animal models, the SDT rat (32) and the db/db mouse (47), although the type II diabetic Goto-Kakizaki (GK) rat did not show increased void volume or frequency even at 70 wk of age (40). Bladder dysfunction tends to progress from a state of bladder compensation to decompensation, when decreased bladder contractility results in increased residual urine volumes (15). Based on the voiding behavior and contractility results, we suggest that at 27 wk of age, the bladders from type II diabetic rats remain in the compensated state. It should be pointed out, however, that the metabolic cages contained wire bottoms; thus trapping of small volumes of urine is a potential problem that could result in an underestimation of voiding volume.
Our studies examining smooth muscle contractility and sensitivity to exogenous pharmacological stimulation were performed at 16 and 27 wk of age. The maximal tension in response to various agonists was not changed in bladders of diabetic animals at 16 wk, except in response to the cumulative addition of carbachol. There was increased contractility in response to ATP at multiple concentrations in diabetic animals, and an increased sensitivity to KCl as measured by a lower EC50 value. These results combined with the increased frequency of voiding but decreased void volume in diabetic rats at 16 wk suggest the bladder may not have reached the compensated stage at this age in diabetic rats.
Our studies in 27-wk-old rats show an increased maximal tension in response to ATP and a similar trend in response to carbachol and bethanechol. There was increased contractility to both the cumulative and noncumulative addition of ATP. The sensitivity of bladder smooth muscle from diabetic rats to ATP and bethanechol was also increased, as measured by decreased EC50 values. These results combined with the increased void volume and void frequency all suggest that the diabetic animals were in the compensated bladder stage at 27 wk of age. In the GK rat, Ueda's group (50) similarly found increased responses to carbachol and ATP but no change in response to KCl, and Satoh's group (40) also found increased carbachol responses in the same model. Interestingly, in models of type I diabetes two groups found increased KCl-induced contractility during the compensated state of bladder function (15, 26). It appears that the bladder smooth muscle during the compensated state in type I models responds differently to activators of muscle contraction than in bladder smooth muscle from type II models. Leiria et al. (26) showed that bladder smooth muscle from a type 1 diabetic mouse model had increased contractile response to carbachol, ATP, KCl, and EFS. In contrast, our results show that only the ATP-induced contractile response was increased in the compensated state. Gonulalan et al. (22) showed that the sensitivity of the purinergic response during EFS stimulation of bladder smooth muscle from STZ-induced type 1 diabetic rats was similar to the nondiabetic controls, consistent with results published by Danesghari's group (14, 15). On the other hand, using an alloxan-induced type I rabbit, Mumtaz et al. (34) demonstrated that the purinergic component of EFS was enhanced. Our results using the type II diabetes rat model show that the purinergic portion of EFS is nearly absent in animals in the compensated state. The results of these studies suggest that alterations in ATP responsiveness of bladder smooth muscle are not a generalized response to a pathophysiological state and more importantly may in fact be specific to type II diabetes and not a generalized response to diabetes.
While enhanced contractility to muscarinic agonists has been shown in many type II diabetic models (15, 26, 35, 40, 50), others have shown decreased contractility (30, 43). There may be many explanations for the differences seen in these studies (species, diabetes induction methodology, and animal age), but it is interesting to note that both studies showing decreased contractility to muscarinic agonists were performed in animals with a longer duration of diabetes. The spontaneously diabetic biobreeding rats were 1 yr of age and the alloxan-induced diabetic rabbits were 4 mo post-diabetes induction when they showed decreased contractility (30, 43). This suggests a link between the duration of diabetes and state of bladder dysfunction.
One of our most important observations was the change in ATP-induced bladder smooth muscle contraction from diabetic animals. Many studies have shown increased levels of NANC-induced contractions in models of bladder dysfunction. Normally, there is little NANC-induced contraction in the bladder, especially in the normal human bladder (3, 36, 37, 42). We determined the effects of ATP through both exogenous application of ATP and pharmacological inhibition or desensitization of P2X receptors. Exogenous ATP-induced contraction was enhanced in bladders from diabetic animals. However, the purinergic component of an EFS-induced bladder contraction was either reduced or not changed in the bladders of diabetic animals. It is generally accepted that P2X receptors mediate contraction of smooth muscle while P2Y receptors mediate relaxation (2, 7). In experiments where P2X receptors were desensitized through repeated applications of α,β-methylene ATP, the purinergic component of an EFS-induced contraction was decreased in bladders of diabetic animals compared with control animals. However, when P2X receptors were blocked by the antagonist PPADS (or suramin) there was no difference in the purinergic component of an EFS-induced contraction between bladders from diabetic and control animals. While α,β-methylene ATP desensitizes primarily P2X1 and P2X3 receptors, PPADS inhibition is known to affect a broader variety of P2X receptors (51). P2X1 and P2X2 receptors are the purinergic receptors primarily expressed on smooth muscle cells (8, 39). Therefore, we examined the presence of P2X1 receptors in bladder smooth muscle and found no difference in P2X1 expression levels between control and diabetic animals. There are many possible explanations for the disparate results between the exogenous application of ATP and EFS-induced contractile experiments. These include receptor internalization after desensitization by α,β-methylene ATP or different levels of ATP in the neuromuscular junction after EFS-induced release.
Bhetwal et al. (5) showed significant differences in both intracellular signaling and magnitude of contraction in response to the exogenous addition of an agonist compared with EFS-induced contractions. They suggested that the exogenous addition of an agonist “floods” the tissue whereas EFS provides a more physiologically relevant response mediated via the enteric nervous system and interstitial cells of Cajal (ICC)-dependent fundus smooth muscle contraction. This is consistent with our differential responses to the exogenous addition of agonist and EFS, although we did not examine a potential role of ICC. Differential signaling in response to activation of muscarinic and purinergic receptors was shown by Tsai et al. (45) during EFS-induced contractions. They demonstrated that the initial phase of a murine bladder smooth muscle contraction was more dependent on purinergic receptor-mediated activation of the myosin light chain (MLC) kinase and MLC phosphorylation whereas the sustained contraction relies more on muscarinic receptor activation and phosphorylation of the Ca2+-sensitizing protein CPI-17. This is consistent with our results using bladders from control animals in which inhibition of both muscarinic and purinergic receptors almost abolished EFS-induced force, demonstrating a role for both receptor types in contraction. However, in bladders from diabetic animals, desensitization of purinergic receptors did not decrease EFS-induced force and in fact increased force.
While diabetes modified the purinergic component of EFS-induced contraction, it did not affect the total EFS-induced contraction or the cholinergic portion of the neurogenic contraction. However, there was a residual neurogenic component of the contraction resistant to both cholinergic and purinergic inhibition that was increased in bladders from diabetic animals. This neurogenic residual component could be due to activation of P2X receptors not affected by α,β-methylene ATP or PPADS. Other studies have also shown residual components of EFS-induced contractions in diabetic animal models (4, 24, 27). There was also an increased TTX-resistant component to EFS-induced contraction in diabetic animals. These increased responses were blocked by removal of extracellular calcium or inhibition of voltage-gated calcium channels by nifedipine, suggesting they were due to direct muscle stimulation. It is possible this TTX-resistant component was increased due to an increased instability of the smooth muscle membrane in the bladder of diabetic animals, resulting in enhanced calcium influx.
In selected experiments, while determining the effect of purinergic receptor inhibition on EFS-induced contractions, we noticed an increase in spontaneous activity after addition of either α,β-methylene ATP or PPADS. This increase in spontaneous activity was only present in muscle from diabetic animals. When the activity was stimulated by α,β-methylene ATP, it was removed by washout. The addition of PPADS also increased spontaneous activity, but the effect was only partially reversible by washout. The spontaneous activity was TTX resistant, but sensitive to nifedipine. Because P2X receptors are ligand-gated ion channels, these results may suggest the effect is due to dysregulation of cation channels, most likely calcium channels in the bladder smooth muscle of diabetic animals. There was no difference in expression levels of P2X1 receptors, consistent with the suggestion that receptor function and not numbers may account for the spontaneous activity. This result is consistent with the TTX-resistant, but nifedipine-sensitive component of the EFS contraction.
In summary, bladders from male type II diabetic animals reached the compensated stage of diabetic bladder dysfunction by 27 wk of age. This was shown by an increased void volume, increased voiding frequency, and increased contractility to both exogenous carbachol and ATP. Some of these changes were seen at 16 wk of age, but were enhanced at 27 wk of age. Similar to vascular smooth muscle from hypertensive animals (6), our results suggest the possibility that diabetes results in membrane instability and possibly enhanced calcium influx through nifedipine-sensitive calcium channels. Although exogenous ATP-induced contraction was enhanced in diabetic animals, endogenous ATP-induced contraction was significantly reduced. These results support the thesis put forth by Sun and Chai (44) that purinergic receptors are a critical part of the cellular mechanisms leading to bladder dysfunction.
GRANTS
This study was funded in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK 85734. D. M. Kendig was funded by a Drexel University College of Medicine Aging Initiative predoctoral fellowship.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: D.M.K. and R.S.M. provided conception and design of research; D.M.K., H.K.E., and R.S.M. performed experiments; D.M.K., H.K.E., and R.S.M. analyzed data; D.M.K., H.K.E., and R.S.M. interpreted results of experiments; D.M.K. and H.K.E. prepared figures; D.M.K. and H.K.E. drafted manuscript; D.M.K., H.K.E., and R.S.M. edited and revised manuscript; D.M.K., H.K.E., and R.S.M. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address for D. M. Kendig: Dept. of Biology, Loyola University of Maryland, Baltimore, MD 21210.
REFERENCES
- 1.American Diabetes Association. Statistics About Diabetes [Online]. http://www.diabetes.org/diabetes-basics/statistics/ [25 February 2015].
- 2.Aronsson P, Andersson M, Ericsson T, Giglio D. Assessment and characterization of purinergic contractions and relaxations in the rat urinary bladder. Basic Clin Pharmacol Toxicol 107: 603–613, 2010. [DOI] [PubMed] [Google Scholar]
- 3.Bayliss M, Wu C, Newgreen D, Mundy AR, Fry CH. A quantitative study of atropine-resistant contractile responses in human detrusor smooth muscle, from stable, unstable and obstructed bladders. J Urol 162: 1833–1839, 1999. [PubMed] [Google Scholar]
- 4.Benkó R, Lázár Z, Pórszász R, Somogyi GT, Barthó L. Effect of experimental diabetes on cholinergic, purinergic and peptidergic motor responses of the isolated rat bladder to electrical field stimulation or capsaicin. Eur J Pharmacol 478: 73–80, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Bhetwal BP, Sanders KM, An C, Trappanese DM, Moreland RS, Perrino BA. Ca2+ sensitization pathways accessed by cholinergic neurotransmission in the murine gastric fundus. J Physiol 591: 2971–2986, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bohr DF, Webb RC. Vascular smooth muscle membrane in hypertension. Annu Rev Pharmacol Toxicol 28: 389–409, 1988. [DOI] [PubMed] [Google Scholar]
- 7.Bolego C, Pinna C, Abbracchio MP, Cattabeni F, Puglisi L. The biphasic response of rat vesical smooth muscle to ATP. Br J Pharmacol 114: 1557–1562, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burnstock G. Therapeutic potential of purinergic signalling for diseases of the urinary tract. BJU Int 107: 192–204, 2011. [DOI] [PubMed] [Google Scholar]
- 9.Burnstock G, Dumsday B, Smythe A. Atropine resistant excitation of the urinary bladder: the possibility of transmission via nerves releasing a purine nucleotide. Br J Pharmacol 44: 451–461, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen D, Wang MW. Development and application of rodent models for type 2 diabetes. Diabetes Obes Metab 7: 307–317, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Cheng JT, Yu BC, Tong YC. Changes of M3-muscarinic receptor protein and mRNA expressions in the bladder urothelium and muscle layer of streptozotocin-induced diabetic rats. Neurosci Lett 423: 1–5, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Christ GJ, Bushman W, Fraser MO. Impact of diabetes on the prostate and urethra: implications to improved bladder dysfunction understanding and treatment. J Urol 182, Suppl 6: S38–S44, 2009. [DOI] [PubMed] [Google Scholar]
- 13.Daneshgari F, Leiter EH, Liu G, Reeder J. Animal models of diabetic uropathy. J Urol 182: S8–S13, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daneshgari F, Liu G, Birder L, Hanna-Mitchell AT, Chacko S. Diabetic bladder dysfunction: current translational knowledge. J Urol 182: S18–S26, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daneshgari F, Liu G, Imrey PB. Time dependent changes in diabetic cystopathy in rats include compensated and decompensated bladder function. J Urol 176: 380–386, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Daneshgari F, Moores C, Frinjari H, Babineau D. Patient related risk factors for recurrent stress urinary incontinence surgery in women treated at a tertiary care center. J Urol 176: 1493–1499, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Eika B, Levin RM, Longhurst PA. Comparison of urinary bladder function in rats with hereditary diabetes insipidus, streptozotocin-induced diabetes mellitus, and nondiabetic osmotic diuresis. J Urol 151: 496–502, 1994. [DOI] [PubMed] [Google Scholar]
- 18.Eika B, Levin RM, Longhurst PA. Modulation of urinary bladder function by sex hormones in streptozotocin-diabetic rats. J Urol 152: 537–543, 1994. [DOI] [PubMed] [Google Scholar]
- 19.Etgen GJ, Oldham BA. Profiling of Zucker diabetic fatty rats in their progression to the overt diabetic state. Metabolism 49: 684–688, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Fan EW, Chen LJ, Cheng JT, Tong YC. Changes of urinary bladder contractility in high-fat diet-fed mice: the role of tumor necrosis factor-α. Int J Urol 21: 831–835, 2014. [DOI] [PubMed] [Google Scholar]
- 21.Gasbarro G, Lin DL, Vurbic D, Quisno A, Kinley B, Daneshgari F, Damaser MS. Voiding function in obese and type 2 diabetic female rats. Am J Physiol Renal Physiol 298: F72–F77, 2010. [DOI] [PubMed] [Google Scholar]
- 22.Gonulalan U, Kosan M, Hafez G, Arioglu E, Akdemir O, Ozturk B, Gur S, Cetinkaya M. The effect of diabetes mellitus on α-1-adrenergic receptor subtypes in the bladder of rats. Urology 80: 951–956, 2012. [DOI] [PubMed] [Google Scholar]
- 23.Ioanid CP, Noica N, Pop T. Incidence and diagnostic aspects of the bladder disorders in diabetics. Eur Urol 7: 211–214, 1981. [DOI] [PubMed] [Google Scholar]
- 24.Kennedy C, Tasker PN, Gallacher G, Westfall TD. Identification of atropine- and P2X1 receptor antagonist-resistant, neurogenic contractions of the urinary bladder. J Neurosci 27: 845–851, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King AJ. The use of animal models in diabetes research. Br J Pharmacol 166: 877–894, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leiria L, Mónica F, Carvalho F, Claudino M, Franco-Penteado C, Schenka A, Grant A, De Nucci G, Antunes E. Functional, morphological and molecular characterization of bladder dysfunction in streptozotocin-induced diabetic mice: evidence of a role for L-type voltage-operated Ca2+ channels. Br J Pharmacol 163: 1276–1288, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leiria LO, Sollon C, Calixto MC, Lintomen L, Monica FZ, Anhe GF, De Nucci G, Zanesco A, Grant AD, Antunes E. Role of PKC and CaV1.2 in detrusor overactivity in a model of obesity associated with insulin resistance in mice. PLoS One 7: e48507, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu G, Daneshgari F. Alterations in neurogenically mediated contractile responses of urinary bladder in rats with diabetes. Am J Physiol Renal Physiol 288: F1220–F1226, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Liu G, Daneshgari F. Diabetic bladder dysfunction. Chin Med J (Engl) 127: 1357–1364, 2014. [PMC free article] [PubMed] [Google Scholar]
- 30.Longhurst PA. Urinary bladder function 6 months after the onset of diabetes in the spontaneously diabetic BB rat. J Urol 145: 417–422, 1991. [DOI] [PubMed] [Google Scholar]
- 31.Longhurst PA, Belis JA, O'Donnell JP, Galie JR, Westfall DP. A study of the atropine-resistant component of the neurogenic response of the rabbit urinary bladder. Eur J Pharmacol 99: 295–302, 1984. [DOI] [PubMed] [Google Scholar]
- 32.Matsumoto Y, Torimoto K, Matsuyoshi H, Hirayama A, Fujimoto K, Yoshimura N, Hirao Y. Long-term effects of diabetes mellitus on voiding function in a new model of type 2 diabetes mellitus, the spontaneously diabetic Torii (SDT) rat. Biomed Res 30: 331–335, 2009. [DOI] [PubMed] [Google Scholar]
- 33.Mokdad AH, Ford ES, Bowman BA, Nelson DE, Engelgau MM, Vinicor F, Marks JS. The continuing increase of diabetes in the US. Diabetes Care 24: 412, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Mumtaz FH, Lau DH, Siddiquui EJ, Morgan RJ, Thompson CS, Mikhailidis DP. Changes in cholinergic and purinergic neurotransmission in the diabetic rabbit bladder. In Vivo 20: 1–4, 2006. [PubMed] [Google Scholar]
- 35.Nobe K, Yamazaki T, Tsumita N, Hashimoto T, Honda K. Glucose-dependent enhancement of diabetic bladder contraction is associated with a rho kinase-regulated protein kinase C pathway. J Pharmacol Exp Ther 8: 940–950, 2009. [DOI] [PubMed] [Google Scholar]
- 36.O'Reilly BA, Kosaka AH, Chang TK, Ford AP, Popert R, McMahon SB. A quantitative analysis of purinoceptor expression in the bladders of patients with symptomatic outlet obstruction. BJU Int 87: 617–622, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Palea S, Artibani W, Ostardo E, Trist DG, Pietra C. Evidence for purinergic neurotransmission in human urinary bladder affected by interstitial cystitis. J Urol 150: 2007–2012, 1993. [DOI] [PubMed] [Google Scholar]
- 38.Peterson RGSW, Neel MA, Little LA, Eichberg J. Zucker diabetic fatty rat as a model for non-insulin-dependent diabetes mellitus. ILAR J 32: 16–19, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruggieri MR. Mechanisms of disease: role of purinergic signaling in the pathophysiology of bladder dysfunction. Nat Clin Pract Urol 3: 206–215, 2006. [DOI] [PubMed] [Google Scholar]
- 40.Saito M, Okada S, Kazuyama E, Satoh I, Kinoshita Y, Satoh K. Pharmacological properties, functional alterations and gene expression of muscarinic receptors in young and old type 2 Goto-Kakizaki diabetic rat bladders. J Urol 180: 2701–2705, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Seidell JC. Obesity, insulin resistance, and diabetes: a worldwide epidemic. Br J Nutr 83: 5–8, 2007. [DOI] [PubMed] [Google Scholar]
- 42.Sjogren C, Andersson KE, Husted S, Mattiasson A, Moller-Madsen B. Atropine resistance of transmurally stimulated isolated human bladder muscle. J Urol 128: 1368–1371, 1982. [DOI] [PubMed] [Google Scholar]
- 43.Su X, Changolkar A, Chacko S, Moreland RS. Diabetes decreases rabbit bladder smooth muscle contraction while increasing levels of myosin light chain phosphorylation. Am J Physiol Renal Physiol 287: F690–F699, 2004. [DOI] [PubMed] [Google Scholar]
- 44.Sun Y, Chai TC. Role of purinergic signaling in voiding dysfunction. Curr Bladder Dysfunc Rep 5: 219–224, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsai MH, Kamm KE, Stull JT. Signalling to contractile proteins by muscarinic and purinergic pathways in neurally stimulated bladder smooth muscle. J Physiol 590: 5107–5121, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Unger RH, Orci L. Diseases of liporegulation: new perspective on obesity and related disorders. FASEB J 15: 312–321, 2001. [DOI] [PubMed] [Google Scholar]
- 47.Vadhavkar M, Golbidi S, Sea J, Longpre M, Stothers L, Laher I. Exercise improves bladder function in diabetic mice. Neurourol Urodyn 30: 174–182, 2011. [DOI] [PubMed] [Google Scholar]
- 48.Van Den Eeden SK, Sarma AV, Rutledge BN, Cleary PA, Kusek JW, Nyberg LM, McVary KT, Wessells H. Effect of intensive glycemic control and diabetes complications on lower urinary tract symptoms in men with type 1 diabetes: diabetes control and complications trial/epidemiology of diabetes interventions and complications (DCCT/EDIC) study. Diabetes Care 32: 664–670, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature 444: 875–880, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Yono M, Latifpour J, Yoshida M, Ueda S. Age-related alterations in the biochemical and functional properties of the bladder in type 2 diabetic GK rats. J Recept Signal Transduct Res 25: 147–157, 2005. [DOI] [PubMed] [Google Scholar]
- 51.Ziganshin AU, Hoyle CH, Bo X, Lambrecht G, Mutschler E, Baumert HG, Burnstock G. PPADS selectively antagonizes P2X-purinoceptor-mediated responses in the rabbit urinary bladder. Br J Pharmacol 110: 1491–1495, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]