

Doxorubicin treatment for cancer can induce cardiomyopathy later in life. In the present study we utilized Sirt3 knockout (Sirt3.KO) and Sirt3 transgenic (Sirt3.tg) mice to demonstrate the critical role of Sirt3 in negatively regulating doxorubicin-induced cardiac hypertrophy in mice by upregulating oxoguanine-DNA glycosylase-1 (OGG1) levels and preventing mitochondrial DNA damage.
Keywords: Sirt3, doxorubicin, mitochondria, ROS
Abstract
Doxorubicin (Doxo) is a chemotherapeutic drug widely used to treat variety of cancers. One of the most serious side effects of Doxo is its dose-dependent and delayed toxicity to the heart. Doxo is known to induce cardiac mitochondrial damage. Recently, the mitochondrial sirtuin SIRT3 has been shown to protect mitochondria from oxidative stress. Here we show that overexpression of SIRT3 protects the heart from toxicity of Doxo by preventing the drug-induced mitochondrial DNA (mtDNA) damage. Doxo treatment caused depletion of Sirt3 levels both in primary cultures of cardiomyocytes and in mouse hearts, which led to massive acetylation of mitochondrial proteins. Doxo-induced toxicity to cardiomyocytes was associated with increased reactive oxygen species (ROS) production, mitochondrial fragmentation, and cell death. Overexpression of SIRT3 helped to attenuate Doxo-induced ROS levels and cardiomyocyte death. Sirt3 knockout (Sirt3.KO) mice could not endure the full dose of Doxo treatment, developed exacerbated cardiac hypertrophy, and died during the course of treatment, whereas Sirt3 transgenic (Sirt3.tg) mice were protected against Doxo-induced cardiotoxicity. Along with Sirt3, we also observed a concomitant decrease in levels of oxoguanine-DNA glycosylase-1 (OGG1), a major DNA glycosylase that hydrolyzes oxidized-guanine (8-oxo-dG) to guanine. Depletion of OGG1 levels was associated with increased mtDNA damage. Sirt3.KO mice and Doxo-treated mice showed increased 8-oxo-dG adducts in DNA and corresponding increase in mtDNA damage, whereas, 8-oxo-dG adducts and mtDNA damage were markedly reduced in Sirt3 overexpressing transgenic mice hearts. These results thus demonstrated that Sirt3 activation protects the heart from Doxo-induced cardiotoxicity by maintaining OGG1 levels and protecting mitochondria from DNA damage.
NEW & NOTEWORTHY
Doxorubicin treatment for cancer can induce cardiomyopathy later in life. In the present study we utilized Sirt3 knockout (Sirt3.KO) and Sirt3 transgenic (Sirt3.tg) mice to demonstrate the critical role of Sirt3 in negatively regulating doxorubicin-induced cardiac hypertrophy in mice by upregulating oxoguanine-DNA glycosylase-1 (OGG1) levels and preventing mitochondrial DNA damage.
anthracyclines are among one of the most effective drugs available today for cancer chemotherapy (39). These are effective against a wide variety of cancers including leukemia, Hodgkin's lymphoma, thyroid cancer, soft tissue sarcoma, multiple myeloma, and others (18). Doxorubicin (Doxo), also known as adriamycin, is the drug of choice for the treatment of many types of cancers (18). However, the clinical utility of Doxo is limited by its dose-dependent and delayed toxicity to the heart causing potentially irreversible cardiomyopathic changes leading to congestive heart failure (21).
Doxo-induced cardiac damage is considered primarily through the overt production of reactive oxygen species (ROS), which is generated predominantly through mitochondria (6, 8). Doxo-induced activation of ROS is attained through several mechanisms. Mitochondrial enzymes such as NADH dehydrogenase and cytochrome P-450 reductase undergo redox cycling upon interaction with Doxo leading to generation of ROS (3, 6). This is exacerbated by the reduced activity of several ROS scavenging enzymes including catalase, glutathione peroxidase, and MnSOD (9, 16). In addition, several ROS-independent mechanisms have also been proposed for the cardiac toxicity of Doxo. Pommier et al. (26) have shown that Doxo interacts with DNA and inhibits the progression of topoisomerase-IIβ (Top2β), thereby preventing DNA unwinding and ultimately protein synthesis. With Top2β, Doxo forms a highly stabilized cleavable DNA enzyme complex, which prevents ligation of DNA strands after double strand breakage (41). Correspondingly, deletion of Top2β in mouse heart was found to be cardioprotective against Doxo (41). Another mechanism implicated in Doxo-induced cardiotoxicity is systemic accumulation of iron (24). A mouse deficient in Hfe-gene (a model of human hereditary hemochromatosis in which iron absorption from the intestine is increased) was shown to have greater sensitivity to Doxo than wild-type animals (22). This idea was further supported by the observation that dexrazoxane an iron chelator was capable of reducing cardiac toxicity of Doxo (36). In the same vein overexpression of a mitochondrial iron exporter protein, ABCB8, decreased mitochondrial iron levels and rescued the heart from Doxo-induced cardiac damage (14). One of the major limitations of these hypotheses is that they do not adequately explain the mysterious development of cardiomyopathy that appears years after the completion of the therapy. One hypothesis that could explain the delayed onset of cardiomyopathy is the development of mitochondrial DNA (mtDNA) damage, occurring during the acute phase of the administration of Doxo, and the molecular determinants that help to perpetuate this damage over time to manifest as cardiomyopathy (15).
Human mtDNA is a double-stranded circular 16.5-kb DNA containing 37 genes, all of which are essential for normal mitochondrial function. Due to its proximity to electron transport chain, mtDNA is more vulnerable to damage than nuclear DNA (19). Mutations in the mitochondrial genome cause deleterious consequences to overall health of the organism since bulk of ATP is produced by oxidative phosphorylation and mtDNA encodes major components of the mitochondrial respiratory complexes (1). mtDNA damage is implicated in the pathology of many diseases including cancer, metabolic syndromes, type 2 diabetics, and cardiovascular diseases (29).
A major defect in the integrity of DNA occurs by oxidation of the guanine residue. During oxidative stress guanine residues are oxidized to 7,8-dihydro-8-oxoguanine (8-oxo-dG), and addition of these adduct to DNA causes DNA lesions. In humans 8-oxo-dG levels in mtDNA were found to accumulate over the lifespan of an individual (23). Mitotically fixed and aerobically active tissues including brain and hearts were shown to have increased accumulation of 8-oxo-dG adducts with aging (2). The enzyme that hydrolyzes 8-oxo-dG from DNA is 8-oxoguanine-DNA glycosylase-1 (OGG1), a DNA repair enzyme (33). mtDNA isolated from OGG1 null mice were shown to have 20-fold more 8-oxo-dG than mtDNA from wild-type animals (7). Furthermore, cardiac-specific overexpression of OGG1 was shown to protect mtDNA and reduce cardiac fibrosis following trans-aortic constriction. Down regulation of mitochondrial OGG1 has been also shown to promote brain aging (38). Moreover, a mouse carrying a mutant OGG1 gene was shown to have threefold increase in 8-oxo-dG adducts in the liver at 9 wk of age, which increased to sevenfold at 14 wk of age, suggesting that reduced activity of the OGG1 gene leads to aging associated accumulation of 8-oxo-dG adducts and DNA damage (38). By the same token Doxo treatment was also shown to increase 8-oxo-dG levels in several models of cellular toxicity.
One of the enzymes that have been shown to reduce cellular damage occurring with aging is SIRT3 deacetylase. SIRT3 is a class III histone deacetylase which is present primarily in mitochondria along with other sirtuins, SIRT4 and SIRT5 (20). Among these, only SIRT3 possess robust deacetylase activity (17). Several mitochondrial targets of SIRT3, that are critical for metabolism, apoptosis and mitochondrial detoxification have been identified (10). Also, SIRT3 can regulate fusion-fission dynamics of mitochondria, which is necessary for maintaining fitness of mitochondrial population. These studies suggest that by coordinately deacetylating enzymes involved in multiple mitochondrial processes, SIRT3 fine tunes the mitochondrial functions. A role of SIRT3 mediated deacetylation is also considered a critical mechanism mediating the benefits of calorie restriction, an intervention that can prolong lifespan of a variety of organisms (13).
In this study we show that Doxo treatment downregulates SIRT3 levels in mice, which are associated with decreased levels of OGG1 and accumulation of 8-oxo-dG in the heart mitochondria. Cardiac-specific overexpression of SIRT3 helped to maintain OGG1 levels, reduced mtDNA damage, and protected the heart from cardiotoxicity of Doxo. Since SIRT3 levels have been shown to be reduced with aging, these studies may explain why there is delayed occurrence of Doxo toxicity to the heart.
MATERIALS AND METHODS
Cardiomyocyte culture.
Primary cultures of cardiac myocytes were prepared from neonatal rat hearts. In brief, hearts were removed from 1- to 3-day-old pups (Sprague-Dawley rats, either sex) and kept in cold DMEM. Ventricles were cut into four to six evenly sized pieces using small scissors and digested using collagenase type II (Worthington). The digested solution was collected with the cannula syringe avoiding the tissue chunks and was added to one of the already aliquoted 10 ml FBS (100%). These steps were repeated six to seven times until no tissue chunks are visible. Tissue digest was spun and pellet was dissolved in DMEM with 5% FBS. Cells were preplated for 1 h to remove fibroblasts, and unattached cardiomyocytes in suspension were collected and plated in fibronectin-coated culture plates. Cardiomyocytes cultures were used after 24 h of plating.
ROS detection.
ROS levels were detected using CM-H2DCFDA reagent (Invitrogen) as per the manufacturer's instructions. In brief, primary cultures of cardiomyocytes were infected with Ad.SIRT3 or Ad.empty virus (10 multiplicity of infection). Twenty-four hours after infection cells were treated with Doxo for 15 min. Cells were stained with CM-H2DCFDA, acquired by FACSCalibur, and analyzed with use of FlowJo. The mean fluorescence intensity of cells positive for CM-H2DCFDA staining was determined.
Cell death assay.
Cardiomyocytes were cultured in six-well plates were treated with 10 μM Doxo for 24 h. Cells were harvested from tissue culture plates and centrifuged at 1,000 rpm for 5 min at 4°C. Supernatant was removed, and cells were washed twice with cold PBS. Cells were then resuspended in 100 μl of cold 1× binding buffer, and 20 μl of 7AAD (BD) were added. Samples were incubated for 15 min in the dark at room temperature. A total of 400 μl of 1× binding buffer was added and analyzed by flow cytometry using a FacScan analyzer (Becton-Dickinson, San Jose, CA). Results were processed using FlowJo software.
Tetramethyl rhodamine methyl ester uptake.
To monitor mitochondrial membrane potential (ΔΨm), tetramethyl rhodamine methyl ester (TMRM; Invitrogen), a ΔΨm-dependent cationic dye, was used. In brief, primary cultures of cardiomyocytes were infected with Ad.SIRT3 or Ad.empty virus. Twenty-four hours after infection cells were treated with Doxo for another 24 h. Cells were stained with TMRM, acquired by FACSCalibur, and analyzed with use of FlowJo. The mean fluorescence intensity of cells positive for TMRM staining was determined.
mtDNA damage assay.
Genomic DNA was isolated using Qiagen Genomic-tip 20/G and Qiagen DNA Buffer Set (Qiagen, Gaithersburg, MD) per the manufacturer's instruction. Eluted DNA was incubated with isopropanol overnight at −80°C and centrifuged 12,000 g for 60 min. DNA was washed with 70% ethanol and dissolved in TE buffer. PCR was performed using Ex-taq (Clonetech, Mountain View, CA). Primer sequences for long PCR were forward: 5′-CCCAGCTACTACCATCATTCAAGTAG-3′ and reverse: 5′-GAGAGATTTTATGGGTGTAATGCGGTG-3′. Short PCR was performed using forward primer sequence 5′-GCAAATCCATATTCATCCTTCTCAAC-3′ and the reverse primer sequence was the same as long PCR. Resultant PCR products were quantified using Pico-green (Life Technologies). Values obtained from the long fragments were normalized using values from short fragments. The lesion frequency per amplicon was then calculated as λ = −ln(AD/AO), where AD/AO is the ratio of the amplification of the treated samples (AD) to the amplification of the control samples (AO).
8-Oxo-dG level estimation.
8-Oxo-dG levels were estimated using 8-oxo-dG ELISA kit (Trevigen) as per manufacturer's instructions.
Antibodies, immunoblotting, and immunoprecipitation analyses.
The OGG1 antibody was purchased from Santa Cruz Biotechnology and Novus Biologicals. The actin antibody was from Santa Cruz Biotechnology. Anti-acetyl lysine antibody was from cell signaling. MnSOD antibody was from Millipore. Cell or heart ventricular tissue lysates were prepared in radio immunoprecipitation assay (RIPA) buffer [50 mM Tris·HCl (pH 7.5), 0.1% Nonidet P-40, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 100 mM phenylmethylsulfonyl fluoride (PMSF), 5 mM sodium orthovanadate, 10 mM β-glycerol phosphate, and 20 mM NaF and Sigma protease inhibitors]. Typically, 20–50 μg of protein lysates was used for immunoblots. Immunoprecipitation experiments using 500-1,000 μg of total protein lysates were carried out using standard protocols.
Real-time PCR analysis of mRNA levels.
Total RNA was isolated from mouse hearts by using Trizol Reagent (Invitrogen). The residual genomic DNA was digested by incubating the RNA preparation with 0.5 units of RNase-free DNase-1 per microgram of RNA in 1× reaction buffer for 15 min at room temperature, followed by heat inactivation at 90°C for 5 min. Two micrograms of DNase-treated RNA were reverse transcribed by use of Fermentas, RevertAid First Strand cDNA Synthesis Kit. The resultant cDNA was diluted 10-fold before PCR amplification. A reverse transcriptase minus reaction served as a negative control. The mRNA levels were measured by SYBR green real-time PCR. Primer sequences were ANF forward: 5′-TCGTCTTGGCCTTTTGGCT-3′ and reverse: 5′-TCCAGGTGGTCTAGCAGGTTCT-3′, and collagen-1: forward: 5′-AAACCCGAGGTATGCTTGATCTGTA- 3′ and reverse: 5′-GTCCCTCGACTCCTACATCTTCTGA-3′.
Imaging of cardiac fibroblasts.
Cardiac fibroblasts on 12-mm coverslips were transfected with SIRT3 or empty vector. Twenty-four hours after infection cells were treated with 50 nM Doxo for 48 h. Cells were washed with PBS and fixed with 3.7% formaldehyde in PBS for 15 min followed by permeabilization with 0.1% Triton X-100 for 5 min. They were then blocked with 10% BSA in PBS followed by incubation with primary antibody overnight at 4 °C. Thereafter, cells were incubated with a secondary antibody conjugated with either Alexa Fluor 594 or FITC for 1 h. Cells were washed and mounted in ProLong Gold antifade reagent with DAPI. Cells were visualized using a Leica SP2 laser scanning microscope.
Doxo treatment of mice.
Sirt3 knockout (KO) mice were generously provided by F. W. Alt. Sirt3.tg mice were generated and characterized similar to the method described earlier (34). Doxo reconstituted in 0.85% sterile sodium chloride was administered by intraperitoneal injection. Control animals were also treated simultaneously with identical volume of 0.85% sterile NaCl. Sirt3-Tg mice and their respective controls were treated with 5 mg/kg Doxo every 15 days for a total of three doses (cumulative dose: 15 mg/kg body wt). Sirt3.KO mice and their respective controls received two doses of Doxo (cumulative dose: 10 mg/kg body wt). Fifteen days after the last dose of Doxo cardiac hypertrophy and heart function of mice were studied. All the mice used in this study were males and 6 mo of age. All animal protocols were reviewed and approved by the University of Chicago Institutional Animal Care and Use Committee.
Measurement of mouse heart functions.
Chest hairs of mice were removed with a topical depilatory agent and transthoracic echocardiography was performed while mice were under inhaled isoflurane (∼1%) for anesthesia, delivered via nose cone. Limb leads were attached for electrocardiogram gating, and the animals were imaged in the left lateral decubitus position with a VisualSonics Vevo 770 machine, using a 30-MHz high-frequency transducer. Body temperature was maintained using a heated imaging platform and warming lamps. Two-dimensional images were recorded in parasternal long- and short-axis projections, with guided M-mode recordings at the midventricular level in both views. Left ventricle (LV) cavity size and wall thickness were measured in at least three beats from each projection and averaged. LV wall thickness [interventricular septum (IVS) and posterior wall (PW) thickness] and internal dimensions at diastole and systole (LVIDd and LVIDs, respectively) were measured. LV fractional shortening [(LVIDd − LVIDs)/LVIDd] and relative wall thickness [(IVS thickness + PW thickness)/LVIDd] were calculated from the M-mode measurements. Stroke volume was measured by catheterization of left coronary artery of mice using a sensor tip catheter (1F; Millar PV System).
Statistical analysis.
Statistical differences among groups were determined with either Student's t-test (for two groups) or one-way ANOVA. P < 0.05 was considered significant.
RESULTS
SIRT3 protects cardiomyocytes and fibroblasts from Doxo-induced damage.
SIRT3 is the only mitochondrial deacetylase with robust deacetylase activity (17). To test the effect of Doxo on mitochondrial acetylation, we treated cardiomyocytes with 1 or 2 μM Doxo for 24 h. Western blotting analysis using a pan acetyl lysine antibody revealed significantly increased mitochondrial protein acetylation levels following Doxo treatment (Fig. 1A). Since SIRT3 is the major deacetylase in mitochondria, we analyzed SIRT3 levels in Doxo-treated cells. We found that increased mitochondrial acetylation was associated with reduced levels of SIRT3 (Fig. 1B). Doxo treatment is known to induce ROS production in cardiomyocytes. To test the cardioprotective effect of SIRT3 on Doxo-induced damage, we first examined the ability of SIRT3 to block ROS levels induced by Doxo. Cultures of cardiomyocytes were overexpressed with adenovirus expressing SIRT3 (Ad.SIRT3) or empty adenovirus (Ad.mock) and treated with different doses of Doxo. Cells were stained with CM-H2DCFDA, a nonfluorescent dye that fluoresces upon oxidation by ROS. We found that cells overexpressed with SIRT3 suppressed Doxo-induced ROS levels (Fig. 1, C and D). To support these findings we also performed a cell-death experiment. Cardiomyocytes overexpressing SIRT3 showed significantly reduced cell death compared with nonexpressing cells, suggesting that SIRT3 expression protects cardiomyocytes from Doxo-induced cell death (Fig. 1, E and F). To further substantiate these findings we treated Ad.SIRT3 or empty adenovirus overexpressing cells with TMRM, a dye that measures the mitochondrial membrane potential and indicates functional or uncoupled mitochondria. Doxo-treated cardiomyocytes showed reduced uptake of TMRM whereas cardiomyocytes overexpressed with SIRT3 showed increased TMRM uptake, suggesting that activation of SIRT3 protects cardiomyocytes from Doxo-induced mitochondrial damage (Fig. 1, G and H).
Fig. 1.

SIRT3 overexpression protects cardiomyocytes from doxorubicin (Doxo)-mediated injury. A: primary cultures of rat neonatal cardiomyocytes were treated with 1 or 2 uM Doxo. Mitochondrial lysate was prepared and analyzed for lysine acetylation using anti-acetyl lysine antibody. B: primary cultures of cardiomyocytes were treated with Doxo for 24 h. Cell lysates were prepared and subjected to immunostaining for SIRT3. For loading control, the blot was probed with anti-actin antibody. C: primary cultures of cardiomyocytes were infected with adenovirus vectors expressing SIRT3 or the empty (mock) adenovirus (Ad.mk). Following 24 h of infection, cells were treated with different doses of Doxo. Cells were stained with CM-H2DCFDA and reactive oxygen species (ROS) levels were measured by fluorescence-activated cell sorter. D: quantification of mean fluorescence intensity (MFI) in different groups of cells. All values are means ± SE; n = 4. *P < 0.05, compared with cell infected with the mock virus. E: primary cultures of cardiomyocytes were infected with Ad.SIRT3 or Ad.mk virus. Following 24 h of infection, cells were treated with 10 uM Doxo for 24 h. Extent of apoptosis was measured by estimating the percentage of 7AAD-positive cells by FACS analysis. F: quantification of cell death in different group of cells. All values are means ± SE; n = 5. G: cardiomyocytes overexpressed with SIRT3 were treated with 1 uM Doxo for 24 h. Cells were stained with tetramethyl rhodamine methyl ester (TMRM) and mitochondrial dye incorporation was measured by FACS analysis. H: quantification of MFI of TMRM staining in different groups of cells. All values are means ± SE; n = 5. I: cardiac fibroblasts were overexpressed with SIRT3 tagged with Flag or control vector. Cells were treated with 50 nM Doxo for 48 h and then stained for α-smooth muscle actin (α-SMA), DAPI, and Flag. Note, SIRT3 overexpressing cells (white arrow) show markedly reduced expression of α-SMA, whereas cells negative for SIRT3 show robust expression of α-SMA; Scale = 10 μm.
Since Doxo-induced cardiotoxicity is associated with increased fibrosis, we also tested whether SIRT3 expression can block Doxo-mediated activation of cardiac fibroblasts. Primary cultures of cardiac fibroblasts transfected with SIRT3 tagged with Flag or control vector for 24 h were treated with Doxo (50 nM) for 48 h. Fibroblast to myofibroblast transformation was assessed by staining for the expression of α-SMA using confocal microscopy. Stimulation of fibroblasts with Doxo resulted in marked increase in stress fiber formation as evidenced by increased α-SMA staining. This was blocked in all the cells overexpressed with SIRT3 (Fig. 1I). These data show that SIRT3 is capable of attenuating the Doxo-induced transformation of cardiac fibroblasts to myofibroblasts.
Sirt3.tg mice are protected from Doxo-mediated cardiac hypertrophy and fibrosis.
To test the cardioprotective effect of SIRT3 in vivo, we generated transgenic mice overexpressing murine Sirt3 in the heart, under the control of α-myosin heavy chain promoter. Mice were treated with three doses of Doxo (5 mg/kg) over a 15-day interval. Cardiac functions were assessed 15 days after the last injection. Doxo injection resulted in 25% increase in heart weight-to-tibia length (HW/TL) ratio in control mice, whereas Sirt3.tg mice showed no noticeable increase in HW/TL ratio (Fig. 2A). Additionally Sirt3.tg mice showed preserved cardiac function as measured by increased LV fractional shortening and increased stroke volume in mice treated with Doxo compared with nontransgenic controls (Fig. 2, B and C). Consistent with this, Sirt3.tg mice also showed significantly reduced cardiac fibrosis and reduced activation of fetal gene program (ANF) following Doxo injection, compared with nontransgenic mice (Fig. 2, D–F). Electron microscopic examination of the heart sections revealed mitochondrial deformities with loss of structural integrities in Doxo-treated nontransgenic mice, while Sirt3.tg mice exhibited minimal mitochondrial damage (Fig. 2G). These results suggested that overexpression of Sirt3 is capable of protecting the heart from Doxo-induced ultrastructural damage and development of cardiac hypertrophy in mice.
Fig. 2.

Sirt3 overexpressing transgenic mice are protected from Doxo-induced cardiac damage. A–C: Sirt3.tg and nontransgenic (ntg) mice were treated with either vehicle or Doxo as described in methods. Left ventricular (LV) fractional shortening (FS) and stroke volume (SV) were measured as described in materials and methods. Mouse HW/TL ratio was determined after death. All values are means ± SE; n = 5–7. D: representative heart sections stained with Masson's trichrome for determining fibrosis in Sirt3.tg and ntg mouse hearts after Doxo treatment. Scale = 20 μm. E: quantification of cardiac fibrosis in different group of mice. All values are means ± SE; n = 5. F: collagen and ANF mRNA levels in the hearts of different treatment groups of mice. All values are means ± SE; n = 4. G: representative electron microscopy images of hearts from control (ntg) and Sirt3.tg mice after treatment with saline or Doxo. Note: hearts from Sirt3.tg mice treated with Doxo show well-aligned mitochondria.
Sirt3.KO mice show exacerbated cardiac hypertrophic response.
To investigate the requirement of Sirt3 in blocking Doxo-induced cardiac damage, we studied the effect of Doxo treatment in Sirt3.KO mice, along with their wild-type controls. Sirt3.KO mice were unable to withstand three doses of Doxo treatment. All the mice died within the first week following the third dose of Doxo injection (Fig. 3A). Therefore, in this group we assessed cardiac function in all the mice at 15 days following the second injection of Doxo. At the basal level, Sirt3.KO mice showed an increased HW/TB ratio, compared with their respective controls, and Doxo injection further magnified this difference (Fig. 3B). We also observed reduced LV fractional shortening and reduced stroke volume in Sirt3.KO mice, which got significantly worse following Doxo injection (Fig. 3, C and D). Additionally, Sirt3.KO mice showed increased fibrosis compared with wild-type controls, and Doxo injection further increased the interstitial fibrosis in Sirt3.KO mice, as revealed by Masson's trichrome staining and increased collagen mRNA expression (Fig. 4, A–C). Moreover, Sirt3.KO mice showed increased ANF expression, which was further elevated following Doxo injection (Fig. 4C). We also performed ultrastructural examination of the heart. Sirt3.KO hearts showed fragmentation and clustering of mitochondria compared with wild-type controls, consistent with our previous results (30). Even though Doxo-induced ultrastructural damage both in wild-type and Sirt3.KO mice, the latter showed extensive loss of sarcomeres as well as swollen and disrupted mitochondria (Fig. 4D). Along with these changes, we also observed increased acetylation of mitochondrial proteins in Doxo-treated hearts and this change was further aggravated when Sirt3.KO mice were infused with Doxo, suggesting that lack of Sirt3 exacerbated Doxo-induced mitochondrial damage (Fig. 4E). Collectively, these data indicated that Sirt3 is required for protecting the heart from Doxo-induced toxicity.
Fig. 3.

Sirt3 knockout (Sirt3.KO) mice do not endure full dose of Doxo treatment and developed exacerbated cardiac hypertrophy and deterioration of cardiac function after treatment. A, top: schematic representation of Doxo treatment protocol to mice. A, bottom: Kaplan-Meier survival analysis of wild-type (WT) and Sirt3.KO mice treated with Doxo. B–D: WT and Sirt3.KO mice were treated with vehicle or Doxo and their heart weight-to-tibia length (HW/TL) ratio (B), LV fractional shortening (C), and SV (D) were determined as described in materials and methods. All values are means ± SE; n = 5–7.
Fig. 4.

Sirt3-deficiency promotes Doxo-induced cardiac injury. A: representative heart sections stained with Masson's trichrome for determining fibrosis in WT and Sirt3.KO mice treated with Doxo. B: quantification of cardiac fibrosis in different group of mice treated or not treated with Doxo. C: collagen and ANF mRNA levels in the hearts of WT and Sirt3.KO mice treated with vehicle or Doxo. Values are means ± SE; n = 5. D: representative electron microscopy images of the heart from WT and Sirt3.KO mice treated with Doxo or vehicle. Scale bar = 2 μm. E: total heart lysate from different group of mice was analyzed by Western blotting using pan acetyl lysine (Ac.K) and MnSOD antibody.
SIRT3 protects cells from Doxo-induced mtDNA damage.
Increased mtDNA damage has been suggested to be one of the mechanisms leading to the aging process. Although, SIRT3 has been shown to improve mitochondrial functions and reduce many aging associated diseases, its effect on preventing mtDNA damage has never been studied in animal models. Our in vitro results suggested that overexpression of SIRT3 can reduce Doxo-induced ROS levels. Several studies have established that increased ROS levels are associated with increased mtDNA damage. We therefore tested the hypothesis whether SIRT3 protects cells by blocking Doxo-mediated mtDNA damage. An indicator of intact DNA is its ability to support PCR amplification since DNA damage can hamper the ability of DNA polymerase to extend the DNA strand, resulting in nonamplification during PCR. By utilizing this technique, we assessed whether the absence of SIRT3 enhances Doxo-induced mtDNA damage. Cardiac fibroblasts from wild-type and Sirt3.KO mice were treated with two different doses of Doxo for 24 h, and mtDNA damage was estimated. Compared with wild-type controls Sirt3 deficient cells showed significantly increased mtDNA damage, which was further augmented by Doxo treatment, suggesting that Sirt3 is needed for efficient mtDNA damage repair (Fig. 5A). We next sought to investigate whether overexpression of SIRT3 can protect cells from Doxo-induced mtDNA damage. Rat neonatal cardiomyocytes infected with Ad.SIRT3 or the empty vector were treated with Doxo at different doses and DNA damage was estimated by PCR. The results showed that overexpression of SIRT3, but not the empty vector, mitigated Doxo-induced mtDNA damage (Fig. 5B).
Fig. 5.
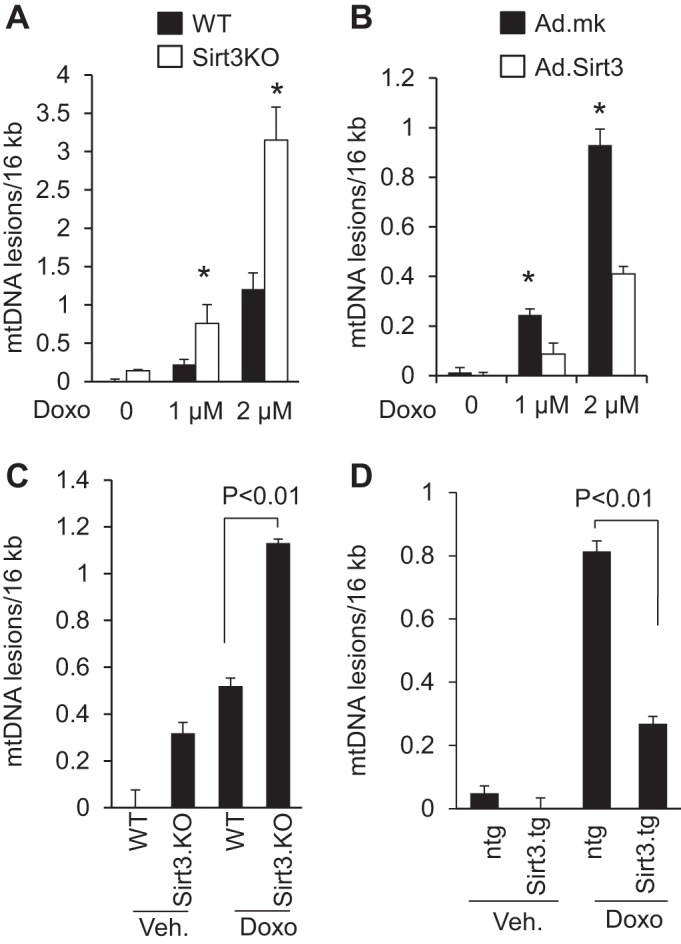
SIRT3 overexpression mitigates Doxo-induced mitochondrial (mt)DNA damage. A: primary cultures of cardiac fibroblasts obtained from WT and Sirt3.KO mice were treated with Doxo for 24 h. mtDNA damage was assessed by quantitative PCR analysis. *Significantly different (P < 0.01) from fibroblasts of WT mice. B: primary cultures of rat neonatal cardiomyocytes were infected with adenovirus expressing SIRT3 (Ad.SIRT3) or empty adenovirus (Ad.mk). Following 24 h of infection, cells were treated with different doses of Doxo as indicated and mtDNA damage was assessed. *Significantly different (P < 0.01) from cells infected with empty virus. C: mitochondrial DNA damage was assessed in whole heart of WT and Sirt3.KO mice treated with saline or Doxo. D: mtDNA damage was assessed in whole heart of control (ntg) and Sirt3.tg mice treated with saline or Doxo. All values are means ± SE; n = 5–7.
We extended these studies to our in vivo mouse models lacking Sirt3 or having Sirt3 overexpressed in the heart. Mice lacking Sirt3 showed increased mtDNA damage compared with their wild-type controls, and injection of Doxo in Sirt3.KO mice further exacerbated mtDNA damage by nearly twofold, compared with vehicle-treated animals (Fig. 5C). To determine the protective effect of Sirt3 on mitochondrial damage in vivo, we infused Doxo in nontransgenic and Sirt3.tg mice. Sirt3 overexpressing mice showed significantly reduced mtDNA lesions after exposure to Doxo, compared with nontransgenic control mice (Fig. 5D). These results suggested that Sirt3 plays a critical role in protecting the mtDNA from Doxo-mediated damage.
SIRT3 repairs mtDNA damage by increasing activity of OGG1.
A previous study has shown that human OGG1, a DNA repair enzyme present in mitochondria, is subject to modification by lysine acetylation and deacetylation. SIRT3 by deacetylation prevents OGG1 degradation in a glioma cell line (4). To test whether Sirt3 can bind to OGG1 in cardiomyocytes, we infected rat cardiomyocytes with adenovirus expressing SIRT3 or empty adenovirus vector for 24 h. Cell lysates were immunoprecipitated with SIRT3 and subjected to Western blotting with use of anti-OGG1 antibody. We found that OGG1 was coprecipitated with SIRT3 in cardiomyocytes (Fig. 6A). We next sought to find whether OGG1 was acetylated during Doxo treatment of cells. OGG1 was immunoprecipitated from cardiomyocytes treated or not treated with Doxo and subjected to Western blotting with use of anti-acetyl lysine antibody. The results indicated that OGG1 was highly acetylated upon treatment with Doxo (Fig. 6, B and C). We then assessed if SIRT3 can deacetylate OGG1. To this end we immunoprecipitated OGG1 from cardiomyocytes overexpressed with SIRT3 or with Ad.empty vector. Western blotting analysis showed that OGG1 was completely deacetylated in Sirt3 overexpressing cells (Fig. 6D). These data suggested that SIRT3 can physically bind to and deacetylate OGG1. Lysine acetylation of OGG1 has been shown to reduce its protein stability. To evaluate the consequence of Sirt3-mediated deacetylation of OGG1 in the heart, we determined Sirt3 and OGG1 levels in the mouse heart subjected to Doxo injection. We found reduced levels of OGG1 in Sirt3.KO hearts (Fig. 6E). Treatment of mice with Doxo reduced both Sirt3 and OGG1 levels of the heart (Fig. 6, E and F). To test if overexpression of Sirt3 can maintain OGG1 levels after Doxo treatment, we analyzed heart lysates of control and Sirt3.tg mice infused with Doxo or saline by Western analysis using anti-OGG1 and anti-Sirt3 antibodies. Our results indicated that overexpression of Sirt3 helped to maintain the OGG1 levels in the mouse heart, suggesting that Sirt3-mediated deacetylation promotes stability of the enzyme (Fig. 6, G and H).
Fig. 6.

Sirt3 deacetylates and prevents Doxo-mediated degradation of oxoguanine-DNA glycosylase-1 (OGG1) in the mouse heart. A: cell lysate of cardiomyocytes overexpressed with SIRT3 was subjected to immunoprecipitation (IP) with indicated antibodies. The resulting beads were analyzed by immunoblotting with anti-OGG1 or anti-SIRT3 antibodies. Note; SIRT3 binds to OGG1 in cardiomyocytes. B: primary cultures of rat cardiomyocytes were treated with 1 uM Doxo for 24 h. Cells were harvested and cell lysate was subjected to immunoprecipitation with anti-OGG1 antibody. The resulting beads were analyzed by immunoblotting with anti-acetyl lysine antibody. C: quantification of relative acetylated OGG1 levels in vehicle and Doxo-treated cardiomyocytes. D: heart lysates of Sirt3.tg and control (ntg) mice were subjected to immunoprecipitation with anti-OGG1 antibody. The resulting beads were analyzed by immunoblotting with anti-acetyl lysine antibody. Note: Sirt3 deacetylates OGG1 in the heart. E: heart lysate of WT and Sirt3.KO mice treated with vehicle or Doxo were subjected to immunoblotting with indicated antibodies. G: heart lysate of control, ntg, and Sirt3.tg mice treated with vehicle or Doxo were subjected to immunoblotting with indicated antibodies. Results are shown for 2 animals in each group. F and H: quantification of OGG1 levels in different group of hearts. Values are means ± SE; n = 5.
OGG1 is the major enzyme involved in the hydrolysis of 8-oxo-dG adducts and impairment in OGG1 function results in the accumulation of 8-oxo-dG adducts and DNA damage. To understand the effect of Doxo-induced depletion of Sirt3 and OGG1 levels, we estimated the 8-oxo-dG levels in the DNA. Compared with wild-type controls, Sirt3.KO cells exhibited higher 8-oxo-dG levels, which got further elevated when treated with Doxo. Consistent with this finding, adenovirus-mediated overexpression of SIRT3 helped to reduce the accumulation of 8-oxo-dG adducts in cells treated with Doxo (Fig. 7, A and B). To further test the inhibitory effect of Sirt3 on the formation of 8-oxo-dG adducts in vivo, we isolated total DNA from Sirt3.KO and wild-type hearts treated or not treated with Doxo and 8-oxo-dG levels were measured. Sirt3.KO mice showed elevated levels of 8-oxo-dG, and compared with wild type, Doxo treatment elevated levels of 8-oxo-dG adducts more in Sirt3.KO hearts, thus suggesting that absence of Sirt3 favors the formation of 8-oxo-dG adducts (Fig. 7C). We next determined if overexpression of Sirt3 can prevent formation of 8-oxo-dG adducts in vivo. Total DNA was isolated from Sirt3.tg or nontransgenic mouse hearts treated or not treated with Doxo and 8-oxo-dG levels were measured. Sirt3-Tg mice infused with Doxo showed significantly lower levels of 8-oxo-dG, compared with its nontransgenic controls, suggesting that Sirt3 plays an important role in regulating the activity of OGG1 and formation of 8-oxo-dG adduct and thereby protecting the mtDNA from oxidative-stress mediated damage (Fig. 7D).
Fig. 7.

Sirt3 overexpression decreases 8-oxo-dG levels. A: primary cultures of mouse cardiac fibroblasts obtained from WT and Sirt3.KO mice were treated with Doxo for 24 h and 8-oxo-dG content in total DNA was measured. *Significantly higher (P < 0.01) compared with cells from WT mice. B: primary cultures of cardiomyocytes were infected with adenovirus expressing SIRT3 or mock adenovirus (Ad.mk). Following 24 h of infection, cells were treated with different doses of Doxo as indicated and 8-oxo-dG content in total DNA was measured. *Significantly higher (P < 0.01) compared with cells overexpressed with Sirt3. C: 8-oxo-dG content in the DNA of whole heart of WT and Sirt3.KO mice treated with saline or Doxo. D: 8-oxo-dG content in the DNA of whole heart of ntg control and Sirt3.tg mice treated with saline or Doxo. All values are means ± SE; n = 5. E: model illustrating how Sirt3 protects the heart from Doxo-induced cardiac injury. Doxo treatment downregulates Sirt3 levels in the heart resulting in decreased levels of OGG1 and increased levels of ROS in mitochondria, both contributing to mtDNA damage. Doxo can also cause DNA damage by directly binding to DNA. Activation of Sirt3 leads to increased OGG1 levels and decreased ROS production, thereby mitigating mtDNA damage by decreasing the oxidative damage as well by increasing the efficiency of DNA repair. These changes together with the ability of Sirt3 to promote mitochondrial fusion enhance the overall health of mitochondrial population, and thereby protecting cardiac myocytes from death and the development of cardiac failure.
DISCUSSION
This study was designed to investigate the role of Sirt3 in the development of Doxo-induced cardiomyopathy. By using Sirt3.tg and Sirt3.KO mice we demonstrated that Sirt3 expression protects the heart from Doxo-mediated cardiac injury. Chronic Doxo treatment led to reduced SIRT3 levels both in vitro and in vivo models of cardiac injury. Overexpression of SIRT3 mitigated Doxo-induced ROS production, mtDNA damage, and cell death. We also found that Sirt3 activation helps to maintain levels of the DNA repair enzyme OGG1. Consequently, we observed a reduced level of 8-oxo-dG adducts and mtDNA damage in Sirt3.tg mice, compared with their nontransgenic controls, treated with Doxo. Together, our studies have revealed that SIRT3 is a negative regulator of Doxo-induced cardiac damage.
Lysine acetylation is a widespread protein modification that occurs in mitochondria. Sirt3.KO mice show global hyperacetylation of mitochondrial proteins, suggesting that it is a major deacetylase regulating reversible acetylation of mitochondrial proteins (17). Acetylation compromises the enzymatic activity of several mitochondrial proteins (10). In the present study we observed significantly increased acetylation of mitochondrial proteins following Doxo treatment, which is associated with a concomitant reduction in SIRT3 levels. Reduced SIRT3 levels can increase ROS levels by several mechanisms. Sirt3-mediated deacetylation can increase the activity of MnSOD, an enzyme involved in the conversion of superoxide to hydrogen peroxide, implying that deficiency of SIRT3 can hamper the activity of MnSOD, thereby increasing the ROS levels (27, 37). Another mechanism by which Sirt3 can regulate ROS levels is by deacetylating and activating isocitrate dehydrogenase 2 (IDH-2), an enzyme that consumes NADPH (40). During the course of this study Cheung et al. (5) reported that Doxo can also reduce SIRT3 levels in H9C2 cells and promote mitochondrial ROS production. In accordance with these observations we found that treatment with Doxo increases oxidative stress in primary cardiomyocytes and overexpression of SIRT3 helps to contain the increased ROS induced by Doxo.
Increased ROS levels are also positively correlated with increased apoptosis. Other than regulating ROS levels, SIRT3 can regulate apoptosis by targeting other substrates. In cardiomyocytes, SIRT3 has been shown to act as an antiapoptotic molecule. Sirt3-mediated deacetylation of Ku70 augments its interaction with the proapoptotic protein Bax, thereby preventing the proapoptotic translocation of Bax to mitochondria (35). Yet another mechanism by which SIRT3 can protect against Doxo-induced cell death is through OPA1, a GTPase anchored to the inner mitochondrial membrane that contributes to mitochondrial fusion (30). In agreement with these findings we found that overexpression of SIRT3 can rescue cardiomyocytes from Doxo-induced cell death and it is capable of containing Doxo-induced toxicity to the heart.
Previous studies have shown that SIRT3 is required to prevent cardiac hypertrophy, and deficiency of SIRT3 makes heart highly sensitive to hypertrophic stimuli (11, 34). In line with these findings, we found that Doxo-induced cardiac toxicity is associated with reduced levels of SIRT3. Sirt3-deficient mice show exacerbated Doxo-induced cardiac hypertrophy, compared with respective controls. Deficiency of SIRT3 has also been shown to promote activation of cardiac fibroblasts to transform into myofibroblasts leading to development of fibrosis (25). In the present study, we found that treatment of cardiac fibroblasts with Doxo induces fibroblast to myofibroblast conversion and overexpression of SIRT3 suppresses this transformation. Therefore, increased fibrosis observed in our mice subjected to Doxo treatment could be again attributed to decreased levels of SIRT3 in the heart.
One of the consequences of ROS attack is the oxidation of cellular proteins, lipids, and DNA (12). Among the four bases of DNA, guanine is more susceptible to oxidative damage, leading to formation of 8-oxo-dG, which is the most abundant DNA lesion upon oxidative exposure (28). OGG1 is the major DNA glycosylase that is involved in the hydrolysis of 8-oxo-dG from DNA (7). Downregulation of OGG1 is reported in different types of cancers, metabolic syndromes, neurologic disorders, aging-associated diseases, and cardiac hypertrophy (31, 32). Here, we found that deficiency of SIRT3 is associated with increased mtDNA damage and lack of SIRT3 exacerbates mtDNA damage when exposed to Doxo. Additionally, overexpression of SIRT3 attenuated mtDNA damage induced by Doxo, suggesting that SIRT3 plays a crucial role in mtDNA damage repair. Consistent with our findings it has been reported recently that OGG1 is subject to modification of reversible lysine acetylation and acetylated OGG1 is vulnerable to degradation by calpain, a calcium-dependent cysteine protease (4). We found that SIRT3 can bind to and deacetylate OGG1 in cardiomyocytes. Doxo treatment induced OGG1 acetylation and overexpression of SIRT3 resulted in deacetylation and stabilization of OGG1. Depletion of SIRT3 due to Doxo treatment also depleted OGG1 levels, suggesting that increased mtDNA damage and formation of 8-oxo-dG adducts observed due to Doxo treatment could be the consequence of OGG1 depletion as a result of low SIRT3 levels. Decreased OGG1 activity is also confirmed by observing high levels of 8-oxo-dG adducts in Sirt3.KO and Doxo-treated samples. Although the mechanism associated with the regulation of OGG1 levels was demonstrated in vitro, the present study for the first time shows that similar mechanism operates in vivo for models of cardiac hypertrophy (4). Thus our data suggest that accumulation of persistent mtDNA damage could be an important factor contributing to development of Doxo-induced cardiac hypertrophy in mice (Fig. 7E).
In summary, our results described here demonstrate that Doxo toxicity to the heart is associated with reduced SIRT3 levels and mtDNA damage. Because SIRT3 levels are also reduced with aging of humans, it is tempting to believe that the delayed toxicity of this drug therapy could be partly linked with reduced cardiac SIRT3 levels with age. Therefore, strategies targeted to maintain mitochondrial SIRT3 levels should be able to protect to the heart from Doxo-meditated toxicity.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants R01-HL-117041 and R01-HL-111455 (to M. P. Gupta).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: V.B.P. and M.P.G. conception and design of research; V.B.P., S.B., Y.H.F., G.H.K., M.G., and S.S. performed experiments; W.W.S. analyzed data; M.P.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. F. W. Alt for providing SIRT3-KO mice. We also thank Dr. Vytas Bindokas, Dr. Christine Labno, Shirley Bond, and Yimei Chen for helping in microscopy work.
REFERENCES
- 1.Alexeyev M, Shokolenko I, Wilson G, LeDoux S. The maintenance of mitochondrial DNA integrity–critical analysis and update. Cold Spring Harb Perspect Biol 5: a012641, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnheim N, Cortopassi G. Deleterious mitochondrial-DNA mutations accumulate in aging human tissues. Mutat Res 275: 157–167, 1992. [DOI] [PubMed] [Google Scholar]
- 3.Berlin V, Haseltine WA. Reduction of adriamycin to a semiquinone-free radical by NADPH cytochrome P-450 reductase produces DNA cleavage in a reaction mediated by molecular oxygen. J Biol Chem 256: 4747–4756, 1981. [PubMed] [Google Scholar]
- 4.Cheng Y, Ren X, Gowda ASP, Shan Y, Zhang L, Yuan YS, Patel R, Wu H, Huber-Keener K, Yang JW, Liu D, Spratt TE, Yang JM. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis 4: e731, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung KG, Cole LK, Xiang B, Chen K, Ma X, Myal Y, Hatch GM, Tong Q, Dolinsky VW. Sirtuin-3 (SIRT3) protein attenuates doxorubicin-induced oxidative stress and improves mitochondrial respiration in H9c2 cardiomyocytes. J Biol Chem 290: 10981–10993, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem 261: 3060–3067, 1986. [PubMed] [Google Scholar]
- 7.de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, Klungland A, Bohr VA. Repair of 8-oxodeoxyguanosine lesions in mitochondrial DNA depends on the oxoguanine DNA glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial DNA of OGG1-defective mice. Cancer Res 61: 5378–5381, 2001. [PubMed] [Google Scholar]
- 8.Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem 261: 3068–3074, 1986. [PubMed] [Google Scholar]
- 9.Doroshow JH, Locker GY, Myers CE. Enzymatic defenses of the mouse heart against reactive oxygen metabolites: alterations produced by doxorubicin. J Clin Invest 65: 128–135, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finley LW, Haigis MC. Metabolic regulation by SIRT3: implications for tumorigenesis. Trends Mol Med 18: 516–523, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albnay, NY) 2: 914–923, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halliwell B. Oxygen and nitrogen are pro-carcinogens. Damage to DNA by reactive oxygen, chlorine and nitrogen species: measurement, mechanism and the effects of nutrition. Mutat Res 443: 37–52, 1999. [DOI] [PubMed] [Google Scholar]
- 13.Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, Westphall MS, Pagliarini DJ, Prolla TA, Assadi-Porter F, Roy S, Denu JM, Coon JJ. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell 49: 186–199, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ichikawa Y, Ghanefar M, Bayeva M, Wu RX, Khechaduri A, Prasad SV, Mutharasan RK, Naik TJ, Ardehali H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest 124: 617–630, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lebrecht D, Setzer B, Ketelsen UP, Haberstroh J, Walker UA. Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation 108: 2423–2429, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Li TM, Singal PK. Adriamycin-induced early changes in myocardial antioxidant enzymes and their modulation by probucol. Circulation 102: 2105–2110, 2000. [DOI] [PubMed] [Google Scholar]
- 17.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV Jr, Weissman S, Verdin E, Schwer B. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 27: 8807–8814, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malta S, Niraula NP, Singh B, Liou K, Sohng JK. Limitations in doxorubicin production from Streptomyces peucetius. Microbiol Res 165: 427–435, 2010. [DOI] [PubMed] [Google Scholar]
- 19.Mandavilli BS, Santos JH, Van Houten B. Mitochondrial DNA repair and aging. Mutat Res 509: 127–151, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell 16: 4623–4635, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 56: 185–229, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Miranda CJ, Makui H, Soares RJ, Bilodeau M, Mui J, Vali H, Bertrand R, Andrews NC, Santos MM. Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood 102: 2574–2580, 2003. [DOI] [PubMed] [Google Scholar]
- 23.Palmeira CM, Serrano J, Kuehl DW, Wallace KB. Preferential oxidation of cardiac mitochondrial DNA following acute intoxication with doxorubicin. Biochim Biophys Acta 1321: 101–106, 1997. [DOI] [PubMed] [Google Scholar]
- 24.Panjrath GS, Patel V, Valdiviezo CI, Narula N, Narula J, Jain D. Potentiation of Doxorubicin cardiotoxicity by iron loading in a rodent model. J Am Coll Cardiol 49: 2457–2464, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Pillai VB, Samant S, Sundaresan NR, Raghuraman H, Kim G, Bonner MY, Arbiser JL, Walker DI, Jones DP, Gius D, Gupta MP. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun 6: 6656, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pommier Y, Leo E, Zhang HL, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 17: 421–433, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu XL, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab 12: 662–667, 2010. [DOI] [PubMed] [Google Scholar]
- 28.Radak Z, Boldogh I. 8-Oxo-7,8-dihydroguanine: links to gene expression, aging, and defense against oxidative stress. Free Radic Biol Med 49: 587–596, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci USA 108: 4135–4140, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Samant SA, Zhang HJ, Hong ZG, Pillai VB, Sundaresan NR, Wolfgeher D, Archer SL, Chan DC, Gupta MP. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol 34: 807–819, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sampath H, McCullough AK, Lloyd RS. Regulation of DNA glycosylases and their role in limiting disease. Free Radic Res 46: 460–478, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sampath H, Vartanian V, Rollins MR, Sakumi K, Nakabeppu Y, Lloyd RS. 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS One 7: 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh SK, Szulik MW, Ganguly M, Khutsishvili I, Stone MP, Marky LA, Gold B. Characterization of DNA with an 8-oxoguanine modification. Nucleic Acids Res 39: 6789–6801, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest 119: 2758–2771, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol 28: 6384–6401, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swain SM, Vici P. The current and future role of dexrazoxane as a cardioprotectant in anthracycline treatment: expert panel review. J Cancer Res Clin Oncol 130: 1–7, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tao RD, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang HY, Kim HS, Flynn CR, Hill S, McDonald WH, Olivier AK, Spitz DR, Gius D. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 40: 893–904, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian F, Tong TJ, Zhang ZY, McNutt MA, Liu XW. Age-dependent down-regulation of mitochondrial 8-oxoguanine DNA glycosylase in SAM-P/8 mouse brain and its effect on brain aging. Rejuv Res 12: 209–215, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Weiss RB. The anthracyclines–will we ever find a better doxorubicin. Semin Oncol 19: 670–686, 1992. [PubMed] [Google Scholar]
- 40.Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem 287: 14078–14086, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang S, Liu XB, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 18: 1639–1642, 2012. [DOI] [PubMed] [Google Scholar]